

Abstract
Several lines of research support the hypothesis that migraine is a spectrum of illness, with clinical symptoms that vary along a continuum from episodic migraine to chronic migraine. Physiologic changes may result in episodic migraine evolving into chronic migraine over months to years in susceptible individuals. With chronification, headache frequency increases, becoming more disabling and less responsive to therapy. Neurophysiologic and functional imaging research has reported that chronic migraine may be associated with severity‐specific metabolic, functional, and structural abnormalities in the brainstem. Without longitudinal studies, it is unclear whether these changes may represent a continuum of individual progression and/or are reversible. Furthermore, chronic migraine is associated with larger impairments in cortical processing of sensory stimuli when compared with episodic migraine, possibly caused by more pronounced cortical hyperexcitability.
Progressive changes in nociceptive thresholds and subsequent central sensitization due to recurrent migraine attacks in vulnerable individuals contribute to the chronic migraine state. This may result in changes to baseline neurologic function between headache attacks, evident in both electrophysiological and functional imaging research. Patients experiencing migraine chronification may report increased non‐headache pain, fatigue, psychiatric disorders (eg, depression, anxiety), gastrointestinal complaints, and other somatic conditions associated with their long‐term experience with migraine pain.
Recent research provides a foundation for differentiating episodic and chronic migraine based on neurophysiologic and neuroimaging tools. In this literature review, we consider these findings in the context of models designed to explain the physiology and progression of episodic migraine into chronic migraine, and consider treatment of chronic migraine in susceptible individuals. Advances in pharmacotherapy provide treatment options for chronic migraine. Of the currently available treatment options, only onabotulinumtoxinA and topiramate have received regulatory approval and have demonstrated efficacy in patients with chronic migraine, although the exact mechanisms of action are not fully elucidated.
Keywords: chronic migraine, episodic migraine, pathophysiology, onabotulinumtoxinA, topiramate, literature review
Abbreviations
- BOLD fMRI
blood oxygen level–dependent functional magnetic resonance imaging
- CAMERA
Cerebral Abnormalities in Migraine, an Epidemiological Risk Analysis
- CGRP
calcitonin gene‐related peptide
- fMRI
functional magnetic resonance imaging
- GABA
γ‐aminobutyric acid
- MEG
magnetoencephalography
- MRI
magnetic resonance imaging
- MSPA
magnetic suppression of perceptual accuracy
- PET
positron emission tomography
- PREEMPT
Phase III REsearch Evaluating Migraine Prophylaxis Therapy
- SNAP‐25
synaptosomal‐associated protein
- SNARE
soluble N‐ethylmaleimide–sensitive factor attachment protein receptor
- TMS
transcranial magnetic stimulation
- TRPA1
transient receptor potential cation channel ankyrin subfamily member 1
- TRPV1
transient receptor potential cation channel vanilloid subfamily member 1
- VAMP
vesicle‐associated membrane protein
INTRODUCTION
Migraine is understood to be a spectrum of illness, consisting of episodic and chronic forms. Although chronic migraine typically progresses from episodic migraine, emerging epidemiologic evidence supports unique underlying physiology of the two migraine states.1, 2, 3 In addition, regulatory agencies consider episodic and chronic migraine as unique indications, requiring separate regulatory approval.
In this review, we provide an update of a previous literature review on the physiologic mechanisms underlying chronic migraine, focusing on newly published research retrieved using several key search terms (eg, migraine, pathophysiology, mechanism of action; full list available upon request).4 First, we examine new neurophysiologic and functional imaging studies that have revealed differences between episodic and chronic migraine, and provide insight into the underlying physiology of migraine in the brain. This research reaffirms our previous findings that the biology of migraine recapitulates the concept of a spectrum disorder with variations along the continuum.
We then consider these findings in the context of models designed to explain the progression of episodic migraine into chronic migraine. We also briefly discuss new research on the mechanisms of action of two approved migraine prophylactic therapies, onabotulinumtoxinA for chronic migraine, and topiramate for migraine, and consider how these may impact migraine physiology and reduce the burden of illness for the patient. Although novel prophylactic treatments for chronic migraine are currently in development (eg, calcitonin gene‐related peptide [CGRP] ligand and receptor targets), this review focuses on those with regulatory approval and demonstrated efficacy. These emerging treatments have been the subject of several recent reviews.5, 6
MIGRAINE: THE CLINICAL SPECTRUM DISORDER
As initially suggested by Mathew et al over 30 years ago,7 migraine is currently conceptualized as a continuum from episodic to chronic forms of migraine headache, whereby “chronic” indicates severity or patient‐specific symptomatic burden, as opposed to duration of disease. Based on the current classification guidelines,8 on one end of this continuum is episodic migraine and on the other end is chronic migraine, with variations in headache‐day frequency and symptoms along the continuum (Fig. 1).8, 15, 16
Figure 1.
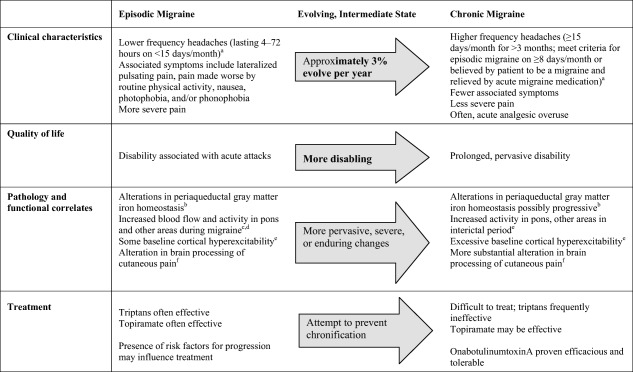
Overview of features associated with episodic and chronic migraine.8 Of note, the ICHD‐3b criteria cited here do not differ substantially from the ICHD‐2 criteria, which many of the studies cited herein used to define migraine and chronic migraine.9, 10, 11, 12, 13 Source: Adapted and updated from Aurora.14
Chronic migraine is the most common type of chronic daily headache seen by headache specialists.17 Globally, approximately 2% of the population experiences chronic migraine; prevalence is 2.5‐ to 6.5‐fold higher in women (1.7%–4.0%) than in men (0.6%–0.7%).18 Approximately 3% of people with episodic migraine progress to chronic migraine each year;19 the clinical progression typically occurs gradually, with increasing attack frequency over time.19, 20, 21 However, in some patients, the progression may be abrupt.
Risk Factors for Clinical Progression
Several studies have characterized factors associated with migraine progression. Higher risk is reportedly associated with nonmodifiable (eg, female sex, lower socioeconomic status, unmarried)20 and modifiable (eg, acute headache medication use, caffeine intake, obesity, other pain syndromes, previous head or neck injury, snoring, stressful life events) risk factors.20 Headache frequency is also an important risk factor for progression.22 Individuals with ≥4 headache days per month have an exponential increase in the risk of transformation from episodic to chronic migraine.23 Furthermore, some clinicians have suggested that high‐frequency episodic migraine may represent a “pre‐chronic migraine” state, which creates an opportunity for identification and early treatment in an effort to prevent further transition into chronic migraine.24
The risk of developing migraine may be influenced by genetics, although an association between known migraine‐related single‐nucleotide polymorphisms and chronification has not been established.25 Mutations have been identified in genes coding for calcium channels and sodium‐potassium pumps among those with familial hemiplegic migraine and episodic ataxia type‐2;26, 27 these mutations may cause 5‐HT receptor dysfunction and increased synaptic glutamate concentrations, contributing to neuronal hyperexcitability26 and subsequent cortical spreading depression. Findings from preclinical models28, 29 and human studies suggest that cortical spreading depression is a component of the migraine with aura experience.30, 31 No definitive relation of cortical spreading depression with migraine pain or migraine without aura has yet been demonstrated.29 While the association between genetic mutations and chronic migraine remains inconclusive, these studies provide support for the general concept of migraine hyperexcitability observed with chronic migraine, as we discuss further in studies of the physiology of visual suppression.
Indicators of Progression to Chronic Migraine
Neurological changes have been implicated in migraine progression in two models of migraine.19, 32 Cady et al view neurological changes underlying progression from the perspective of increased levels of anxiety, depression, fatigue, gastrointestinal disorders, and nonheadache pain between migraines, which occur after years of episodic migraine attacks.32 This is not surprising, considering the widespread central impact of migraine. Bigal and Lipton propose that reduced nociceptive thresholds and changes in pain pathways that underlie allodynia and central sensitization may represent physiologic correlates of progression, while stroke and radiographic white matter lesions may provide anatomic signs of progression.19 In support of these models, chronic migraine has been associated with frontal lobe neuropsychologic dysfunction,33 altered cortical pain processing,34 and brainstem vascular malformations.35 Furthermore, Noseda et al mapped single trigeminovascular neurons that project to the posterior, lateral posterior, and lateral dorsal thalamic nuclei and found that they ultimately connected with auditory, ectorhinal, insular, parietal‐association, retrosplenial, somatosensory, and visual cortices, which influence affect, memory, motor control, sensory perception (ie, auditory, olfactory, visual), and spatial orientation.36
Chronic stimulation of central pain pathways during repeated migraine attacks may increase central sensitization by decreasing nociceptive thresholds.13, 19, 37 As Bigal and Lipton's model proposes, cutaneous allodynia may serve as an indicator of migraine progression, as it is believed to signify central sensitization, wherein second‐order brainstem trigeminal neurons are increasingly sensitive to innocuous input.38
Cutaneous allodynia is common in people with migraine. Burstein et al reported that non‐noxious stimuli during a migraine attack produced a pain reaction among 79% of patients with migraine seen at a medical center; all patients in his study experienced episodic migraine attacks 1–6 times per month for at least the previous 3 years and some also experienced frequent tension‐type headaches.38 Later studies confirmed this finding in individuals with episodic as well as chronic migraine;37, 39, 40 however, one study reported greater allodynia severity among individuals with chronic migraine than those with episodic migraine,39 and another found that individuals who met the criteria for episodic migraine with aura or chronic migraine showed a higher frequency of cutaneous allodynia than people with episodic migraine without aura.40 One interpretation for these differences is that individuals who experience aura exhibit more persistent or severe central sensitization than those with episodic migraine without aura. Zappaterra et al found that patients with chronic headache (defined as headache >15 days per month, including chronic tension‐type headache and medication overuse headache that started as transformed migraine) show higher rates of acute and interictal allodynia and increases in pathological mean cutaneous pain threshold scores relative to those with episodic headache (including episodic migraine with and without aura and episodic tension‐type headache).41 Together with observations of lower pain thresholds in individuals with chronic migraine (as opposed to episodic migraine)13 and atypical cortical processing of cutaneous nociceptive input,34, 42 these findings support the hypothesis of physiologic progression involving disrupted central pain mechanisms.
Clinically Important Differences between Episodic and Chronic Migraine
Important distinctions exist along the continuum between episodic and chronic migraine (Fig. 1). Chronic migraine imparts a substantially greater burden with disability scores nearly twice as high among individuals with chronic versus those with episodic migraine.1, 43 Patients with chronic migraine experience higher rates of comorbidities, including impaired sleep, mental health disorders (especially anxiety and depression), and gastrointestinal dysfunction.20, 44, 45, 46, 47, 48 Chronic migraineurs also experience an increased frequency of emergency department visits,49 greater economic burden,50 and suffer greater detriments to their work, school, home, social, and leisure activities.1, 43 This biological disability is often further confounded by some pharmacotherapies that may be accompanied by intolerable side effects;51 treatment with more tolerable drugs, which may be successful in episodic migraine, is frequently unsuccessful in patients with chronic migraine.
MIGRAINE NEUROPHYSIOLOGY AND IMAGING FINDINGS
Emerging evidence supports the existence of both structural52, 53, 54, 55 and functional brain alterations in migraine. Important advances into the pathogenesis and pathophysiology of migraine have been afforded by neurophysiologic tests (eg, magnetic suppression of perceptual accuracy [MSPA], magnetoencephalography [MEG], transcranial magnetic stimulation [TMS]), functional imaging (eg, functional magnetic resonance imaging [fMRI], blood oxygen level–dependent [BOLD] fMRI, positron emission tomography [PET], perfusion weighted imaging), and structural imaging (eg, magnetic resonance imaging [MRI]). The research summarized below discusses some insights from these approaches (Table 1).
Table 1.
Neurophysiological and Functional Imaging in Migraine
| Technique | Episodic Migraine | Chronic Migraine | Interpretation/Implication |
|---|---|---|---|
| Neurophysiological Techniques | |||
| Magnetic Suppression of Perceptual Accuracy (MSPA) | Letter reporting accuracy decreased by magnetic pulse, but not as much as in controls † | Letter reporting accuracy not decreased by magnetic pulse, in contrast to episodic migraine and controls † | Intracortical inhibitory mechanisms may be more impaired in chronic migraine than episodic migraine, leading to a greater increase in baseline cortical excitability † |
| Magnetoencephalography (MEG) | Intermittent excitability associated with migraine attack ‡‡ | Persistent excitability during and between attacks ‡‡ | Different pathophysiologic mechanisms underlie episodic and chronic migraine ‡‡ |
| Functional Imaging Techniques | |||
| Positron Emission Tomography (PET) | Increased activity in brainstem (pons) and selected cortical areas during migraine‡,§ | Increased activity in pons, right temporal cortex; decreased activity in selected cortical areas, caudate nuclei; all findings in the interictal period † | Certain brain regions (eg, pons, rostral medulla) may be overactive during attacks‡,§ of episodic migraine but continuously overactive † in chronic migraine |
| Activity in dorsal rostral pons, anterior cingulate cortex, and cuneus correlated with pain scores; activity in anterior cingulate cortex and pulvinar correlated with paresthesia scores ¶ | Pulvinar, cingulate and cuneus activity likely linked to affective component of pain; pons activity may be associated with migraine pathophysiology ¶ | ||
| Magnetic Resonance Imaging (MRI; relaxation rates R2, R2' and R2*) | Significant increase in R2' and R2* values in periaqueductal gray matter vs controls, not different from chronic migraine †† | Significant increase in R2' and R2* values in periaqueductal gray matter vs controls, not different from episodic migraine †† | Iron homeostasis in the periaqueductal gray may be persistently impaired in migraineurs, perhaps caused by repeated attacks †† |
| No differences in R2' and R2* values in red nucleus and substantia nigra vs controls †† | Significant decrease in R2' and R2* values in periaqueductal gray matter, red nucleus, and substantia nigra compared with the episodic migraine and controls †† | May be due to hyperoxia associated with head pain during an attack †† | |
Neurophysiologic Studies
Cortical Hyperexcitability
MSPA utilizes TMS to investigate cortical excitability in migraine.58 During the MSPA test, a series of 3 letters (trigram) is flashed briefly on a computer screen. Each trigram is followed by a 40‐ to 190‐ms interval, after which a single high‐intensity magnetic pulse is delivered to the occipital skull via a stimulation coil. Participants are asked to report which letters were flashed in each trigram.
In people without migraine, response profiles show a U‐shaped function, in which letter‐reporting accuracy is high at short (40 ms) and long (190 ms) intervals, but no better than chance for mid‐range (100 ms) intervals (Fig. 2). Among migraineurs, marked differences in perceptual accuracy are reported between those with episodic and chronic migraine, with chronic migraineurs demonstrating no measurable difference in perceptual accuracy throughout the range of TMS pulse intervals (Fig. 2), and episodic migraineurs showing decreased letter‐reporting accuracy at the mid‐range intervals (most pronounced at 100 ms), though less pronounced than those without migraine.12, 58 The neural basis of these results may be attributed to inhibitory neurons that are activated at the mid‐range intervals59 in people without migraine, impairing perceptual accuracy; however, increased baseline cortical excitability caused by impaired intracortical inhibitory mechanisms may make perceptual suppression more difficult in those individuals with migraine.60 These findings suggest a continuum of cortical excitability, wherein people with episodic migraine exhibit increased cortical excitability over those without migraine, and people with chronic migraine exhibit an even greater degree of cortical excitability over episodic migraineurs and nonmigraineurs.12, 58
Figure 2.
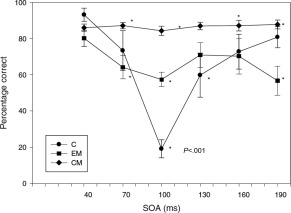
Cortical excitability as measured by magnetic suppression of perceptual accuracy. *P < .001. Stimulus onset accuracy is the time between the appearance of the letter trigram and the delivery of the TMS pulse. Bars show standard errors. Source: Aurora et al.12
Cortical excitability has also been studied using MEG. Patients with chronic migraine demonstrated persistent cortical excitability between migraine episodes. This differs from the intermittent excitability and potentiation observed in episodic migraine and suggests different, or more extreme, effects in the patient with chronic migraine.57, 58, 61 This finding, combined with MSPA research, suggests central inhibitory dysfunction in chronic migraine, with increased cortical hyperexcitability as a significant factor underlying the transformation of episodic migraine into chronic migraine.
Functional Imaging Studies
Activation During and Between Migraine Episodes
Several PET studies have examined cerebral activation during migraine attacks.10, 11 Weiller et al found increased blood flow in the cingulate, auditory and visual association cortices, and brainstem; injection of the acute migraine medication sumatriptan relieved headache pain and light and sound sensitivity, and only the brainstem remained activated.10 In a PET study of migraine evoked via glyceryl trinitrate infusion, Afridi et al noted increased pons activation during and after an episode compared with the baseline premigraine state; other structures (eg, cingulate, insula, cerebellum, prefrontal cortex, putamen) were active during the attack, but not after it was fully controlled with sumatriptan.11 They also reported concordance between the side of pons activation and the laterality of the migraine, and suggested that pain lateralization during a migraine may be attributed to lateralized pons dysfunction.11 In view of the central role of the trigeminal pathway in chronic migraine, pontine activation is not unexpected. Another PET study among chronic migraineurs reported that interictal glucose metabolism increased in the pons and right temporal cortex, but decreased bilaterally in the medial frontal, parietal, and somatosensory cortices, as well as caudate nuclei.12 These results suggest that the normal inhibitory capacity of the cortex is reduced in people with chronic migraine,12 although no such activation of the pons between attacks has been reported among those with episodic migraine.
Brainstem Activation
Recent evidence implicates rostral brainstem activation during migraine episodes. Several rostral brainstem nuclei (eg, periaqueductal gray, raphe nucleus, locus coeruleus) are known to modulate sensory information; therefore, dysfunction in these nuclei has been proposed to contribute to sensory abnormalities observed during migraine episodes (eg, throbbing headache, sensitivity to light and sound).62 Chronic dysfunction among these nuclei may contribute to increased headache frequency, and potentially migraine chronification.62
Studies using fMRI have identified activity in the red nucleus and substantia nigra during spontaneous migraine‐related visual aura and visually triggered migraine.63, 64 Although these areas are best known for their roles in motor function, they are also involved in sensory processing and pain.65, 66, 67 Another study using BOLD‐fMRI has also associated spontaneous visual aura with changes consistent with cortical spreading depression, beginning in the extrastriate cortex and progressing to the occipital cortex.68 This result is supported by a MEG clinical trial in which visually triggered aura was associated with MEG‐direct current shifts typical of those observed during cortical spreading depression.69
Structural Observations
Dysmodulation of the pain system in the brainstems of migraineurs has been demonstrated using neurophysiologic and functional imaging studies, which indicate that chronic migraine is associated with progressive abnormalities in the periaqueductal gray matter,61 and possibly other brain regions as well.70, 71 Convincing evidence is mounting that suggests that repeated migraines are associated with iron accumulation in periaqueductal gray matter,9 globus pallidus,70, 71 red nucleus, and putamen.71 All of these areas are involved in central pain processing and proposed migraine physiology. Iron accumulation during an attack may catalyze free radical injury, which may be increasingly impactful with repeated attacks. High‐resolution MRI was used to map iron homeostasis as an indicator of brain function in the periaqueductal gray matter during migraine episodes among individuals with chronic migraine/chronic daily headache and between attacks in individuals with episodic migraine.9 Results showed significant impairment in iron homeostasis in the periaqueductal gray of both migraine groups compared with the control group (nonmigraineurs), but no difference between the episodic and chronic migraineurs.9 These results support the notion that repeated migraine attacks may impair periaqueductal gray function, resulting in elevated iron concentrations, and that this structure may contribute to migraine episodes through dysregulation of the trigeminovascular nociceptive system.9 Another MRI study showed increased iron deposits in people with migraine, but not controls, in 3 deep nuclei, putamen, globus pallidus, and red nucleus, suggesting a disturbed central antinociceptive neuronal network.71 Further evidence of iron accumulation in migraine has been identified in the basal ganglia; this study showed that people with chronic migraine displayed more iron accumulation than those with episodic migraine when evaluated by T2 MRI.70
Mainero et al demonstrated that repeated migraine attacks increase functional connectivity between the periaqueductal gray and brain regions involved in pain modulation (eg, prefrontal cortex, anterior cingulate, amygdala), potentially reducing the ability to inhibit pain response, and increasing hyperexcitability.72 Volumetric MRI (voxel‐based morphometry) studies have demonstrated a reduction in the gray matter in pain network structures, and increased density of brainstem structures in patients with chronic migraine.52, 73 Kim et al reported a positive correlation between reductions in gray matter volume and increased headache duration and lifetime headache frequency.52 A separate study identified the sites of gray matter reduction to be involved in pain circuitry.73
The MRI CAMERA (Cerebral Abnormalities in Migraine, an Epidemiological Risk Analysis) cross‐sectional study provided further evidence for structural anomalies in the brains of people with migraine. In this study population of 295 migraineurs and 140 age‐ and sex‐matched controls, those with migraine had higher incidence of lesions (eg, subclinical infarcts in the cerebellum/posterior circulation, brainstem hyperintense lesions) and experienced frequent syncope and orthostatic insufficiency/intolerance.53, 74 An updated analysis from this study found that frequent syncope and orthostatic intolerance were independent risk factors for subclinical lesions in both migraineurs and controls (ie, independent of migraine status).74 Nonetheless, female migraineurs without frequent syncope or orthostatic intolerance still remained at 2‐fold higher risk for substantial deep white matter hyperintense lesion load.74 CAMERA also found an increased risk of brain lesions and iron accumulation in those with a longer disease duration or higher migraine frequency, although longitudinal studies are needed to determine whether these lesions have relevant functional correlates and gradually accumulate over time.53 In sum, the investigators in CAMERA are re‐evaluating their study population 8 years after the initial study to determine how these physiologic changes modulate over time.53
These studies suggest that migraine is associated with progressive structural abnormalities in the periaqueductal gray matter and associated deep nuclei, and the affected structures may be functionally impaired in migraine and/or central to the dysregulated trigeminal nociceptive network. Although the role of iron accumulation in the development of chronic migraine remains unclear, several studies have suggested that increased duration since diagnosis and headache frequency may influence overall iron deposition,70, 71 or alternatively be an epiphenomenon. Nevertheless, it is not clear whether these findings correlate with attack frequency, chronicity, and/or migraine‐specific symptomatology (eg, aura), and whether the iron signals are dynamic and potentially reversible with therapy.
Summary of Neurophysiologic, Functional, and Structural Imaging Findings
Taken together, findings from studies using a range of techniques suggest persistent changes in certain brain structures among chronic migraineurs, while fewer or transient changes occur in individuals with episodic migraine, supporting the spectrum model of migraine. It is not clear whether any of these changes were present before clinical symptoms and reflect a fundamental biology of susceptibility, or alternatively, are the signature of consequent abnormalities in pain signaling. Nevertheless, whether pathophysiologic epiphenomenon or etiologic, clear abnormalities are observed on a population basis, suggesting that migraine itself is associated with abnormalities and that, in particular, episodic migraine and chronic migraine may form a continuum. A goal of future research would be to further elucidate how these differences manifest between episodic and chronic migraine, including identifying if any of these changes to brain structures are reversible. In the long run, future research should identify means of preventing the transition to chronic migraine in susceptible individuals.
Insights into the Physiology of Migraine
Our view is that migraine is a “system disorder” associated with both peripheral and central dysfunction;75 linear constructs are over‐simplistic in describing the complex disorder we know clinically as migraine. The nidus of attack onset has not been established, and is likely multifactorial within individuals and heterogeneous across the affected populations. Several possible scenarios suggest extracranial origins of intracranial pain, as well as intracranial origins of extracranial pain, both resulting in local release of proinflammatory mediators.76 The imaging and neurophysiology findings support the premise that cortical dysfunction and hyperexcitability are important components of migraine, which may be caused by peripheral sensitization of the trigeminal nerve and the upper cervical afferents.14, 28, 29 Repeated stimulation of trigeminal fibers may cause increased release of nociceptive neurotransmitters and neuropeptides (eg, CGRP, glutamate, substance P, neurokinin A),28, 29 and/or upregulation of ion channels or sensory receptors (eg, transient receptor potential cation channel ankyrin subfamily member 1 [TRPA1], transient receptor potential cation channel vanilloid subfamily member 1 [TRPV1]) on nociceptive nerve endings.76, 77 These events can sensitize peripheral neurons (ie, meningeal and dural trigeminal sensory afferents) and promote central sensitization,76, 78 which is pervasive in chronic migraine.14
In chronic migraine, central sensitization, observable by the development of cutaneous allodynia, is associated with dysfunctional activity of central trigeminal sensory neurons,76 including spontaneous firing, firing in response to innocuous stimuli, and a reduced firing threshold.19, 32, 78 This aberrant activity may be mediated by increased descending facilitation, impaired descending inhibition of nociceptive activity within the trigeminal cervical complex from key brainstem pain modulatory centers,29 or heightened peripheral nociceptive input (ie, peripheral sensitization). Central sensitization likely contributes to maintaining the pain referral patterns in the trigeminal nerve and the upper cervical afferents29 as well as the persistent pain of chronic migraine.19, 28 Individuals with chronic migraine display persistent cortical hyperexcitability,14 often resulting in cutaneous allodynia (ie, central sensitization) even during the interictal period.39, 41
Careful immunohistochemistry tractology studies revealed that the trigeminal nucleus has direct, single neuronal connections to thalamus, hypothalamus, amygdala, and other deep nuclei.31, 79, 80, 81 Through these pathways and further projections to cortex, peripheral trigeminal stimulation via a sensitized trigeminal nucleus, may contribute to autonomic, limbic and other migraine‐associated dysfunction, in addition to cortical spreading depression. Emerging evidence supports a neurolimbic pain model of migraine, in which ascending and descending connections between the periaqueductal gray and limbic system influence the occurrence of migraine attacks.75 Indeed, evidence suggests that the periaqueductal gray also regulates mood and emotion along with the limbic system, and this interaction may explain the common occurrence of psychiatric comorbidities (eg, depression, anxiety) in people with migraine, especially those with chronic migraine.75
TREATMENT OPTIONS
OnabotulinumtoxinA82 is the only prophylactic treatment globally approved specifically for chronic migraine, and has demonstrated efficacy for up to 56 weeks in the large‐scale PREEMPT (Phase III REsearch Evaluating Migraine Prophylaxis Therapy) trials.83, 84, 85, 86 Although not specifically licensed for chronic migraine, orally administered topiramate87 is an effective prophylactic treatment for patients with migraine,47, 88, 89 and may be effective in patients with chronic migraine.90 To understand how these therapies may affect the underlying migraine physiology, we have summarized their mechanisms of action.
Onabotulinumtoxin A Mechanism of Action
Normally, neuronal stimulation initiates a series of intracellular events that leads to a neuropeptide‐containing vesicle fusing with the nerve cell membrane. This process is facilitated by interaction between proteins on the vesicle (ie, vesicle‐associated membrane protein [VAMP/synaptobrevin]) and on the internal membrane surface (eg, synaptosomal‐associated protein [SNAP‐25]), which together form the soluble N‐ethylmaleimide–sensitive factor attachment protein receptor (SNARE) complex.76, 91, 92 The SNARE complex is fundamental to vesicular trafficking and fusion of the vesicle with the membrane. The molecular biological mechanism of action of onabotulinumtoxinA is well established, whereby it inhibits fusion of intracellular vesicles with the nerve membrane93 by cleaving SNAP‐25.94 By impairing intraneuronal vesicular fusion, onabotulinumtoxinA modulates neuropeptide release and downregulates receptors and ion channels important in nociception.76, 92 This mechanism is important in the current notion of migraine prevention via disrupting the cascade of events that leads to peripheral and central sensitization, as described below.
In chronic migraine, maladaptive pain responses to peripheral chemical or mechanical stimuli result in the peripheral release of neurotransmitters and neuropeptides (eg, CGRP, glutamate, substance P) and/or activation/upregulation of ion channels and receptors on peripheral meningeal nociceptors.76, 91 Peripheral stimulation may also result in the release of proinflammatory cytokines, which activate mast cells and contribute to regional neuroinflammation. The effect of receptor upregulation is to lower the nociceptor threshold for stimulation (the hallmark of peripheral sensitization). As peripheral sensitization builds in chronic migraine, it contributes to central sensitization and the development of cutaneous allodynia or hyperalgesia.76, 95
OnabotulinumtoxinA blocks the release of inflammatory neuropeptides from stimulated trigeminal sensory neurons.76, 77, 91, 96, 97 In vitro, onabotulinumtoxinA inhibits substance P release98 and reduces stimulated, but not basal, release of CGRP.99 A preclinical bladder pain model demonstrated that onabotulinumtoxinA inhibits CGRP release from afferent nerve terminals and significantly reduces pain responses after exposure to acetic acid.100 Receptors such as TRPV1 have been shown to mediate CGRP release, thus leading to neuronal hyperexcitability.101 In the rat formalin‐pain model, onabotulinumtoxinA demonstrated inhibition of several of the neurophysiologic and neurochemical effects of formalin (eg, glutamate release, Fos‐like immunoreactivity, evoked activity of wide dynamic‐range neurons), which are regarded as measures of nociceptive processing.102 A rat migraine model demonstrated that onabotulinumtoxinA administered into craniofacial muscles decreased the effects of glutamate by reducing the mechanical sensitivity of temporalis muscle nociceptors and decreasing blood perfusion, and may attenuate the provoked release of CGRP from muscle nociceptors.103 An in vitro study demonstrated that onabotulinumtoxinA prevents and reverses mechanosensitization in C‐type, but not Aδ‐type, meningeal nociceptors.76 The authors suggest that onabotulinumtoxinA accomplishes this effect either by associating with select C‐type meningeal nociceptor ion channels or by reducing cell surface expression of these channels and/or associated receptors, such as TRPV1 and TRPA1.76 A follow‐up study in an animal model of chronic migraine showed that extracranial administration of onabotulinumtoxinA reduces intracranial responsiveness to TRPV1 and TRPA1 activation 7 days after administration, presumably by inhibiting docking of synaptic vesicles containing TRPV1 and TRPA1 receptors thereby preventing their membrane insertion.104 Surface expression of other receptors, such as P2X3, may also be inhibited by onabotulinumtoxinA.105 Recent clinical data demonstrating a peripheral action of anti‐CGRP monoclonal antibodies106, 107, 108 and CGRP receptor antagonists109 support the notion that onabotulinumtoxinA neuromodulates pain through a peripheral mechanism. In fact, onabotulinumtoxinA has recently been shown to reduce serum CGRP concentration in patients with chronic migraine (pretreatment median, 74.1 pg/mL; 1 month post‐treatment median, 51.9 pg/mL, P < .001).110 Interestingly, one month after treatment, CGRP levels significantly decreased in patients defined as onabotulinumtoxinA responders (pretreatment median, 76.9 pg/mL; post‐treatment median, 52.5 pg/mL; P = .003) but not nonresponders (pretreatment median, 50.5 pg/mL; post‐treatment median, 51.9 pg/mL; P > .05).
Topiramate Mechanism of Action
It is thought that topiramate has dual effects on neurotransmission – enhancing inhibitory effects while minimizing excitatory effects, both of which are implicated in migraine physiology. The pharmacologic mechanisms underlying this antimigraine activity may include regulation of cell membrane ion channels (voltage‐gated sodium and calcium channel blockage, potassium channel activation), modulation of neurotransmitter release (inhibition of glutamate, enhancement of γ‐aminobutyric acid [GABA]‐evoked currents), and inhibition of some carbonic anhydrase isozymes and kainate‐evoked currents.111, 112 Studies have demonstrated topiramate's inhibitory effect on excitability in motor and visual cortices.113, 114, 115 Based on this broad mechanism of action, topiramate may prevent the development of cortical spreading depression by reducing nociceptive transmission and generally inhibiting neuronal hyperexcitability.116 Similarly, topiramate has demonstrated cognitive adverse events, which are likely a reflection of the central inhibitory effects. Pooled analyses of clinical trial results suggest that preventive topiramate treatment in patients with episodic migraine may reduce the risk of headache‐day increase, which in some cases may prevent migraine chronification.23
In summary, the antinociceptive effect of onabotulinumtoxinA has been demonstrated to directly inhibit peripheral sensitization by preventing the release of neurotransmitters and neuropeptides as well as inhibiting membrane expression of relevant ion channels/receptors in the periphery, thereby inhibiting the development of, or attenuating, central sensitization.76, 77, 96 Support for this mechanism of action comes from clinical studies demonstrating suppression of cutaneous allodynia, an indicator of central sensitization, after onabotulinumtoxinA injection in the periorbital skin.117, 118 Topiramate is postulated to both enhance inhibitory and minimize excitatory neurotransmission, and may reduce the risk of progression to chronic migraine; however, this drug is not currently approved for use specifically in people with chronic migraine.
Implications
Clinical, neurophysiologic, and functional imaging studies increasingly support the hypothesis that enduring and pervasive alterations can occur in the brains of individuals with chronic migraine, whereas the changes underlying episodic migraine are predominantly intermittent, occurring during migraine attacks. Migraine should be considered a spectrum disorder with chronic migraine being considered unique from episodic migraine, although its relationship (primarily as a predisposing condition) is acknowledged. Evidence is building that the neuroplastic changes observed during the transition from episodic to chronic migraine also occur in other forms of chronic pain (eg, fibromyalgia, low back pain).119
Given the disability associated with chronic migraine and the substantial interference of this condition with everyday activities, patients with episodic migraine who have risk factors for progression (eg, frequent attacks, acute headache medication use, obesity, snoring, stressful life events) should be monitored closely for headache frequency and chronification.120 Without effective treatment, continued migraine attacks can be associated with structural changes.119 The risk of progression may potentially be reduced through a combined treatment approach in patients at risk of chronification (eg, acute migraine treatments to reduce migraine attack severity, prophylactic medications to decrease migraine frequency). Clinically, treatment of migraine‐related inflammatory symptoms may reverse peripheral and central sensitization; however, medication (eg, triptans) overuse may also increase sensitivity to triggers, potentially increasing the likelihood of sensitization.119 Furthermore, switching triptan regimens may be associated with increased headache‐related disability in some cases.121 Therefore, the concept of appropriate interventional therapy forms the basis of the notion that effective treatment of chronic migraine has the promise of reversing the underlying pathophysiology. Some evidence suggests that modifying some risk factors122, 123, 124 can move a patient from a chronic migraine pattern to an episodic migraine pattern. However, modification of risk factors, when possible, or the use of effective therapy, has not been prospectively demonstrated to prevent chronification.
Patients with chronic migraine require effective, tolerable treatments that provide pain relief, while avoiding serious or intolerable side effects. Treatments that are effective for patients with episodic migraine are not necessarily effective for those with chronic migraine (and vice versa), which may be expected based on the pathophysiologic differences in these conditions noted in this and other articles.125 Abortive medications (eg, triptans) are often effective in patients with episodic migraine, as is the prophylactic medication topiramate, which in one pooled analysis showed reduced risk of increase in headache days, potentially preventing chronification in some patients (see above).23 Consistent with this notion, prophylaxis with an effective treatment may revert a patient's headache frequency down the continuum from chronic to episodic migraine; this was recently demonstrated in a 2‐year open‐label prospective study of patients receiving headache prophylaxis with 195 U of onabotulinumtoxinA every 3 months (±1 week) during a 2‐year period.126 OnabotulinumtoxinA has been shown to be an effective prophylactic treatment for patients with chronic migraine (≥15 headache days per month) in double‐blind, placebo‐controlled clinical trials,83, 86 a prospective real‐life data analysis,127 and a 2‐year open‐label prospective study,126 and may reduce healthcare visits (ie, emergency department, urgent care, hospitalizations) associated with chronic migraine.128 Evidence‐based guidelines concluded that episodic migraine (<15 headache days per month) was not responsive to onabotulinumtoxinA treatment;129 however, the treatment paradigm utilized in the registration clinical trials (PREEMPT) has not been systematically assessed in patients with <15 headache days per month. Although the classification of episodic and chronic migraine is dichotomized at 15 headache days per month, this distinction is primarily empiric, and the pathophysiology and likely treatment responsiveness may be ambiguous at the intersection,24 as would be expected in a complex biologic system disorder. The next phase of research should aim to assess whether treatments for chronic migraine work in individuals with high‐frequency episodic migraine, to more precisely define a headache‐day frequency threshold for the pathophysiologic shift.
CONCLUSION
As a complex spectrum disorder, the recurring migraine clinical and pathophysiological features may evolve over time, due to decreased nociceptive thresholds in vulnerable individuals, the hallmark of peripheral and central sensitization. Neurophysiological and functional imaging studies show changes in baseline neurologic function between migraine attacks in people with chronic (as opposed to episodic) migraine, and suggest that chronic migraine is associated with progressive brain dysfunction.
Differences in physiology between episodic and chronic migraineurs highlight the need for state‐specific and effective treatments for these patient populations. Topiramate is an effective option for patients with migraine, and has been suggested to prevent chronification.23 For those who have already progressed to chronic migraine, onabotulinumtoxinA is an effective prophylactic option that can interrupt the clinical signs attributed to the pathological cascade of events causing peripheral sensitization, via direct inhibition of the peripheral release of neurotransmitters and neuropeptides, and surface expression of relevant membrane receptors, which then indirectly blocks central sensitization. The clinical trial data have shown that, for some patients, onabotulinumtoxinA reduces the number of headache days per month into the range seen with episodic migraine,83, 84, 85, 86 and it is appealing to consider that by effectively decreasing the frequency of chronic migraine to the range of episodic migraine, this prophylactic treatment might mitigate the chronification pathophysiology seen in patients with chronic migraine. Notwithstanding ambiguity at the intersection, further research into underlying differences between episodic and chronic migraine will continue to advance science, and likely provide further effective treatments.
Acknowledgments
Writing and editorial assistance was provided to the authors by Amanda M. Kelly, MPhil, MSHN, of Complete Healthcare Communications, LLC (Chadds Ford, PA), a CHC Group company, and Kristine W. Schuler, MS, and funded by Allergan plc (Dublin, Ireland).
Conflicts of Interest: Dr. Aurora has received consulting fees/honoraria from Allergan, eNeura, Merck, and Teva, and speaker's bureau participation for Allergan. Dr. Brin is an employee of Allergan plc, and receives stock or stock options in Allergan.
Funding: This review was sponsored by Allergan plc (Dublin, Ireland). Both authors met the ICMJE authorship criteria. Neither honoraria nor payments were made for authorship.
Dr. Aurora (first author) expressed a desire to update a previous review article written by her on this topic and suggested a collaboration with Dr. Brin (Allergan). Allergan (Dr. Brin) provided topic ideas on this manuscript to Complete Healthcare Communications (Chadds Ford, PA). With the exception of Dr. Brin, other individuals at Allergan were not involved in the development of the manuscript with the author or the vendor. Allergan had the opportunity to review the final version of the manuscript and provide comments; however, the authors maintained complete control over the content of the paper.
REFERENCES
- 1. Bigal ME, Rapoport AM, Lipton RB, Tepper SJ, Sheftell FD. Assessment of migraine disability using the Migraine Disability Assessment (MIDAS) questionnaire: A comparison of chronic migraine with episodic migraine. Headache. 2003;43:336‐342. [DOI] [PubMed] [Google Scholar]
- 2. Buse DC, Manack AN, Fanning KM, et al. Chronic migraine prevalence, disability, and sociodemographic factors: Results from the American Migraine Prevalence and Prevention Study. Headache. 2012;52:1456‐1470. [DOI] [PubMed] [Google Scholar]
- 3. Adams AM, Serrano D, Buse DC, et al. The impact of chronic migraine: The Chronic Migraine Epidemiology and Outcomes (CaMEO) Study methods and baseline results. Cephalalgia. 2015;35:563–578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Aurora SK, Kulthia A, Barrodale PM. Mechanism of chronic migraine. Curr Pain Headache Rep. 2011;15:57‐63. [DOI] [PubMed] [Google Scholar]
- 5. Wrobel Goldberg S, Silberstein SD. Targeting CGRP: A new era for migraine treatment. CNS Drugs. 2015;29:443‐452. [DOI] [PubMed] [Google Scholar]
- 6. Mitsikostas DD, Rapoport AM. New players in the preventive treatment of migraine. BMC Med. 2015;13:279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Mathew NT, Stubits E, Nigam MP. Transformation of episodic migraine into daily headache: Analysis of factors. Headache. 1982;22:66‐68. [DOI] [PubMed] [Google Scholar]
- 8. Headache Classification Committee of the International Headache Society . The International Classification of Headache Disorders, 3rd edition (beta version). Cephalalgia . 2013;33:629‐808. [DOI] [PubMed] [Google Scholar]
- 9. Welch KM, Nagesh V, Aurora SK, Gelman N. Periaqueductal gray matter dysfunction in migraine: Cause or the burden of illness? Headache. 2001;41:629‐637. [DOI] [PubMed] [Google Scholar]
- 10. Weiller C, May A, Limmroth V, et al. Brain stem activation in spontaneous human migraine attacks. Nat Med. 1995;1:658‐660. [DOI] [PubMed] [Google Scholar]
- 11. Afridi SK, Matharu MS, Lee L, et al. A PET study exploring the laterality of brainstem activation in migraine using glyceryl trinitrate. Brain. 2005;128:932‐939. [DOI] [PubMed] [Google Scholar]
- 12. Aurora SK, Barrodale PM, Tipton RL, Khodavirdi A. Brainstem dysfunction in chronic migraine as evidenced by neurophysiological and positron emission tomography studies. Headache. 2007;47:996‐1003. discussion 1004‐1007. [DOI] [PubMed] [Google Scholar]
- 13. Kitaj MB, Klink M. Pain thresholds in daily transformed migraine versus episodic migraine headache patients. Headache. 2005;45:992‐998. [DOI] [PubMed] [Google Scholar]
- 14. Aurora SK. Is chronic migraine one end of a spectrum of migraine or a separate entity? Cephalalgia. 2009;29:597‐605. [DOI] [PubMed] [Google Scholar]
- 15. Olesen J, Bousser MG, Diener HC, et al. New appendix criteria open for a broader concept of chronic migraine. Cephalalgia. 2006;26:742‐746. [DOI] [PubMed] [Google Scholar]
- 16. Olesen J, Steiner TJ. The International classification of headache disorders, 2nd edn (ICDH‐II). J Neurol Neurosurg Psychiatry. 2004;75:808‐811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Dodick DW. Clinical practice. Chronic daily headache. N Engl J Med. 2006;354:158‐165. [DOI] [PubMed] [Google Scholar]
- 18. Natoli JL, Manack A, Dean B, et al. Global prevalence of chronic migraine: A systematic review. Cephalalgia. 2010;30:599‐609. [DOI] [PubMed] [Google Scholar]
- 19. Bigal ME, Lipton RB. Concepts and mechanisms of migraine chronification. Headache. 2008;48:7‐15. [DOI] [PubMed] [Google Scholar]
- 20. Scher AI, Midgette LA, Lipton RB. Risk factors for headache chronification. Headache. 2008;48:16‐25. [DOI] [PubMed] [Google Scholar]
- 21. Bigal ME, Rapoport AM, Sheftell FD, Tepper SJ, Lipton RB. Chronic migraine is an earlier stage of transformed migraine in adults. Neurology. 2005;65:1556‐1561. [DOI] [PubMed] [Google Scholar]
- 22. Scher AI, Stewart WF, Ricci JA, Lipton RB. Factors associated with the onset and remission of chronic daily headache in a population‐based study. Pain. 2003;106:81‐89. [DOI] [PubMed] [Google Scholar]
- 23. Limmroth V, Biondi D, Pfeil J, Schwalen S. Topiramate in patients with episodic migraine: Reducing the risk for chronic forms of headache. Headache. 2007;47:13‐21. [DOI] [PubMed] [Google Scholar]
- 24. Lipton RB, Penzien DB, Turner DP, Smitherman TA, Houle TT. Methodological issues in studying rates and predictors of migraine progression and remission. Headache. 2013;53:930‐934. [DOI] [PubMed] [Google Scholar]
- 25. Louter M, Fernandez‐Morales J, de Vries B, et al. Candidate‐gene association study searching for genetic factors involved in migraine chronification. Cephalalgia. 2015;35:500–507. [DOI] [PubMed] [Google Scholar]
- 26. Ophoff RA, Terwindt GM, Vergouwe MN, et al. Familial hemiplegic migraine and episodic ataxia type‐2 are caused by mutations in the Ca2+ channel gene CACNL1A4. Cell. 1996;87:543‐552. [DOI] [PubMed] [Google Scholar]
- 27. De Fusco M, Marconi R, Silvestri L, et al. Haploinsufficiency of ATP1A2 encoding the Na+/K+ pump alpha2 subunit associated with familial hemiplegic migraine type 2. Nat Genet. 2003;33:192‐196. [DOI] [PubMed] [Google Scholar]
- 28. Pietrobon D. Migraine: New molecular mechanisms. Neuroscientist. 2005;11:373‐386. [DOI] [PubMed] [Google Scholar]
- 29. Pietrobon D, Moskowitz MA. Pathophysiology of migraine. Annu Rev Physiol. 2013;75:365‐391. [DOI] [PubMed] [Google Scholar]
- 30. Lauritzen M. Pathophysiology of the migraine aura. The spreading depression theory. Brain. 1994;117(Pt 1):199‐210. [DOI] [PubMed] [Google Scholar]
- 31. Noseda R, Burstein R. Migraine pathophysiology: Anatomy of the trigeminovascular pathway and associated neurological symptoms, cortical spreading depression, sensitization, and modulation of pain. Pain. 2013;154(Suppl.1):S44‐53. [DOI] [PubMed] [Google Scholar]
- 32. Cady RK, Schreiber CP, Farmer KU. Understanding the patient with migraine: The evolution from episodic headache to chronic neurologic disease. A proposed classification of patients with headache. Headache. 2004;44:426‐435. [DOI] [PubMed] [Google Scholar]
- 33. Mongini F, Keller R, Deregibus A, Barbalonga E, Mongini T. Frontal lobe dysfunction in patients with chronic migraine: A clinical‐neuropsychological study. Psychiatry Res. 2005;133:101‐106. [DOI] [PubMed] [Google Scholar]
- 34. de Tommaso M, Losito L, Difruscolo O, Libro G, Guido M, Livrea P. Changes in cortical processing of pain in chronic migraine. Headache. 2005;45:1208‐1218. [DOI] [PubMed] [Google Scholar]
- 35. Obermann M, Gizewski ER, Limmroth V, Diener HC, Katsarava Z. Symptomatic migraine and pontine vascular malformation: Evidence for a key role of the brainstem in the pathophysiology of chronic migraine. Cephalalgia. 2006;26:763‐766. [DOI] [PubMed] [Google Scholar]
- 36. Noseda R, Jakubowski M, Kainz V, Borsook D, Burstein R. Cortical projections of functionally identified thalamic trigeminovascular neurons: Implications for migraine headache and its associated symptoms. J Neurosci. 2011;31:14204‐14217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Cooke L, Eliasziw M, Becker WJ. Cutaneous allodynia in transformed migraine patients. Headache. 2007;47:531‐539. [DOI] [PubMed] [Google Scholar]
- 38. Burstein R, Yarnitsky D, Goor‐Aryeh I, Ransil BJ, Bajwa ZH. An association between migraine and cutaneous allodynia. Ann Neurol. 2000;47:614‐624. [PubMed] [Google Scholar]
- 39. Bigal ME, Ashina S, Burstein R, et al. Prevalence and characteristics of allodynia in headache sufferers: A population study. Neurology. 2008;70:1525‐1533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Lovati C, D'Amico D, Rosa S, et al. Allodynia in different forms of migraine. Neurol Sci. 2007;28(Suppl.2):S220‐221. [DOI] [PubMed] [Google Scholar]
- 41. Zappaterra M, Guerzoni S, Cainazzo MM, Ferrari A, Pini LA. Basal cutaneous pain threshold in headache patients. J Headache Pain. 2011;12:303‐310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. de Tommaso M, Valeriani M, Guido M, et al. Abnormal brain processing of cutaneous pain in patients with chronic migraine. Pain. 2003;101:25‐32. [DOI] [PubMed] [Google Scholar]
- 43. Bigal ME, Serrano D, Reed M, Lipton RB. Chronic migraine in the population: Burden, diagnosis, and satisfaction with treatment. Neurology. 2008;71:559‐566. [DOI] [PubMed] [Google Scholar]
- 44. Penzien DB, Rains JC, Lipton RB. Introduction to the special series on the chronification of headache: Mechanism, risk factors, and behavioral strategies aimed at primary and secondary prevention of chronic headache. Headache. 2008;48:5‐6. [Google Scholar]
- 45. Ferrari A, Leone S, Vergoni AV, et al. Similarities and differences between chronic migraine and episodic migraine. Headache. 2007;47:65‐72. [DOI] [PubMed] [Google Scholar]
- 46. Lipton RB. Tracing transformation: Chronic migraine classification, progression, and epidemiology. Neurology. 2009;72:S3‐7. [DOI] [PubMed] [Google Scholar]
- 47. Silberstein SD, Dodick D, Freitag F, et al. Pharmacological approaches to managing migraine and associated comorbidities–clinical considerations for monotherapy versus polytherapy. Headache. 2007;47:585‐599. [DOI] [PubMed] [Google Scholar]
- 48. Adams AM, Serrano D, Buse DC, et al. The impact of chronic migraine: The Chronic Migraine Epidemiology and Outcomes (CaMEO) Study methods and baseline results. Cephalalgia. 2015;35:536–578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Freitag FG, Kozma CM, Slaton T, Osterhaus JT, Barron R. Characterization and prediction of emergency department use in chronic daily headache patients. Headache. 2005;45:891‐898. [DOI] [PubMed] [Google Scholar]
- 50. Munakata J, Hazard E, Serrano D, et al. Economic burden of transformed migraine: Results from the American Migraine Prevalence and Prevention (AMPP) study. Headache. 2009;49:498‐508. [DOI] [PubMed] [Google Scholar]
- 51. Goadsby PJ. Advances in the understanding of headache. Br Med Bull. 2005;73‐74:83‐92. [DOI] [PubMed] [Google Scholar]
- 52. Kim JH, Suh SI, Seol HY, et al. Regional grey matter changes in patients with migraine: A voxel‐based morphometry study. Cephalalgia. 2008;28:598‐604. [DOI] [PubMed] [Google Scholar]
- 53. Kruit M, van BM, Launer L, Terwindt G, Ferrari M. Migraine is associated with an increased risk of deep white matter lesions, subclinical posterior circulation infarcts and brain iron accumulation: The population‐based MRI CAMERA study. Cephalalgia. 2010;30:129‐136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Kurth T, Mohamed S, Maillard P, et al. Headache, migraine, and structural brain lesions and function: Population based Epidemiology of Vascular Ageing‐MRI study. BMJ. 2011;342:c7357‐ [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Granziera C, DaSilva AF, Snyder J, Tuch DS, Hadjikhani N. Anatomical alterations of the visual motion processing network in migraine with and without aura. PLoS Med. 2006;3:e402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Matharu MS, T Bartsch, N Ward, RS Frackowiak, R Weiner, PJ Goadsby. Central neuromodulation in chronic migraine patients with suboccipital stimulators: a PET study. Brain. 2004;127:220‐30. [DOI] [PubMed] [Google Scholar]
- 57. Chen WT, Wang SJ, Fuh JL, Lin CP, Ko YC, Lin YY. Persistent ictal‐like visual cortical excitability in chronic migraine. Pain. 2011;152:254‐258. [DOI] [PubMed] [Google Scholar]
- 58. Aurora SK, Barrodale P, Chronicle EP, Mulleners WM. Cortical inhibition is reduced in chronic and episodic migraine and demonstrates a spectrum of illness. Headache. 2005;45:546‐552. [DOI] [PubMed] [Google Scholar]
- 59. Moliadze V, Zhao Y, Eysel U, Funke K. Effect of transcranial magnetic stimulation on single‐unit activity in the cat primary visual cortex. J Physiol. 2003;553:665‐679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Mulleners WM, Chronicle EP, Palmer JE, Koehler PJ, Vredeveld JW. Suppression of perception in migraine: Evidence for reduced inhibition in the visual cortex. Neurology. 2001;56:178‐183. [DOI] [PubMed] [Google Scholar]
- 61. Aurora SK. Spectrum of illness: Understanding biological patterns and relationships in chronic migraine. Neurology. 2009;72:S8‐13. [DOI] [PubMed] [Google Scholar]
- 62. Srikiatkhachorn A. Towards the better understanding about pathogenesis of chronic daily headache. J Med Assoc Thai. 2006;89(Suppl. 3):S234‐243. [PubMed] [Google Scholar]
- 63. Welch KM, Cao Y, Aurora S, Wiggins G, Vikingstad EM. MRI of the occipital cortex, red nucleus, and substantia nigra during visual aura of migraine. Neurology. 1998;51:1465‐1469. [DOI] [PubMed] [Google Scholar]
- 64. Cao Y, Aurora SK, Nagesh V, Patel SC, Welch KM. Functional MRI‐BOLD of brainstem structures during visually triggered migraine. Neurology. 2002;59:72‐78. [DOI] [PubMed] [Google Scholar]
- 65. Chudler EH, Dong WK. The role of the basal ganglia in nociception and pain. Pain. 1995;60:3‐38. [DOI] [PubMed] [Google Scholar]
- 66. Brown LL, Schneider JS, Lidsky TI. Sensory and cognitive functions of the basal ganglia. Curr Opin Neurobiol. 1997;7:157‐163. [DOI] [PubMed] [Google Scholar]
- 67. Iadarola MJ, Berman KF, Zeffiro TA, et al. Neural activation during acute capsaicin‐evoked pain and allodynia assessed with PET. Brain. 1998;121 (Pt 5):931‐947. [DOI] [PubMed] [Google Scholar]
- 68. Hadjikhani N, Sanchez Del Rio M, Wu O, et al. Mechanisms of migraine aura revealed by functional MRI in human visual cortex. Proc Natl Acad Sci U S A. 2001;98:4687‐4692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Bowyer SM, Aurora KS, Moran JE, Tepley N, Welch KM. Magnetoencephalographic fields from patients with spontaneous and induced migraine aura. Ann Neurol. 2001;50:582‐587. [DOI] [PubMed] [Google Scholar]
- 70. Tepper SJ, Lowe MJ, Beall E, et al. Iron deposition in pain‐regulatory nuclei in episodic migraine and chronic daily headache by MRI. Headache. 2012;52:236‐243. [DOI] [PubMed] [Google Scholar]
- 71. Kruit MC, Launer LJ, Overbosch J, van Buchem MA, Ferrari MD. Iron accumulation in deep brain nuclei in migraine: A population‐based magnetic resonance imaging study. Cephalalgia. 2009;29:351‐359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Mainero C, Boshyan J, Hadjikhani N. Altered functional magnetic resonance imaging resting‐state connectivity in periaqueductal gray networks in migraine. Ann Neurol. 2011;70:838‐845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Valfre W, Rainero I, Bergui M, Pinessi L. Voxel‐based morphometry reveals gray matter abnormalities in migraine. Headache. 2008;48:109‐117. [DOI] [PubMed] [Google Scholar]
- 74. Kruit MC, Thijs RD, Ferrari MD, Launer LJ, van Buchem MA, van Dijk JG. Syncope and orthostatic intolerance increase risk of brain lesions in migraineurs and controls. Neurology. 2013;80:1958‐1965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Maizels M, Aurora S, Heinricher M. Beyond Neurovascular: Migraine as a dysfunctional neurolimbic pain network. Headache. 2012;52:1553–1565. [DOI] [PubMed] [Google Scholar]
- 76. Burstein R, Zhang X, Levy D, Aoki KR, Brin MF. Selective inhibition of meningeal nociceptors by botulinum neurotoxin type A: Therapeutic implications for migraine and other pains. Cephalalgia. 2014;34:853‐869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Aoki KR, Francis J. Updates on the antinociceptive mechanism hypothesis of botulinum toxin A. Parkinsonism Relat Disord. 2011;17:S28‐33. [DOI] [PubMed] [Google Scholar]
- 78. Dodick D, Silberstein S. Central sensitization theory of migraine: Clinical implications. Headache. 2006;46(Suppl. 4):S182‐191. [DOI] [PubMed] [Google Scholar]
- 79. Burstein R, Yamamura H, Malick A, Strassman AM. Chemical stimulation of the intracranial dura induces enhanced responses to facial stimulation in brain stem trigeminal neurons. J Neurophysiol. 1998;79:964‐982. [DOI] [PubMed] [Google Scholar]
- 80. Malick A, Burstein R. Cells of origin of the trigeminohypothalamic tract in the rat. J Comp Neurol. 1998;400:125‐144. [DOI] [PubMed] [Google Scholar]
- 81. Malick A, Strassman RM, Burstein R. Trigeminohypothalamic and reticulohypothalamic tract neurons in the upper cervical spinal cord and caudal medulla of the rat. J Neurophysiol. 2000;84:2078‐2112. [DOI] [PubMed] [Google Scholar]
- 82. BOTOX (onabotulinumtoxinA for injection, for intramuscular, intradetrusor, or intradermal use). Irvine, CA: Full Prescribing Information, Allergan, Inc; 2013. [Google Scholar]
- 83. Aurora SK, Dodick DW, Turkel CC, et al. OnabotulinumtoxinA for treatment of chronic migraine: Results from the double‐blind, randomized, placebo‐controlled phase of the PREEMPT 1 trial. Cephalalgia. 2010;30:793‐803. [DOI] [PubMed] [Google Scholar]
- 84. Aurora SK, Winner P, Freeman MC, et al. OnabotulinumtoxinA for treatment of chronic migraine: Pooled analyses of the 56‐week PREEMPT clinical program. Headache. 2011;51:1358‐1373. [DOI] [PubMed] [Google Scholar]
- 85. Diener HC, Dodick DW, Aurora SK, et al. OnabotulinumtoxinA for treatment of chronic migraine: Results from the double‐blind, randomized, placebo‐controlled phase of the PREEMPT 2 trial. Cephalalgia. 2010;30:804‐814. [DOI] [PubMed] [Google Scholar]
- 86. Dodick DW, Turkel CC, DeGryse RE, et al. OnabotulinumtoxinA for treatment of chronic migraine: Pooled results from the double‐blind, randomized, placebo‐controlled phases of the PREEMPT clinical program. Headache. 2010;50:921‐936. [DOI] [PubMed] [Google Scholar]
- 87. Topamax (topiramate) tablets or sprinkle capsules for oral use Full Prescribing Information, Titusville, NJ: Janssen Pharmaceuticals, Inc; 2012. [Google Scholar]
- 88. Diener HC, Bussone G, Van Oene JC, Lahaye M, Schwalen S, Goadsby PJ. Topiramate reduces headache days in chronic migraine: A randomized, double‐blind, placebo‐controlled study. Cephalalgia. 2007;27:814‐823. [DOI] [PubMed] [Google Scholar]
- 89. Mathew NT, Jaffri SF. A double‐blind comparison of onabotulinumtoxina (BOTOX) and topiramate (TOPAMAX) for the prophylactic treatment of chronic migraine: A pilot study. Headache. 2009;49:1466‐1478. [DOI] [PubMed] [Google Scholar]
- 90. Cady RK, Schreiber CP, Porter JA, Blumenfeld AM, Farmer KU. A multi‐center double‐blind pilot comparison of onabotulinumtoxinA and topiramate for the prophylactic treatment of chronic migraine. Headache. 2011;51:21‐32. [DOI] [PubMed] [Google Scholar]
- 91. Aoki KR. Evidence for antinociceptive activity of botulinum toxin type A in pain management. Headache. 2003;43:S9‐15. [DOI] [PubMed] [Google Scholar]
- 92. Whitcup SM, Turkel CC, DeGryse RE, Brin MF. Development of onabotulinumtoxinA for chronic migraine. Ann N Y Acad Sci. 2014;1329:67‐80. [DOI] [PubMed] [Google Scholar]
- 93. Simpson LL. The origin, structure, and pharmacological activity of botulinum toxin. Pharmacol Rev. 1981;33:155‐188. [PubMed] [Google Scholar]
- 94. Blasi J, Chapman ER, Link E, et al. Botulinum neurotoxin A selectively cleaves the synaptic protein SNAP‐25. Nature. 1993;365:160‐163. [DOI] [PubMed] [Google Scholar]
- 95. Dolly JO, Aoki KR. The structure and mode of action of different botulinum toxins. Eur J Neurol. 2006;13(Suppl.4):1‐9. [DOI] [PubMed] [Google Scholar]
- 96. Aoki KR. Review of a proposed mechanism for the antinociceptive action of botulinum toxin type A. Neurotoxicology. 2005;26:785‐793. [DOI] [PubMed] [Google Scholar]
- 97. Hargreaves R. New migraine and pain research. Headache. 2007;47:S26‐S43. [DOI] [PubMed] [Google Scholar]
- 98. Welch MJ, Purkiss JR, Foster KA. Sensitivity of embryonic rat dorsal root ganglia neurons to Clostridium botulinum neurotoxins. Toxicon. 2000;38:245‐258. [DOI] [PubMed] [Google Scholar]
- 99. Durham PL, Cady R. Regulation of calcitonin gene‐related peptide secretion from trigeminal nerve cells by botulinum toxin type A: Implications for migraine therapy. Headache. 2004;44:35‐42. discussion 42‐33. [DOI] [PubMed] [Google Scholar]
- 100. Chuang YC, Yoshimura N, Huang CC, Chiang PH, Chancellor MB. Intravesical botulinum toxin A administration produces analgesia against acetic acid induced bladder pain responses in rats. J Urol. 2004;172:1529‐1532. [DOI] [PubMed] [Google Scholar]
- 101. Meng J, Ovsepian SV, Wang J, et al. Activation of TRPV1 mediates calcitonin gene‐related peptide release, which excites trigeminal sensory neurons and is attenuated by a retargeted botulinum toxin with anti‐nociceptive potential. J Neurosci. 2009;29:4981‐4992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Cui M, Khanijou S, Rubino J, Aoki KR. Subcutaneous administration of botulinum toxin A reduces formalin‐induced pain. Pain. 2004;107:125‐133. [DOI] [PubMed] [Google Scholar]
- 103. Gazerani P, Au S, Dong X, Kumar U, Arendt‐Nielsen L, Cairns BE. Botulinum neurotoxin type A (BoNTA) decreases the mechanical sensitivity of nociceptors and inhibits neurogenic vasodilation in a craniofacial muscle targeted for migraine prophylaxis. Pain. 2010;151:606‐616. [DOI] [PubMed] [Google Scholar]
- 104. Zhang X, Strassman AM, Novack V, Brin MF, Burstein R. Extracranial injections of botulinum neurotoxin type A inhibit intracranial meningeal nociceptors' responses to stimulatin of TRPV1 and TRPA1 channels: Are we getting closer to solving this puzzle? Cephalalgia. 2016;36:875‐886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Apostolidis A, Popat R, Yiangou Y, et al. Decreased sensory receptors P2X3 and TRPV1 in suburothelial nerve fibers following intradetrusor injections of botulinum toxin for human detrusor overactivity. J Urol. 2005;174:977‐982. discussion 982‐973. [DOI] [PubMed] [Google Scholar]
- 106. Dodick DW, Goadsby PJ, Spierings EL, Scherer JC, Sweeney SP, Grayzel DS. Safety and efficacy of LY2951742, a monoclonal antibody to calcitonin gene‐related peptide, for the prevention of migraine: A phase 2, randomised, double‐blind, placebo‐controlled study. Lancet Neurol. 2014;13:885‐892. [DOI] [PubMed] [Google Scholar]
- 107. Dodick DW, Goadsby PJ, Silberstein SD, et al. Safety and efficacy of ALD403, an antibody to calcitonin gene‐related peptide, for the prevention of frequent episodic migraine: A randomised, double‐blind, placebo‐controlled, exploratory phase 2 trial. Lancet Neurol. 2014;13:1100‐1107. [DOI] [PubMed] [Google Scholar]
- 108. Bigal ME, Edvinsson L, Rapoport AM, et al. Safety, tolerability, and efficacy of TEV‐48125 for preventive treatment of chronic migraine: A multicentre, randomised, double‐blind, placebo‐controlled, phase 2b study. Lancet Neurol. 2015;14:1091‐1100. [DOI] [PubMed] [Google Scholar]
- 109. Ho TW, Ferrari MD, Dodick DW, et al. Efficacy and tolerability of MK‐0974 (telcagepant), a new oral antagonist of calcitonin gene‐related peptide receptor, compared with zolmitriptan for acute migraine: A randomised, placebo‐controlled, parallel‐treatment trial. Lancet. 2008;372:2115‐2123. [DOI] [PubMed] [Google Scholar]
- 110. Cernuda‐Morollon E, Ramon C, Martinez‐Camblor P, Serrano‐Pertierra E, Larrosa D, Pascual J. OnabotulinumtoxinA decreases interictal CGRP plasma levels in patients with chronic migraine. Pain. 2015;156:820‐824. [DOI] [PubMed] [Google Scholar]
- 111. Shank RP, Gardocki JF, Streeter AJ, Maryanoff BE. An overview of the preclinical aspects of topiramate: Pharmacology, pharmacokinetics, and mechanism of action. Epilepsia. 2000;41(Suppl. 1):S3‐9. [PubMed] [Google Scholar]
- 112. Silberstein SD. Topiramate in migraine prevention. Headache. 2005;45(Suppl. 1):S57‐65. [DOI] [PubMed] [Google Scholar]
- 113. Silberstein SD, Goadsby PJ. Migraine: Preventive treatment. Cephalalgia. 2002;22:491‐512. [DOI] [PubMed] [Google Scholar]
- 114. Aurora SK, Barrodale PM, Vermaas AR, Rudra CB. Topiramate modulates excitability of the occipital cortex when measured by transcranial magnetic stimulation. Cephalalgia. 2010;30:648‐654. [DOI] [PubMed] [Google Scholar]
- 115. Artemenko AR, Kurenkov AL, Filatova EG, Nikitin SS, Kaube H, Katsarava Z. Effects of topiramate on migraine frequency and cortical excitability in patients with frequent migraine. Cephalalgia. 2008;28:203‐208. [DOI] [PubMed] [Google Scholar]
- 116. Ruiz L, Ferrandi D. Topiramate in migraine progression. J Headache Pain. 2009;10:419‐422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Gazerani P, Pedersen NS, Staahl C, Drewes AM, Arendt‐Nielsen L. Subcutaneous botulinum toxin type A reduces capsaicin‐induced trigeminal pain and vasomotor reactions in human skin. Pain. 2009;141:60‐69. [DOI] [PubMed] [Google Scholar]
- 118. Gazerani P, Staahl C, Drewes AM, Arendt‐Nielsen L. The effects of botulinum toxin type A on capsaicin‐evoked pain, flare, and secondary hyperalgesia in an experimental human model of trigeminal sensitization. Pain. 2006;122:315‐325. [DOI] [PubMed] [Google Scholar]
- 119. Puretic MB, Demarin V. Neuroplasticity mechanisms in the pathophysiology of chronic pain. Acta Clin Croat. 2012;51:425‐429. [PubMed] [Google Scholar]
- 120. Lipton RB, Bigal ME. Looking to the future: Research designs for study of headache disease progression. Headache. 2008;48:58‐66. [DOI] [PubMed] [Google Scholar]
- 121. Serrano D, Buse DC, Kori SH, et al. Effects of switching acute treatment on disability in migraine patients using triptans. Headache. 2013;53:1415‐1429. [DOI] [PubMed] [Google Scholar]
- 122. Calhoun AH, Ford S. Behavioral sleep modification may revert transformed migraine to episodic migraine. Headache. 2007;47:1178‐1183. [DOI] [PubMed] [Google Scholar]
- 123. Calhoun A, Ford S. Elimination of menstrual‐related migraine beneficially impacts chronification and medication overuse. Headache. 2008;48:1186‐1193. [DOI] [PubMed] [Google Scholar]
- 124. Rains JC. Chronic headache and potentially modifiable risk factors: Screening and behavioral management of sleep disorders. Headache. 2008;48:32‐39. [DOI] [PubMed] [Google Scholar]
- 125. Mathew NT. Pathophysiology of chronic migraine and mode of action of preventive medications. Headache. 2011;51(Suppl. 2):84‐92. [DOI] [PubMed] [Google Scholar]
- 126. Negro A, Curto M, Lionetto L, Martelletti P. A two years open‐label prospective study of onabotulinumtoxinA 195 U in medication overuse headache: A real‐world experience. J Headache Pain. 2015;17:1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127. Khalil M, Zafar HW, Quarshie V, Ahmed F. Prospective analysis of the use of onabotulinumtoxinA (BOTOX) in the treatment of chronic migraine; real‐life data in 254 patients from Hull, U.K. J Headache Pain. 2014;15:54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128. Rothrock JF, Bloudek LM, Houle TT, Andress‐Rothrock D, Varon SF. Real‐world economic impact of onabotulinumtoxinA in patients with chronic migraine. Headache. 2014;54:1565‐1573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129. Naumann M, So Y, Argoff CE, et al. Assessment: Botulinum neurotoxin in the treatment of autonomic disorders and pain (an evidence‐based review): Report of the Therapeutics and Technology Assessment Subcommittee of the American Academy of Neurology. Neurology. 2008;70:1707‐1714. [DOI] [PubMed] [Google Scholar]