

Abstract
Tetrabromobisphenol A (TBBPA) is used in a diverse array of products to improve fire safety. The National Toxicology Program (NTP) recently completed a 2‐year bioassay for TBBPA. The objective of the present study was to develop a cancer‐based and a non‐cancer based toxicity value and to compare such to appropriate estimates of human exposure. Data from the NTP 2‐year and 13‐week studies were selected to develop candidate toxicity values. Benchmark dose modeling and subsequent evaluation of candidate values resulted in selection of an oral reference dose (RfD) of 0.6 mg kg−1 day−1 based on uterine hyperplasia in rats and an oral cancer slope factor (OSF) of 0.00315 per mg kg−1 day−1 based on an increased incidence of uterine tumors in rats. Lifetime average daily dose (LADD) estimates ranged from 2.2 E−7 to 3.9 E−6 mg kg−1 day−1 based on age‐adjusted exposures to TBBPA via breast milk consumption, dietary intake, soil/dust ingestion and drinking water ingestion in infants, young children, older children and adults. Average daily dose (ADD) estimates ranged from 3.2 E −7 to 8.4 E−5 mg kg−1 day−1. Resulting margin of exposure (MOE) values were > 800 000 for non‐cancer endpoints and > 32 000 000 for cancer‐based endpoints. These data collectively indicate a low level of health concern associated with exposures to TBBPA based on current data. It is anticipated that the exposure estimates, along with the toxicity values described within, should be informative for understanding human health hazards associated with TBBPA. Copyright © 2015. The Authors. Journal of Applied Toxicology Published by John Wiley & Sons Ltd.
Keywords: TBBPA, flame retardant, toxicity value, RfD, cancer slope factor, margin of exposure
Short abstract
Data from the National Toxicology Program (NTP) were utilized to develop an oral reference dose (RfD) and oral cancer slope factor (OSF) for tetrabromobisphenol A (TBBPA). Comparison of exposure estimates based on breast milk consumption, dietary intake, soil/dust ingestion and drinking water ingestion, resulted in margins of exposure > 800 000 in infants, young children, older children and adults. These data collectively indicate a low level of health concern associated with exposures to TBBPA.
Introduction
Tetrabromobisphenol A (TBBPA) is the most widely produced and used brominated flame retardant, primarily because of its effectiveness and low hazard profile (BSEF, 2012). It is used to improve fire safety in a wide variety of consumer products. TBBPA‐containing polymers are used in epoxy and polycarbonate resins, as well as in acrylonitrile‐butadiene‐styrene (ABS) and phenolic resins, which are ultimately used in products such as printed circuit boards, communications and electronics equipment, appliances, transportation devices, sports and recreation equipment, automotive parts, pipes and fittings (Birnbaum and Staskal, 2004; BSEF, 2012). TBBPA is primarily used as a reactive component, as well as an additive flame retardant in a limited number of applications. Although TBBPA is generated by the bromination of bisphenol A (BPA), it is important to note that this bromination results in a compound with very different chemical and physical properties, as well as different toxicities than BPA. Additionally, there is currently no evidence of dehalogenation of TBBPA to BPA in vivo; recent toxicokinetic studies of TBBPA do not report on BPA as a measurable metabolite of TBBPA (Knudsen et al., 2014). TBBPA can be released to the environment via various mechanisms, including during manufacture and production, use of TBBPA‐containing products and recycling of TBBPA‐containing products. Once in the environment, TBBPA generally distributes to the soil and sediment; it has low to moderate water solubility, a low vapor pressure and a moderately high octanol/water partition coefficient (de Wit, 2002).
TBBPA has been detected in human serum samples in both occupational and non‐occupational settings, as well as in breast milk, demonstrating that the compound is absorbed in humans after exposure (Jakobsson et al., 2002; Sjodin et al., 2003; Shi et al. 2013). A large number of studies have reported TBBPA in media associated with human exposure, including soil, foodstuffs and, to a lesser extent, water and air (EU, 2006; Health Canada, 2013; Colnot et al., 2014). As a result of the potential for widespread exposure, and evidence of potentially increasing trends, there has been increasing interest in characterizing potential hazards. Toxicity data in humans are limited to dermal irritation studies (NTP, 2002) and a recent report of a weak correlation between serum concentrations and thyroid hormones in a cross‐sectional evaluation (Kim and Oh, 2014).
Many laboratory studies have been conducted with TBBPA and key findings reported in these studies include the following: (1) a lack of reproductive and developmental toxicity in a guideline‐based two‐generation study (including developmental neurotoxicity), (2) a lack of adverse findings in a guideline‐based 90‐day study, and (3) a lack of genotoxicity and mutagenicity in standard assays (Schroeder, 2002a, 2002b, 2003; EU, 2006; Williams and DeSesso, 2010; NTP, 2013; Health Canada, 2013). However, a number of repeated dose studies in the peer‐reviewed literature have reported associations between TBBPA exposure and hepatotoxicity, body weight changes, endocrine disruption, nephrotoxicity, neurotoxicity and developmental toxicity in rodents (Sato et al., 1996; Szymanska et al., 2000; Fukuda et al., 2004; Germer et al., 2006; Tada et al., 2006, 2007; Lilienthal et al., 2008; Van der Ven et al., 2008; Imai et al., 2009; Saegusa et al., 2009, 2012; Decherf et al., 2010; Watanabe et al., 2010; Zatecka et al., 2013). Most recently, the National Toxicology Program (NTP) released findings from a 2‐year animal bioassay for TBBPA, that included both cancer and non‐cancer data in rats and mice (NTP, 2013). Data from this bioassay indicated that chronic administration of TBBPA at very high doses (up to 1000 mg kg−1 day−1) resulted in uterine tumors in female rats (classified as equivocal evidence) and liver tumors in male mice (classified as some evidence), as well as a number of non‐neoplastic effects (e.g. hyperplasia).
Currently, there is only a single toxicity value available for TBBPA – the UK Committee on Toxicity (COT) developed a tolerable daily intake of 1 mg kg−1 day−1 in 2004 as part of an assessment in which the COT concluded that TBBPA did not raise specific toxicological concerns (COT, 2004). Three other agencies have conducted health‐based assessments that utilized theee margin of exposure (MOE) approach for evaluating TBBPA, although these agencies did not develop toxicity or health‐based values as part of these efforts (EU, 2006; EFSA, 2011; Health Canada, 2013). Health Canada, the European Food Safety Authority (EFSA) and the European Union (EU) reviewed available toxicity data, selected critical effect levels from laboratory studies, and then compared such to modeled or calculated estimates of human exposure. The findings of all three regulatory assessments were similar, acceptable MOE values were obtained, regardless of exposure scenario and receptor (e.g. infant and adult). Recently, Colnot et al. (2014) published findings of an independent evaluation of TBBPA toxicity and exposure. Similar to the approach used by EFSA, Health Canada and the EU, these authors conducted an MOE assessment, the results of which indicated that exposure were below the derived‐no‐effect‐levels for endpoints of potential concern in REACH. Notably, these assessments all relied on data on non‐cancer endpoints as there were no data characterizing carcinogenicity at the time that these assessments were conducted.
Given that a number of relevant toxicity studies have become available since the development of the tolerable daily intake (TDI) by the COT almost a decade ago, including carcinogenicity data recently released by the NTP, the first objective of the current study was to review the available toxicity data to develop both cancer and non‐cancer toxicity values for TBBPA. The second objective was to quantitatively characterize potential consumer exposures to TBBPA. And finally, the third objective was to conduct a margin of exposure (MOE) assessment. Specifically, we compared the points of departure (PODs) selected for use in the development of the cancer and non‐cancer toxicity values to conservative estimates of potential exposure for infants, young children, older children and adults. Margin of safety (MOS) values are also presented. It is anticipated that the exposure estimates, along with the toxicity values described herein, should be informative for risk assessors and regulators interested in characterizing human health hazards associated with TBBPA.
Materials and methods
Development of toxicity values
Toxicity values were developed for both cancer and non‐cancer endpoints associated with chronic, oral exposure to TBBPA as described below.
Toxicity data selection
As no human data are available to characterize the toxicity of TBBPA, published peer‐reviewed studies and select unpublished studies reporting findings in laboratory animals were used to develop toxicity values. A literature search was conducted to identify relevant publications. To be considered, a study had to have a quality and reliability rating equivalent to a Klimisch score of K1 or K2 (Klimisch et al., 1997), and the study design had to incorporate the following minimum parameters: in vivo study, multiple dose levels, repeated dosing, mammalian species and relevant route of exposure. Only studies which specifically evaluated the toxicity of TBBPA were considered in this assessment; studies focused on the potential toxicity of metabolites were not considered. Copies of the unpublished studies were provided directly by the study sponsors (note: these data are also summarized by EU 2006; Colnot et al., 2014). Data from the chronic NTP bioassay were obtained directly from the study report (NTP, 2013). Key studies considered by the EU, EFSA and Health Canada (EU, 2006; EFSA, 2011; Health Canada, 2013) were also included in the data selection process. A database was generated to summarize key study information from these various sources, such as dose levels, effects evaluated and most sensitive findings These study data were then thoroughly reviewed to identify studies with the most robust, consistent, as well as the most sensitive, findings related to cancer and non‐cancer. Specific datasets were subsequently selected for use in the development of PODs and toxicity factors for cancer and non‐cancer endpoints
Dose–response modeling and POD development
Dose–response modeling was conducted on selected cancer and non‐cancer datasets using US EPA's Benchmark Dose Software (BMDS) v.2.4. The standard software suites for continuous and dichotomous models were used for dose–response analysis. For dichotomous datasets, a benchmark response (BMR) of 10% extra risk was used to obtain benchmark dose (BMD10) values along with the 95% lower confidence limits (BMDL10), consistent with US EPA recommendations (USEPA, 2012). For continuous datasets, the BMR was set to 1 standard deviation in order to obtain (BMD1SD) and (BMDL1SD) values (USEPA, 2012). Model fits were judged acceptable using the criteria of a P‐value, visual inspection and scaled residuals. Afterwards, acceptable models were compared using the Akaike information criterion (AIC), where the lowest AIC was favored. The resulting BMDL10 and BMDL1SD values were identified as PODs for the respective datasets. Where necessary, the exposure concentrations were adjusted for duration of exposure prior to dose–response modeling.
Toxicity value derivation
Oral cancer slope factor (OSF) values were derived for cancer‐based endpoints using allometrically scaled BMDL10 values (USEPA, 2005a, 2005b). Considering the limited MOA data available at this time, only linear multistage cancer models were utilized per US EPA recommendations (USEPA, 2012).
Reference dose (RfD) values were derived for non‐cancer endpoints by adjusting each BMDL value to a human equivalent dose (HED) by allometric scaling. Each HED value was subsequently divided by applicable uncertainty factors (as appropriate) consistent with typical US EPA recommendations (USEPA, 2002):
Equation 1
RfD Derivation
(1)
where,
- RfD
= Reference dose (mg kg−1 day−1)
- HED
= Human Equivalent Dose (mg kg−1 day−1)
- UFA
= uncertainty factor for interspecies variation (unitless)
- UFH
= uncertainty factor for intraspecies variation (unitless)
- UFS
= uncertainty factor for subchronic‐to‐chronic extrapolation (unitless)
- UFL
= uncertainty factor for LOAEL‐to‐NOAEL extrapolation (unitless); and
- UFD
= uncertainty factor for database deficiencies (unitless).
Development of estimates of potential exposure
Exposure to TBBPA was characterized by calculating a total daily intake for consumers (i.e. non‐occupational) based on oral exposure to TBBPA via the diet, infant breast milk consumption, drinking water and soil/dust ingestion. Dermal exposure was not assessed as data are limited for this endpoint, and previous assessments have demonstrated that intake associated with dermal exposure is negligible (EU, 2006). Estimates of intake were calculated using concentrations of TBBPA in these media in standard intake equations for three scenarios: (1) central tendency, (2) upper bound and (3) regulatory default. These scenarios were selected to demonstrate a range of possible exposure estimates that reflect exposures from the most plausible scenario for the general consumer population (central tendency), a plausible upper‐end for the general consumer population (upper bound) and a reasonable worst‐case exposure (regulatory default). The central tendency and upper bound scenarios are generally based on reasonable media concentrations and the most up‐to‐date exposure parameters, where as the regulatory default scenario is based on regulatory default exposure parameters (e.g. USEPA default consumption rates), and maximum media concentrations (where reported). For each of the exposure scenarios, adult, older children, young child and infant age groups were evaluated, and intake estimates were presented both as an average daily dose and a lifetime average daily dose (for use in non‐cancer and cancer comparisons, respectively).
Media concentration data selection
Concentrations of TBBPA in the diet, breast milk, water, and soil/dust were characterized using data from the published literature and government documents. Summary data presented by the EU (2006) and Health Canada (2013) were used as a preliminary guide to characterizing media concentrations, followed by a comprehensive literature search to identify additional relevant publications published through to August 2013. To be considered for inclusion, a study had to be available in English, have a quality and reliability rating equivalent to a K1 or K2 (Klimisch et al., 1997), and had to include an adequate description of sampling locations, methodologies and resulting data (including description of how non‐detect data were handled). Additionally, studies had to be representative of chronic exposure (Benford et al., 2010). A database was generated to summarize key study information from these various sources, such as media type, location, number of samples and range of concentrations. These study data were then reviewed to identify key studies for use in developing relevant, conservative (although still plausible) and worst‐case media concentrations for use in developing exposure estimates for TBBPA.
Daily intake calculations
Daily intake was calculated using two approaches. Per standard practice, an average daily dose (ADD) was generated for use in non‐cancer evaluations and a lifetime average daily dose (LADD) was generated for use in cancer evaluations (EFSA, 2011; USEPA, 1991, 1992). This allowed for assessment of various age groups separately in the non‐cancer assessment, whereas the cancer‐based evaluations were based on an age‐adjusted scenario with exposures combined across age groups. Table 1 provides the exposure parameters used in the calculation ADD and LADD for the various scenarios. The equations for infant breast milk exposures were based on those used by the EU (2006) to calculate the average daily uptake for a breastfeeding infant aged 0–3 months, 4–12 months, as well as 0–12 month average. Drinking water, soil/dust and age‐adjusted equations were based ingestion equations used by the USEPA (2013) in developing regional screening levels. Equations used in the calculation of the ADD, LADD and age‐adjusted values are provided in the Supporting Information.
Table 1.
Exposure parameters
| Parameter | Central Tendency | Reference | Upper‐Bound | Reference | Regulatory Default | Reference |
|---|---|---|---|---|---|---|
| Averaging Time | ||||||
| Averaging time, adult, non‐carcinogenic | 5,110 d | USEPA 2013 | 5,110 d | USEPA 2013 | 5,110 d | USEPA 2013 |
| Averaging time, older child non‐carcinogenic | 3,650 d | USEPA 2013 | 3,650 d | USEPA 2013 | 3,650 d | USEPA 2013 |
| Averaging time, young child, non‐carcinogenic | 1,825 d | USEPA 2013 | 1,825 d | USEPA 2013 | 1,825 d | USEPA 2013 |
| Averaging time, carcinogenic | 28,470 d | USEPA 2011 (assumes 78 yr lifetime) | 28,470 d | USEPA 2011 (assumes 78 yr lifetime) | 25,550 d | USEPA 1991 |
| Averaging time, infant (0–3 months) | 91 d | USEPA 2013 | 91 d | USEPA 2013 | 91 d | USEPA 2013 |
| Averaging time, infant (4–12 months) | 274 d | USEPA 2013 | 274 d | USEPA 2013 | 274 d | USEPA 2013 |
| Body Weight | ||||||
| Body weight, adult | 70 kg | USEPA 2013 | 70 kg | USEPA 2013 | 70 kg | USEPA 1991 |
| Body weight, older child (6 to <16 years) | 44 kg | USEPA 2011 | 44 kg | USEPA 2011 | 45 kg | USEPA 2000 |
| Body weight, young child (1 to <6 years) | 17 kg | USEPA 2011 | 17 kg | USEPA 2011 | 15 kg | USEPA 1991 |
| Body weight, infant (0–3 months) | 6 kg | USEPA 2011 | 6 kg | USEPA 2011 | 6 kg | USEPA 2011 |
| Body weight, infant (4–12 months) | 9 kg | USEPA 2011 | 9 kg | USEPA 2011 | 9 kg | USEPA 2011 |
| Breast Milk Consumption Rate | ||||||
| Consumption rate of breast milk (0–3 months) | 0.68 kg/d | USEPA 2011 | 1.01 kg/d | USEPA 2011 | 1.01 kg/d | USEPA 2011 |
| Consumption rate of breast milk (4–12 months) | 0.68 kg/d | USEPA 2011 | 1.03 kg/d | USEPA 2011 | 1.03 kg/d | USEPA 2011 |
| Consumption rate of breast milk, age‐adjusted | 0.01 kg‐yr/kg‐d | Based on infant 0–3 mos and infant 4–12 mos | 0.01 kg‐yr/kg‐d | Based on infant 0–3 mos and infant 4–12 mos | 0.01 kg‐yr/kg‐d | Based on infant 0–3 mos and infant 4–12 mos |
| Drinking Water Consumption Rate | ||||||
| Consumption rate of drinking water, adult | 1.04 L/d | USEPA 2011 | 2.96 L/d | USEPA 2011 | 2 L/d | USEPA 1989 |
| Consumption rate of drinking water, older child | 0.47 L/d | USEPA 2011 | 1.57 L/d | USEPA 2011 | 2 L/d | USEPA 1997 |
| Consumption rate of drinking water, young child | 0.31 L/d | USEPA 2011 | 0.92 L/d | USEPA 2011 | 1 L/d | USEPA 2000 |
| Consumption rate of drinking water, age‐adjusted | 0.41 L‐yr/kg‐d | Based on adult, adolescent, and child | 1.22 L‐yr/kg‐d | Based on adult, adolescent, and child | 1.18 L‐yr/kg‐d | Based on adult, adolescent, and child |
| Soil and Dust Consumption Rate | ||||||
| Consumption rate of soil and dust, adult | 50 mg/d | USEPA 2011 | 50 mg/d | USEPA 2011 | 100 mg/d | USEPA 1991 |
| Consumption rate of soil and dust, older child | 32.8 mg/d | Kirman et al., 2011 | 92.2 mg/d | Kirman et al., 2011 | 200 mg/d | USEPA 1991 |
| Consumption rate of soil and dust, young child | 32.8 mg/d | Kirman et al., 2011 | 92.2 mg/d | Kirman et al., 2011 | 200 mg/d | USEPA 1991 |
| Consumption rate of soil and dust, age‐adjusted | 38.2 mg/yr‐kg‐d | Based on adult, adolescent, child, and infant 4–12 mos | 63.6 mg/yr‐kg‐d | Based on adult, adolescent, child, and infant 4–12 mos | 136 mg/yr‐kg‐d | Based on adult, adolescent, child, and infant 4–12 mos |
| Exposure Duration | ||||||
| Exposure duration, adult, non‐ carcinogenic | 14 yr | USEPA 2013 | 14 yr | USEPA 2013 | 14 yr | USEPA 2013 |
| Exposure duration, older child, non‐carcinogenic | 10 yr | USEPA 2013 | 10 yr | USEPA 2013 | 10 yr | USEPA 2013 |
| Exposure duration, young child, non‐carcinogenic | 5 yr | USEPA 2013, a | 5 yr | USEPA 2013, a | 5 yr | USEPA 2013, a |
| Exposure duration, infant (0–3 months) | 0.25 yr | USEPA 2013 | 0.25 yr | USEPA 2013 | 0.25 yr | USEPA 2013 |
| Exposure duration, infant (4–12 months) | 0.75 yr | USEPA 2013 | 0.75 yr | USEPA 2013 | 0.75 yr | USEPA 2013 |
| Exposure Frequency | ||||||
| Exposure frequency | 365 d/yr | USEPA 2003 | 365 d/yr | USEPA 2003 | 365 d/yr | USEPA 2003 |
| Other | ||||||
| Absorbed fraction of ingested TBBPA | 1 | Default | 1 | Default | 1 | EU 2006 |
| Fraction of fat in breast milk | 0.04 kg fat/kg milk | USEPA 2011 Table 15‐1 | 0.04 kg fat/kg milk | USEPA 2011 Table 15‐1 | 0.04 kg fat/kg milk | USEPA 2011 Table 15‐1 |
| Total dietary intake, age‐adjusted | 46 yr | Based on adult, adolescent, and child | 46 yr | Based on adult, adolescent, and child | 48 yr | Based on adult, adolescent, and child |
Young child exposure duration changed from 6 years (USEPA 2013) to 5 years herein since infant is evaluated separately.
CTE, central tendency exposure; d, day; kg, kilogram; kg/d, kilogram per day; kg‐yr/kg‐d, kilogram‐year per kilogram‐day, l, liter, :l/d, liter per day; l‐yr/kg‐d, liter‐year per kilogram‐day; mg, milligram; mg/d, milligram per day; mg‐yr/kg‐d, milligram‐year per kilogram‐day; mos, months; RME, reasonable maximum exposure; yr, year.
ADD estimates for each scenario (central tendency, upper bound, and regulatory default) were generated for 0‐ to 3‐month‐old infants, 4‐ to 12‐month‐old infants, 0‐ to 12‐month‐old infants, young children (1‐<6 years), older children (6 ‐ <16 years) and adults. The 0‐ to 3‐month‐old ADD was based solely on exposure to TBBPA via breast milk. The 4‐ to 12‐month old infant ADD was based on exposure to TBBPA via breast milk and soil/dust (assumes child is crawling). Owing to the low limits of detection of TBBPA in food, combined with the low intake of meat and fish relative to vegetables, fruits and grain products by infants and the lack of data characterizing concentrations of TBBPA in baby food, the authors chose to exclude the potential exposure of infants to dietary sources on the basis given the general lack of sufficient data. The young child, older child and adult exposures are based on the cumulative exposure to TBBPA in the diet, soil/dust and drinking water. LADD estimates were generated for each scenario (central tendency, upper bound and regulatory default) based on an age‐adjusted, combined exposure to TBBPA from breast milk, soil/dust, diet and drinking water.
Margin of exposure and margin of safety calculations
Margin of exposure (MOE) and margin of safety (MOS) estimates were generated using standard approaches. By definition, the MOE is a quantitative measure between the dose associated with a small increase in adverse effect and the level of exposure. MOE estimates were derived by dividing the points of departure for cancer and non‐cancer endpoints by the LADD or ADD, respectively. MOS is often associated with variable definitions; in this paper, the MOS is similar to MOE, except exposure is compared with doses associated with the toxicity values (Eqn 2), which are inherently calculated to represent safe levels of exposure associated with cancer and non‐cancer effects. For the cancer assessment, the MOS was evaluated by comparing the risk specific dose (RSD) associated with 10−6, 10−5 and 10−4 risk levels (i.e. acceptable risk levels in a regulatory framework) to the estimates of potential exposure, as represented by the LADD. For the non‐cancer assessment, MOS was evaluated by comparing the RfD to the estimates of potential exposure, as represented by the ADD.
| (2) |
Results
Toxicity data selection‐
Approximately 20 studies [were thoroughly reviewed and considered for use as critical studies in the development of toxicity values for TBBPA (Szymanska, 1995; Sato et al., 1996; Szymanska et al., 2000; Schroeder, 2002a, 2002b, 2003; Fukuda et al., 2004; Germer et al., 2006; Tada et al., 2006, 2007; Verwer et al., 2007; Lilienthal et al., 2008; van der Ven et al., 2008; Imai et al., 2009; Kang et al., 2009; Saegusa et al., 2009, 2012; Decherf et al., 2010; NTP, 2013)]. These represented studies from the peer review literature, unpublished guideline studies and data from the recent NTP 2‐year bioassay. Five of these studies were guideline studies or otherwise equivalent to a Klimisch quality and reliability score of K1 (Klimisch et al., 1997; Schroeder 2002a, 2002b, 2003; Verwer et al., 2007; Van der Ven et al., 2008; NTP, 2013). The remaining studies included in the database were assigned a Klimisch score of K2 (see Supplemental Table 1 provided as supporting information for scoring rationale). Several studies in the literature were not included for consideration as the study design and/or reporting did not meet minimum criteria. For example, Zatecka et al. (2013) was initially reviewed, but was not selected for inclusion in the database owing to significant limitations in study design (e.g. single dose, uncertainty in dose estimation, non‐traditional exposure paradigm, etc.).
The peer‐review and unpublished studies reviewed represented various routes of administration (i.e. oral gavage, diet, water and intraperitoneal), a wide range in durations of exposure (e.g. short exposure during a specific developmental window, 2 years etc.), and a diversity of endpoints. Exposure to TBBPA in these laboratory studies resulted in reports of neurotoxicity (primarily developmental neurotoxicity), reproductive and developmental toxicity, renal toxicity, hepatic toxicity, endocrine disruption and carcinogenicity (note: conflicting findings were observed for several of these effects across studies).
After consideration of all of the available data, it was determined that the recent NTP Toxicological Review of TBBPA was of the highest quality and relevance for the characterization of toxicity and development of chronic toxicity values for cancer and non‐cancer endpoints due to the robustness of the study design and duration of exposure. In the 2‐year study (the only such study conducted to date), rats and mice of both sexes were exposed via oral gavage to 0, 250, 500 and 1000 mg kg−1 day−1. Endpoints assessed included body weight, survival, general clinical observations, neoplastic lesions and non‐neoplastic lesions. NTP also conducted a 13‐week study that evaluated many of the same endpoints as the 2‐year bioassay and also included an evaluation of thyroid hormones as part of a clinical chemistry panel. Thus, endpoints carried forward for further evaluation as candidate endpoints in the development of cancer and non‐cancer PODs and toxicity factors included all lesions associated with a statistically significant, positive dose response relationship in the NTP studies. As further discussed below, non‐neoplastic effects included forestomach lesions in male and female mice, renal tubule and liver lesions in male mice, and uterine hyperplasia and rete ovarian cysts in female rats, as well as decreases in T4 in male and female rats. Neoplastic lesions further evaluated included liver tumors in male mice and uterine tumors in female rats.
Developmental and reproductive toxicity data reported by Fukuda et al. (2004), Tada et al. (2006) and Schroeder (2002b, 2003) were also carefully reviewed with respect to selection of a critical endpoint to characterize non‐cancer toxicity of TBBPA, as such adversities could indicate the potential for a sensitive window of exposure. Fukuda et al. (2004) reported polycystic lesions associated with dilation of the renal tubules in newborn rats after exposure to high doses of TBBPA via gavage from postnatal day (PND) 4–21; although in a further investigation of the renal tubule dilation by the study authors, 5‐week old rats exposed to 0, 2000 or 6000 mg kg−1 day−1 for 18 days exhibited no histopathological alterations in the kidney. Tada et al. (2006) reported renal and hepatic toxicity in murine offspring after pre‐ and postnatal maternal exposures to TBBPA in the diet (GD0 – PND 21, estimated doses ranging from 16 to 4156 mg kg−1 day−1); although no treatment‐related effects were observed for reproductive endpoints. When these findings were considered along with those from a guideline two‐generation study in rats (Schroeder 2002b, 2003), the data collectively indicate that developmental exposures to TBBPA do not result in functional adversities. In the two‐generation study, no histopathological effects were observed in the kidneys of the adult F0 and F1 animals, and no treatment‐related effects were observed in F1 or F2 pups (i.e. body weight, clinical findings, sex ratios, survival to weaning, macroscopic findings or organ weight data). As such, the developmental toxicity data reported by Fukuda et al. (2004) and Tada et al. (2006) were not carried forward as critical datasets for consideration in the development of a non‐cancer toxicity factor.
Cancer‐based points of departure and toxicity value
In the NTP (2013) 2‐year bioassay (the only such study available), TBBPA was associated with an increased incidence of uterine tumors in Wistar Han rats and an increase in the incidence of hepatoblastoma in male B6C3F1/N mice. The NTP study authors characterized the level of evidence for these two tumor types as ‘clear evidence’ and ‘some evidence’, respectively. Upon detailed review of the hepatoblastoma data reported in male mice, it was observed that the overall dose–response was weak, as evidenced by the marginal significance of the trend test (P = 0.07). In addition, the NTP report indicated that the hepatoblastomas ’were often found adjacent to, or arising from, hepatocellular adenomas and carcinomas‘ (NTP, 2013). This is notable considering that the numbers of male mice with hepatocellular adenomas or carcinomas did not differ between treated and control animals (Table 2). Importantly, the NTP study authors also noted that hepatocellular adenoma, hepatocellular carcinoma and hepatoblastoma are ’considered to represent a biological and morphological continuum’ (NTP, 2013). In fact, a review article co‐authored by several NTP authors (Turusov et al., 2002) stated: ’Because hepatoblastomas frequently appear to arise within hepatocellular adenomas and hepatocellular carcinomas, it is reasonable to combine the incidence of mice with hepatoblastomas with the incidence of mice with hepatocellular adenomas and hepatocellular carcinomas in an overall evaluation for hazard identification studies’.
Table 2.
Summary of liver tumors observed in male mice (NTP, 2013)
| Liver Tumor Type | 0 mg/kg | 250 mg/kg | 500 mg/kg |
|---|---|---|---|
| Hepatocellular adenoma or carcinoma | 39/50 | 39/50 | 43/50 (P=0.2)a |
| Hepatoblastoma | 2/50 | 11/50 (P=0.007) | 8/50 (P=0.05) |
| Hepatocellular adenoma, hepatocellular carcinoma, or hepatoblastoma | 39/50 | 42/50 (P=0.22) | 43/50 (0.15) |
P‐values for one‐sided Fisher's Exact Test.
Other sources also support combining hepatoblastomas, hepatocellular adenomas and hepatocellular carcinomas (e.g. Brix et al., 2010). We, therefore, examined the individual animal data in the NTP (2013) report to score the incidence of the three aforementioned tumor types – treating each tumor type as if it were a single type (thus not double counting). The resulting incidences of the combined tumors were 39/50, 42/50 and 43/50, respectively, in the 0, 250 and 500 mg kg−1 groups (note: the NTP did not consider findings from the highest dose group owing to a significant decrease in survival) (Table 2). These findings indicate a lack of treatment‐related effect, as further supported by the lack of statistical significant when evaluated relative to controls (Table 2). As such, liver tumors were not further considered as a critical endpoint in the derivation of a cancer‐based toxicity value.
Uterine tumors were assessed by the NTP using two pathology review processes; data from both review processes combined were used for dose–response modeling as they provide the most comprehensive and thorough evaluation of the neoplastic lesions in the uterus. The combined incidence of uterine adenomas, adenocarcinomas and malignant mixed Müllerian tumors is provided in Table 3, and the results of the dose–response modeling are shown in Fig. 1A. The multistage model provided the best overall fit to these data (i.e. lowest AIC; P‐value = 0.75). The BMD10 and BMDL10 values were 195.3 and 126.6 mg kg−1 day−1, respectively. A HED of 31.7 mg kg−1 day−1 was obtained by allometric scaling of the BMDL10. The resulting human oral cancer slope factor (OSF) was determined to be 0.00315 per mg kg−1 day−1 (i.e. 0.1/31.7). This OSF is associated with a risk‐specific dose (RSD) of 0.00032 mg kg−1 day−1 at the 10−6 risk level (traditionally acceptable risk range is 10−4 to 10−6). Accordingly, the RSDs at the 10−5 and 10−4 risk levels are 0.0032 and 0.032 mg kg−1 day−1, respectively.
Table 3.
Incidence of combined uterine adenomas, adenocarcinomas, and malignant mixed Müllerian tumors observed in female rats (NTP, 2013)
| Study Dose (mg/kg/day) | Duration Adjusted Dose | N | Combined Uterine Tumors | P‐valuea |
|---|---|---|---|---|
| 0 | 0 | 50 | 6 | ‐‐ |
| 250 | 178.6 | 50 | 11 | 0.168 |
| 500 | 357.1 | 50 | 16 | 0.007 |
| 1000 | 714.3 | 50 | 19 | 0.002 |
Poly‐3 test.
Figure 1.
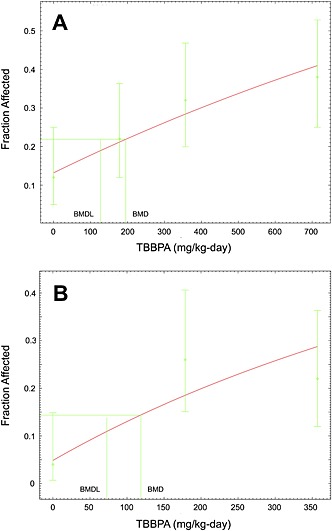
Benchmark dose modeling results of uterine effects in female rats. (A) Incidence of combined uterine tumors from the National Toxicology Program (NTP, 2013). (B) Incidence of uterine hyperplasia (NTP, 2013). Note: the highest dose group was dropped for modeling uterine hyperplasia in order to improve model fit.
Non‐cancer‐based points of departure and toxicity values
In the NTP 2‐year bioassay on TBBPA, body weight was decreased in Wistar Han male rats (500 and 1000 mg kg−1 day−1 dose groups) and in female mice (1000 mg kg−1 day−1 dose group). No significant non‐neoplastic effects were observed in male rats. In female rats, uterine endometrial atypical hyperplasia and ovarian rete cysts were found to be associated with exposure to TBBPA. In mice, non‐neoplastic effects were observed in the forestomach of both males and females, and included ulcers, mononuclear cell cellular infiltration, inflammation, and epithelial hyperplasia. Additionally, in male mice, there was evidence of an increased incidence of renal tubule cytoplasmic alterations, hepatic clear cell foci and hepatic eosinophilic foci. Findings in the 13‐week study were generally unremarkable; however, decreased total thyroxine (T4) was observed in the 500 and 1000 mg kg−1 treatment groups (and to lesser extent in the 100 mg kg−1 group) of male and female F344 rats. Levels of T4 were not assessed in the chronic NTP bioassay. It should be noted that NTP used F344 rats in the 13‐week subchronic study and Wistar Han rats in the 2‐year chronic bioassay.
Thus, non‐neoplastic effects considered from the NTP studies included forestomach lesions, uterine hyperplasia, rete ovarian cysts, renal tubule cytoplasmic alterations, hepatic foci and decreased T4. Prior to characterizing dose–response relationships and establishing a non‐cancer POD and corresponding toxicity value, it was important to first determine that each of these candidate endpoints was in fact adverse, relevant to humans, and biologically associated with a non‐cancer effect.
Endpoints determined to be unsuitable for characterization of human non‐cancer effects
Liver lesions were only observed in male mice, and included clear cell foci and eosinophilic foci. Notably, clear cell foci in the liver have been considered by EPA to be a pre‐neoplastic lesion when it is observed in animals that also develop liver tumors (USEPA, 2013; 1‐4‐dioxane). As described above, male mice developed liver tumors in the TBBPA bioassay and thus it was determined that clear cell foci should be considered a pre‐neoplastic lesion. With respect to eosinophilic foci, the incidence rate was high in the male control group (40%), similar to that observed for the incidence of liver tumors discussed previously. Eosinophilic foci are among a group of cellular alterations (including clear cell foci) in the liver that are often considered to be clonal expansions of initiated cells (Greaves, 2012). Considering that clear cell foci and eosinophilic foci only occurred in male mice, and that male mice were the only animals to develop liver tumors in the NTP (2013) bioassay, these endpoints were both considered preneoplastic and thus not appropriate for non‐cancer assessment (USEPA 2013).
The incidence of renal tubule cytoplasmic alterations in male mice increased with dose of TBBPA; NTP characterized this effect as a ’reduction or loss of normal vacuoles in the cortical proximal tubules in male mice‘ (NTP, 2013). NTP further characterized this lesion and the associated lysosomal–vacuolar system in mice as being sexually dimorphic. It has been shown that orchiectomized male mice exhibit a reduced vacuolization pattern in the proximal tube that is more consistent with female mice, and that administration of testosterone to female mice results in an expanded vacuolization pattern more consistent with male mice (Koenig et al., 1980). It was also shown that male mice, as well as female mice, treated with testosterone, have increased expression of lysosomal proteins and increased protein levels in urine (Koenig et al., 1980). In the NTP study, male mice exposed to TBBPA exhibited a significant decrease in renal tube vacuolization as well as a significant decrease in kidney nephropathy (i.e. kidney damage). Thus, the changes in renal tubular vacuolization appear to be a trait specific to male mice, and the reduced vacuolization was associated with reduced nephropathy (i.e. reduced adverse effects). As a result, the cytoplasmic alteration was both not relevant to humans and not adverse, and thus was not considered as a suitable endpoint for non‐cancer assessment.
In the 13‐week study conducted by the NTP, a dose‐dependent decrease in total serum T4 was observed in male and female F344 rats, with no significant changes in serum T3, TSH, thyroid weight or thyroid histopathology. Also, no changes were observed in the thyroid gland after administration of TBBPA to either Wistar–Han rats or B6C3F1 mice for 2 years (NTP, 2013). Based on the lack of consistent and concordant changes in T4, T3 and TSH serum levels, as well as lack of adverse effects associated with this decreased T4 reported both in the NTP study as well as in the literature (EU, 2006; Schroeder 2002a, 2002b) the toxicological significance of this endpoint is uncertain. As such, this endpoint was not considered to be adverse, and thus was not further considered as a critical effect for non‐cancer assessment. Notably, both Health Canada and the European Union also concluded that reductions in T4 were not considered adverse in the absence of any other relevant thyroid‐related effects (EU, 2006; Health Canada, 2013).
Dose–response assessment of relevant non‐cancer endpoints
In rats, exposure to TBBPA was associated with uterine hyperplasia and ovarian cysts (NTP, 2013). Notably, NTP characterized the uterine hyperplasia as a potential preneoplastic lesion. If indeed this lesion is pre‐neoplastic, it would not be suitable as a non‐cancer endpoint for the same reasons described above for clear cell and eosinophilic foci in the male mouse liver (USEPA, 2013). However, given the uncertainty as to whether this uterine hyperplasia is indeed a pre‐neoplastic lesion, we considered this endpoint for non‐cancer assessment. Modeling the incidence of uterine hyperplasia initially resulted in poor model fits. As such, the highest dose was omitted (consistent with US EPA guidance), resulting in more reasonable model fits. The P‐value for the model fit was 0.08, which is only slightly below EPA's recommendation that P‐values be ≥ 0.1 (USEPA, 2012). Notably, however, EPA does accept P‐values of ≥ 0.05 when modeling cancer data, and thus it was determined that the P‐value of 0.08 was sufficient for modeling uterine hyperplasia. Visual inspection of the model indicates a reasonable fit of the data (Fig. 1B); in addition, the scaled residual nearest the BMD meets EPA's recommendation of being ≤/2/ (USEPA, 2012). It is also worth noting the BMDL10 value of 72.8 mg kg−1 day−1 (Table 4) is lower, and thus more health‐protective, than the corresponding LOAEL value of 178.6 8 mg kg−1 day−1. Moreover, the BMD10/BMDL10 ratio was 1.6, which is below the ratio of 5 that is considered high and indicative of increased uncertainty in the BMD10 and BMDL10 estimates (Wignall et al., 2014).
Table 4.
Non‐cancer points of departure (POD) and Reference dose (RfD) Array
| Species/Sex | Endpoint | BMD10 mg/kg‐day | POD (BMDL10) mg/kg‐day | HED mg/kg‐day | UFa Unitless | RfD mg/kg‐day |
|---|---|---|---|---|---|---|
| Mice | Forestomach | |||||
| Female | Hyperplasia | 88.7 | 70.5 | 11.6 | 30 | 0.4 |
| Ulcer | 102.2 | Not Calculated | ||||
| Infiltration | 106.3 | Not Calculated | ||||
| Inflammation | 105.5 | Not Calculated | ||||
| Mice Male | Hyperplasia | 103 | Not Calculated | |||
| Ulcer | 190.5 | Not Calculated | ||||
| Infiltration | 175.6 | Not Calculated | ||||
| Inflammation | 218.1 | Not Calculated | ||||
| Rat, Female | Uterine endometrial atypical hyperplasia | 118.7 | 72.8 | 18.2 | 30 | 0.6 |
| Rat, Female | Rete ovarian cysts | 596.7 | 355.4 | 88.9 | 30 | 3.0 |
UFA=3; UFH=10.
The incidence of ovarian rete cysts were 1/50, 0/49, 6/50 and 6/49. When modeled, this endpoint led to the highest BMDL10 value, viz. 355 8 mg kg−1 day−1 (Table 4).
Both male and female mice administered TBBPA by oral gavage developed ulceration, infiltration, inflammation and hyperplasia of the forestomach (NTP, 2013). BMDL10 values for these eight endpoints (four lesions in each sex) ranged from 88.7 to 218.1 8 mg kg−1 day−1 (Table 4). From these, the lowest BMDL10 value (88.7 8 mg kg−1 day−1; forestomach hyperplasia in female mice) was selected for derivation of a candidate RfD. Although forestomach hyperplasia is likely a downstream event to ulceration and inflammation, the PODs were essentially the same for all forestomach lesions (varying only ~2‐fold; Table 4). It is also likely the case that the dose spacing and histopathological evaluations do not allow for resolution about which event(s) came first. As such, we selected the most conservative endpoint for the forestomach, and did not attempt to parse out which lesion preceded the others.
Consistent with typical US EPA risk assessment practices (USEPA, 2002), candidate POD and RfD values were derived for the critical effects associated with TBBPA exposure (Table 4). As these endpoints were observed in the 2‐year bioassay, the HED values were divided by three‐fold to account for potential interspecies differences in pharmacodynamics (UFA) and 10‐fold to account for potential intraspecies variability (UFH). A database uncertainty factor (UFD) of 1 was selected owing to availability of chronic oral exposure studies in both rats and mice, as well as the availability of a 2‐generation reproductive and developmental toxicity study (including developmental neurotoxicity) for TBBPA that found no evidence of adverse effect in the F0, F1 or F2 generations (Schroeder 2002b, 2003). An uncertainty factors for extrapolation from a lowest observed adverse effect level (UFL) were not needed as the BMD approach was utilized; similarly, an uncertainty factor for extrapolation from subchronic to chronic (UFS) was not needed as the data were obtained from a chronic bioassay.
The lowest candidate RfD was one of the three candidate RfD values derived from the forestomach lesions; however, selection of this value is associated with uncertainty given its questionable relevance to humans. Unlike humans, rodents have both a stomach and a forestomach; the forestomach serves as a storage compartment that releases minimally digested food into the glandular stomach in response to energy demands (Greaves, 2012). In the 2‐year study, there was increased mortality in the 1000 mg kg−1 dose group owing to ’gastrointestinal toxicity’, yet no indication of gastrointestinal (including forestomach) cancer was observed (NTP, 2013), indicating that these lesions posed no carcinogenic risk. It seems reasonable, therefore, that the lesions could have been induced by the high concentrations of corn oil‐solubilized TBBPA stored in the forestomach. It is also notable that both humans and rodents have a glandular stomach, and no TBBPA‐induced lesions were reported in the glandular stomachs of mice or rats in the 2‐year bioassay (NTP, 2013).
In contrast to the biological uncertainty associated with the use of forestomach lesions for a human critical effect, the non‐cancer toxicity of TBBPA to the rat uterus indicates that this may be the most sensitive target organ. There was a weak dose–response pattern for uterine endometrial atypical hyperplasia (2/50, 13/50, 11/50 and 13/50) although the incidence of uterine hyperplasia was consistently elevated. Because of such, this endpoint posed some challenges for dose–response modeling (see above); however, a reasonable fit to the data was achieved (Fig. 1B). The resulting POD and RfD were in the middle of the arrayed values (Table 4). Although there remains some uncertainty as to whether uterine hyperplasia represents a non‐neoplastic or pre‐neoplastic lesion, as well as uncertainty regarding the relevance of such effects in humans given the large disparity in the doses administered in the study compared to human exposures, the lesion is associated with a higher level of confidence with respect to characterization of non‐cancer effects in humans as compared with the forestomach lesions (though both endpoints result in similar toxicity values). Thus, it is proposed that uterine hyperplasia serve as the basis for an oral RfD for TBBPA, and accordingly, the proposed RfD for oral exposure to TBBPA is 0.6 mg kg−1 day−1.
Exposure
Media concentrations
There is a wealth of data on concentrations of TBBPA in food/diet, breast milk, water and soil/dust. These data were collected and analyzed by research groups from across the globe and represent different methods of collection, analyzes, and interpretation, and often, were not directly relevant to consumer exposures. As such, careful consideration was given to the studies and datasets used in the estimation of intake. The selection process also considered study quality and relevance, representativeness of chronic consumer exposure, as well as consistency of the data relative to other studies. No preference was given to the country of location where samples were obtained; however, the location and type of samples collected were considered relative to the media type and representativeness of consumer exposure. Additionally, because TBBPA was often below the analytical limits of detection, the use of non‐detect data in the analysis and interpretation of such were also carefully considered when selecting representative datasets.
Drinking water
No studies were identified that reported direct measurements of TBBPA in actual drinking water. Information was limited to a single abstract that suggested TBBPA formation via bromination of BPA in drinking water if the water was stored in polycarbonate containers and sanitized with bromine and ozone, although such a scenario was considered negligible as water supplies are not routinely brominated (Peterman et al., 2000). As such, environmental water samples were used as a surrogate, which is an extremely conservative approach, particularly considering that even in environmental sampling, TBBPA is not measured at concentrations above detection limits. Four key studies were selected for potential consideration, representing water samples collected in France, China and the UK (Harrad et al., 2009; Labadie et al., 2010; Yang et al., 2012; He et al., 2013). While Yang et al. (2012) collected samples from a main watershed lake over a course of three sampling periods, only maximum water concentrations were provided in the manuscript (other data provided graphically in figures, but levels could not be accurately distinguished). Data from Labadie et al. (2010) and He et al. (2013) were not utilized as both studies measured river water samples in locations associated with suspected or known sources of BFRs, and thus were not considered to be representative of typical consumer exposures in drinking water. Data collected by Harrad et al. (2009) as part of an environmental monitoring program in the UK were determined to be the most representative of the available data. These data were collected from nine freshwater lakes, each with three sampling events, and an average concentration by lake provided in the manuscript. The average concentrations of TBBPA by lake were utilized in the intake equations; the maximum average concentration reported was used for the regulatory default scenario, whereas mean and 95th percentile values were derived assuming a normal distribution across the average of the nine lakes and use in the central tendency and upper‐bound scenarios, respectively (Table 5).
Table 5.
Media concentrations used in the exposure assessment calculations
| Media | Central Tendency | Upper‐Bound | Regulatory Default | Units | Ref |
|---|---|---|---|---|---|
| CMilkfat | 0.0001 | 0.00128 | 0.01246 | mg/kg | Shi et al., 2013a |
| CSoil/Dust | 0.11 | 0.46 | 1.4 | mg/kg | Harrad et al. (2010)b |
| CDW | 0.00000096 | 0.000001008 | 0.000003200 | mg/L | Harrad et al. (2008)c |
| Total Dietary Intake | 0.000000256 | 0.00000028 | 0.00000028 | mg/kg‐d | Shi et al. (2009)d |
Median, 95th percentile, and maximum concentrations, respectively; concentrations are lipid adjusted; % lipid accounted for in the intake calculations.
Median, 95th percentile, and maximum concentrations, respectively.
Maximum concentration reported used for the regulatory default scenario; mean and 95th percentile values were derived assuming a normal distribution across the average of the nine lakes and use in the central tendency and upper‐bound scenarios, respectively.
Medium bound intake used for central tendency, upper‐bound intake used for upper‐bound and regulatory default.
Breast milk
There were several studies available in the published literature that reported concentrations of TBBPA in breast milk. It is notable that across these studies, a large percentage of samples evaluated reported that TBBPA was not present at a concentration above the detection limit. Studies for potential inclusion were narrowed based on year of collection (recent data preferred), robustness of data, and quality of data evaluation and reporting. Three studies, representing samples collected from Chinese, German and French women, were selected as key studies (Kemmlein 2000 as cited by EU 2006; Cariou et al., 2008; Shi et al., 2013). The most recent data published by Shi et al. (2013) were selected for use in the intake equations (note: these data were not available at the time Health Canada, EFSA, and the EU conducted their analysis). These data were collected from Chinese women in 2011 as part of a well‐designed exposure study. Data reporting included concentrations of TBBPA in breast milk by percentile, and also included incorporation of non‐detect samples in the derivation of such (TBBPA was detected in only 55% of the samples analyzed). The median, 95th percentile and maximum concentrations were utilized in the intake equations (Table 5).
The data published by Cariou et al. (2008) characterizing breast milk concentrations in samples collected in French women between 2004 and 2006 were carefully reviewed as the maximum concentration reported in this study was utilized by both Health Canada (2013) and EFSA (2011) in their health assessments for TBBPA. However, these data were part of a very short publication that appeared to be associated with an extended meeting abstract, and thus it is not clear if the publication was subject to a traditional peer review. This was highlighted by major shortcomings in data reporting, and potentially data analysis, such as the mean, median, minimum and maximum concentrations presented in the paper did not account for non‐detect samples. This finding is critical as TBBPA was not detected in 43 of the 77 samples, and thus the concentrations reported by the study authors did not accurately reflect the concentrations measured in French women. The German data (Kemmlein 2000 as cited by EU 2006) were initially selected as key data despite the unavailability of an English translation of the study because these data were utilized by the EU in their assessment (EU, 2006) of TBBPA; however, further review of these data indicate that it is limited to a single sample collected in the Faroe Islands over a decade ago, and thus was not selected for use in the intake assessment.
Soil/Dust
There were many datasets characterizing TBBPA concentrations in soil and dust available in the published literature. A key criterion used to identify relevant datasets was relevance of the sample to typical exposure, with consideration for conservative (higher) concentrations for some of the exposure scenarios. For example, soil/sediment samples collected near a chemical manufacturing plant or recycling plant were not considered relevant, nor were dust samples collected from inside a television set considered relevant.
When the literature was surveyed, the concentrations in dust were generally higher than soil, and thus studies reporting dust concentrations were further reviewed and two papers were ultimately selected for potential use based on the levels reported (i.e. highest concentrations of TBBPA in dust). A recent paper by Ni and Zeng (2013) reported data for 56 samples collected from air conditioning filters in Chinese office buildings. However, the maximum TBBPA concentration was the highest concentration reported in the literature (by several orders of magnitude), and was also very high relative to the other samples collected in the same study based on comparison to the mean and standard deviation concentrations provided (and thus is not considered to be generally representative). Given the inconsistency in this single data point relative to other data, as well as the lack of representativeness of the sample collection technique (e.g. represented accumulated levels that were not subject to standard fate and transport properties), these data were not selected for use in the exposure estimates. Rather, data collected in schools and daycares in the UK as published by Harrad et al. (2010) were selected. This dataset was utilized by Health Canada (2013) in their assessment of TBBPA, and was supported by previous investigations of TBBPA in dust in homes, offices and cars by the same authors (Abdallah et al., 2008; Harrad et al., 2009). Notably, the concentrations in dust from schools were higher than levels reported in cars and offices (Harrad et al., 2010) and were ultimately selected as the dust concentrations for use in the exposure assessment. Further, this study was judged to be of good quality and relevance based on the use of a well‐described sampling procedure and relatively robust analytical techniques. The median, 95th percentile and maximum concentrations were utilized in the intake equations (Table 5).
Diet
Two approaches were considered for characterizing dietary intake to TBBPA based on the data available. The first option was to calculate intake for individual food types that had measured concentrations and the second option was to utilize dietary intake estimates from total diet studies. The latter option was determined to be more robust and appropriate for use in the current study given that the data were already in the form of a total daily intake, and that the estimates were generated based on consideration of data from total diet studies (or similar). And while these data are often specific to a particular population or region, they were judged to be of greater quality and relevance as compared with the option of calculating intake only for specific food types, which would be associated with a high level of variability and uncertainty owing to the range and/or lack of media concentrations, consumption rates, and inability to capture all food types.
Six total diet studies or comprehensive evaluations of dietary intake were identified in the published literature (de Winter‐Sorkina et al., 2003; EU, 2006; Driffield et al., 2008; Shi et al., 2009; Food Safety Authority of Ireland, 2010; EFSA, 2011). However, dietary intake in the majority of these studies was based on estimates of TBBPA because TBBPA was consistently not measured at levels above the detection limit, and thus the regulatory/health agencies instead conservatively assumed that TBBPA was present at a concentration equal to the detection limit when developing dietary exposure estimates (de Winter‐Sorkina et al., 2003; Driffield et al., 2008; Food Safety Authority of Ireland, 2010; EFSA, 2011). The study by Shi et al. (2009) was ultimately selected as the basis for the dietary intake estimates used in this current assessment as TBBPA was detected in approximately 70% of the whole samples evaluated (Table 5). Shi et al. (2009) evaluated TBBPA in four food groups of animal origin (eggs and egg products, aquatic foods, milk and milk products, meat and meat products) and then utilized the data to develop lower, medium and upper‐bound intakes using different proxy values for the non‐detect samples. Notably, the Shi et al. (2009) study was also utilized by Health Canada in their exposure assessment.
Daily intake estimates
Lifetime average daily dose (LADD) estimates are provided in Table 6. For the scenarios evaluated, LADD estimates ranged from 2.2 E−7 to 3.9 E−6 mg kg−1 day−1 for the three different scenarios considered in this assessment (central tendency, upper‐bound and regulatory default). Exposure to TBBPA via soil/dust ingestion was the largest contributor, followed by dietary intake (includes both exposure via breast milk and foodstuffs), and to a lesser extent, exposure via drinking water.
Table 6.
Lifetime average daily dose (LADD) and cancer‐based margin of exposure (MOE) and margin of safety (MOS)
| Route of Exposure | Intake (mg/kg‐day) | ||
|---|---|---|---|
| Central Tendency | Upper‐Bound | Regulatory Default | |
| Total Dietary Intakea | 1.6E‐07 | 2.5E‐07 | 1.1E‐06 |
| Drinking Water | 5.0E‐09 | 1.6E‐08 | 5.4E‐08 |
| Soil/Dust | 5.4E‐08 | 3.7E‐07 | 2.7E‐06 |
| Total Dose | 2.2E‐07 | 6.4E‐07 | 3.9E‐06 |
| MOE b | 5.8E+08 | 2.0E+08 | 3.3E+07 |
| MOS c | 1.5E+03 | 5.0E+02 | 8.3E+01 |
Includes breast milk and food consumption as appropriate to the receptor.
Calculated using a POD of 126.6 mg/kg‐day.
Calculated using a RSD of 0.00032 mg/kg‐day (10−6 risk level).
Average daily dose estimates (ADD) varied by scenario and receptor (Table 7). The lowest estimates of ADD were calculated for adults in the Central Tendency scenario (3.2 E −7 mg kg−1 day−1), and the highest estimates calculated for infants aged 0 to 3 months in the Regulatory Default Scenario (8.4 E−5 mg kg−1 day−1). In infants, the soil/dust pathway was the exposure route that contributed the most to the overall ADD for the Central Tendency scenarios, whereas the percent contribution of exposure via breast milk was significantly greater in the Upper Bound and Regulatory Default scenarios, 68% and 87%, respectively. Although currently available data indicates that ingestion TBBPA in foodstuffs in not an exposure pathway of concern (based on data demonstrating that TBBPA has only been detected at very low in fruits, vegetables, and grain products commonly consumed by infants), the authors recognize that the lack of data available to characterize TBBPA concentrations in all potentially relevant foodstuffs consumed by infants aged 1 year or less is an uncertainty in this analysis,. Additional analyzes may be warranted when data appropriate for characterizing exposure to TBBPA in all potentially relevant foodstuffs become available. Further, intake estimates for infants did not include drinking water; however, exposures via this route are not anticipated to be significant (based on comparisons to intake of such in adults). In young children, older children and adults, the exposure estimates were driven by dietary intake in the Central Tendency scenario, but by soil/dust exposures in the Upper Bound and Regulatory Default scenarios.
Table 7.
Average daily dose (ADD) and non‐cancer based margin of error (MOE) and margin of safety (MOS)
| Scenario/Route | Average Daily Dose (mg/kg‐day) | |||||
|---|---|---|---|---|---|---|
| 0–3 mos | 4‐12 mos | 0‐12 mos (weighted average) | Young Child | Older Child | Adult | |
| Central Tendency Scenario | ||||||
| Total Dietary Intake | 6.9E‐07 | 6.5E‐07 | 6.6E‐07 | 9.7E‐07 | 3.6E‐07 | 2.3E‐07 |
| Drinking Water | ‐‐ | ‐‐ | ‐‐ | 1.8E‐08 | 1.0E‐08 | 1.4E‐08 |
| Soil/Dust | ‐‐ | 1.6E‐06 | 1.6E‐06 | 2.2E‐07 | 8.1E‐08 | 7.9E‐08 |
| Total ADD | 6.9E‐07 | 2.3E‐06 | 2.3E‐06 | 1.2E‐06 | 4.6E‐07 | 3.2E‐07 |
| MOE a | 1.1E+08 | 3.2E+07 | 3.2E+07 | 6.0E+07 | 1.6E+08 | 2.3E+08 |
| MOS b | 8.7E+05 | 2.7E+05 | 2.6E+05 | 5.0E+05 | 1.3E+06 | 1.9E+06 |
| Upper Bound Scenario | ||||||
| Total Dietary Intake | 8.6E‐06 | 5.8E‐06 | 6.5E‐06 | 1.1E‐06 | 4.0E‐07 | 2.5E‐07 |
| Drinking Water | ‐‐ | ‐‐ | ‐‐ | 5.6E‐08 | 3.6E‐08 | 4.3E‐08 |
| Soil/Dust | ‐‐ | 3.1E‐06 | 3.1E‐06 | 2.6E‐06 | 9.6E‐07 | 3.3E‐07 |
| Total ADD | 8.6E‐06 | 8.9E‐06 | 9.6E‐06 | 3.7E‐06 | 1.4E‐06 | 6.2E‐07 |
| MOE a | 8.5E+06 | 8.2E+06 | 7.6E+06 | 2.0E+07 | 5.2E+07 | 1.2E+08 |
| MOS b | 7.0E+04 | 6.8E+04 | 6.3E+04 | 1.6E+05 | 4.3E+05 | 9.6E+05 |
| Regulatory Default Scenario | ||||||
| Total Dietary Intake | 8.4E‐05 | 5.7E‐05 | 6.3E‐05 | 1.2E‐06 | 3.9E‐07 | 2.5E‐07 |
| Drinking Water | ‐‐ | ‐‐ | ‐‐ | 2.1E‐07 | 1.4E‐07 | 9.1E‐08 |
| Soil/Dust | ‐‐ | 9.3E‐06 | 9.3E‐06 | 1.9E‐05 | 6.2E‐06 | 2.0E‐06 |
| Total ADD | 8.4E‐05 | 6.6E‐05 | 7.3E‐05 | 2.0E‐05 | 6.8E‐06 | 2.3E‐06 |
| MOE a | 8.7E+05 | 1.1E+06 | 1.0E+06 | 3.6E+06 | 1.1E+07 | 3.1E+07 |
| MOS b | 7.2 E+03 | 9.1E+03 | 8.2E+03 | 3.0E+04 | 8.9E+04 | 2.6E+05 |
Calculated using a POD of 72.8 mg/kg‐day.
Calculated using a RfD of 0.6 mg/kg‐day.
Margin of exposure and margin of safety estimates
For the cancer‐based MOE, the POD used in the development of the OSF was the BMDL of 126.6 mg kg−1 day−1. This BMDL was compared with the LADD estimates, resulting in margins of exposure greater than 32 000 000 for each scenario evaluated (Table 6 and Fig. 2). Similarly, in the non‐cancer‐based comparison of exposure and toxicity (POD of 72.8 mg kg−1 day−1), the resulting MOEs were large (>800 000) for each scenario evaluated (Table 7 and Fig. 2).
Figure 2.
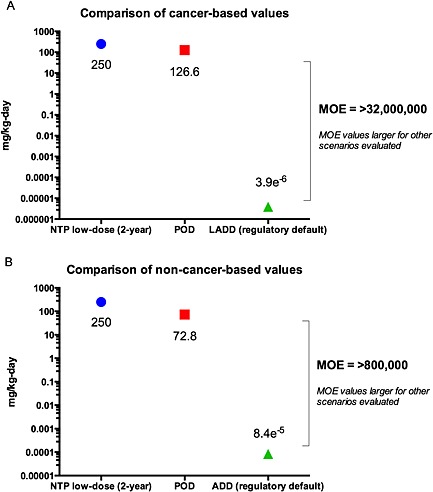
Comparison of cancer (A) and non‐cancer (B) toxicity values to the associated POD and to the lowest dose tested in the National Toxicology Program (NTP) 2‐year study. Note: exposure estimates shown are based on most conservative exposure scenario (regulatory default).
Margin of safety (MOS) estimates were also sufficiently large. Using the most conservative RSD of 0.00032 mg kg−1 day−1 (i.e. dose at the 10−6 risk level) associated with the OSF, the resulting cancer‐based MOS estimate for the regulatory default scenario (i.e. reasonable worst case) was ~80. Thus, the total lifetime average daily exposure would have to be increased ~80 times or greater to reach a risk level of 10−6 for the lowest – highly conservative – MOS identified in this study. Notably, a margin of safety > 1500 was derived for the most plausible exposure scenario. Non‐cancer‐based MOS estimates ranged from >7000 to > 1 000 000.
Discussion
Robust data of both high quality and relevance were available to characterize both cancer and non‐cancer endpoints associated with chronic, oral exposures to TBBPA, as well as to characterize reproductive and developmental endpoints. Supporting data were available to characterize a diversity of endpoints, including: body/organ weight, organ histopathology, reproductive/developmental toxicity, neurotoxicity, nephrotoxicity, hepatotoxicity, cardiotoxicity, endocrine disruption, carcinogenicity, and hematology and serum biochemistry. The GLP, guideline‐based NTP 2‐year and 13‐week studies were determined the highest quality and relevance for the characterization of toxicity and development of chronic toxicity values for cancer and non‐cancer endpoints owing to the robustness of the study design and duration of exposure. Multiple datasets from the NTP 2‐year studies were selected for dose response modeling. From these datasets, a human OSF of 0.00315 per mg kg−1 day−1 was calculated, based on an increased incidence of uterine tumors in rats, and an oral RfD of 0.6 mg kg−1 day−1 based on uterine hyperplasia in rats was selected from an array of candidate RfD values. The PODs underlying these specific toxicity factors were used to generate MOE estimates for infants, young children, older children and adults. Even when maximum concentrations of TBBPA in the diet, breast milk, soil/dust and water were used (i.e. in the regulatory default scenario, representing the reasonable worst case), resulting exposures were many orders of magnitude below PODs, regardless of receptor (MOE values > 800 000).
Data in the published literature indicate that TBBPA is not genotoxic in either well‐conducted bacterial and yeast mutagenicity assays or in an in vitro chromosomal aberration study in human lymphocytes (EU, 2006; Health Canada, 2013; NTP, 2013). Thus, it is highly unlikely that TBBPA is acting through a genotoxic or mutagenic MOA to elicit the carcinogenic effects observed in the NTP bioassay. Rather, the data suggest that the toxicities observed at high doses may potentially be the result of disruption of endocrine parameters. Although a full evaluation of such was not conducted in this assessment, it is notable that a number of studies have reported associations between exposure to TBBPA and decreased levels of T4 in laboratory animals, including the studies judged to be of high quality and relevance (Schroeder, 2002a, 2002b, 2003; van der Ven et al., 2008; NTP, 2013). Associations between TBBPA and T4 have also been investigated in humans. Recently, Kim and Oh (2014) reported that TBBPA serum concentrations correlated weakly with thyroid hormones in humans based on the observation of a positive relationship for free T4, although a negative relationship was observed for T3. When considered collectively, these data generally indicate that other effects commonly associated with thyroid hormone disruption (e.g. changes in T3, TSH, thyroid weight thyroid histopathology) do not consistently accompany the decreased levels of T4 (Schroeder, 2002a,2002b, 2003; van der Ven et al., 2008; NTP, 2013). Further, decreases in serum T4 levels have not been associated with adverse effects in reproductive and developmental toxicity studies that included neurobehavioral and neuropathology assessments (Schroeder, 2002b, 2003; Williams and Desesso, 2010). Taken together, these data indicate that decreased serum concentrations of T4 appear to have little adverse impact on parameters associated with a disruption in thyroid homeostasis in rat. This conclusion is similar to that reached by the EU (2006) and Health Canada (2013), as well as Colnot et al. (2014).
Aside from perturbations in T4, a number of other endocrine‐related effects have been reported in the literature for TBBPA. For example, binding and activity related to androgenic, and particularly estrogenic, compounds have been reported (though are somewhat contradictory) (Hamers et al., 2006; Kitamura et al., 2010; Li et al., 2010; Gosavi et al., 2013). It is also notable that some of the non‐neoplastic lesions observed in the NTP study were also associated with potential disruption of endocrine parameters. For example, the decreased vacuolization in the male mice renal tubules may be related to interference of testosterone, as inhibiting testosterone reduces vacuolization in male mice and administration of testosterone increases vacuolization in female mice (Koenig et al., 1980). Additionally, the ovarian rete cysts observed in rats have been observed in humans with endocrine dystrophies, although the cysts were not associated with a single hormone abnormality (Sommers, 1953). Thus, although a clear relationship remains to be elucidated (particularly at human relevant exposure doses), there are a number of studies indicating the potential for an association between TBBPA and disruption of endocrine parameters (although these occur primarily at high doses). As such, the selection of an oral RfD that is consistent with this pathway seems to provide the most biological plausibility based on existing data, thus lending support to the selection of the uterine hyperplasia as the basis for establishing a non‐cancer toxicity value. The uterus is clearly responsive to hormonal changes; for example, both age‐related excess of estrogens and xenobiotic‐related estrogenic effects can induce endometrial hyperplasia (Greaves, 2012).
The collective nature of these endocrine‐related effects are also notable when considering a potential underlying mode of action (MOA) associated with the development of uterine tumors, as well as in considering the relevance of such to human exposures. Even the lowest dose used in the NTP study (250 mg kg−1 day−1) is more than five orders of magnitude higher than the highest estimates of exposure generated in this study (Fig. 2), and is also orders of magnitude higher than the intake estimates generated by regulatory bodies (EU, 2006; EFSA, 2011; Health Canada, 2013). Without data characterizing the same endpoints at lower, more environmentally relevant doses, it is often difficult to make such extrapolations; and in particular, it is difficult to differentiate which effects are relevant to human exposure versus which effects may be owing to the impact of high doses on physiological function and saturation of protective mechanisms. It is well accepted that there is a high likelihood that key steps in any mechanistic pathway can become overwhelmed, and as a result new modes of toxicity are associated with effects observed at higher doses (Slikker et al., 2004). Notably, no effects were observed in animals administered 10 or 50 mg kg−1 day−1 in the NTP 13‐week study (NTP, 2013), nor were effects observed in a 90‐day study at doses ranging from 100 to 1000 mg kg−1 day−1 TBBPA [Schroeder, 2002a, EU, 2006], suggesting that the high doses utilized in the 2‐year study may have saturated protective mechanisms or otherwise impacted normal physiology.
The findings presented in this study demonstrate that the non‐cancer‐based MOE was several orders of magnitude regardless of exposure scenario evaluated. This finding is consistent with the conclusions reached by the European Union (2006), the European Food Safety Authority (2011), Health Canada (2013) and Colnot et al. (2014). The EU evaluated multiple exposure scenarios, including a number of occupational and non‐occupational scenarios. For consumers, the EU concluded that their assessment indicated no health effects of potential concern to adults, and given that consumer exposures were negligible, there were no concerns in relation to any toxicological endpoint (EU, 2006). In their evaluation of infants, the EU utilized measured levels of TBBPA in breast milk to estimate exposure (rather than using a model as was done for other scenarios), and compared the time‐weighted average daily uptake in a breast‐feeding infant (0.024 × 10−3 mg kg−1 day−1) to a NOAEL of 40 mg kg−1 day−1, resulting in a margin of safety (MOS) of 1.7 × 106. Similarly, the health assessment reported by Health Canada (2013) was based on the comparison of an upper‐bound intake in breastfed infants relative to a LOAEL of 140 mg kg−1 day−1, resulting in a MOE of 7.2 × 105, thus leading to the conclusion that the margin of exposure was adequate to address uncertainties in available data. Notably, the POD established in the current study for non‐cancer effects was 72.8 mg kg−1 day−1, a value which is within the range of those utilized by the EU (2006) and Health Canada (2013).
The European Food Safety Authority (EFSA) developed a MOE by comparing a BMDL10 of 16 mg kg−1 day−1 (van der Ven et al., 2008) to daily intake estimates for adult fish consumers and infants (EFSA, 2011). It is notable, however, that in developing exposure estimates, EFSA requested data characterizing levels of TBBPA in food, but only received data for a single food group (fish and other seafood). Further, all of the TBBPA concentrations in the data obtained for fish and other seafood samples were non‐detect, a finding that was similarly observed in the current study when evaluating concentrations of TBBPA in foodstuffs. As a result, EFSA developed a worst‐case intake estimate for adult, high fish consumers of 2.6 ng kg−1 day−1 using the analytical limit of quantification (LOQ) as a proxy of TBBPA concentrations in fish for all non‐detected results. When this estimate of exposure was compared to the POD of 16 mg kg−1 day−1 (which is lower than that established in the current study), EFSA concluded that current dietary exposure to TBBPA did not raise a health concern. Similar conclusions were reached based on an assessment of infant exposure via breast milk. EFSA calculated daily exposures ranging from 0.41 to 257 ng kg−1 day−1 for high milk consuming infants, resulting in MOE estimates ranging from 4 × 107 to 6 × 104 (even larger MOEs reported for infants with average milk consumption), thus leading to the conclusion that exposure via human milk did not raise a health concern. These MOE estimates are similar to the MOEs generated in the current study for infants (8.7 × 105 to 1.1 × 108). And finally, although not directly related to food, EFSA developed an MOE based on exposures to dust in homes, classrooms and cars. The resulting MOE of 1.3 × 107 indicated a lack of concern for children exposure to TBBPA from dust; a finding similar to that of the current study (i.e. MOE estimates associated with soil/dust exposure ranged from 3.3 × 106 to 6 × 107 in children). And most recently, Colnot et al. (2014) presented derived‐no‐effect‐levels (DNELs) ranging from 0.16 to 10 mg kg−1 day−1 for the general population – which the authors noted were several orders of magnitude higher than current exposure levels.
When the EU, Health Canada and EFSA conducted their assessments, no carcinogenicity data were available, and thus evaluations of carcinogenicity were limited to qualitative characterizations based on available genotoxicity and mutagenicity data (all of which were negative). The results of the current study suggest that the MOE for cancer is > 32 000 000. Even the MOS associated with the most conservative exposure scenario (reasonable worst case) and extremely conservative linear low‐dose extrapolation is >80 at a risk level of 10−6. The OSF for TBBPA was derived using a default, linear approach even although data clearly indicate that TBBPA is not genotoxic or mutagenic and supporting data indicate that TBBPA is likely associated with a threshold‐based mode of action involving perturbation of endocrine parameters. However, departure from default approaches in the US often requires sufficient evidence for a defined MOA in order to utilize a threshold‐based approach for evaluating cancer (USEPA, 2005a, 2005b). As discussed above, available data indicate that TBBPA may be acting through disruption of endocrine function at high doses, which would be consistent with a threshold‐based response. If the tumors were the result of a threshold‐based MOA, an RfD protective of uterine cancer would be developed and compared with other non‐cancer endpoints in order to propose the most protective RfD. However, more data are required to characterize key events in a MOA for TBBPA prior to the application of such non‐linear dose response modeling. Although it is likely that uterine hyperplasia is a precursor event, there remains some uncertainty as to whether it is non‐neoplastic or preneoplastic. If it is indeed non‐neoplastic, then the RfD presented here for uterine hyperplasia would be considered protective of uterine tumors. However, evaluation of the MOA and human relevance for the tumors observed in the NTP study after chronic exposure to very high doses of TBBPA is beyond the scope of this article.
As with any assessment, there are a number of uncertainties inherent in both the toxicity and exposure evaluation. With respect to the toxicity evaluation, scientific judgment was used to determine the endpoints associated with the most biological and human relevance, as well as determinations regarding adversity. Although such decisions were made based on regulatory precedence and supported in the peer‐review literature, the selection of critical endpoints to serve as the basis of the toxicity values clearly impacts the resulting assessment (note: selection of other candidate RfD values would result in similar MOE findings). The daily estimates of intake exposure for TBBPA are also associated with uncertainty, though the approaches employed were generally conservative. For example, the regulatory default scenario relies primarily on the input of maximum media concentrations. Another example is the use of environmental monitoring data as a proxy for drinking water concentrations is highly conservative. Thus, while such selections introduce uncertainty, they were clearly conservative with respect to characterizing consumer exposure; that is to say, actual exposures are likely to be lower than those presented here, thus resulting in even higher margins of exposure and safety.
Additionally, while the intake estimates were not country specific, there is uncertainty in application of such to any specific population. Both dietary and breast milk media concentrations came from Chinese studies, an area that is associated with a high level of TBBPA usage (Shi et al., 2009; BSEF, 2012). Upon reviewing available data, it was observed that media concentrations in China tended to be more often detected, or more often tended to be associated with higher concentrations, relative to those reported in studies from other parts of the world, thus suggesting that the daily intake estimates would be additionally conservative for consumers in other parts of the world.
The hazard identification, dose–response modeling, and subsequent development of an oral reference dose and cancer slope factor presented in this study provide critical information needed for the quantitative assessment of cancer risk and non‐cancer hazard for TBBPA. These toxicity values represent state‐of‐the science values as they consider data quality, were based on most robust dataset available, and were developed using sophisticated benchmark dose modeling techniques (Benford et al., 2010). Similarly, exposure estimates were generated for several scenarios and various receptors in an effort to characterize the range of potential consumer exposures, thereby capturing variability in exposures to TBBPA. As new exposure and toxicity data become available, it should be incorporated for continued improvement in the characterization of human health hazards associated with TBBPA. In the interim, it is anticipated that the exposure estimates, along with the toxicity values described herein, should be informative for risk assessors and regulators interested in characterizing human health hazards associated with TBBPA. Nonetheless, even considering the range of conservative exposures assessed in this study, the resulting margins of exposure, as well as margins of safety, indicate a low level of health concern.
Supporting information
Supporting info item
Acknowledgments
This work was funded by the North American Flame Retardant Alliance (NAFRA) Panel of the American Chemistry Council (ACC). The funders were given the opportunity to review the draft manuscript; the purpose of such review was to allow input on the clarity of the science presented but not in interpretation of the research findings. The researchers’ scientific conclusions and professional judgments were not subject to the funders’ control; the contents of this manuscript reflect solely the view of the authors. The authors employment affiliation is as shown on the cover page. ToxStrategies is a private consulting firm providing services to private and public organizations on toxicology and risk assessment issues.
Wikoff, D. , Thompson, C. , Perry, C. , White, M. , Borghoff, S. , Fitzgerald, L. , and Haws, L. C. (2015) Development of toxicity values and exposure estimates for tetrabromobisphenol A: application in a margin of exposure assessment. J. Appl. Toxicol., 35: 1292–1308. doi: 10.1002/jat.3132.
References
- Abdallah MA, Harrad S, Covaci A. 2008. Hexabromocyclododecanes and tetrabromobisphenol‐A in indoor air and dust in Birmingham, U.K: implications for human exposure. Environ. Sci. Technol. 42: 6855–6861. [DOI] [PubMed] [Google Scholar]
- Benford D, Bolger PM, Carthew P, Coulet M, DiNovi M, Leblanc JC, Renwick AG, Setzer W, Schlatter J, Smith B, Slob W, Williams G, Wildemann T. 2010. Application of the Margin of Exposure (MOE) approach to substances in food that are genotoxic and carcinogenic. Food Chem. Toxicol. 48(Suppl 1): S2–24. [DOI] [PubMed] [Google Scholar]
- Birnbaum LS, Staskal DF. 2004. Brominated flame retardants: cause for concern? Environ. Health Perspect. 112: 9–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brix AE, Hardisty JF, McConnell EE. 2010. Combining neoplasms for evaluation of rodent carcinogenesis studies In Cancer Risk Assessment, Hsu CH, Stedeford T. (eds). John Wiley & Sons: New Jersey; 699–715. [Google Scholar]
- Bromine Science and Environmental Forum (BSEF) . 2012. TBBPA Factsheet ‐ Tetrabromobisphenol A for printed circuit boards and ABS plastics.
- Cariou R, Antignac JP, Zalko D, Berrebi A, Cravedi JP, Maume D, Marchand P, Monteau F, Riu A, Andre F, Le Bizec B. 2008. Exposure assessment of French women and their newborns to tetrabromobisphenol‐A: occurrence measurements in maternal adipose tissue, serum, breast milk and cord serum. Chemosphere 73: 1036–1041. [DOI] [PubMed] [Google Scholar]
- Colnot T, Kacew S, Dekant W. 2014. Mammalian toxicology and human exposures to the flame retardant 2,2',6,6'‐tetrabromo‐4,4'‐isopropylidenediphenol (TBBPA): implications for risk assessment. Arch. Toxicol. 88: 553–573. [DOI] [PubMed] [Google Scholar]
- Committee on Toxicity of Chemicals in Food, Consumer Products and the Environment (COT) . 2004. Tetrabromobisphenol A – Review of the toxicological data.
- Decherf S, Seugnet I, Fini JB, Clerget‐Froidevaux MS, Demeneix BA. 2010. Disruption of thyroid hormone‐dependent hypothalamic set‐points by environmental contaminants. Mol. Cell. Endocrinol. 323: 172–182. [DOI] [PubMed] [Google Scholar]
- Driffield M, Harmer N, Bradley E, Fernandes AR, Rose M, Mortimer D, Dicks P. 2008. Determination of brominated flame retarndants in food by LC‐MS/MS: diastereoisomer‐specific hexabromocyclododecanda dn tetrabromobisphenol A. Food Addit. Contam. Part A Chem. Anal. Control Expo. Risk Assess. 25: 895–903. [DOI] [PubMed] [Google Scholar]
- European Food Safety Administration (EFSA) . 2011. Scientific Opinion on Tetrabromobisphenol A (TBBPA) and its derivatives in food. EFSA J. 9: 2477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- European Union (EU) . 2006. Risk Assessment Report: 2,2’,6,6’‐TETRABROMO‐4,4’‐ISOPROPYLIDENEDIPHENOL (TETRABROMOBISPHENOL‐A or TBBP‐A), Part II – Human Health. Volume 63.
- Food Safety Authority of Ireland (FSAI) . 2010. Investigation into levels of chlorinated and brominated organic pollutants in carcass fat, offal, eggs and milk produced in Ireland.
- Fukuda N, Ito Y, Yamaguchi M, Mitumori K, Koizumi M, Hasegawa R, Kamata E, Ema M. 2004. Unexpected nephrotoxicity induced by tetrabromobisphenol A in newborn rats. Toxicol. Lett. 150: 145–155. [DOI] [PubMed] [Google Scholar]
- Germer S, Piersma AH, van der Ven L, Kamyschnikow A, Fery Y, Schmitz HJ, Schrenk D. 2006. Subacute effects of the brominated flame retardants hexabromocyclododecane and tetrabromobisphenol A on hepatic cytochrome P450 levels in rats. Toxicology 218: 229–236. [DOI] [PubMed] [Google Scholar]
- Gosavi RA, Knudsen GA, Birnbaum LS, Pedersen LC. 2013. Mimicking of estradiol binding by flame retardants and their metabolites: a crystallographic analysis. Environ. Health Perspect. 121: 1194–1199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greaves P. 2012. Histopathology of Preclinical Toxicity Studies. Elsevier‐Academic Press: London. [Google Scholar]
- Hamers T, Kamstra JH, Sonneveld E, Murk AJ, Kester MH, Andersson PL, Legler J, Brouwer A. 2006. In vitro profiling of the endocrine-disrupting potency of brominated flame retardants. Toxicol Sci. 92(2):157–173. [DOI] [PubMed] [Google Scholar]
- Harrad S, Abdallah MA, Rose NL, Turner SD, Davidson TA. 2009. Current‐use brominated flame retardants in water, sediment, and fish from English lakes. Environ. Sci. Technol. 43: 9077–9083. [DOI] [PubMed] [Google Scholar]
- Harrad S, Goosey E, Desborough J, Abdallah M, Roosens L, Covaci A. 2010. Dust from U.K. primary school classrooms and daycare centers: The significance of dust as a pathway of exposure of young U.K. children to brominated flame retardants and polychlorinated biphenyls. Environ. Sci. Technol. 44: 4198–4202. [DOI] [PubMed] [Google Scholar]
- He MJ, Luo XJ, Yu LH, Wu JP, Chen SJ, Mai BX. 2013. Diasteroisomer and enantiomer‐specific profiles of hexabromocyclododecane and tetrabromobisphenol A in an aquatic environment in a highly industrialized area, South China: vertical profile, phase partition, and bioaccumulation. Environ. Pollut. 179: 105–110. [DOI] [PubMed] [Google Scholar]
- Health Canada/ Environment Canada . 2013. Screening Assessment Report: Phenol, 4,4'‐(1‐methylethylidene) bis[2,6‐dibromo‐ (Chemical Abstracts Service Registry Number 79‐94‐7), Ethanol, 2,2'‐[(1‐methylethylidene)bis[(2,6‐dibromo‐4,1‐phenylene)oxy]]bis (Chemical Abstracts Service Registry Number 4162‐45‐2), Benzene, 1,1'‐(1methylethylidene)bis[3,5‐dibromo‐4‐(2‐ propenyloxy)‐ (Chemical Abstracts Service Registry Number 25327‐89‐3).
- Imai T, Takami S, Cho YM, Hirose M, Nishikawa A. 2009. Modifying effects of prepubertal exposure to potassium perchlorate and tetrabromobisphenol A on susceptibility to N‐bis(2‐hydroxypropyl)nitrosamine‐ and 7,12‐dimethylbenz(a)anthracene‐induced carcinogenesis in rats. Toxicol. Lett. 185: 160–167. [DOI] [PubMed] [Google Scholar]
- Jakobsson K, Thuresson K, Rylander L, Sjodin A, Hagmar L, Bergman A. 2002. Exposure to polybrominated diphenyl ethers and tetrabromobisphenol A among computer technicians. Chemosphere 46: 709–716. [DOI] [PubMed] [Google Scholar]
- Kang MJ, Kim JH, Shin S, Choi JH, Lee SK, Kim HS, Kim ND, Kang GW, Jeong HG, Kang W, Chun YJ, Jeong TC. 2009. Nephrotoxic potential and toxicokinetics of tetrabromobisphenol A in rat for risk assessment. J. Toxicol. Environ. Health A 72: 1439–1445. [DOI] [PubMed] [Google Scholar]
- Kim UJ, Oh JE. 2014. Tetrabromobisphenol A and hexabromocyclododecane flame retardants in infant‐mother paired serum samples, and their relationships with thyroid hormones and environmental factors. Environ. Pollut. 184: 193–200. [DOI] [PubMed] [Google Scholar]
- Kirman C, Budinsky RA, Yost L, Baker BF, Zabik JM, Rowlands JC, Long TF, Simon T. 2011. Derivation of Soil Clean‐Up Levels for 2,3,7,8‐Tetrachloro‐dibenzo‐p‐dioxin (TCDD) Toxicity Equivalence (TEQD/F) in Soil Through Deterministic and Probabilistic Risk Assessment of Exposure and Toxicity. Hum. Ecol. Risk Assess. 17: 125–158. [Google Scholar]
- Kitamura S1, Suzuki T, Sanoh S, Kohta R, Jinno N, Sugihara K, Yoshihara S, Fujimoto N, Watanabe H, Ohta S. 2010. Comparative study of the endocrine-disrupting activity of bisphenol A and 19 related compounds. Toxicol Sci. 84(2): 249–259. [DOI] [PubMed] [Google Scholar]
- Klimisch HJ, Andreae M, Tillmann U. 1997. A systematic approach for evaluating the quality of experimental toxicological and ecotoxicological data. Regul. Toxicol. Pharmacol. 25: 1–5. [DOI] [PubMed] [Google Scholar]
- Knudsen GA, Sanders JM, Sadik AM, Birnbaum LS. 2014. Disposition and kinetics of tetrabromobisphenol A in female Wistar Han rats. Toxicol. Rep. 1: 214–223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koenig H, Goldstone A, Blume G, Lu CY. 1980. Testosterone‐mediated sexual dimorphism of mitochondria and lysosomes in mouse kidney proximal tubules. Science 209: 1023–1026. [DOI] [PubMed] [Google Scholar]
- Labadie P, Tlili K, Alliot F, Bourges C, Desportes A, Chevreuil M. 2010. Development of analytical procedures for trace‐level determination of polybrominated diphenyl ethers and tetrabromobisphenol A in river water and sediment. Anal. Bioanal. Chem. 396: 865–875. [DOI] [PubMed] [Google Scholar]
- Li J, Ma M, Wang Z. 2010. In vitro profiling of endocrine disrupting effects of phenols. Toxicol. In Vitro 24: 201–207. [DOI] [PubMed] [Google Scholar]
- Lilienthal H, Verwer CM, van der Ven LT, Piersma AH, Vos JG. 2008. Exposure to tetrabromobisphenol A (TBBPA) in Wistar rats: neurobehavioral effects in offspring from a one‐generation reproduction study. Toxicology 246: 45–54. 10.1016/j.tox.2008.01.007. [DOI] [PubMed] [Google Scholar]
- Ni HG, Zeng H. 2013. HBCD and TBBPA in particulate phase of indoor air in Shenzhen, China. Sci. Total Environ. 458–460: 15–19. [DOI] [PubMed] [Google Scholar]
- National Toxicology Program (NTP) . 2002. Tetrabromobisphenol A [79‐94‐7]: Review of Toxicological Literature.
- National Toxicology Program (NTP) . 2013. NTP Technical Report on the toxicology studies of tetrabromobisphenol A (CAS no. 79‐94‐7) in F344/NTac rats and B6C3F1/N mice and toxicology and carcinogenesis studies of tetrabromobisphenol A in Wistar Han [Crl:Wi(Han)] rats and B6C3F1/N mice. NIH Publication no. 14–5929.
- Peterman PH, Orazio CE, Gale RW. 2000. Detection of tetrabromobisphenol A and formation of brominated 13C‐bisphenol A's in commercial drinking water stored in reusable polycarbonate containers. Div. Environ. Chem. Preprints Extended Abstr. 40: 431–433. As cited in Health Canada 2013 [Google Scholar]
- Saegusa Y, Fujimoto H, Woo GH, Inoue K, Takahashi M, Mitsumori K, Hirose M, Nishikawa A, Shibutani M. 2009. Developmental toxicity of brominated flame retardants, tetrabromobisphenol A and 1,2,5,6,9,10‐hexabromocyclododecane, in rat offspring after maternal exposure from mid‐gestation through lactation. Reprod. Toxicol. 28: 456–467. [DOI] [PubMed] [Google Scholar]
- Saegusa Y, Fujimoto H, Woo GH, Ohishi T, Wang L, Mitsumori K, Nishikawa A, Shibutani M. 2012. Transient aberration of neuronal development in the hippocampal dentate gyrus after developmental exposure to brominated flame retardants in rats. Arch. Toxicol. 86: 1431–1442. [DOI] [PubMed] [Google Scholar]
- Sato T, Watanabe K, Nagase H, Kito H, Niikawa M. 1996. Toxicity of the brominated flame retardant (tetrabromobisphenol‐A). Toxicol. Environ. Chem. 55: 159–171. [Google Scholar]
- Schroeder R. 2002a. A 90‐day oral toxicity study of tetrabromobisphenol A in rats with a recovery group. Study Number: 474–006 MPI Research, Inc: Mattawan, MI. [Google Scholar]
- Schroeder R. 2002b. An oral two generation reproductive, fertility, and developmental neurobehavioral study of tetrabromobisphenol A in rats. Study Number: 474–004 MPI Research, Inc: Mattawan, MI. [Google Scholar]
- Schroeder R. 2003. Amendment to the final report: An oral two generation reproductive, fertility, and developmental neurobehavioral study of tetrabromobisphenol A in rats. Study Number: 474–004. MPI Research, Inc: Mattawan, MI. [Google Scholar]
- Shi Z, Jiao Y, Hu Y, Sun Z, Zhou X, Feng J, Li J, Wu Y. 2013. Levels of tetrabromobisphenol A, hexabromocyclododecanes and polybrominated diphenyl ethers in human milk from the general population in Beijing, China. Sci. Total Environ. 452–453: 10–18. [DOI] [PubMed] [Google Scholar]
- Shi ZX, Wu YN, Li JG, Zhao YF, Feng JF. 2009. Dietary exposure assessment of Chinese adults and nursing infants to tetrabromobisphenol‐A and hexabromocyclododecanes: occurrence measurements in foods and human milk. Environ. Sci. Technol. 43: 4314–4319. [DOI] [PubMed] [Google Scholar]
- Sjodin A, Patterson DG, Jr , Bergman A. 2003. A review on human exposure to brominated flame retardants‐‐particularly polybrominated diphenyl ethers. Environ. Int. 29: 829–839. [DOI] [PubMed] [Google Scholar]
- Slikker W, Anderson ME, Bogdanffy MS, Bus JS, Cohen SD, Conolly RB, David RM, Doerrer HG, Dorman DC, Gaylor DW, Hattis D, Rogers JM, Setzer RW, Swenbery JA, Wallace K. 2004. Dose‐dependent transitions in mechanisms of toxicity: case studies. Toxicol. Appl. Pharmacol. 201: 226–294. [DOI] [PubMed] [Google Scholar]
- Sommers SC. 1953. Ovarian Rete Cysts. Am. J. Pathol. 29: 853–859. [PMC free article] [PubMed] [Google Scholar]
- Szymanska, JA . 1995. Comparison of Hepatotoxicity of Monobromobenzene, Dibromobenzenes, Hexabromobenzene and Tetrabromobisphenol A In Advances in Organobromine Chemistry II, Desmurs J‐R, Gerard B, Goldstein MJ. (eds). 7; 1–428. [Google Scholar]
- Szymanska JA, Piotrowski JK, Frydrych B. 2000. Hepatotoxicity of tetrabromobisphenol‐A: effects of repeated dosage in rats. Toxicology 142: 87–95. [DOI] [PubMed] [Google Scholar]
- Tada Y, Fujitani T, Ogata A, Kamimura H. 2007. Flame retardant tetrabromobisphenol A induced hepatic changes in ICR male mice. Environ. Toxicol. Pharmacol. 23: 174–178. 10.1016/j.etap.2006.08.007. [DOI] [PubMed] [Google Scholar]
- Tada Y, Fujitani T, Yano N, Takahashi H, Yuzawa K, Ando H, Kubo Y, Nagasawa A, Ogata A, Kamimura H. 2006. Effects of tetrabromobisphenol A, brominated flame retardant, in ICR mice after prenatal and postnatal exposure. Food Chem. Toxicol. 44: 1408–1413. [DOI] [PubMed] [Google Scholar]
- Turusov VS, Torii M, Sills RC, Willson GA, Herbert RA, Hailey JR, Haseman JK, Boorman GA. 2002. Hepatoblastomas in mice in the US National Toxicology Program (NTP) studies. Toxicol. Pathol. 30: 580–591. [DOI] [PubMed] [Google Scholar]
- USEPA . 1989. Risk Assessment Guidance for Superfund: Volume I – Human Health Evaluation Manual (Part A). Office of Solid Waste and Emergency Response. EPA/540/1‐89/002a. December.
- USEPA . 1991. OSWER DIRECTIVE: 9285.6‐03. Risk Assessment Guidance for Superfund Volume I: Human Health Evaluation Manual, Supplemental Guidance, " Standard Default Exposure Factors" , Interim Final, Office of Emergency and Remedial Response, Toxics Integration Branch. March.
- USEPA . 1992. Guidelines for Exposure Assessment. Risk Assessment Forum. Washington DC. EPA/600/Z‐92/001. May. [Google Scholar]
- USEPA . 1997. Exposure Factors Handbook. Office of Research and Development, National Center for Environmental Assessment: Washington DC. EPA/600/P‐95/002Fa, −002Fb, and ‐002Fc. August. [Google Scholar]
- USEPA . 2000. Supplemental Guidance to RAGS: Region 4 Bulletins, Human Health Risk Assessment Bulletins. EPA Region 4.
- USEPA . 2002. A review of the reference dose and reference concentration processes. Risk Assessment Forum, Washington, DC: [Google Scholar]
- USEPA . 2003. Example Exposure Scenarios. National Center for Environmental Assessment: Washington, DC. EPA/6001R‐03/036. [Google Scholar]
- USEPA . 2005a. Guidelines for Carcinogen Risk Assessment. U.S. Environmental Protection Agency: Washington, DC, EPA/630/P‐03/001F, 2005. [Google Scholar]
- USEPA . 2005b. Human Health Risk Assessment Protocol for Hazardous Waste Combustion Facilities, Final. Office of Solid Waste and Emergency Response. EPA530‐R‐05‐006. September.
- USEPA . 2011. Exposure Factors Handbook: 2011 Edition. Office of Research and Development, National Center for Environmental Assessment: Washington DC: EPA/600/R‐090/052F. September. [Google Scholar]
- USEPA . 2012. Benchmark Dose Technical Guidance. Risk Assessment Forum, U.S. Environmental Protection Agency: Washington, DC. [Google Scholar]
- USEPA . 2013. Regional Screening Table. http://www.epa.gov/reg3hwmd/risk/human/rb‐concentration_table/index.htm [February 2014]
- Van der Ven LT, Van de Kuil T, Verhoef A, Verwer CM, Lilienthal H, Leonards PE, Schauer UM, Canton RF, Litens S, De Jong FH, Visser TJ, Dekant W, Stern N, Hakansson H, Slob W, Van den Berg M, Vos JG, Piersma AH. 2008. Endocrine effects of tetrabromobisphenol‐A (TBBPA) in Wistar rats as tested in a one‐generation reproduction study and a subacute toxicity study. Toxicology 245: 76–89. [DOI] [PubMed] [Google Scholar]
- Verwer CM, van der Ven LT, van den Bos R, Hendriksen CF. 2007. Effects of housing condition on experimental outcome in a reproduction toxicity study. Regul. Toxicol. Pharmacol. 48: 184–193. [DOI] [PubMed] [Google Scholar]
- Watanabe W, Shimizu T, Sawamura R, Hino A, Konno K, Hirose A, Kurokawa M. 2010. Effects of tetrabromobisphenol A, a brominated flame retardant, on the immune response to respiratory syncytial virus infection in mice. Int. Immunopharmacol. 10: 393–397. [DOI] [PubMed] [Google Scholar]
- Wignall JA, Shapiro AJ, Wright FA, Woodruff TJ, Chiu WA, Guyton KZ, Rusyn I. 2014. Standardizing benchmark dose calculations to improve science‐based decisions in human health assessments. Environ. Health Perspect. 122: 499–505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams AL, DeSesso JM. 2010. The potential of selected brominated flame retardants to affect neurological development. J. Toxicol. Environ. Health B Crit. Rev. 13: 411–448. [DOI] [PubMed] [Google Scholar]
- de Winter‐Sorkina R, Bakker MI, van Donkersgoed G, van Klaveren JD. 2003. Dietary intake of brominated flame retardants by the Dutch population. National Institute of Public Health and the Environment (RIVM), RIVM Report no. 310305001.
- de Wit CA. 2002. An overview of brominated flame retardants in the environment. Chemosphere 46: 583–624. [DOI] [PubMed] [Google Scholar]
- Yang S, Wang S, Liu H, Yan Z. 2012. Tetrabromobisphenol A: tissue distribution in fish, and seasonal variation in water and sediment of Lake Chaohu, China. Environ. Sci. Pollut. Res. Int. 19: 4090–4096. [DOI] [PubMed] [Google Scholar]
- Zatecka E, Ded L, Elzeinova F, Kubatova A, Dorosh A, Margaryan H, Dostalova P, Peknicova J. 2013. Effect of tetrabrombisphenol A on induction of apoptosis in the testes and changes in expression of selected testicular genes in CD1 mice. Reprod. Toxicol. 35: 32–39. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting info item