

Abstract
Objective
Innate lymphoid cells (ILCs) are emerging mediators of immunity, and accumulation of inflammatory ILC populations can occur in inflammatory‐mediated conditions. Since early lymph node (LN) activation has been shown in rheumatoid arthritis (RA), we aimed to investigate the frequency and distribution of ILCs in LN biopsy specimens obtained during the earliest phases of RA.
Methods
Twelve patients with early RA, 12 individuals with IgM rheumatoid factor and/or anti–citrullinated protein antibodies without arthritis (RA risk group), and 7 healthy controls underwent ultrasound‐guided inguinal LN biopsy. ILC subsets and the expression of vascular cell adhesion molecule (VCAM) and intercellular adhesion molecule (ICAM) by LN endothelial cells and fibroblasts were analyzed by flow cytometry.
Results
Although no differences in the frequencies of total ILCs (Lin−CD45+/lowCD127+) were found, the distribution of the ILC subpopulations differed among groups. RA patients showed lower numbers of lymphoid tissue–inducer (LTi) cells (c‐Kit+NKp44− ILCs) and increased ILC1 (c‐Kit−NKp44− ILCs) and ILC3 (c‐Kit+NKp44+ ILCs) numbers compared with controls (P < 0.001, P < 0.050, and P < 0.050, respectively). Individuals at risk of RA exhibited an increased frequency of ILC1 compared with controls (P < 0.01). LTi cells paralleled the expression of adhesion molecules on endothelial cells and fibroblasts.
Conclusion
Our findings indicate that during the at‐risk and earliest phases of RA, the ILC distribution in LN changes from a homeostatic profile toward a more inflammatory profile, thereby providing evidence of a role for ILCs in RA pathogenesis.
In recent years, there has been growing interest in the biology of innate lymphoid cells (ILCs), since they have been reported to be crucial mediators involved in immunity and tissue remodeling. ILCs share 3 main characteristics: the absence of somatically rearranged antigen receptors, a lack of myeloid and dendritic cell phenotype markers, and lymphoid morphology 1. ILCs are not a single population, but 3 main subsets can be identified on the basis of their extracellular markers as well as their transcription factors and cytokine profile. Group 1 ILCs comprise classic natural killer (NK) cells and interferon‐γ–producing ILC1, group 2 ILCs are producers of Th2 cytokines, and group 3 ILCs include lymphoid tissue–inducer (LTi) cells and interleukin‐22 (IL‐22)– and IL‐17–producing ILC3 1.
These subsets are not totally stable cell populations and a certain degree of functional plasticity among them has been described, mainly in response to inflammatory stimuli 2. ILCs can orchestrate inflammation, innate and adaptive responses, and homeostatic processes throughout the body 3. LTi cells are pivotal players in lymphoid tissue development and homeostasis, since they can closely interact with stromal cells, leading to up‐regulation of adhesion molecules on stromal cells and the production of homeostatic chemokines, thereby promoting the attraction and retention of additional cell types 4. However, although LTi cells are present and capable of secreting different mediators in adult lymphoid tissues 5, little is known about their function in adult tissues. In addition, whether LTi cells are altered in inflammatory conditions remains unknown. In contrast, ILC1, ILC2, and ILC3 have been reported to promote immunity to infection in mucosal and surface barriers (for review, see refs. 1 and 3). Excessive accumulation and/or activation of specific ILC populations has been related to the pathogenesis of a number of diseases, such as inflammatory bowel disease, psoriasis, and asthma 2, 3, 6. Some evidence suggests that experimental targeting of these subsets leading to decreased ILC counts results in alleviation of disease 7, thereby supporting their role in these conditions.
We hypothesize that an altered distribution of ILC subsets may play a role in the pathogenesis of chronic immune‐mediated autoimmune diseases, such as rheumatoid arthritis (RA). Since the production of autoantibodies precedes the development of signs and symptoms of clinical disease in RA 8, we have set up a system to study the earliest phases of RA by selecting individuals who are positive for autoantibodies but have no clinically apparent disease. Such individuals have recently been defined as having a systemic autoimmunity associated with RA (RA risk) as recommended by the Study Group for Risk Factors for RA under the auspices of the European League Against Rheumatism (EULAR) Standing Committee of Investigative Rheumatology 9. Since the production of autoantibodies is initiated in lymphoid tissue, it is hypothesized that lymph node (LN) activation precedes synovial tissue inflammation. We studied the frequency and distribution of ILC subsets in LN biopsy specimens obtained from individuals at risk of RA, patients with RA, and healthy volunteers. Overall, our results show an inflammatory‐biased ILC profile, which may be associated with an impaired LN microenvironment in the earliest phases of RA.
PATIENTS AND METHODS
Study subjects
We included 12 individuals with systemic autoimmunity associated with RA (RA risk), which was defined as the presence of IgM rheumatoid factor (IgM‐RF) and/or anti–citrullinated protein antibodies (ACPAs) in subjects with arthralgia but without any evidence of arthritis 9. IgM‐RF was measured using an IgM‐RF enzyme‐linked immunosorbent assay (ELISA) from Sanquin with an upper limit of normal (ULN) of 12.5 IU/ml until December 2009, and using an IgM‐RF ELISA from Hycor Biomedical with a ULN of 49 IU/ml thereafter. ACPA was measured using anti–cyclic citrullinated peptide 2 ELISA CCPlus (Euro‐Diagnostica; ULN 25 kAU/liter). After a median follow‐up time of 38.9 months (interquartile range [IQR] 12.0–51.4 months), none of these at‐risk individuals had yet developed RA. We also included 12 patients with early RA who met the 2010 American College of Rheumatology/EULAR criteria 10, had not received disease‐modifying antirheumatic drugs (DMARDs) or biologic agents, and had a disease duration (defined by having arthritis in any joint) of <1 year. Patients with RA and individuals at risk of RA were compared with 7 seronegative healthy controls. Healthy controls had no history of recent viral infection, autoimmunity, or malignancy, and had never received DMARDs, biologic agents, or experimental drugs. The study was approved by the institutional review board of the Academic Medical Center, and all study subjects gave written informed consent. Table 1 shows the demographic characteristics of the subjects.
Table 1.
Demographic and clinical characteristics of the study subjects*
| Healthy controls (n = 7) | Individuals at risk of RA (n = 12) | RA patients (n = 12) | |
|---|---|---|---|
| Sex, no. (%) female | 5 (71) | 8 (67) | 10 (83) |
| Age, years | 31 (28–40) | 44 (34–54) | 54 (35–60) |
| IgM‐RF positive, no. (%) | 0 (0) | 5 (42) | 9 (75) |
| IgM‐RF level, kU/liter | 1 (1–1) | 19 (4–144) | 79 (17–434) |
| ACPA positive, no. (%) | 0 (0) | 7 (58) | 7 (58) |
| ACPA level, kAU/liter | 3 (1–4) | 97 (3–389) | 81 (1–686) |
| ESR, mm/hour | – | 5.0 (2.0–8.0) | 13 (9.3–21) |
| CRP, mg/liter | 0.6 (0.3–1.4) | 1.9 (0.6–5.7) | 6.4 (1.1–21.2) |
| Tender joint count (28 joints assessed) | – | 2 (1–2) | 4 (1–21) |
| Swollen joint count (28 joints assessed) | – | 0 (0) | 8 (4–10) |
| DAS28 | – | – | 4.9 (3.9–5.9) |
Except where indicated otherwise, values are the median (interquartile range). RA = rheumatoid arthritis; IgM‐RF = IgM rheumatoid factor; ACPA = anti–citrullinated protein antibody; ESR = erythrocyte sedimentation rate; CRP = C‐reactive protein; DAS28 = Disease Activity Score in 28 joints.
Sample processing and flow cytometric analysis
All study subjects underwent ultrasound‐guided inguinal LN needle biopsy as previously described 11. Freshly collected LN biopsy specimens were put through a 70‐μm cell strainer (BD Falcon) to obtain a single‐cell suspension. Then cells were washed in PBA buffer (phosphate buffered saline containing 0.01% NaN3 and 0.5% bovine serum albumin [both from Sigma‐Aldrich]) and subjected to extracellular staining for 30 minutes at 4°C in PBA with directly labeled antibodies. For ILCs we used the following antibodies: fluorescein isothiocyanate (FITC)–conjugated anti‐CD3 (clone CLB‐T3/2, 16A9; Sanquin), FITC‐conjugated anti‐CD14 (clone CLB‐mon/1, 8G3; Sanquin), FITC‐conjugated anti‐CD19 (clone HIB19; eBioscience), FITC‐conjugated anti‐CD34 (clone 8G12; BD Biosciences), allophycocyanin (APC)–eFluor780–conjugated anti‐CD127 (clone eBioRDR5; eBioscience), anti‐CD45 V500 (clone HI30; BD Biosciences), phycoerythrin‐conjugated anti‐CD117/c‐kit (clone 104D2; eBioscience), and Alexa Fluor 647–conjugated anti‐CD336/NKp44 (clone P44‐8; BioLegend). For LN fibroblasts and endothelial cells, we used anti‐CD45 V500, Alexa Fluor 647–conjugated antipodoplanin (gp38) (clone NC‐08; BioLegend), APC–eFluor780–conjugated anti‐CD31 (platelet endothelial cell adhesion molecule 1) (clone WM‐59; eBioscience), PerCP‐conjugated anti–intercellular adhesion molecule 1 (anti–ICAM‐1; CD54) (clone 1H4; EXBIO), and anti–vascular cell adhesion molecule 1 (anti–VCAM‐1; CD106) (clone STA; eBioscience). Cells were measured using a FACSCanto II flow cytometer (BD Biosciences), and data were analyzed using FlowJo software.
Statistical analysis
Non‐normally distributed data are presented as the median and IQR. Differences between study groups were analyzed using the Kruskal‐Wallis test followed by Dunn's post hoc test. GraphPad Prism software was used for statistical analysis. P values less than 0.05 were considered statistically significant.
RESULTS
To investigate the main ILC subsets, LN cell suspensions were analyzed by flow cytometry following a gating strategy based on phenotype markers that characterize ILC subpopulations 1 (Figure 1). First, lineage‐negative events were gated and their expression of CD45 and CD127 was assessed, after which the total ILC population was defined as Lin−CD45+/lowCD127+. Next, the ILC subsets were distinguished based on their differential expression of c‐Kit and NKp44, with LTi cells defined as c‐Kit+NKp44−, and ILC3 defined as c‐Kit+ NKp44+. Finally, c‐Kit/NKp44 double‐negative cells were considered to be ILC1. Lymphoid morphology was confirmed for each subpopulation. Then, CD69 expression was analyzed in total ILCs as well as in individual ILC subsets, using the Lin−CD45− population as the negative control.
Figure 1.
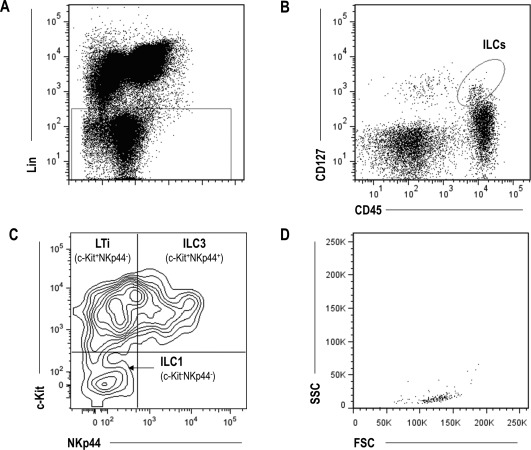
Gating strategy for analysis of innate lymphoid cells (ILCs). Lymph node biopsy specimens were processed to obtain a single‐cell suspension that was immediately stained for ILC markers. Results from a representative healthy control are shown. A, Gating of lineage‐negative (CD3−, CD14−, CD19−, and CD34−) events. B, Determination of expression of CD45 and CD127 to identify the total ILC population (Lin−CD45+/lowCD127+). C, Identification of different ILC subsets (lymphoid tissue–inducer [LTi] cells, ILC3, and ILC1) based on their expression of NKp44 and c‐Kit. D, Backgating to test the lymphoid morphology in each ILC subpopulation.
First, we observed that the frequency of total ILCs of Lin− events did not differ among the 3 study groups (median 0.19% [IQR 0.03–0.28%] in healthy controls, 0.06% [IQR 0.02–0.20%] in individuals at risk of RA, and 0.02% [IQR 0.01–0.17%] in patients with RA; P = 0.301). Also, no difference in the Lin− population was found (P = 0.134). Since variability in the sizes of the LN biopsy specimens was noted as an important factor contributing to the variability of ILC number, the frequency of total ILCs within the total number of cells collected (taking into account the frequency of Lin− events) was calculated, revealing no differences among groups (P = 0.510).
Next, we analyzed the relative frequencies of the different ILC subsets and, interestingly, we found that their distribution within the total ILC population differed between controls, individuals at risk of RA, and RA patients (Figure 2A). The population of LTi cells, the most abundant ILC subset, was significantly decreased in RA patients compared with healthy controls and individuals at risk of RA, exhibiting a progressive decline according to disease status. In contrast, the ILC1 population was significantly increased in both those at risk of RA and RA patients, and the ILC3 population was significantly increased in RA patients compared with individuals at risk of RA (Figure 2A). The frequencies of ILC subsets were not related to clinical or demographic parameters (data not shown). Additionally, due to the role of CD69 in cell activation and retention as well as its relevance to ILC subsets 12, with higher CD69 associated with enhanced functionality, we evaluated the expression of CD69 on these subsets. A higher frequency of CD69+ ILCs was found in healthy controls (Figure 2B). When individual ILC subsets were examined, this pattern was only observed in LTi cells, while ILC1 and ILC3 showed no significant differences. Overall, these results support the notion that the relative distribution of ILC subsets and their expression of CD69 is altered in LN during the at‐risk phase and earliest phase of RA.
Figure 2.
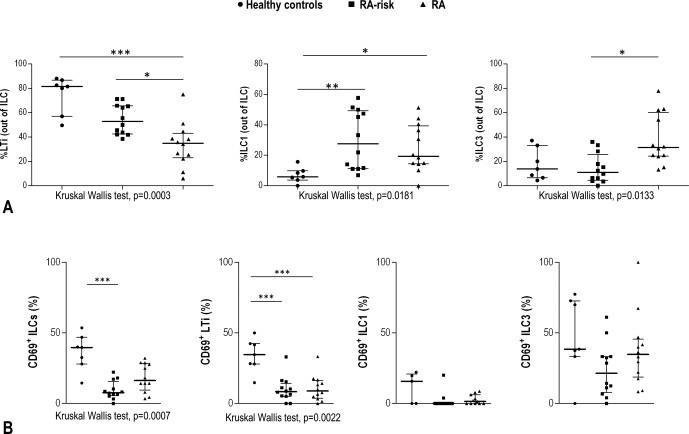
Analysis of innate lymphoid cell (ILC) subsets in lymph node biopsy specimens from healthy controls, individuals at risk of rheumatoid arthritis (RA), and patients with RA. A, Percentages of lymphoid tissue–inducer (LTi) cells, ILC1, and ILC3 among total ILCs. B, Frequency of CD69 in total ILCs and in ILC subsets. Symbols represent individual samples; horizontal lines and error bars show the median and interquartile range. Differences between the 3 groups were assessed by Kruskal‐Wallis test (P values shown at the bottom of each panel); Dunn's post hoc test was then used to compare differences between 2 groups. ∗ = P < 0.050; ∗∗ = P < 0.010; ∗∗∗ = P < 0.001.
Finally, since LTi cells have been described to influence other, nonlymphoid populations 3, 13, we aimed to study whether changes in LTi cell numbers may be related to changes in endothelial and fibroblast subsets present within the LN. Therefore, VCAM and ICAM expression, quantified as the percentage of positive cells for each marker compared with unstained cells, by endothelial cells (CD45−CD31+) and fibroblastic reticular cells (CD45−CD31−gp38+) was assessed in a subgroup of individuals (4 healthy controls, 2 individuals at risk of RA, and 4 RA patients). The group of individuals with paired samples did not differ from the initial group in age (P = 0.700), sex (P = 0.825), or disease status (P = 0.419). Interestingly, the frequency of LTi cells was positively associated with the expression of VCAM (r = 0.766, P = 0.015) and ICAM (r = 0.593, P = 0.070) on LN endothelial cells. Similarly, LTi cell numbers correlated with VCAM expression on fibroblastic reticular cells (r = 0.740, P = 0.014). LTi cell numbers were not related to the frequency of endothelial or fibroblastic reticular cell populations (both P > 0.050).
DISCUSSION
Our data revealed that the distribution of ILCs changes within the LN compartment during the at‐risk and earliest phases of RA. To the best of our knowledge, this is the first study to demonstrate ILC disturbances within the LN in RA. Although none of the at‐risk individuals developed RA during follow‐up, these individuals produced autoantibodies specific for RA and thus displayed features of systemic autoimmunity associated with RA, hence suggesting a role for ILCs during RA development. Moreover, the frequency of LTi cells was associated with the expression of adhesion molecules by stromal cells, which may suggest potential cross‐talk between ILCs and the stromal cell compartment. Since the pool of total ILCs was unchanged among the different study groups, our results highlight a role for an ILC imbalance in LN as an early event in RA pathogenesis.
During systemic autoimmunity associated with RA, ILCs within the LN microenvironment seem to exhibit a shift from a more “homeostatic” profile, characterized by a higher frequency of LTi cells, toward a more “inflammatory/activated” one, characterized by an increased frequency of potentially proinflammatory cytokine–producing ILC subsets (ILC1 and ILC3). Taking into account that NKp44 can be considered an activation marker, and the fact that RA patients exhibit features of an active adaptive immune response, a link between LN activation and ILC imbalance is supported. Interestingly, dysregulation of ILC subsets and increased ILC3 counts have recently been reported in skin biopsy specimens from psoriasis patients 14. Similarly, increased frequencies of ILC1 and ILC3 have been reported in mucosal or surface barriers in other diseases 2, 3. Taken together, these lines of evidence suggest a role for ILC accumulation as a pathogenic mechanism in tissue damage in the end stages of the disease. Of interest, our findings revealed that skewed ILC distribution is an early event in RA pathogenesis.
Moreover, biased ILC distribution is found in the LN, a place where adaptive immunity is initiated, thus revealing an ILC alteration in a tissue other than the target tissue. An increased frequency of cytokine‐producing ILCs has been associated with pathogenic outcomes 2, 7, 15, whereas their selective inhibition was found to be related to disease abrogation 7, 16. Similarly, excessive LN activation may lead to LN damage, which can be restored by LTi cells 13. Of interest, recent evidence suggests that LTi cells may regulate immune cell homeostasis within LNs 17, 18, including maintenance of T cell memory 19. Thus, the ILC profile may be a pivotal player for regulation of immune homeostasis and function 20. It is challenging to determine the underlying mechanisms driving this ILC imbalance, although changes in cytokine production within the LN microenvironment as a consequence of LN activation preceding RA development 21 may be key in the orchestration of ILC differentiation 2. Interestingly, although evidence is limited, some cytokines altered during LN activation 21, 22 are related to certain ILC subsets.
It is difficult to ascertain whether the altered ILC distribution can have an impact on the function of the LN microenvironment from a mechanistic point of view. However, our paired analysis of endothelial and fibroblast subsets provides some insight. One of the main functions of LTi cells is the production of lymphotoxin α1β2 (LTα1β2), which promotes the expression of adhesion molecules on stromal cells 2, 4, thereby promoting migration and colonization of hematopoietic cells during LN development or after injury. Accordingly, our results highlight an association between the frequency of LTi cells and the expression of VCAM and ICAM on endothelial and stromal cells. This finding may be consistent with current evidence linking LTi cells with LN homeostasis, where LTi cell–derived LTα1β2 plays a pivotal role. Abrogation of LTα1β2 signaling leads to decreased expression of homeostatic chemokines and survival factors by stromal cells, which is essential for LN reestablishment after infections 13. Thus, a disturbed capacity to resolve an infection might lead to activation of autoreactive lymphocytes. Therefore, it may be speculated that the reduced LTi cell counts in RA patients might reflect an impaired function of LN remodeling. Hence, relatively low LTi cell numbers may aid an autoimmune‐prone LN microenvironment through their effects on stromal cells; however, this warrants further functional studies.
It is interesting to note that not only was the frequency of LTi cells decreased, but CD69 expression was also lower in those at risk of RA and in RA patients. Interestingly, a high frequency of CD69+ ILCs in blood has been related to a better outcome upon hematopoietic stem cell transplantation 12, thus suggesting a link between CD69 expression on ILCs and protective/reparative mechanisms. The decreased LTi cell numbers and lower CD69 expression is consistent with the proposed role of CD69 as a marker for cell retention 23. However, what triggers LTi cells to down‐regulate CD69 and potentially cause the egress of LTi cells remains to be determined.
Since we did not analyze ILC2, it could be that ILC frequencies, especially that of ILC1, are overestimated. Another point that should be addressed is the ILC nomenclature adopted in this manuscript. Since there is no consensus about whether adult LTi cells resemble their fetal counterparts, some authors have proposed alternative terms (LTi‐like or natural cytotoxicity triggering receptor–negative ILC3). However, we preferred to follow the recent proposal for uniform ILC nomenclature by leading experts 1. The observed association between LTi cell frequency and the expression of adhesion molecules on stromal cells, the central role for LTi cells, supports our decision. ILC functionality in vitro may not be representative of that in vivo, and the small sample size of the LN needle biopsy specimens hampers isolation of low‐frequency ILC subsets, which makes functional studies rather challenging. However, our findings warrant further additional studies of ILC functionality. Overall, the present study is the first to show an imbalance between homeostatic and inflammatory ILC subsets in LN biopsy specimens obtained during the at‐risk and earliest phases of RA, which may underlie the earliest steps in RA pathogenesis.
AUTHOR CONTRIBUTIONS
All authors were involved in drafting the article or revising it critically for important intellectual content, and all authors approved the final version to be published. Dr. van Baarsen had full access to all of the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis.
Study conception and design
Gerlag, Tak, van Baarsen.
Acquisition of data
Rodríguez‐Carrio, Hähnlein, Ramwadhdoebe, Semmelink, Choi, van Lienden, Maas, Gerlag, Tak, Geijtenbeek, van Baarsen.
Analysis and interpretation of data
Rodríguez‐Carrio, Hähnlein, Ramwadhdoebe, Semmelink, Geijtenbeek, van Baarsen.
ADDITIONAL DISCLOSURES
Authors Gerlag and Tak are currently employees of GlaxoSmithKline, but were employed by Academic Medical Center, University of Amsterdam, during the time the study was conducted.
ACKNOWLEDGMENTS
We especially thank the study participants. We thank the staff of the Radiology Department at the Academic Medical Center for lymph node sampling, the staff of the flow cytometry facility at the Hematology Department at the Academic Medical Center, and the Academic Medical Center Rheumatology Department technicians for sample processing. We would like to thank Professors Reina Mebius and Yotam Bar‐Ephraim (VU University Medical Center, Amsterdam, The Netherlands) for critical review of the manuscript.
Supported by the IMI‐funded European Union project BeTheCure (nr115142), the Seventh Framework HEALTH Programme (Euro‐TEAM grant FP7‐HEALTH‐F2‐2012‐305549), the Dutch Arthritis Foundation (grant 11‐1‐308), and The Netherlands Organisation for Health Research and Development (Veni project 916.12.109). Dr. Rodríguez‐Carrio's work was supported by the FPU program (Spanish Ministry of Education grant AP2010‐1614) and an EFIS‐IL short‐term program grant.
REFERENCES
- 1. Spits H, Artis D, Colonna M, Diefenbach A, di Santo JP, Eberl G, et al. Innate lymphoid cells: a proposal for uniform nomenclature. Nat Rev Immunol 2013;13:145–9. [DOI] [PubMed] [Google Scholar]
- 2. Bernink JH, Peters CP, Munneke M, te Velde AA, Meijer SL, Weijer K, et al. Human type 1 innate lymphoid cells accumulate in inflamed mucosal tissues. Nat Immunol 2013;14:221–9. [DOI] [PubMed] [Google Scholar]
- 3. Sanati G, Aryan Z, Barbadi M, Rezaei N. Innate lymphoid cells are pivotal actors in allergic, inflammatory and autoimmune diseases. Expert Rev Clin Immunol 2015;11:885–95. [DOI] [PubMed] [Google Scholar]
- 4. Vondenhoff MF, Greuter M, Goverse G, Elewaut D, Dewint P, Ware CF, et al. LTβR signaling induces cytokine expression and up‐regulates lymphangiogenic factors in lymph node anlagen. J Immunol 2009;182:5439–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Lane PJ, Gaspal FM, McConnell FM, Withers DR, Anderson G. Lymphoid tissue inducer cells: pivotal cells in the evolution of CD4 immunity and tolerance? Front Immunol 2012;3:1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Bernink JH, Germar K, Spits H. The role of ILC2 in pathology of type 2 inflammatory diseases. Curr Opin Immunol 2014;31:115–20. [DOI] [PubMed] [Google Scholar]
- 7. Powell N, Walker AW, Stolarczyk E, Canavan JB, Gökmen MR, Marks E, et al. The transcription factor T‐bet regulates intestinal inflammation mediated by interleukin‐7 receptor+ innate lymphoid cells. Immunity 2012;37:674–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Rantapää‐Dahlqvist S, de Jong BA, Berglin E, Hallmans G, Wadell G, Stenlund H, et al. Antibodies against cyclic citrullinated peptide and IgA rheumatoid factor predict the development of rheumatoid arthritis. Arthritis Rheum 2003;48:2741–9. [DOI] [PubMed] [Google Scholar]
- 9. Gerlag DM, Raza K, van Baarsen LG, Brouwer E, Buckley CD, Burmester GR, et al. EULAR recommendations for terminology and research in individuals at risk of rheumatoid arthritis: report from the Study Group for Risk Factors for Rheumatoid Arthritis. Ann Rheum Dis 2012;71:638–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Aletaha D, Neogi T, Silman AJ, Funovits J, Felson DT, Bingham CO III, et al. 2010 rheumatoid arthritis classification criteria: an American College of Rheumatology/European League Against Rheumatism collaborative initiative. Arthritis Rheum 2010; 62:2569–81. [DOI] [PubMed] [Google Scholar]
- 11. De Hair MJ, Zijlstra IA, Boumans MJ, van de Sande MG, Maas M, Gerlag DM, et al. Hunting for the pathogenesis of rheumatoid arthritis: core‐needle biopsy of inguinal lymph nodes as a new research tool. Ann Rheum Dis 2012;71:1911–2. [DOI] [PubMed] [Google Scholar]
- 12. Munneke JM, Björklund AT, Mjösberg JM, Garming‐Legert K, Bernink JH, Blom B, et al. Activated innate lymphoid cells are associated with a reduced susceptibility to graft‐versus‐host disease. Blood 2014;124:812–21. [DOI] [PubMed] [Google Scholar]
- 13. Scandella E, Bolinger B, Lattmann E, Miller S, Favre S, Littman DR, et al. Restoration of lymphoid organ integrity through the interaction of lymphoid tissue‐inducer cells with stroma of the T cell zone. Nat Immunol 2008;9:667–75. [DOI] [PubMed] [Google Scholar]
- 14. Villanova F, Flutter B, Tosi I, Grys K, Sreeneebus H, Perera GK, et al. Characterization of innate lymphoid cells in human skin and blood demonstrates increase of NKp44+ ILC3 in psoriasis. J Invest Dermatol 2014;134:984–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Fuchs A, Vermi W, Lee JS, Lonardi S, Gilfillan S, Newberry RD, et al. Intraepithelial type 1 innate lymphoid cells are a unique subset of IL‐12‐ and IL‐15‐responsive IFN‐γ‐producing cells. Immunity 2013;38:769–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Perry JS, Han S, Xu Q, Herman ML, Kennedy LB, Csako G, et al. Inhibition of LTi cell development by CD25 blockade is associated with decreased intrathecal inflammation in multiple sclerosis. Sci Transl Med 2012;4:145ra106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Dummer W, Ernst B, LeRoy E, Lee D, Surh C. Autologous regulation of naive T cell homeostasis within the T cell compartment. J Immunol 2001;166:2460–8. [DOI] [PubMed] [Google Scholar]
- 18. Lane PJ, Gaspal FM, McConnell FM, Withers DR, Anderson G. Lymphoid tissue inducer cells: pivotal cells in the evolution of CD4 immunity and tolerance? Front Immunol 2012;3:24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Withers DR, Gaspal FM, Mackley EC, Marriott CL, Ross EA, Desanti GE, et al. Cutting edge: lymphoid tissue inducer cells maintain memory CD4 T cells within secondary lymphoid tissue. J Immunol 2012;189:2094–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Xu W, di Santo JP. Taming the beast within: regulation of innate lymphoid cell homeostasis and function. J Immunol 2013;191:4489–96. [DOI] [PubMed] [Google Scholar]
- 21. Li J, Kuzin I, Moshkani S, Proulx ST, Xing L, Skrombolas D, et al. Expanded CD23+CD21hi B cells in inflamed lymph nodes are associated with the onset of inflammatory‐erosive arthritis in TNF‐transgenic mice and are targets of anti‐CD20 therapy. J Immunol 2010;184:6142–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Kuzin II, Bouta EM, Schwarz EM, Bottaro A. TNF signals are dispensable for the generation of CD23+ CD21/35‐high CD1d‐high B cells in inflamed lymph nodes. Cell Immunol 2015;296:133–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Shiow LR, Rosen DB, Brdicková N, Xu Y, An J, Lanier LL, et al. CD69 acts downstream of interferon‐α/β to inhibit S1P1 and lymphocyte egress from lymphoid organs. Nature 2006;440:540–4. [DOI] [PubMed] [Google Scholar]