

Summary
Inducible promoters such as Plac are of limited usability for industrial protein production with Pseudomonas putida. We therefore utilized cell density‐dependent auto‐inducible promoters for recombinant gene expression in P. putida KT2440 based on the RoxS/RoxR Quorum Sensing (QS) system of the bacterium. To this end, genetic regions upstream of the RoxS/RoxR‐regulated genes ddcA (PR ox132) and PP_3332 (PR ox306) were inserted into plasmids that mediated the expression of superfolder green fluorescent protein (sfGFP) and surface displayed mCherry, confirming their promoter functionalities. Mutation of the Pribnow box of PR ox306 to the σ70 consensus sequence (PR ox3061) resulted in a more than threefold increase of sfGFP production. All three promoters caused cell density‐dependent expression, starting transcription at optical densities (OD 578) of approximately 1.0 (PR ox132, PR ox306) or 0.7 (PR ox3061) as determined by RT‐qPCR. The QS dependency of PR ox306 was further shown by cultivating P. putida in media that had already been used for cultivation and thus contained bacterial signal molecules. The longer P. putida had grown in these media before, the earlier protein expression in freshly inoculated P. putida appeared with PR ox306. This confirmed previous findings that a bacterial compound accumulates within the culture and induces protein expression.
Introduction
To date, biotechnology mostly uses inducible promoters to regulate the expression of recombinant proteins. However, especially in the context of industrial applications their use is connected to some disadvantages. The frequently employed T7 RNA polymerase‐dependent expression system, for example requires the addition of isopropyl‐β‐d‐1‐thiogalactopyranoside (IPTG), making a production process expensive (Nocadello and Swennen, 2012). The need for monitoring the optical density (OD578) of the cell culture and determining the optimal induction point makes an automatization of protein expression more difficult when using inducible promoters (Briand et al., 2016). Additionally, induction usually takes place in the exponential growth phase of the bacterial culture. In this phase, overexpression of proteins increases the metabolic burden of each cell and proteins which are detrimental for bacterial growth cannot be produced in some cases (Jaishankar and Srivastava, 2017). These disadvantages could be circumvented by using auto‐inducing expression strategies for recombinant protein production.
The bacterial cell‐to‐cell communication mechanism of Quorum Sensing (QS), which is based on the production, release and detection of hormone‐like signal molecules (auto‐inducers), offers the possibility to link population‐wide gene expression to a particular bacterial cell density (Waters and Bassler, 2005). In the last years, QS‐based auto‐inducible promoter systems were established for recombinant gene expression in both Gram‐negative and Gram‐positive bacteria (Anderson et al., 2006; Tsao et al., 2010; Nocadello and Swennen, 2012; Guan et al., 2015). In Pseudomonas putida, a Gram‐negative soil bacterium with increasing importance for biotechnological applications, the QS‐dependent RhlRI‐promoter from Pseudomonas aeruginosa was used to express genes for rhamnolipid production (Cha et al., 2008; Cao et al., 2012). However, no cell density‐dependent promoter system for recombinant protein expression in P. putida relying on a native QS‐system has been available so far.
Previously, Espinosa‐Urgel and Ramos (2004) identified the so far only known QS‐system of P. putida KT2440, RoxS/RoxR, a two‐component system formed by a sensor histidine kinase (RoxS) and a response regulator (RoxR). It was shown that RoxS/RoxR regulates a large number of genes, responsible, for example for cytochrome oxidase activity and redox signalling, or, like the gene ddcA, involved in seed colonization. Expression of ddcA was directly dependent on cell density. A gene encoding for a yet unknown cytochrome c‐type protein, PP_3332, was also shown to be controlled by the RoxS/RoxR system (Espinosa‐Urgel and Ramos, 2004; Fernández‐Piñar et al., 2008). For induction of expression, RoxS is believed to interact with tetradecanoic acid and fatty acids of similar chain lengths and subsequently phosphorylates RoxR (Fernández‐Piñar et al., 2012). Phosphorylated RoxR regulates the expression of cell density‐dependent genes by binding to a putative RoxR recognition element (RoxR RE) (Fernández‐Piñar et al., 2008).
To enable this QS‐system for recombinant protein production in P. putida, we utilized the regions upstream of the RoxS/RoxR‐regulated genes ddcA and PP_3332 that are expected to contain all necessary functionalities for protein expression under control of RoxS/RoxR and inserted these into plasmids to mediate the auto‐induced and cell density‐dependent protein expression. To detect and quantify protein expression, we used the two fluorescent proteins superfolder green fluorescent protein (sfGFP) and mCherry. While sfGFP was expressed intracellularly, mCherry was expressed in the form of the autotransporter‐fusion protein MATE‐mCherry in order to evaluate the RoxS/RoxR system in the context of a more complex secretion pathway. Proteins expressed as MATE (maximized autotransporter mediated expression) fusion proteins are translocated to the cell surface via type Va secretion (Sichwart et al., 2015), which has recently also been demonstrated with P. putida as host (Tozakidis et al., 2016; Schulte et al., 2017).
We could show that both regions not only function as promoters for recombinant protein expression in P. putida KT2440, but also that the expression is auto‐induced and cell density‐dependent. The promoter strength could be enhanced by mutating the Pribnow box to the consensus sequence of the major σ‐factor of the σ70‐family. This study complements the P. putida toolbox by a valuable promoter system with the perspective for industrial protein production.
Results and discussion
Construction of plasmids for QS‐dependent recombinant gene expression
To generate cell density‐dependent promoters for recombinant sfGFP expression in P. putida, a 132 bp part of the upstream genetic region of ddcA (PP_4615) was amplified from the chromosomal DNA of P. putida KT2440 (Fig. 1A, top panel). This region, termed PRox132, includes the RoxR RE, −10 (Pribnow box) and −35 regions and the ribosome binding site (RBS) of ddcA. Second, the 306 bp long complete intergenic region of genes PP_3331 (uncharacterized protein) and PP_3332 (putative cytochrome c‐type protein), termed PRox306, was amplified (Fig. 1A, bottom panel). To increase the promoter strength of PRox306, the Pribnow box was identified as TAGACT using the online software BPROM (Solovyev and Salamov, 2011), and two positions were mutated to obtain the consensus sequence of the primary σ‐factor of the σ70 family, TATAAT (Fig. 1B). This essential σ‐factor is dominantly present in bacteria (Paget and Helmann, 2003). The mutated PRox306 was termed PRox3061. They were inserted into plasmid DNA upstream of the start codon of superfolder green fluorescent protein (sfGFP), resulting in plasmids pPRox132‐sfGFP, pPRox306‐sfGFP and pPRox3061‐sfGFP.
Figure 1.

A. Genetic origins of PRox promoters. Top panel: PRox132 is part of the upstream genetic region of ddcA (light grey). It starts with the RoxR recognition element (Rox RE), the regulatory regions (−35 and −10) and a ribosome binding site (RBS), which is located in the 5′ untranslated region (white). Bottom panel: PRox306 consists of the whole intergenic region between P. putida KT2440 genes PP_3332 and PP_3331.
B. DNA sequences of PRox132, PRox306 and PRox3061. The −35 and −10 regions predicted by BPROM (Solovyev and Salamov, 2011) are depicted in bold. Experimentally determined transcriptional start sites are marked with + 1. In PRox3061, the −10 region was mutated to the consensus sequence of the major σ‐factor of the σ70 family, TATAAT. The initiation codon of the regulated gene is underlined. The sequence of the Rox RE is shaded, the putative RBS sequence depicted in italics.
The start of the transcripts produced under control of these promoters were determined by the SMART 5′ RACE technique (Sherwood et al., 2009). As depicted in Fig. 1B, the transcript started for PRox132 with the sequence GGC in correct distance to the −10 region, and for PRox306 as well as for PRox3061 with the sequence TCC in correct distance to the −10 region. However, for PRox132, additional transcriptional start sites besides GGC were identified. All of them were located upstream of the −35 region of PRox132. This could have been due to the upstream located replication gene of the plasmid. It cannot be excluded that this gene has been read through without termination and thereby overlapped with the gene transcripts controlled by PRox132.
Expression of sfGFP
To test the functionality of PRox132, PRox306 and PRox3061 as promoters, P. putida KT2440 cells were transformed with the abovementioned plasmids. Cell growth based on optical density at 578 nm (OD578) as well as fluorescence intensity (FI) were monitored throughout cultivation of 100 ml cultures (Fig. 2A–C). As control, P. putida without plasmid was cultivated likewise. For comparison with established promoters, P. putida pPGAP‐sfGFP and P. putida pPBAD‐sfGFP were also cultivated. In pPGAP‐sfGFP, sfGFP is constitutively expressed under control of PGAP, the promoter of glyceraldehyde‐3‐phosphate dehydrogenase from Zymomonas mobilis (Conway et al., 1987), which has been shown to be functional in P. putida in experiments before (data not shown). In pPBAD‐sfGFP, expression of sfGFP is controlled by the widely applied arabinose‐inducible promoter (Guzman et al., 1995). Arabinose was added to this culture upon reaching an OD578 of 0.5. In addition to lysogeny broth medium (LB, Fig. 2A–B), PRox3061 was also tested in minimal MOPS medium (Fig. 2C).
Figure 2.

Expression of sfGFP by P. putida (A) under control of PRox132, PRox306 and PRox3061 and (B) under control of the constitutive promoter PGAP and the inducible promoter PBAD.
C. Comparison of sfGFP expression under control of PRox3061 in LB and MOPS medium.
D. Quantitative comparison of sfGFP accumulation within the cells with different promoters. P. putida cells containing plasmids for expression of sfGFP under control of the different promoters were cultivated in 500 ml shake flasks in LB or MOPS medium at 30°C, and OD 578 and fluorescence intensity (FI, excitation: 485 nm, emission: 510 nm) were monitored. After reaching stationary phase, the cells were lysed, their proteins separated by SDS‐PAGE and sfGFP detected by means of anti‐sfGFP and secondary HRP‐conjugated antibodies. The detected band intensities were quantified and provided relative to the band intensity of PRox3061. The data are derived from one representative experiment, with the mean of three technical replicates shown. The error bars represent the standard deviation.
All strains with PRox promoters showed an increase in FI/OD578 during growth and hence expressed sfGFP. Therefore, both the PRox132 and PRox306 regions appeared to contain a functional promoter, and PRox306 was not corrupted when mutated to PRox3061. A truncated version of PRox306, starting at the RoxR RE and devoid of the region upstream, was also tested, but did not lead to expression of sfGFP (data not shown). This indicates that PRox306 contains elements important for RoxS/RoxR signalling that are not identified so far.
In LB medium, PBAD produced the highest sfGFP levels with final FI/OD578 values of over 400 after reaching the stationary phase. PRox3061 induced expression with similar strength as PGAP (FI/OD578 = 143 vs. 132), which is approximately five times as high as with the unmutated PRox306 (FI/OD578 = 29). However, PRox3061 produced only roughly a third the amount of sfGFP compared to PBAD. PRox132 exerted a much smaller final sfGFP yield (FI/OD578 = 8). While the strain with PGAP controlled expression already started to show fluorescence from the beginning of cultivation, the strains with PRox promoters did not show considerable fluorescence up to an OD578 of approximately 1.7, giving a first indication for its cell density dependency. In minimal medium, the protein production by PRox3061 was lower than in LB medium (FI/OD578 = 42 vs. 143), whereas induction of protein expression was induced at a similar OD578.
The accumulated amount of sfGFP within the cells was determined by a semi‐quantitative Western blot. To this end, cells of each strain were harvested after reaching stationary phase, lysed, and their proteins were separated by SDS‐PAGE. After blotting, sfGFP was labelled with specific antibodies and secondary HRP‐conjugated antibodies and the intensities of the detected signals were analysed with ImageJ (Rueden et al., 2017) (Fig. 2D). The amount of sfGFP within the bacterial cells was consistent with the fluorescence intensities observed in the cultivation experiments.
To exclude an effect of different translational efficiencies on the amount of sfGFP produced under control of PRox132 and PRox306, the Ribosome Binding Site (RBS) Calculator (Salis, 2011) was used. For PRox132, translation efficiency of the RBS was given as nearly threefold higher than for PRox306 (307 vs. 140 arbitrary units). This means that the observed higher sfGFP yield with PRox306 was not an effect of the different RBS, but rather of different promoter strengths.
Surface display of mCherry
The applicability of the RoxS/RoxR QS‐system was also tested for expression of a larger protein that is subjected to secretion, in the present case by the autotransporter (type Va) pathway. To this end, plasmids were constructed in which the DNA sequence for sfGFP from the abovementioned plasmids was replaced by the sequence for MATE‐mCherry (Fig. 3A). This protein was described in detail before (Sichwart et al., 2015). In brief, MATE proteins are translocated into the periplasm of the host and subsequently incorporated into the outer membrane to display the so‐called passenger domain, in the case as presented here mCherry, on the cell surface. Surface display and functionality of MATE proteins with P. putida as a host have been shown before (Tozakidis et al., 2016; Schulte et al., 2017). The plasmids pPRox132‐MATE‐mCherry, pPRox306‐MATE‐mCherry and pPRox3061‐MATE‐mCherry were inserted into P. putida, and FI was monitored throughout cultivation in a microtiter plate (MTP) reader (Fig. 3B–C). An increasing FI/OD578 could be observed during growth of all strains except the control strain. Interestingly, in contrast to sfGFP expression, the promoter strength of PRox132 and PRox306 seemed to be similar, as can be seen by the final FI/OD578 values of approximately 1500 in both strains after 24 h. This is contradictory to the previous results, in which PRox306 mediated a much stronger sfGFP expression than PRox132. At this point, there is no verifiable explanation for this observation. It can only be speculated that the complex secretion mechanism of MATE‐mCherry distorts the expression strength in a way that the amount of fluorescent mCherry on the cell surface does not reflect the strength of the promoters. In contrast, the mCherry yield was approximately twofold higher with PRox3061 than with PRox306 after 24 h, as similarly observed in the sfGFP‐experiments.
Figure 3.
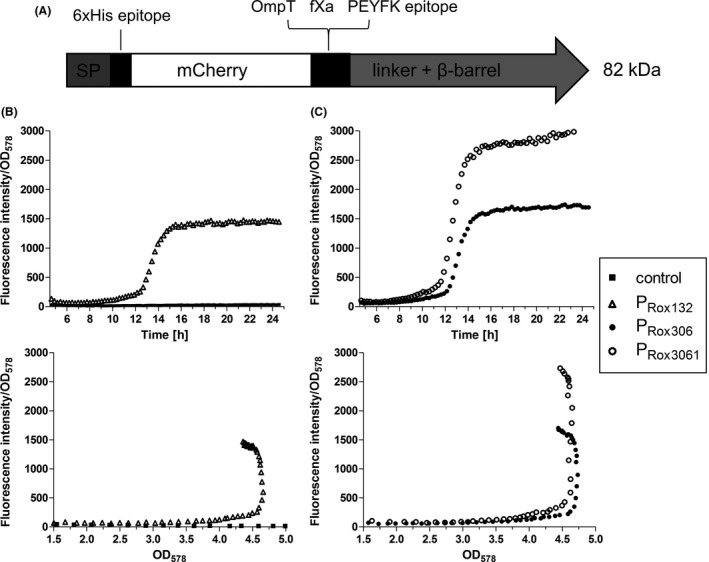
A. Scheme of the unprocessed MATE‐mCherry fusion protein. After cleavage of the signal peptide (SP), the protein consists of a 6xHis epitope, the passenger mCherry, OmpT and fXa cleavage sites, a PEYFK epitope and the EhaA linker and β‐barrel (Sichwart et al., 2015).
B,C. Expression of MATE‐mCherry under control of PRox132, PRox306 and PRox3061. P. putida without plasmid and with plasmid pPRox132‐MATEmCherry (B), pPRox306‐MATE‐mCherry and pPRox3061‐MATE‐mCherry (C) were cultivated in LB medium in a 24‐well MTP at 30°C. OD 578 and fluorescence intensity (FI, excitation: 580 nm, emission: 620 nm) were monitored. These data are mean values of biological triplicates. Error bars are not visible due to small standard deviations that are covered by the icons.
To verify surface display of mCherry, P. putida cells expressing MATE‐mCherry and sfGFP under control of PRox132 were analysed via fluorescence microscopy (Fig. 4). While sfGFP‐fluorescence was located within the cytoplasm as expected (Fig. 4B), mCherry‐fluorescence was localized at the cell membrane of P. putida cells (Fig. 4D). Cells expressing MATE‐mCherry were slightly smaller (1.4–1.9 μm in length) (Fig. 4C) than cells expressing sfGFP (Fig. 4A, 2.5–2.8 μm in length). It is likely that the expression of membrane‐anchored MATE‐mCherry caused morphological changes in P. putida KT2440 as described previously, for example for Escherichia coli as host (Wagner et al., 2007; Gubellini et al., 2011).
Figure 4.
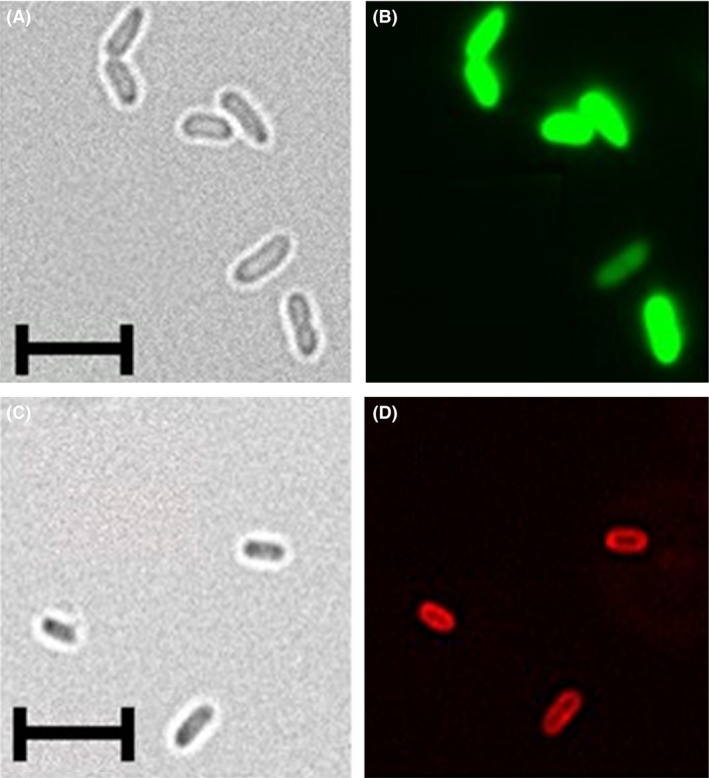
Analysis of P. putida pPRox3061‐sfGFP (A,B), and pPRox132‐MATE‐mCherry (C,D) via fluorescence microscopy. The strains were cultivated at 30°C, 200 rpm in LB medium for 8 h and 24 h respectively. 2.5 × 106 cells were washed three times with PBS and fixed on a microscope slide with DABCO/Mowiol. The samples were analysed with the 100× oil immersion lens of a BZ‐9000 fluorescence microscope (Keyence, Neu‐Isenburg, Germany).
A,C. Brightfield pictures. (B) GFP‐filter, excitation 472/30 nm, emmission 593/40 nm.
D. TexasRed‐filter, excitation 560/40 nm, emmission 630/75 nm. The length of the scales corresponds to 5 μm.
Cell density dependency of PRox132 and PRox306 on transcriptional level
In order to investigate if protein expression with PRox promoters is indeed depending on the cell density of P. putida, transcription levels of MATE‐mCherry as a function of OD578 were determined (Fig. 5). To this end, P. putida strains expressing MATE‐mCherry under control of PRox132, PRox306 and PRox3061 as described above were cultivated, samples were taken at different time points and the OD578 was determined. MATE‐mCherry mRNA transcripts of these samples were quantified by means of reverse transcription quantitative PCR (RT‐qPCR) taking transcription of rpoD as reference. The gene rpoD, encoding for a transcription factor of the σ70‐factor family, has been shown before to be transcribed in equal amounts at all bacterial growth phases (Savli et al., 2003; Wang and Nomura, 2010). P. putida expressing MATE‐mCherry under control of the constitutive promoter PBG35 (Zobel et al., 2015) was used as control. PBG35 was used here instead of PGAP because cells transformed with pPGAP‐MATE‐mCherry were not viable, perhaps because protein expression was too high. While PBG35 showed gene expression levels independent of the growth phase of the bacteria (Fig. 5A), PRox promoters showed a sharp increase in relative gene expression at a particular cell density. In case of PRox132, gene expression was upregulated at an OD578 of 1.0 5‐fold in comparison with the initial gene expression level (Fig. 5B). In case of PRox306, gene expression was upregulated threefold at the same OD578 of approximately 1 (Fig. 5C). With PRox3061, a sevenfold increase of MATE‐mCherry mRNA transcripts was observed at an OD578 of 0.7, further increasing up to a factor of 10 at an OD578 of 1.0 (Fig. 5D). These experiments confirmed that PRox promoters are auto‐induced and dependent on the cell density of P. putida KT2440.
Figure 5.

RT‐qPCR analysis of MATE‐mCherry expression under control of PBG35 (A), PRox132 (B), PRox306 (C) and PRox3061 (D). P. putida cells containing plasmids for expression of MATE‐mCherry under control of different promoters were cultivated in LB medium at 30°C, 200 rpm. At different time points, 2.5 × 108 cells were removed from the cultures and the total amount of RNA was isolated. 1000 ng of total RNA was reversely transcribed to cDNA. 50 ng cDNA was amplified and analysed with specific primers in a qPCR cycler. The gene expression of MATE‐mCherry was determined relative to the gene expression of the reference gene rpoD (Fujita et al., 1995). The measured Cq values were analysed by applying a model of Pfaffl (2001). All RT‐qPCR analyses were performed as biological triplicates, each of them conducted as technical triplicates. Error bars indicate the standard deviation.
Effect of conditioned medium on MATE‐mCherry expression
To further substantiate the cell density‐dependent regulation of the PRox promoters, P. putida pPRox306‐MATE‐mCherry was cultivated in the presence of conditioned medium (CM), that is medium that was used for P. putida cultivation before, subsequently harvested and released from the bacteria. CM should therefore contain all signal molecules (in addition to waste and other metabolites) secreted by P. putida. CM has been used to assess cell density‐dependence of ddcA expression before (Espinosa‐Urgel and Ramos, 2004). In CM, the concentration of auto‐inducers is supposed to be the higher the longer P. putida has been cultivated therein. 24 h CM (final OD578 of the culture: 5.0) should have a higher auto‐inducer concentration than 12 h CM (final OD578 = 3.0) and 8 h CM (final OD578 = 2.3) due to the increasing cell number and the longer time the cells produced auto‐inducers. The higher the concentration of auto‐inducers, the earlier the induction of QS‐regulated promoters should consequently take place as the induction threshold is reached earlier due to the auto‐inducer concentration provided by CM. These considerations were confirmed by cultivation experiments with CM (Fig. 6). When cultivated in LB medium supplied with 20% (v/v) 24 h CM, P. putida KT2440 pPRox306‐MATE‐mCherry showed increasing fluorescence after 7 h of cultivation. In contrast, when cultivated in LB medium without CM (fresh medium), the cells started to show fluorescence after 9 h (Fig. 6A). Cultures in LB medium supplemented with 20% (v/v) 12 h CM started to increase FI after 7.5 h (Fig. 6B). Hence, the induction of PRox306 was shown to be cell density‐dependent. Signalling molecules, enriched in CM during cultivation of P. putida KT2440 cells, are assumed to be responsible for the earlier increase in FI/OD578 of P. putida KT2440 pPRox306‐MATE‐mCherry cultivated in presence of different CM. Fernández‐Piñar et al. (2012) identified tetradecanoic acid and fatty acids of similar chain lengths to be the auto‐inducers of the RoxS/RoxR QS‐system. Whether these molecules were present in CM as applied here was not investigated. Nevertheless, further studies would be very interesting as the production of the relevant signal molecules could be modulated and adapted to individual needs.
Figure 6.
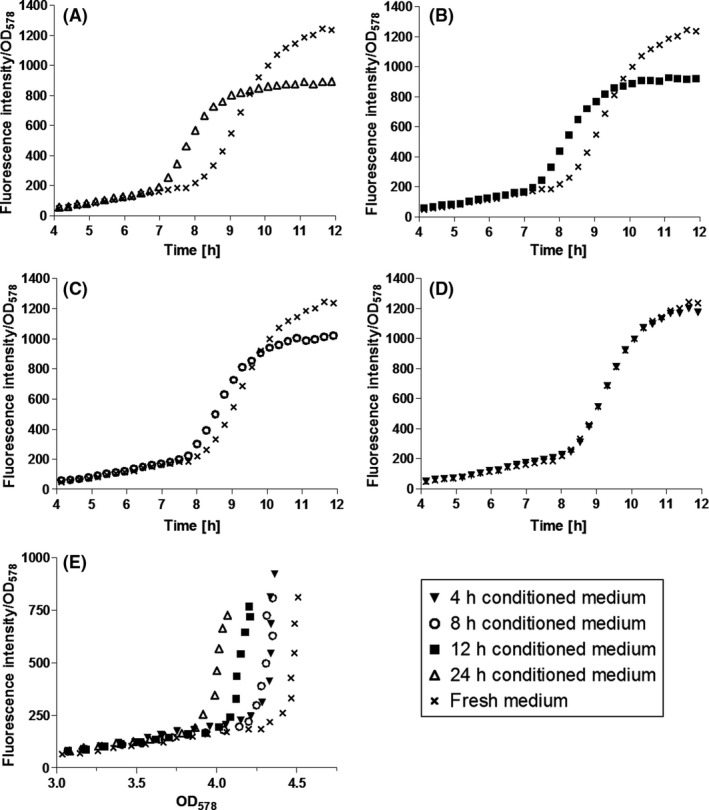
Influence of conditioned medium (CM) on expression of MATE‐mCherry. P. putida pPRox306‐MATE‐mCherry was cultivated in LB medium to an OD 578 = 0.8. Five millilitres of the cell suspension were harvested and suspended in mixtures of 4 ml fresh LB and 1 ml of different CM. The cells were then cultivated in a 24‐well MTP for another 12 h while monitoring OD 578 and fluorescence intensity (FI).
A. 24 h CM, (B) 12 h CM, (C) 8 h CM, (D) 4 h CM. These data are mean values of biological triplicates. Error bars are not visible due to small standard deviations that are covered by the icons.
Conclusions
The RoxS/RoxR QS‐system discovered by Espinosa‐Urgel and Ramos (2004) could successfully be applied to provide new auto‐inducible, cell density‐dependent promoters for the recombinant protein production in P. putida KT2440 in this study. As the underlying RoxS/RoxR QS signalling is native to P. putida, it is sufficient to incorporate the corresponding promoter sequences into a plasmid of choice to mediate protein expression. The mutated version of the PRox306 promoter, PRox3061, leads to stronger expression. Still, this promoter does not reach the overexpression of the strong PBAD promoter. For applications that demand a maximal output of protein, further development will be necessary to increase the promoter strength. The PRox promoters are not restricted to induce expression of cytosolic proteins, but can be used for surface display of recombinant proteins as well, making them interesting tools for the generation of whole‐cell biocatalysts. They will contribute to expand the application of P. putida KT2440 in industrial biotechnology.
Experimental procedures
Bacterial strains and culture conditions
Cloning was carried out in Escherichia coli DH5α (DSM No.: 6897). The cells were cultivated either in lysogeny broth (LB) medium (10 g l−1 tryptone/peptone, 5 g l−1 yeast extract and 10 g l−1 NaCl) at 37°C and 200 rpm or on LB agar plates at 37°C, both containing 50 μg ml−1 kanamycin. P. putida KT2440 (DSM No.: 6125) was cultivated either in LB medium, MOPS medium (LaBauve and Wargo, 2012) or on LB agar plates, with 50 μg ml−1 kanamycin when necessary, at 30°C. Main cultures were inoculated to OD578 of 0.05 from an overnight culture and cultivated either in shaking flasks at 30°C and 200 rpm or in 24‐well microtiter plates (MTP) in a MTP reader at 30°C with constant shaking (Infinite M200 Pro, Tecan, Männedorf, Switzerland). To demonstrate cell density‐dependence of PRox306, P. putida KT2440 pPRox306‐MATE‐mCherry was cultivated in LB medium with 20% (v/v) of conditioned medium (CM). CM was prepared by cultivating P. putida KT2440 cells in LB medium for 4, 8, 12 and 24 h respectively. The cells were then separated by centrifugation, and the supernatant was filtrated by a filter with a pore size of 0.22 μm.
Construction of expression plasmids
DNA sequences of PRox132 and PRox306 were amplified directly from the chromosomal DNA of P. putida KT2440 via PCR with specific primers. PGAP was amplified from the chromosomal DNA of Z. mobilis (DSM No.: 3580). Promoter DNA fragments were inserted into pBBR1MCS‐2 plasmids (Kovach et al., 1995) containing sfGFP sequence (Wu et al., 2009) via In‐Fusion Cloning as described by the manufacturer, resulting in pPRox132‐sfGFP, pPRox306‐sfGFP and pPGAP‐sfGFP. pPRox3061‐sfGFP was generated by mutating the PRox306 DNA sequence of pPRox306‐sfGFP via an In‐Fusion Cloning strategy described elsewhere (Raman and Martin, 2014). To generate pPRox132‐MATE‐mCherry, pPRox306‐MATE‐mCherry and pPRox3061‐MATE‐mCherry, promoter sequences were amplified via PCR from the sfGFP‐plasmids and inserted into a pBBR1MCS‐2 plasmid encoding for a MATE‐mCherry fusion protein (Tozakidis et al., 2014; Sichwart et al., 2015). pPBG35‐MATE‐mCherry was prepared analogously. The PBG35‐promoter was synthesized commercially according to the DNA sequence described elsewhere (Zobel et al., 2015). All cloning primers are listed in Table S1. The sequences of all constructed plasmids were verified by Sanger sequencing.
Analysis of fluorescent proteins
Expression of MATE‐mCherry was analysed with the MTP reader. Each cavity of a 24‐well plate was loaded with 1.1 ml of main culture, and cells were cultivated as described before. For measuring sfGFP produced by cultures cultivated in flasks, 200 μl of the culture was transferred to a 96‐well plate, and the OD578 as well as the fluorescence intensity were measured at given timepoints. sfGFP: Excitation 485 nm, emission 510 nm; MATE‐mCherry: Excitation 580 nm, emission 620 nm. The OD578 was determined directly in the MTP and converted by a conversion formula to correct for the non‐linearity of the OD578 at higher cell densities (Meyers et al., 2018).
Fluorescence microscopy
Pseudomonas putida KT2440 pPRox306‐sfGFP and pPRox132‐MATE‐mCherry were cultivated at 30°C, 200 rpm in LB medium for 8 h and 24 h respectively. 1 ml of cell culture was harvested (11 000 g, 1 min) each. The cells were washed three times with 1 ml PBS (137 mM NaCl, 2.7 mM KCl, 10 mM Na2HPO4, 2 mM KH2PO4, pH 7.4) and suspended in 1 ml PBS. 10 μl of each cell suspension was mixed with 10 μl DABCO/Mowiol on a microscope slide and covered with a coverslip. The samples were analysed with the 100× oil immersion lens of a BZ‐9000 fluorescence microscope (Keyence, Neu‐Isenburg, Germany). sfGFP‐fluorescence was excited at 472/30 nm and detected at 593/40 nm with the GFP‐filter. mCherry‐fluorescence was excited at 560/40 nm and detected at 630/75 nm with the TexasRed‐filter.
Reverse transcription quantitative polymerase chain reaction (RT‐qPCR) analysis
Pseudomonas putida KT2440 cells were cultivated in 200 ml LB medium. At different time points during cultivation, the OD578 was determined and the total RNA of 2.5·108 cells was purified (peqGOLD Bacterial RNA Kit and peqGOLD DNase I Digest Kit; PEQLAB Biotechnologie, Erlangen, Germany) according to the manufacturer's protocol. DNase I digestion was performed for 30 min at room temperature. 1000 ng of total RNA was reversely transcribed (qScript™ XLT cDNA SuperMix; Quanta bio, Beverly, MA, USA) according to the manufacturer's description. 50 ng of complementary DNA was amplified with specific primers (Table S2). The qPCR was performed using GreenMasterMix (2×) High ROX (Genaxxon bioscience, Ulm, Germany) and the RotorGene Q 2 Plex HRM qPCR cycler (Qiagen, Hilden, Germany) according to the manufacturer's instructions. The expression level of MATE‐mCherry was analysed relative to the expression level of P. putida reference gene rpoD (Savli et al., 2003; Wang and Nomura, 2010) according to a mathematical model of Pfaffl (2001).
Determination of + 1 transcription site
To identify the transcriptional start site, the technique ‘Switching Mechanism At 5′ end of RNA Transcript Rapid Amplification of cDNA Ends’ (SMART™ RACE cDNA Amplification kit; Takara, Saint‐Germain‐en‐Laye, France) was used according to the manufacturer's description. RNA was isolated as stated above from cells expressing MATE‐mCherry. First‐strand cDNA synthesis was performed following the instructions for the 5′RACE cDNA Amplification with random primers. The RACE‐PCR was performed using a gene‐specific primer (5′‐GATTACGCCAAGCTTCAACTGGCCGCTACCGTCGCGCCAC‐3′) and the provided universal primer mix that anneals to the SMART sequence at the 5′ end of the cDNA. The RACE product was cloned into the provided pRACE vector. The resulting plasmids were sequenced by Sanger sequencing.
Semi‐quantitative Western Blot analysis
Pseudomonas putida KT2440 cells were cultivated in 20 ml LB medium. Afterwards, cells were harvested by centrifugation (3500 g, 5 min; 4°C) and the sediment was suspended in buffer (composed of 5 μl of 1 μg ml−1 aprotinin, 50 μl of 1 mM phenylmethanesulfonyl fluoride and 500 μl of 1 mg ml−1 DNase I solution in 5 ml ddH2O). The OD578 was adjusted to 5. Sodium dodecyl sulphate (SDS) sample buffer (100 mM Tris/HCl, 200 mM dithiothreitol, 4 % (w/v) SDS, 0,2 % (w/v) bromophenol blue, 20 % (v/v) glycerol pH 6.8) was added to the cell suspension in a volume ratio of 1:1 and heated for 15 min at 95°C. Samples were simultaneously separated by SDS‐PAGE in two gels. One of the resulting gels was stained in a Commassie Brilliant Blue solution. The other gel was used for Western blot analysis. Proteins were transferred to a polyvinylidene fluoride membrane by electroblotting (mini‐trans blot; Bio‐Rad, München, Germany). The membrane was blocked with phosphate‐buffered saline with 0.1 % Tween 20 (PBS‐T) and 3 % bovine serum albumin (BSA) for 3 h at room temperature (RT), followed by incubation with the primary antibody (1:500 in PBS‐T containing 0.01 % sodium azide and 5 % BSA, goat‐anti GFP; SICGEN Antibodies, Carcavelos, Portugal) over night at 4°C. The membrane was then washed three times with PBS‐T and incubated with a secondary horseradish peroxidase (HRP) conjugated antibody solution (1:5000 in PBS‐T containing 3% BSA, donkey anti‐rabbit; Santa Cruz Biotechnology, Dallas, TX, USA) for 1 h at RT. After washing the membrane for three times with PBS‐T, it was coated with an Immuno Cruz Western Blot Luminol Reagent (Santa Cruz Biotechnology), and the resulting chemiluminescence was detected with a chemiluminescent reader (ChemoCam ECL imager; Intas, Göttingen, Germany). Chemilumiscence intensity of each band was determined using the program ImageJ (Rueden et al., 2017) and normalized to the overall band intensity of the respective sample in the Coomassie stained gel, which was also determined using ImageJ.
Conflict of interest
None declared.
Supporting information
Table S1. Oligonucleotides used for gene cloning in this study.
Table S2. Oligonucleotides used for qRT‐PCR in this study.
Microbial Biotechnology (2019) 12(5), 1003–1013
Funding Information
Bundesministerium für Bildung und Forschung (031B0002B).
References
- Anderson, J.C. , Clarke, E.J. , Arkin, A.P. , and Voigt, C.A. (2006) Environmentally controlled invasion of cancer cells by engineered bacteria. J Mol Biol 355: 619–627. [DOI] [PubMed] [Google Scholar]
- Briand, L. , Marcion, G. , Kriznik, A. , Heydel, J.M. , Artur, Y. , Garrido, C. , et al (2016) A self‐inducible heterologous protein expression system in Escherichia coli . Sci Rep 6: 33037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao, L. , Wang, Q. , Zhang, J. , Li, C. , Yan, X. , Lou, X. , et al (2012) Construction of a stable genetically engineered rhamnolipid‐producing microorganism for remediation of pyrene‐contaminated soil. World J Microb Biot 28: 2783–2790. [DOI] [PubMed] [Google Scholar]
- Cha, M. , Lee, N. , Kim, M. , Kim, M. , and Lee, S. (2008) Heterologous production of Pseudomonas aeruginosa EMS1 biosurfactant in Pseudomonas putida . Bioresource Technol 99: 2192–2199. [DOI] [PubMed] [Google Scholar]
- Conway, T. , Sewell, G.W. , and Ingram, L.O. (1987) Glyceraldehyde‐3‐phosphate dehydrogenase gene from Zymomonas mobilis: cloning, sequencing, and identification of promoter region. J Bacteriol 169: 5653–5662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Espinosa‐Urgel, M. , and Ramos, J.‐L. (2004) Cell density‐dependent gene contributes to efficient seed colonization by Pseudomonas putida KT2440. Appl Environ Microbiol 70: 5190–5198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernández‐Piñar, R. , Ramos, J.L. , Rodríguez‐Herva, J.J. , and Espinosa‐Urgel, M. (2008) A two‐component regulatory system integrates redox state and population density sensing in Pseudomonas putida . J Bacteriol 190: 7666–7674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernández‐Piñar, R. , Espinosa‐Urgel, M. , Dubern, J.‐F. , Heeb, S. , Ramos, J.L. , and Cámara, M. (2012) Fatty acid‐mediated signalling between two Pseudomonas species. Environ Microbiol Rep 4: 417–423. [DOI] [PubMed] [Google Scholar]
- Fujita, M. , Hanaura, Y. , et al (1995) Analysis of the rpoD gene encoding the principal sigma factor of Pseudomonas putida. Gene 167(1–2): 93–98. [DOI] [PubMed] [Google Scholar]
- Guan, C. , Cui, W. , Cheng, J. , Zhou, L. , Guo, J. , Hu, X. , et al (2015) Construction and development of an auto‐regulatory gene expression system in Bacillus subtilis . Microb Cell Fact 14: 150–164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gubellini, F. , Verdon, G. , Karpowich, N.K. , Luff, J.D. , Boël, G. , Gauthier, N. , et al (2011) Physiological response to membrane protein overexpression in E. coli . Mol Cell Proteomics 10: 1–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guzman, L.M. , Belin, D. , Carson, M.J. , and Beckwith, J. (1995) Tight regulation, modulation, and high‐level expression by vectors containing the arabinose P‐bad promoter. J Bacteriol 177: 4121–4130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaishankar, J. and Srivastava, P. (2017) Molecular basis of stationary phase survival and applications. Front Microbiol 8: 2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kovach, M.E. , Elzer, P.H. , Steven Hill, D. , Robertson, G.T. , Farris, M.A. , Roop, R.M. , and Peterson, K.M. (1995) Four new derivatives of the broad‐host‐range cloning vector pBBR1MCS, carrying different antibiotic‐resistance cassettes. Gene 166: 175–176. [DOI] [PubMed] [Google Scholar]
- LaBauve, A. , and Wargo, M. (2012) Growth and laboratory maintenance of Pseudomonas aeruginosa . Curr Protoc Microbiol 6: Chapter 6: Unit 6E.1.1–6E.1.8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyers, A. , Furtmann, C. , and Jose, J. (2018) Direct optical density determination of bacterial cultures in microplates for high‐throughput screening applications. Enzyme Microb Technol 118: 1–5. [DOI] [PubMed] [Google Scholar]
- Nocadello, S. , and Swennen, E.F. (2012) The new pLAI (lux regulon based auto‐inducible) expression system for recombinant protein production in Escherichia coli . Microb Cell Fact 11: 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paget, M.S.B. and Helmann, J.D. (2003) Protein family review – The sigma(70) family of sigma factors. Genome Biol 4: 203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfaffl, M.W. (2001) A new mathematical model for relative quantification in real‐time RT–PCR. Nucleic Acids Res 29: e45–e45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raman, M. , and Martin, K. (2014) One solution for cloning and mutagenesis: in‐Fusion® HD Cloning Plus. Nat Methods 11: 972.25317449 [Google Scholar]
- Rueden, C.T. , Schindelin, J. , Hiner, M.C. , DeZonia, B.E. , Walter, A.E. , Arena, E.T. , and Eliceiri, K.W. (2017) Image J2: ImageJ for the next generation of scientific image data. Bioinformatics 18: 529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salis, H.M. (2011) The ribosome binding site calculator. Synth Biol, Pt B 498: 19–42. [DOI] [PubMed] [Google Scholar]
- Savli, H. , Karadenizli, A. , Kolayli, F. , Gundes, S. , Ozbek, U. , and Vahaboglu, H. (2003) Expression stability of six housekeeping genes: a proposal for resistance gene quantification studies of Pseudomonas aeruginosa by real‐time quantitative RT‐PCR. J Med Microbiol 52: 403–408. [DOI] [PubMed] [Google Scholar]
- Schulte, M.F. , Tozakidis, I.E.P. , and Jose, J. (2017) Autotransporter‐based surface display of hemicellulases on Pseudomonas putida: whole‐cell biocatalysts for the degradation of biomass. Chemcatchem 9: 3955–3964. [Google Scholar]
- Sherwood, T.A. , Nong, L. , Agudelo, M. , Newton, C. , Widen, R. , and Klein, T.W. (2009) Identification of transcription start sites and preferential expression of select CB2 transcripts in mouse and human B lymphocytes. J Neuroimmune Pharm 4: 476–488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sichwart, S. , Tozakidis, I.E.P. , Teese, M. , and Jose, J. (2015) Maximized Autotransporter‐Mediated Expression (MATE) for Surface display and secretion of recombinant proteins in Escherichia coli . Food Technol Biotechnol 53: 251–260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solovyev, V. , and Salamov, A. (2011) Automatic annotation of microbial genomes and metagenomic sequences In Metagenomics and its Applications in Agriculture, Biomedicine and Environmental Studies. Li R.W. (ed). Hauppauge, NY, USA: Nova Science Publishers, pp. 61–78. [Google Scholar]
- Tozakidis, I.E.P. , Sichwart, S. , Teese, M.G. , and Jose, J. (2014) Autotransporter mediated esterase display on Zymomonas mobilis and Zymobacter palmae . J Biotechnol 191: 228–235. [DOI] [PubMed] [Google Scholar]
- Tozakidis, I.E.P. , Brossette, T. , Lenz, F. , Maas, R.M. , and Jose, J. (2016) Proof of concept for the simplified breakdown of cellulose by combining Pseudomonas putida strains with surface displayed thermophilic endocellulase, exocellulase and beta‐glucosidase. Microb Cell Fact 15: 103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsao, C.‐Y. , Hooshangi, S. , Wu, H.‐C. , Valdes, J.J. , and Bentley, W.E. (2010) Autonomous induction of recombinant proteins by minimally rewiring native quorum sensing regulon of E. coli . Metab Eng 12: 291–297. [DOI] [PubMed] [Google Scholar]
- Wagner, S. , Baars, L. , Ytterberg, A.J. , Klussmeier, A. , Wagner, C.S. , Nord, O. , et al (2007) Consequences of membrane protein overexpression in Escherichia coli . Mol Cell Proteomics 6: 1527–1550. [DOI] [PubMed] [Google Scholar]
- Wang, Q. , and Nomura, C.T. (2010) Monitoring differences in gene expression levels and polyhydroxyalkanoate (PHA) production in Pseudomonas putida KT2440 grown on different carbon sources. J Biosci Bioeng 110: 653–659. [DOI] [PubMed] [Google Scholar]
- Waters, C.M. , and Bassler, B.L. (2005) Quorum sensing: cell‐to‐cell communication in bacteria. Annu Rev Cell Dev Biol 21: 319–346. [DOI] [PubMed] [Google Scholar]
- Wu, X. , Wu, D. , Lu, Z. , Chen, W. , Hu, X. , and Ding, Y. (2009) A novel method for high‐level production of TEV protease by superfolder GFP Tag. J Biomed Biotechnol 2009: 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zobel, S. , Benedetti, I. , Eisenbach, L. , de Lorenzo, V. , Wierckx, N. , and Blank, L.M. (2015) Tn7‐based device for calibrated heterologous gene expression in Pseudomonas putida . ACS Synth Biol 4: 1341–1351. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Oligonucleotides used for gene cloning in this study.
Table S2. Oligonucleotides used for qRT‐PCR in this study.