

Abstract
Many patents for the first biologicals derived from recombinant technology and, more recently, monoclonal antibodies (mAbs) are expiring. Naturally, biosimilars are becoming an increasingly important area of interest for the pharmaceutical industry worldwide, not only for emergent countries that need to import biologic products. This review shows the evolution of biosimilar development regarding regulatory, manufacturing bioprocess, comparability, and marketing. The regulatory landscape is evolving globally, whereas analytical structure and functional analyses provide the foundation of a biosimilar development program. The challenges to develop and demonstrate biosimilarity should overcome the inherent differences in the bioprocess manufacturing and physicochemical and biological characterization of a biosimilar compared to several lots of the reference product. The implementation of approaches, such as Quality by Design (QbD), will provide products with defined specifications in relation to quality, purity, safety, and efficacy that were not possible when the reference product was developed. Actually, the need to prove comparability to the reference product by the biosimilar industry has increased the knowledge about the product and the production‐process associated by the use of powerful analytical tools. The technological challenges to make copies of biologic products while attending regulatory and market demands are expected to help innovation in the direction of attaining more productive manufacturing processes. © 2015 American Institute of Chemical Engineers Biotechnol. Prog., 31:1139–1149, 2015
Keywords: biosimilars, monoclonal antibodies, high‐order structure, bioassays, comparability
Introduction
Biologic medicines, or biologics, are large molecules or complex proteins obtained by genetically engineering eukaryotic or prokaryotic cell lines or through biological sources, such as cells or tissues, and they are used to treat or cure diseases. The first recombinant biologic medicine approved for therapeutic use was human insulin in 1982 in the US.1 Since then, the number of recombinant biologic medicines approved for human use, including monoclonal antibodies (mAbs), has increased considerably. Patents on the first recombinant biologic medicines expired, and companies have developed biosimilars of those products, followed, more recently, with patents expiration on the first approved mAbs.2 The companies planning to produce biosimilars need to reverse‐engineer the complex manufacturing processes used to produce and purify these drugs. Because of their complexity and process specificity, it is impossible to produce biosimilars with exactly the same characteristics of the reference medicine. Furthermore, despite recent advances in technologies used to manufacture, purify, and test biosimilars, laboratory testing is not always sufficient to prove bioequivalence.3 Regulatory agencies, such as the European Medicines Agency (EMA) and US Food and Drug Administration (FDA), along with other regulated countries, require pre‐clinical and clinical comparability studies to demonstrate safety and efficacy, and request additional post‐manufacturing assays to look for immunogenicity of biosimilars.4, 5, 6 A paradigm shift avoiding the use of animals in pre‐clinical studies was recently published by the European Union (EU), through a science‐based approach.7
The first biosimilar product approved in Europe, following the EMA's new approach, was somatropin (2006) followed by erythropoietin (2007) and filgrastim (2008). In 2013, the first mAb biosimilar, infliximab, was approved in Europe.8 The EMA requirements to approve biosimilars vary according to the class of molecule, and decisions occur case by case. The EMA website displays the current market authorization for 19 biosimilars, including follitropin alfa and insulin glargine.8 The FDA began the process of developing regulatory requirements for biosimilars when President Barack Obama signed the Patient Protection and Affordable Care Act (PPAC Act) into law on 23rd March, 2010. The relevant statutory provisions are also referred to as the Biologics Price Competition and Innovation Act (BPCI Act) of 2009.6 By forming regulatory pathways based on a totality‐of‐the‐evidence approach for highly complex drugs, both the FDA and EMA will ensure that patients obtain access to the best care possible while continuing to ensure that safety remains a top priority.9 Regulatory legislation for biosimilars have clarified many aspects of their development, and many countries have adopted their own guidelines based on the EMA and World Health Organization (WHO) publications. However, there are some challenges in areas such as bioanalytics and comparability assay development, the definitions of biological activity under clinical view, the global harmonization of acceptable data, and product commercialization strategies of biosimilars.10 Because of the complexity in the production platform of biologics, very small differences in cell lines or manufacturing processes can have a large impact on the final product, including potential side effects. Immunogenicity, one of the most serious adverse effects, has been observed during the therapeutic use of innovator products, which causes additional safety concerns among biosimilar regulators. Up to now there have been no clear specifications for analytical tests or preclinical and clinical studies to demonstrate biosimilarity in relation to a reference product. Companies developing biosimilars have to develop their own plan to prove biosimilarity.11 In fact, the development of biosimilar bioprocesses is very flexible due to the numerous possibilities and benefits available, such as disposable technology for production, supply chain logistics, and modern in‐process analytical methods for process development and validation.12
Total sales of biologics in the US reached almost $64 billion in 2012, an increase of 18.2% in relation to 2011 sales. The highest selling class of biologics was mAbs, representing approximately 39% of total sales.13 The entry of biosimilars into this market could lead to savings for healthcare systems.
Regulatory Aspects
It is a general consensus that the generic approach for small molecule drugs obtained by chemical synthesis is not appropriate for biosimilars. The approval of “copies” of biologics called biosimilars follows a specific regulation that is based on biosimilarity demonstration regarding quality, safety, and efficacy aspects in relation to a reference product.14 The term “biosimilar” itself is not a consensus by regulatory agencies and each adopts their own term and definition (Table 1). Despite the existence of slight differences in the scope of guidelines, reference product characteristics and datasets required for approval among different regions, the basic principles governing regulatory requirements are very similar.14, 15
Table 1.
Names and Definitions of Biologic Copies According to Different Regulatory Agencies
| Agency | Naming | Definition |
|---|---|---|
| FDA (Food and Drug Administration), USA | Follow‐on Biologic or Biosimilar | “A biological product that is highly similar to a U.S.‐licensed reference biological product notwithstanding minor differences in clinically inactive components, and for which there are no clinically meaningful differences between the biological product and the reference product in terms of the safety, purity, and potency of the product”.16 |
| EMA (European Medicines Agency) | Biosimilar | “A biological medicinal product that contains a version of the active substance of an already authorized original biological medicinal product (reference medicinal product) in the EEA. Similarity to the reference medicinal product in terms of quality characteristics, biological activity, safety and efficacy based on a comprehensive comparability exercise needs to be established”.4 |
| WHO (World Health Organization) | Similar Biotherapeutic Product | “A biotherapeutic product which is similar in terms of quality, safety and efficacy to an already licensed reference biotherapeutic product”.17 |
| PMDA (Pharmaceutical and Medical Devices Agency), Japan | Follow‐on Biologic or Biosimilar | “A biotechnological drug product developed by a different company to be comparable to an approved biotechnology‐derived product (hereinafter “reference product”) of an innovator”.18 |
| Health Canada | Subsequent Entry Biologic | “A biologic product that is similar to and would enter the market subsequent to an approved innovator biologic product”.19 |
| ANVISA (Agência de Vigilância Sanitária), Brazil | Biologic Product | A biologic medicine with known biologic activity that contains no new molecules, already licensed in Brazil and that has gone through all the production steps (including formulation, vialing, freeze drying, labeling, packaging, storage, quality control and biologic product lot release).20 |
The EU pioneered the development of regulatory aspects for biosimilars and also on giving marketing authorization for them, starting from the first generation biologics (somatropin) up to complex molecules such as erythropoietin and mAbs (infliximab). EMA published a general framework guideline for biosimilars in 200521 introducing the principles of biosimilarity that include the very basis of the majority of other guidelines. Technological changes that occurred afterwards added by the experience gained by application reviews led to an updated draft guideline released in 2013 and adopted by the Committee for Medicinal Products for Human Use (CHMP) on October, 2014.4 The reference product for demonstration of biosimilarity continues to be the one approved by the European Economic area (EEA). To promote global development of biosimilars and avoid the repetition of clinical trials, the revised guideline states that alternatively certain clinical studies and in vivo non‐clinical studies could be conducted with non‐EEA authorized reference product providing justification and bridging studies. This reference is acceptable for a product authorized by a regulatory authority with similar scientific and regulatory standards as EMA.4 Health Canada also permits the use of reference products not marketed under their jurisdiction in some circumstances.22 The bridging data should compare the biosimilar candidate, the EEA‐authorized product, and the non‐EEA authorized product in terms of analytical studies and also pharmacokinetic (PK) and pharmacodynamic (PD) data. EMA also developed other guidelines for comparability exercises considering quality, non‐clinical and clinical aspects, and a specific guideline for immunogenicity assessment. EMA has a robust regulatory process and the guidelines are continually revised and updated based on experience obtained with the approval of biosimilars over time.23 Case‐by‐case analysis taking into consideration the class of the biologic under review is the approach used by EU to deal with the diversity and complexity of biologics.14
Following EMA's general guideline, WHO published in 2009 a guideline to present globally acceptable principles to license biosimilars.17 This guideline represented an important step for harmonization on the evaluation and regulation of biosimilars and several countries adopted its principles to elaborate their own guidelines.14
In the US, biologics (i.e., reference products) are licensed under Section 351(a) of Public Health Service Act (PHS Act) and the application for biosimilars is done under section 351(k) of PHS Act and BPCI Act. FDA has published a series of guidance since 2012 to help implementation of BPCI Act that is an abbreviated pathway to approve biologics by demonstration of biosimilarity or interchangeability with reference product in terms of analytical evaluation and also animal studies and clinical trials.24 In 2012, detailed guidance were issued for biosimilarity assessment by analytical methods comparison between the candidate and its reference product24, 25 and also providing answers for common questions regarding the implementation of the BPCI Act.26 To demonstrate biosimilarity FDA recommends the use of a stepwise approach in which the extent of residual uncertainty concerning biosimilarity should be evaluated at each step followed by its identification in the next steps. To evaluate all data that support biosimilarity FDA intends to use totality‐of‐evidence approach.25 FDA has evolved in relation to biologics and biosimilars and launched in 2014 two draft guidance to assist biologic and biosimilar developers and also a list of biologicals called “Purple Book.” Two biosimilars applications under Section 351(k) of PHS Act were filed in 2014, the first for filgrastim (Sandoz)27 and the second for infliximab (Celltrion).28 The first 2014 guidance is related to the demonstration of biosimilarity without clinically meaningful differences based on clinical pharmacology studies by PD and PK analyses. These studies may decrease residual uncertainty, guide subsequent clinical trials to support demonstration of biosimilarity and also support extrapolation of clinical data to additional therapeutic use.29 The second 2014 guidance is related to the Section 351(k)(7) of PHS Act that describes the exclusivity of reference biologics stating that biosimilars may not be licensed before 12 years of approval of the reference product. This guidance determines which data should be provided by biologic developers to the FDA to facilitate the definition of first licensing date of the reference products.30 The “Purple Book” is a reference list of biologicals licensed under Section 351(a) of PHS Act with “Reference Product Exclusivity” and “Expiry Date of First Licensure” data and also lists biosimilars with information of the reference product to which biosimilarity and interchangeability was demonstrated under Section 351(k) of PHS Act. FDA intends to update the list periodically.31 Patent information of the reference product was not included in “Purple Book” due to the fact that many patents are involved in the development of biologics.32 Up to now, FDA has not yet given marketing authorization for any biosimilars under application of Section 351(k) of PHS Act. The recent publication of a series of guidance and reference book clarifies FDA requirements to approve biosimilars which is expected to start soon.
On the occasion of approval of the innovator biologic by the regulatory agencies, its manufacturer receives data exclusivity, a back‐up to the patent system, to delay abbreviated applications for biosimilars and patent challenges by competitors. The data exclusivity for biologics in the EU is the same for chemical products, 10 years, and one additional year when an important new indication is established.33 Biosimilar approval in the US is significantly different from the EU and generic drugs under the Hatch‐Waxman Amendments considering data exclusivity and patent challenges, delaying the approval of biosimilars. According to BPCI Act, the reference product receives 12 years of data exclusivity plus 6 months if pediatric studies were conducted. Biosimilar manufacturer may not submit Section 351(k) application until 4 years of licensure of the reference product. After the biosimilar application's acceptance there is a patent exchange process between the innovator and the biosimilar manufacturers, the innovator manufacturer receives a copy of the application and manufacturing processes and also additional information as requested by the innovator.34 These requirements are target of litigation between both manufacturers. Amgen filed two petitions on October 2014, one requesting the FDA for exchange of information with biosimilar applicants under BPCI Act and another requesting Neupogen (filgrastim) biosimilar application and manufacture processes from Sandoz. Sandoz proposed to provide only application information to Amgen. Up to now, it is not clear if these concerns are mandatory and we will learn by courts decisions with the present and future disputes.35 On 7th January, 2015, FDA granted biosimilarity status for the filgrastim produced by Sandoz.36 The final decision is expected in May; however, patent hurdles with Amgen will delay commercialization of the first biosimilar in the US.37
Besides challenges on the biosimilarity demonstration to the reference product and patent hurdles, there are other regulatory challenges for biosimilars. All guidelines require pharmacovigilance and risk management plan (RMP) for biosimilars when the application is submitted. EMA's RMP should provide detailed information on risks and safety concerns. RMP should be proposed by the manufacturer and then submitted to analysis by the regulatory agencies.38 The RMP for biosimilars should also consider immunogenicity data collection with description of methodology, strategies for monitoring, risk‐minimization, and communication.39 Until now there is no evidence of clinically relevant increase of immunogenicity for approved biosimilars.38 In the US, pharmacovigilance requirements for biosimilars have not been specified but the post marketing reporting is mandatory considering FDA guidance on Good Pharmacovigilance Practice for products with unknown safety risks.40
Other regulatory challenge is related to interchangeability and/or substitution. These terms are often used as synonyms in the US, but not in the EU. According to the European Generic Medicines Association (EGA), interchangeability refers to the prescription of a biosimilar in place of the reference product by prescribers, while substitution means that pharmacists are allowed to dispense a biosimilar.41 EMA does not guarantee interchangeability and established that these aspects are beyond its competence. Therefore, authorities of each Member State should decide after scientific evaluation performed by CHMP and other data submitted to the regulatory agency on support of the request.23 Many countries of the EU, such as Italy, Spain, United Kingdom and France, have opposed the automatic substitution by pharmacists.41 In the US, the determination of interchangeability will be a separate issue from biosimilar's approval and each state will decide on the legislation for substitution, including whether physician or patient would be consulted before pharmacist's dispensation.42 Overall, due to the intense efforts from originator manufacturers concerning health risks and differences of biosimilars in relation to the reference product, giving uncertainty for prescribers and patients, the application of interchangeability and/or substitution is limited.41 The use of biosimilars in the clinic may have a positive impact in the near future, paving the way for adequate decisions.
Extrapolation of indications for biosimilars is another challenge. It is critical to consider that reduced clinical studies conducted by comparability can support extrapolation of indications of biosimilars to other conditions not included in the clinical assessment. The possibility of including approval for all therapeutic indications meant for the original product is one potential advantage to develop biosimilars. The vision of regulatory agencies varies according to the biologic class, and one agency can approve for all indications, while another can approve for few indications.43 One of the reasons for low acceptance of biosimilars by prescribers is the extrapolation of indications without conduction of specific clinical studies. However, the concept of extrapolation of indications has been practiced by manufactures for a long time through introduction of several minor changes after approval of biologics.44 EMA has great experience concerning the extrapolation of indications. Filgrastim is indicated for neutropenia induced by chemotherapy and its biosimilar was approved for all indications of the reference product such as transplantation and peripheral blood progenitor cell mobilization.40, 44 Recently, the filgrastim biosimilar, submitted to FDA in 2014, was unanimously approved for all five indications granted to the reference product Neupogen by Oncologic Drugs Advisory Commitee.36, 45 A biosimilar for erythropoietin was licensed to renal anemia and the approval for cancer derived anemia was a natural consequence of indication extrapolation.40, 44 In 2013, EMA approved the first biosimilar mAb (infliximab) with extrapolation of all indications licensed for the reference product. The same mAb was approved at the same condition in Korea and Japan. However, Canada approved biosimilar of infliximab with extrapolation for some indications but not for Crohn's disease and ulcerative colitis indications based on differences found in certain analytical assays.43, 44
Remarkable advances have occurred in relation to regulatory requirements for biosimilar application and approval. It is expected that the regulatory agencies become more confident in relation to current challenges and update their guidelines with more detailed considerations to ensure the benefits of the biosimilars to healthcare providers and mainly to the patients.
The Impact of the Manufacturing Process
To obtain the approval from the regulatory agencies, biosimilars must go through a rigorous development process and demonstrate no clinically significant differences in safety, purity, and potency compared to the reference product. Considering this complex process, it is important to understand how biosimilars are developed. Biosimilars have to follow the same manufacturing process as their reference biologic product, which is very expensive and time‐consuming. However, there is a potential time and cost savings when comparing the biosimilar development to that of the reference product because some steps are not necessary in the generation of a similar biologic product.46 A timeline scheme is presented in Figure 1, representing the general trend for biosimilars development, in which the discovery/research phase and dose finding studies (Phase II clinical trial) are not required, considering that the biosimilar administration regimen uses the same dosing as the reference product.46, 47, 48 A Phase III study for efficacy equivalence between the biosimilar and its reference product is needed but can be conducted with a smaller number of patients.47 The elimination of one phase of the standard clinical trial protocol is not unique to biosimilar development. Recently FDA has issued guidance for expedited clinical program meant to accelerate approval of breakthrough therapies for life threatening indications, with expected reduction in time by approximately 40%.49, 50 Nonetheless the discovery/research period remains a differential for biosimilar development.
Figure 1.
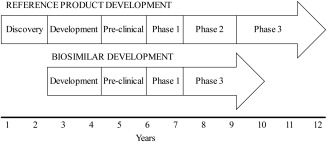
Representative timeline of the steps involved in the development of a biosimilar compared with the reference product (adapted from Hospira website46).
Developers of biosimilars usually do not have access to the details of the manufacturing process and active ingredients used for the reference product development.48 Although following almost the same steps, the inherent variability of the biologic system used and the manufacturing process will not result in a biological product identical to their respective reference product. The only characteristic that will be a copy of the reference product is the amino acid sequence. A comparison of the structural and functional characteristics and the product and process‐related impurities of the biosimilar and its reference product will be necessary.24 Any difference between the products must be justified with regard to the potential impact on the clinical performance of the biosimilar.51
Details in the production of biologic products vary from batch to batch and with any manufacturing change that can occur for different reasons, including scaling‐up the process to address commercial demand, improving the efficiency of the process, and modernizing the process when major equipment needs to be replaced or updated.52, 53, 54 The available comparability protocols allow for these changes to occur, and in the same way, this concept provides support for the biosimilars evolution. The first step in developing a biosimilar is to carefully examine multiple samples of the reference product to determine using analytical techniques how variable this reference is over time and during its shelf life.53 The biosimilar manufacturers are developing and validating powerful analytical tools to compare their products with the originators. An interesting point is that these improved analytical methods that allow for the detection of even small changes also reveal variability between lots of the reference products currently on the market. The analysis of multiple batches of Aranesp, Rituxan/Mabthera, and Enbrel revealed substantial alterations of the glycosylation profile for all the tested products.54 In addition, different lots of Rituxan/Mabthera and Enbrel showed changes in the N‐ and C‐terminal heterogeneity. Rituxan/Mabthera also demonstrated variation in antibody‐dependent cell‐mediated cytotoxicity (ADCC) activity among the batches tested.54 The authors of this study concluded that the observed changes were predicted to not result in an altered clinical profile, and therefore all the analyzed products in the tested timeframe are permitted to remain on the market with unaltered labels according to the health authorities.54
The use of different expression systems in the development of biosimilars compared with the reference drugs may change the post‐translational modifications, such as the glycosylation profile of the protein, which, in turn, could affect the safety or effectiveness of the product. Variations in the glycosylation protein pattern can alter the immunogenicity or clearance of the final product. Moreover, minor modifications in the formulation of biological products that affect inactive ingredients or changes in the primary packaging materials may also alter immunogenicity.55 This situation can be illustrated by the case of the anti‐anemic reference drug Eprex®, in which increased immunogenic responses and elevated rates of pure red cell aplasia were observed following a process change that replaced human serum albumin with polysorbate 80 and glycine as excipients.55, 56, 57 The mechanism by which Eprex® induced pure red cell aplasia is still not fully understood, but it seems to be the result of an increase in the levels of aggregates during storage, although the levels were not reported to have exceeded the specifications.58
The development of biosimilars to replace the biopharmaceuticals for which the patents have expired or are about to expire led to new concepts in manufacturing processes. The production facilities relied on the use of relatively inflexible, hard‐piped equipment, including large stainless steel bioreactors, and tanks to hold product intermediates and buffers, which are now being substituted for single‐use counterparts for the development of biosimilars and also in the development of new products.59 Some advantages for the adoption of single‐use or disposable technologies for the biopharmaceuticals manufacturing are: (1) reduced capital costs for plant construction and commissioning; (2) reduced risk for product cross‐contamination in a multiproduct facility; (3) rapid changeover; (4) lower utility costs due to a reduced need for steaming‐in‐place (SIP); and (5) reduced need for cleaning validation.59 A case study performed by País‐Chanfrau and collaborators (2009) showed that a hybrid plant with significant integration of disposable technology reduced capital costs by up to 40% compared with an exclusive stainless steel facility.60 The need to reduce timelines and initial costs, while relying on multipurpose plants, are all aspects needed for the development of biosimilars. To a great extent, these necessities have helped push the single‐use components industry.61, 62
Comparability
Comparability protocols emerged from the FDA's 1996 guidelines63 and were applied to approved biologics for which sponsors introduced changes to improve the manufacturing process or to implement new equipment or modern analytical assays. There are regulatory requirements to compare the similarity of the products before and after the change without having to apply for a new product development program. Based on this FDA guideline, the International Conference on Harmonization of Technical Requirements for Registration of Pharmaceuticals for Human Use (ICH) Q5E comparability regulatory guidelines were issued which indicated that the comparability protocol could be adopted also during preclinical and clinical studies.64 These two guidelines were the basis for the comparability standards that were applied to biosimilar products being submitted for approval by the regulatory agency. These standards indicate that the product of one manufacturer is compared with the reference drug of another manufacturer.65 Bioprocess manufacturing in the development of biosimilars can promote small changes in the structure of the biologics that can affect their function due to the host cell and the environment in which cells grow. Structural changes occur due to post‐translational modifications such as glycosylation patterning. The glycoform profile of biologics is one of most important aspects and should be well characterized due to their potential impact on clinical outcome.54 In the case of biosimilars, the comparability exercise demonstrates the expected biosimilarity which is verified by a complete analytical characterization of the product in comparison to the reference product. Initially, a detailed characterization of reference product should be performed using orthogonal analytical tools to establish acceptable product attributes, taking into account the multiple batches of the original product. These analytical parameters are the specifications for the biosimilars.52
Biomolecular analyses of biologics are performed for the product characterization, and they are very important factors in the bioprocess. The analyses are composed of three categories: physicochemical, immunological, and biological assays. Overall assay development and validation can cost around US $1.5 million. Physicochemical assays are performed from the beginning until the final step of the bioprocess and account for 66–75% of the product characterization. They can take 400–800 work hours to develop and validate. The immunological properties of the biologics should also be characterized. Bioassays provide information about critical characteristics related to the function of the product and its efficacy. These assays require more time to develop and represent 15% of the total number of assays. Furthermore, bioassays are used for the selection of the drug‐candidate, product release, stability assessment, and comparability studies to support process changes or biosimilarity.66, 67 In relation to mAb biological functions, special attention is necessary because the effector functions should be evaluated even if the product activity does not require these functions. The same procedure should be performed with the biosimilar and its reference product.68 However, it is difficult to evaluate the impact on the safety of biologics by bioassays, and regulatory agencies demand clinical trials and post‐marketing studies to assure the safety and efficacy of biosimilars.3
Biosimilars should be characterized by analytical assays as accurately and thoroughly as possible by comparability studies. Demonstration of the high level of analytical similarity between the biosimilar and the reference product is the first step of these studies. Therefore, all the structural elements of the protein and all the modifications should be evaluated with the capability to detect differences between the biosimilar and the reference products. Based on the observed characteristics of the reference product, the quality target product profile (QTPP) is defined for the biosimilar, including the variability and impurities found among the reference product lots since the early steps of development.68 Analytical techniques to release batches of innovator biologics showed little variability when the product was approved in the beginning of 2000s. However, the current advanced technologies available for biosimilars comparability studies reveal differences in batches even of the originator products manufactured on different geographical places or due to manufacturing changes or simply lot to lot variation. Therefore, the question is how best to describe the similarity of these products. The goal is to perform analytical assays that allow for the analysis of both physicochemical and biological functions of the drugs. It is difficult to determine the number of assays required to fully characterize biosimilars, but it should be sufficient to demonstrate biosimilarity. Furthermore, analytical assays should be developed, qualified, and validated, despite the time‐consuming nature of these.69 A comparability study was performed using standard quality control assays for epoetins (two original products and two biosimilars) and glycosylation profile and potency differences were found among them.70 Recent advances in analytical tools allow the characterization of biosimilars in accurate and robust ways that were not possible when the reference products were developed and approved. State‐of‐art liquid chromatography (LC) and mass spectrometry (MS) were used to compare the identity of a biosimilar mAb and its reference product and verify the presence of sequence variants and post‐translational modifications (PTMs), such as the glycosylation profile.71
Structural information of biologics, such as the conformation stability profile, is very important in ensuring their functional properties. These characteristics for higher order protein structure are used for comparability studies at the early and late steps to demonstrate evidence of similarity.72 Several examples of analytical tools to determine the high order protein structure are 2D NMR (1H/15N) fingerprinting, hydrogen‐deuterium exchange mass spectroscopy (H/DX‐MS), circular dichroism (CD), Fourier transform infra‐red (FTIR), and differential scanning calorimetry. 2D NMR fingerprinting is useful to evaluate the integrity of the protein structure by analyzing the spectra of the biologics. H/DX‐MS assesses the formulation conditions and reflects the solution conformation of the proteins by identifying flexible and rigid domains. CD and FTIR are known methods used to analyze the secondary protein structure, that is, the alpha and beta sheet content. Differential scanning calorimetry evaluates the thermal stability of biologics by monitoring the conformational stability during heating.65 The application of the above‐mentioned higher‐order protein structure analyses gives complementary information about biologics and their reference and can increase the understanding of their mechanisms of action.
Specific guidelines for biosimilar mAbs were issued by the EMA determining that biological activity should be characterized by a comparability study using sensitive immunological methods able to detect differences between a biosimilar and its reference.73 In vitro assays that need to be performed in the testing include binding efficacy to antigen and Fc gamma receptors, neutralization of antigen, and effector function related to Fc (ADCC; complement‐dependent cytotoxicity, CDC). Each mAb has a unique profile in relation to these characteristics, and the assays should account for the properties of the mAb in development. Bioassays usually present great variability, especially when performed with human‐derived material, making the comparison between the biosimilar and the reference mAb still more difficult. The design space should be defined to account for such variability and simultaneously ensure product quality. Some companies have engineered human cell lines to be used in bioassays and thus avoid the use of human blood cells.
Recently the physicochemical characterization, including state‐of the art and higher order structure techniques, of the comparability study for the first biosimilar mAb approved by EMA, Remsima (infliximab), was published. Other studies such as the biological characterization, non‐clinical studies, and clinical trials were performed and showed a high similarity to the reference product.74 Table 2 summarizes the analytical assays used in the comparability studies of Remsima to the reference product. A biosimilar of rituximab (GP2013) was characterized by its physicochemical and biological properties in a comparability study, and it was shown to be highly similar.75 These extensive studies serve as the basis for mAb biosimilar manufacturers to develop comparability studies that are as comprehensive as the ones that led to Remsima's approval.
Table 2.
Physicochemical and Biological Characterizations from the Comparability Studies of the Biosimilar Remsima74
| Characteristic | Attribute | Analytical Tool |
|---|---|---|
| Primary structure | Amino acid sequence | RP‐HPLC, LC‐ESI‐MS, LC‐ESI‐MS peptide mapping |
| Higher order structure | Disulfide structure | LC‐ESI‐MS peptide mapping |
| Free thiol analysis | Elman assay | |
| Secondary and tertiary structure | CD, FTIR, Antibody conformational array, X‐ray crystallography | |
| Thermal stability | DSC | |
| Purity | Monomer content | SEC‐HPLC, SEC‐MALS, SV‐AUC, CE‐SDS |
| Charge heterogeneity/amino acid modification | Charged isoforms | IEF, IEC‐HPLC |
| Deamidation/oxidation/C‐terminal variants | LC‐MS peptide mapping | |
| Glycosylation | N‐glycan analysis | LC‐MS |
| Glycosylation occurrence | CE‐SDS | |
| Oligosaccharide profile | HPLC | |
| Sialic acid analysis | HPAEC‐PAD | |
| Monosaccharide content (fucose, GlcNAc, galactose, and mannose) | HPAEC‐PAD | |
| Potency | Antigen and C1q binding | ELISA |
| FcRn binding | SPR | |
| Antigen neutralization | Cell‐based neutralization assay | |
| Apoptosis | Cell‐based apoptosis assay | |
| CDC | Cell‐based CDC assay |
CD, circular dichroism spectroscopy; CE‐SDS, capillary sodium dodecyl sulfate gel electrophoresis; DSC, differential scanning calorimetry; ELISA, enzyme‐linked immunosorbent assay; FTIR, Fourier transform infrared spectroscopy; GlcNAc, N‐acetylglucosamine; HPAEC‐PAD, anion exchange chromatography with the pulsed amperometric detection; IEF, isoelectric focusing; IEC‐HPLC, ion exchange chromatography; LC‐ESI‐MS, liquid chromatography electrospray ionization mass spectrometry; RP‐HPLC, reversed‐phase high‐performance liquid chromatography; SEC‐HPLC, size‐exclusion chromatography; SEC‐MALS, SEC‐multi angle light scattering; SPR, surface plasmon resonance; SV‐AUC, sedimentation velocity analytical ultracentrifugation.
In addition to the analytical characterizations of biosimilars, comparability studies should include the non‐clinical and clinical studies. The PK evaluation by quantitative assays to measure biosimilar and reference products in the patient serum is mandatory. An overview containing the strategies to demonstrate the PK similarity study using a single analytical method with statistical evaluation was recently described.76
A recent publication by members of the Working Party on Similar Biological Medicinal Products (BMWP) reveals their approach to in vivo testing for biosimilar products.7 Arguments against the use of animal for biosimilar development—and eventual approval—relies on the lack of robust additional information that could be derived from animal studies besides the biological and physicochemical comparison to the reference product performed in vitro. The authors propose a paradigm change based on the low sensitivity of animal testing which makes it difficult to reveal differences between the biosimilar and the reference product. Considering PD, the results obtained through cellular systems, with more precise controls, would not gain by additional animal studies, unless there is an animal model providing information that would predict the outcome in humans. PK is preferable to be analyzed and compared in human volunteers or patients as PK assessment in animals contribute little to the comparability exercise. One of the causes of PK issues comes from formulation components and usually well‐known substances are used for biosimilars development.7 Species difference prevents animal studies to discriminate residual uncertainties related to PK and PD77 and observed differences, whether present, do not prescind the quantitative data only obtained from PK and PD analyses.25, 77 A comprehensive study concludes that animal studies are not suited to detect subtle differences between biosimilars and reference products or to translate them into measurable endpoints by analyzing data of biosimilars submitted to EMA.78
Anti‐drug antibodies (ADA) response is a real concern with unpredictable side effects, the most famous case being the development of pure red cell aplasia after administration of erythropoietin.56 If the intended biosimilar product is not cross‐reactive with the animal endogenous molecules, animal studies would not preclude adverse effects in humans.7 A review by Brinks and collaborators presents the limitations of animal studies to assess immunogenicity.79 FDA also recognizes the limitations of animal testing for immunogenicity prediction in humans. Comparison of immunogenicity between a biosimilar and its reference is rendered more difficult because the number of animal in the studies is not large.25 However, FDA states that animal studies add value to the biosimilar development and any difference could help designing the immunogenicity assessment in humans.25
Animal studies can give relevant information in exceptional situations, for example, when comparative tissue distribution would be needed.7 The FDA considers that the information available on the reference product, the proposed biosimilar and the degree of biosimilarity between the two will dictate the scope and extent of animal toxicity studies. Additional comparative in vitro testing using human cells or tissues can also be used when animal toxicity studies are not warranted.25 The FDA has the ability to waive requirements for in vivo toxicology assessment, depending on the provision of sufficient clinical data.77
An important aspect of the path to demonstrate biosimilarity depends on bioprocesses parameters. Bioprocesses generally involve many parameters that can be interlinked or independent and introduce additional variability, including changes in the raw materials, operators, facilities, and equipment.80 Because of the necessity to ensure product quality for pharmaceutical and biopharmaceutical drugs, in addition to providing an in‐depth understanding of product and process manufacturing, regulatory agencies such as the FDA implement new approaches based on scientific principles to guarantee quality and understanding of the drug and manufacturing processes. The first initiative for the risk‐based approach for current pharmaceutical Good Manufacturing Practices (cGMPs) was introduced by FDA in 2002.81 Then, the process analytical technology (PAT) Guidance for Industry82 was issued to help in designing, developing, and implementing efficient tools during manufacturing and for quality assurance.
The ICH has published quality guidelines, based on GMP risk management for pharmaceutical and biological products since 2005. Q8 and Q11 are guidelines for the development of products and the manufacturing process, respectively.83, 84 These documents represent the basis for the application of the modern quality approach known as Quality by Design (QbD). Briefly, QbD is composed of the following steps: define the QTPP and determine the critical quality attributes (CQAs) for the product and process steps, define the parameters of the process to be controlled to guarantee CQAs, determine the operating ranges of such parameters to consistently yield acceptable product, and, finally, define the design space (manufacturing area that ensures CQAs). Q9, the quality risk management guideline, contains important concepts for the successful implementation of QbD.85 The application of Q9 and sophisticated experimental design, followed by statistical evaluation, allow for the manufacturer to define the critical raw materials and those that have low impact on the process, while also determining critical process parameters to establish the appropriate manufacturing controls. After these studies, it is possible to select the attributes to be monitored and controlled in a design space. Any changes performed inside the design space are acceptable and can be released, but do not request regulatory approval. Q10, the pharmaceutical quality system guideline, complements GMP and plays a critical role in building a robust quality system into the process.86 The parameters of QbD are integrated in the Q10 approach, allowing for process control.87 The application of the QbD concepts into bioprocesses of complex biologics has great advantages for innovator and biosimilar manufacturers to provide high product quality standards. Some advantages are previous in‐depth knowledge of cell culture characteristics, reduced time to set‐up large scale manufacturing, identification of culture batches that do not attend specifications, and a reduction of manufacturing failures.88 Furthermore, QbD helps to ensure homogeneity and quality of the final product. These properties may have impact on the safety and therapeutic effectiveness.
The overall effect of the bioprocess parameters on the quality of a final drug product is difficult to analyze. The use of statistical design of experiments (DoE) methods in the QbD approach aids in understanding the effects of possible multiple combinations and interactions of various parameters on the final drug quality.80 The DoE strategy is scientific‐based and leads to the determination of a design space and strategies for in‐process manufacturing control.67 The FDA initiated a pilot program regarding QbD for small molecules,89 and this experience was very useful and led them to issue a pilot program for biologics.90 In 2009, Genentech and Roche submitted two applications for biologics to the FDA QbD pilot, and both applications obtained approvals in 2013.91 The expectation of the FDA is that the experience gained by these pilot programs will help in the development of specific guidelines for implementing QbD and the risk‐based approach.89 Another expectation is that the positive experiences of both industry and regulators will facilitate the implementation of these high quality approaches since the early stage‐development of biosimilars. Although it necessitates a large investment to develop and implement the project and manage these approaches, when the bioprocess starts working, it will be easier to control or introduce small changes in the manufacturing process because of the previous knowledge of whole product process. This guarantees a time saving, high quality product and, consequently, better efficacy of treatment for the patients.
After the publication of the ICH guidelines, to synchronize and facilitate the implementation of QbD, the FDA and EMA began a pilot program for the parallel assessment of QbD applications for chemical substances in 2011.92 This effort resulted in two question‐and‐answer documents, and in April 2014, a 2‐year‐extension of the program was announced.93 It is expected that a similar pilot program will be extended to implement QbD for biologics. These programs will bring benefits to both biotechnological industries, including biosimilars producers, and regulatory agencies.
Market
Although it requires a large investment of time and money, the development and introduction of biosimilars in the market could provide a cost savings, increased patient access, and promote innovation. Biosimilars are available in the market at 10–30% lower prices compared with their reference products.94 In the EU, the introduction of biosimilars and competition with the new biosimilars has already forced reference drug prices down.95 In the US, it is estimated that the entrance of biosimilars in the market will save $3 to 4.5 billion annually and up to $378 billion over the next 2 decades.95, 96 Another estimate assumes a biosimilar market penetration of 60%, or 4% of total biologics sales over the next decade, amounting to a $44.2 billion savings. However, this potential market is dependent on the forthcoming decisions from the FDA.97 The global biosimilar market is estimated to reach $2.0 billion by 2018 at a compound annual growth rate (CAGR) of more than 20% between 2013 and 2018. The more complex proteins, including more than 50 biosimilar mAbs in the pipeline, suggest a growth rate of 25% by 2018.94 The first pioneering biosimilar is expected to hit the big US market after Novartis' biosimilar filgrastim had its advisory review cleared with a unanimous endorsement. Celltrion expects to reach the US market also, expanding its earnings to some $35 billion a year with biosimilar infliximab sales.98 It is scheduled for 17th March, 2015 a meeting of a public advisory committee of the FDA to discuss the biologics license application (BLA) 125544 for CT‐P13, a proposed biosimilar to Janssen Biotech Inc.'s REMICADE (infliximab), submitted by Celltrion.99
The interest in the biosimilars market is growing in the emerging markets due to a number of factors, including the enormous portion of many government healthcare budgets (50% in Brazil) going toward the high cost of imported branded biologics despite biologics only representing 2–3% of the overall medicines. Brazil has the second largest biologics market among emerging countries, and the Brazilian government is responsible for covering all the healthcare and drug costs for its 205 million inhabitants.100 To reduce the dependence on high‐priced, foreign‐branded biologics with expired patents, the Brazilian government created a program in 2012, updated in 2014 called Product Development Partnerships (PDP)101 to improve its biotechnology and biomanufacturing capabilities and produce its own biosimilars by creating public/private partnerships. The private companies need to fully transfer the technology to the public partners which need to hold a back‐up history of the biosimilar manufacturing processes in case the private companies discontinue its production, whereas the government provides guarantee of purchase for 5 years for provision of the Unified Health System (SUS). In 2013, several PDPs were signed between the Ministry of Health and public institution partners of pharmaceutical companies. To accelerate the development of biosimilars, the Brazilian companies license from and collaborate with foreign companies with drug‐development experience.102 The main focus of the PDPs is the development of biosimilar versions of blockbuster mAbs for cancer and autoimmune diseases, such as Herceptin, Avastin, Enbrel, and others. The approval of these biosimilars follows the tight ANVISA's requirements because Brazil is considered a regulated market.
For the biosimilar companies, the emerging markets such as Brazil, Russia, India, China, and South Korea offer the following advantages over their counterparts in more mature markets: lower labor and goods costs, access to large domestic, and regional market and, in many cases, government support,100 an incentive sought by other countries as well, for example, France.103 Conversely, market approval is easier in non‐regulated countries where other biosimilar mAbs have been launched.104
Concluding Remarks
Biosimilars have been evolving over time; the less complex molecules, some produced in microorganisms, were followed by recombinant mammalian cell derived proteins with varying degrees of complexity until the heavily glycosylated biosimilar of erythropoietin was approved. Now, the time for mAbs has arrived and the market will be boosted by a number of alternatives already in clinical trials. At the same time that the big pharma faces the challenge of losing market exclusiveness, some of these companies are developing biosimilars from other original manufacturers while pursuing new biologics in their development pipeline.98, 105
It is clear that the industry of biosimilars brings benefits for both science and healthcare. Producing copies of a comparable biological medicine to a reference product that is already in use is not easy, especially reducing the production costs to attain market feasibility. Obtaining cell lines with higher productivity for the manufacturing processes, tight control parameters to avoid heterogeneity beyond the reference product are important especially in the biosimilars industry. Understanding lot to lot variability and space design for QbD implementation is critical in the development of powerful analytical tools. The in‐depth analysis of different lots of branded mAbs produced over time has added to the knowledge‐base in the biotech and pharma industries. It is important to consider the link between the manufacturing variability and clinical outcome in the differences found in the lots of marketed mAbs, as not necessarily the differences imply in clinical significance.54
Many contract manufacturing companies were launched on the premise of pursuing biosimilar development and the need to cut time schedules, together with developing new concepts in manufacturing processes by the adoption of the single‐use technology. Biosimilars are here to stay and the biosimilar mAbs are on the cusp to attain approval from quality and safety regulators as well as the confidence of health authorities, doctors, and patients.
Acknowledgments
The authors thank Brazilian Research Council, CNPq, for a productivity fellowship granted to A.M. Moro (311934/2013‐7).
Literature Cited
- 1. Johnson IS. Human insulin from recombinant DNA technology. Science. 1983;219:632–637. [DOI] [PubMed] [Google Scholar]
- 2. Tsiftsoglou AS, Ruiz S, Schneider CK. Development and regulation of biosimilars: current status and future challenges. BioDrugs. 2013;27:203–211. [DOI] [PubMed] [Google Scholar]
- 3. Aagaard AW, Purdy S, Philpott S. Review, approval, and marketing of biosimilars in the United States. BioProcess Int. 2010;8:12–20. [Google Scholar]
- 4. EMA/CHMP/437/04 Rev 1 . Guideline on Similar Biological Medicinal Products. Available at: http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2014/10/WC500176768.pdf. April, 2015. Accessed March 4, 2015.
- 5. EMA/CHMP/BMWP/42832/2005 . Guideline on Similar Biological Medicinal Products Containing Biotechnology‐Derived Proteins as Active Substance: Non‐Clinical and Clinical Issues. Available at: http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2009/09/WC500003920.pdf. June, 2006. Accessed March 4, 2015.
- 6.Biologics Price Competition and Innovation Act of 2009. Section 7002(a)(2)(k)(2)(A)(i)(I)(cc). Available at: www.govtrack.us/congress/bill.xpd?bill=h111‐3590. September, 2009. Accessed December 22, 2014.
- 7. van Aerts LA, De Smet K, Reichmann G, Willem van der Laan J, Schneider CK. Biosimilars entering the clinic without animal studies: a paradigm shift in the European Union. mAbs. 2014;6:1155–1162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.EMA. European public assessment reports (EPAR) for human medicines. Available at: http://www.ema.europa.eu/ema/index.jsp?curl=pages%2Fmedicines%2Flanding%2Fepar_search.jsp&mid=WC0b01ac058001d124&searchTab=searchByAuthType&alreadyLoaded=true&isNewQuery=true&status=Authorised&status=Withdrawn&status=Suspended&status=Refused&keyword=Enter+keywords&searchType=name&taxonomyPath=&treeNumber=&searchGenericType=biosimilars. 2014. Accessed November 12, 2014.
- 9. Weinstein V. Looking at the Recent FDA Biosimilar Guidelines. BioProcess Int. 2012;10:10–14. [Google Scholar]
- 10. McCamish M, Woollett G. Worldwide experience with biosimilar development. MAbs. 2011;3:209–217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Hulse J, Cox C. In vitro functional testing methods for monoclonal antibody biosimilars. BioProcess Int. 2013;11:24–27. [Google Scholar]
- 12. Scott C. A decade of process development. BioProcess Int. 2012;10:72–78. [Google Scholar]
- 13. Aggarwal S. What's fueling the biotech engine—2012 to 2013. Nat Biotechnol. 2014;32:32–39. [DOI] [PubMed] [Google Scholar]
- 14. Wang J, Chow S. On the regulatory approval pathway of biosimilar products. Pharmaceuticals. 2012;5:353–368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Chugh PK, Roy V. Biosimilars: current scientific and regulatory considerations. Curr Clin Pharmacol. 2014;9:53–63. [DOI] [PubMed] [Google Scholar]
- 16.FDA. Information for Consumers (Biosimilars). Available at: http://www.fda.gov/Drugs/DevelopmentApprovalProcess/HowDrugsareDevelopedandApproved/ApprovalApplications/TherapeuticBiologicApplications/Biosimilars/ucm241718.htm. Accessed December 22, 2014.
- 17. WHO Expert Committee on Biological Standardization . Guidelines on evaluation of similar biotherapeutic products (SBPs). Available at: http://www.who.int/biologicals/areas/biological_therapeutics/BIOTHERAPEUTICS_FOR_WEB_22APRIL2010.pdf. October, 2009. Accessed March 4, 2015.
- 18.PDMA. Recent Regulations of Biosimilar in Japan. Available at: http://www.pmda.go.jp/regulatory/file/english_presentation/biologics/B‐E1arato.pdf.2011. Accessed December 22, 2014.
- 19.Health Canada. Fact Sheet: Subsequent Entry Biologics in Canada. Available at: http://www.hc‐sc.gc.ca/dhp‐mps/brgtherap/activit/fs‐fi/fs‐fi_seb‐pbu_07–2006‐eng.php. Accessed December 22, 2014.
- 20.ANVISA. Resolução RDC N° 55 de 16 de dezembro de 2010. Dispõe sobre o registro de produtos biológicos novos e produtos biológicos e dá outras providências. Available at: http://portal.anvisa.gov.br/wps/wcm/connect/73a029004ff7e91d980efe6d6e8afaaa/RDC+N%C2%BA+55,+DE+16+DE+DEZEMBRO+DE+2010.pdf?MOD=AJPERES December, 2010. Accessed December 18, 2014.
- 21. EMA/CHMP/437/04 . Guideline on Similar Biological Medicinal Products. Available at: http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2009/09/WC500003517.pdf. October, 2005. Accessed March 4, 2015.
- 22. Health Canada . Guidance for Sponsors: Information and Submission Requirements for Subsequent Entry Biologics (SEBs). Available at: http://www.hc‐sc.gc.ca/dhp‐mps/alt_formats/pdf/brgtherap/applic‐demande/guides/seb‐pbu/seb‐pbu‐2010‐eng.pdf. March, 2010. Accessed March 4, 2015.
- 23.Consensus Information Paper 2013. What you need to know about Biosimilar Medicinal Products. European Commission. Available at: http://ec.europa.eu/enterprise/sectors/healthcare/files/docs/biosimilars_report_en.pdf. 2013. Accessed January 14, 2015.
- 24.FDA/Center for Drug Evaluation and Research (CDER)/Center for Biologics Evaluation and Research (CBER). Guidance for Industry: Quality Considerations in Demonstrating Biosimilarity to a Reference Protein Product (Draft Guidance). February, 2012.
- 25.FDA/Center for Drug Evaluation and Research (CDER)/Center for Biologics Evaluation and Research (CBER). Guidance for Industry: Scientific Considerations in Demonstrating Biosimilarity to a Reference Product (Draft guidance). February, 2012.
- 26.FDA/Center for Drug Evaluation and Research (CDER)/Center for Biologics Evaluation and Research (CBER). Guidance for Industry. Biosimilars: Questions and Answers Regarding Implementation of the Biologics Price Competition and Innovation Act of 2009 (Draft guidance). February, 2012.
- 27.Sandoz. FDA Accepts Sandoz Application for Biosimilar Filgrastim. Available at: http://www.sandoz.com/media_center/press_releases_news/global_news/2014_07_24_FDA_accepts_Sandoz_application_for_biosimilar_filgrastim.shtml. July, 2014. Accessed December 04, 2014.
- 28.Celltrion. Celltrion files for US FDA approval of Remsima®. Available at: http://www.celltrion.com/en/company/notice_view.asp?idx=456&code=ennews&intNowPage=1&menu_num=&align_year=all. August, 2014. Accessed December 04, 2014.
- 29.FDA/Center for Drug Evaluation and Research (CDER)/Center for Biologics Evaluation and Research (CBER). Guidance for Industry: Clinical Pharmacology Data to Support a Demonstration of Biosimilarity to a Reference Product (Draft guidance). May, 2014.
- 30.FDA/Center for Drug Evaluation and Research (CDER)/Center for Biologics Evaluation and Research (CBER). Guidance for Industry: Reference Product Exclusivity for Biological Products Filed Under Section 351(a) of the PHS Act (Draft guidance). August, 2014.
- 31.FDA. Purple Book: Lists of Licensed Biological Products with Reference Product Exclusivity and Biosimilarity or Interchangeability Evaluations. Available at: http://www.fda.gov/Drugs/DevelopmentApprovalProcess/HowDrugsareDevelopedandApproved/ApprovalApplications/TherapeuticBiologicApplications/Biosimilars/ucm411418.htm. September, 2014. Accessed January 8, 2015.
- 32.Tu EU, Wolfson JA. FDA Throws the (Purple) Book at Biosimilars—Purple v. Orange. Haynes and Boone's Newsroom. Available at: http://www.haynesboone.com/fda‐purple‐book‐v‐orange‐book/. October, 2014. Accessed January 9, 2015.
- 33. Grabowski H, Long G, Mortimer R. Data exclusivity for biologics. Nat Rev Drug Discov. 2011;10:15–16. [DOI] [PubMed] [Google Scholar]
- 34. Mann JS, Mahinka SP. Biosimilars: patent challenges and competitive effects. LMG Life Sci. 2014;September:33–36. [Google Scholar]
- 35.Reichertz P, Kim M. Sandoz and Celltrion Decline the Invitation to Dance: Biosimilars Challenge the Applicability of the BPCIA's Exchange Provisions Before Bringing Suit. Sheppard Mulin. Available at: http://www.fdalawblog.com/2014/11/articles/ip‐and‐technology‐transactions/sandoz‐and‐celltrion‐decline‐the‐invitation‐to‐dance‐biosimilars‐challenge‐the‐applicability‐of‐the‐bpcias‐exchange‐provisions‐before‐bringing‐suit/. November, 2014. Accessed February 10, 2015.
- 36.FDA/Oncologic Drugs Advisory Committee Meeting January 7, 2015. BLA 125553 EP2006, a proposed biosimilar to Neupogen® (filgrastim) Sandoz Inc., a Novartis company. Available at: http://www.fda.gov/downloads/AdvisoryCommittees/CommitteesMeetingMaterials/Drugs/OncologicDrugsAdvisoryCommittee/UCM428780.pdf. January, 2015. Accessed February 12, 2015.
- 37. Ledford H. First biosimilar drug set to enter US market. Nature. 2015;517:253–254. [DOI] [PubMed] [Google Scholar]
- 38. Ebbers HC, Crow SA, Vulto AG, Schellekens H. Interchangeability, immunogenicity and biosimilars. Nat Biotechnol. 2012;30:1186–1190. [DOI] [PubMed] [Google Scholar]
- 39. EMA/CHMP/BMWP/14327/2006 . Guidelines on immunogenicity assessment of biotechnology‐derived therapeutic proteins. Available at: http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2009/09/WC500003946.pdf. April, 2008. Accessed March 4, 2015.
- 40. Tkaczuk KHR, Jacobs IA. Biosimilars in oncology: from development to clinical practice. Semin Oncol. 2014;41:S3–S12. [DOI] [PubMed] [Google Scholar]
- 41. Rovira, J , Espin J, Garcia L, Olry de Labry A. The Impact of Biosimilars' Entry in the EU Market. Andalusian School of Public Health. Available at: http://citeseerx.ist.psu.edu/viewdoc/download?doi=10.1.1.357.2218&rep=rep1&type=pdf. January, 2011. Accessed January 15, 2015.
- 42. Kling J. Fresh from the biotech pipeline—2013. Nat Biotechnol. 2014;32:121–124. [DOI] [PubMed] [Google Scholar]
- 43. Feagan BG, Choquette D, Ghosh S, Gladman DD, Ho V, Meibohm B, Zou G, Xu Z, Shankar G, Sealey DC, Russell AS. The challenge of indication extrapolation for infliximab biosimilars. Biologicals. 2014;42:177–183. [DOI] [PubMed] [Google Scholar]
- 44. Weise M, Kurki P, Wolff‐Holz E, Bielsky MC, Schneider CK. Biosimilars: the science of extrapolation. Blood. 2014;124:3191–3196. [DOI] [PubMed] [Google Scholar]
- 45.Williams A. “No Clinically Meaningful Differences”: The First Accepted Biosimilar Application Has Been Recommend for FDA Approval. Patents Doc. Available at: http://www.patentdocs.org/2015/01/no‐clinically‐meaningful‐differences‐the‐first‐accepted‐biosimilar‐application‐has‐been‐recommend‐fo.html. January, 2015. Accessed February 5, 2015.
- 46.Hospira. Developing Biosimilars. Available at: http://www.hospira.com/en/healthcare_trends/biologics/development_of_biosimilars/. Accessed December 18, 2014.
- 47. Scotté F, Launay‐Vacher V, Rey JB. Colony stimulating factors (CSF) biosimilars. Progress? Target Oncol. 2012;7:S17–S24. [DOI] [PubMed] [Google Scholar]
- 48. Weise M, Bielsky MC, De Smet K, Ehmann F, Ekman N, Narayanan G, Heim HK, Heinonen E, Ho K, Thorpe R, Vleminckx C, Wadhwa M, Schneider CK. Biosimilars‐why terminology matters. Nat Biotechnol. 2011;29:690–693. [DOI] [PubMed] [Google Scholar]
- 49.Subramanian R, Sheppard T, Rubin B, Kramer C. FDA's New Breakthrough Therapy Designation: What does it mean for Pricing and Market Access? OBR Green. Available at: http://obroncology.com/obrgreen/article/FDAs‐New‐Breakthrough‐Therapy‐Designation. September 2013. Accessed February 9, 2015.
- 50.FDA/Center for Drug Evaluation and Research (CDER)/Center for Biologics Evaluation and Research (CBER). Guidance for Industry. Expedited Programs for Serious Conditions—Drugs and Biologics. May, 2014.
- 51. Weise M, Bielsky MC, De Smet K, Ehmann F, Ekman N, Giezen TJ, Gravanis I, Heim HK, Heinonen E, Ho K, Moreau A, Narayanan G, Kruse NA, Reichmann G, Thorpe R, van Aerts L, Vleminckx C, Wadhwa M, Schneider CK. Biosimilars: what clinicians should know. Blood. 2012;120:5111–5117. [DOI] [PubMed] [Google Scholar]
- 52. McCamish M, Woollett G. The state of the art in the development of biosimilars. Clin Pharmacol Ther. 2012;91:405–417. [DOI] [PubMed] [Google Scholar]
- 53. McCamish M, Woollett G. The rise of the biosimilar. Expert Rev Clin Pharmacol. 2012;5:597–599. [DOI] [PubMed] [Google Scholar]
- 54. Schiestl M, Stangler T, Torella C, Cepeljnik T, Toll H, Grau R. Acceptable changes in quality attributes of glycosylated biopharmaceuticals. Nat Biotechnol. 2011;29:310–312. [DOI] [PubMed] [Google Scholar]
- 55. Ahmed I, Kaspar B, Sharma U. Biosimilars: impact of biologic product life cycle and European experience on the regulatory trajectory in the United States. Clin Ther. 2012;34:400–419. [DOI] [PubMed] [Google Scholar]
- 56. Casadevall N, Nataf J, Viron B, Kolta A, Kiladjian JJ, Martin‐Dupont P, Michaud P, Papo T, Ugo V, Teyssandier I, Varet B, Mayeux P. Pure red‐cell aplasia and antierythropoietin antibodies in patients treated with recombinant erythropoietin. N Engl J Med. 2002;346:469–475. [DOI] [PubMed] [Google Scholar]
- 57. Schellekens H. Immunologic mechanisms of EPO‐associated pure red cell aplasia. Best Pract Res Clin Haematol. 2005;18:473–480. [DOI] [PubMed] [Google Scholar]
- 58. Schellekens H. Biosimilar therapeutics‐what do we need to consider? NDT Plus. 2009;2:i27–i36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Shukla AA, Gottschalk U. Single‐use disposable technologies for biopharmaceutical manufacturing. Trends Biotechnol. 2013;31:147–154. [DOI] [PubMed] [Google Scholar]
- 60. País‐Chanfrau JM, Zorrilla K, Chico E. The impact of disposables on project economics in a new antibody plant: a case study. BioPharm Int. 2009;22:62–74. [Google Scholar]
- 61. Russell J. Reducing uncertainty surrounding biosimilar production. Genetic Eng. News. 2012;32:26–30. [Google Scholar]
- 62.PharmaAsia. Rethinking Manufacturing: Toward a Single‐Use Approach. PharmaAsia. Available at: http://www.pharmaasia.com/article/rethinking‐manufacturing‐toward‐a‐single‐use‐approach/10799. September, 2014. Accessed February 6, 2015.
- 63.FDA/Center for Biologics Evaluation and Research (CBER)/Center for Drug Evaluation and Research (CDER) Guidance: Demonstration of Comparability of Human Biological Products, Including Therapeutic Biotechnology‐derived Products; 1996.
- 64.ICH. ICH Harmonised Tripartite Guideline. Comparability of Biotechnological/Biological Products Subject to Changes in their Manufacturing Process Q5E; 2005.
- 65. Federici M, Lubiniecki A, Manikwar P, Volkin DB. Analytical lessons learned from selected therapeutic protein drug comparability studies. Biologicals. 2013;41:131–147. [DOI] [PubMed] [Google Scholar]
- 66. Scott C. Analytical methods for biologics. BioProcess Int. 2007;5:S35–S38. [Google Scholar]
- 67. Scott C. A decade of characterization. BioProcess Int. 2012;10:58–61. [Google Scholar]
- 68. Ridgway A, Ritter N, Schiestl M, Schreitmuller T. Biosimilar products. BioProcess International. Available at: http://www.bioprocessintl.com/manufacturing/biosimilars/biosimilar‐products‐347979/. November, 2013. Accessed August 1, 2014.
- 69. Galbraith D. Biosimilars awaken CROs. BioProcess Int. 2014;12:24–27. [Google Scholar]
- 70. Brinks V, Hawe A, Basmeleh AHH, Joachin‐Rodriguez L, Haselberg R, Somsen GW, Jiskoot W, Schellekens H. Quality of original and biosimilar epoetin products. Pharm Res. 2011;28:386–393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Xie H, Chakraborty A, Ahn J, Yu YQ, Dakshinamoorthy DP, Gilar M, Chen W, Skilton SJ, Mazzeo JR. Rapid comparison of a candidate biosimilar to an innovator monoclonal antibody with advanced liquid chromatography and mass spectrometry Technologies. mAbs. 2010;2:379–394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Wei Z, Shacter E, Schenerman M, Dougherty J, McLeod LD. The role of higher‐order structure in defining biopharmaceutical quality. BioProcess Int. 2011;9:58–66. [Google Scholar]
- 73. EMA/CHMP/BMWP/403543/2010 . Guideline on Similar Biological Medicinal Products Containing Monoclonal Antibodies – Non‐Clinical and Clinical Issues. Available at: http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2012/06/WC500128686.pdf. December, 2012. Accessed March 4, 2015.
- 74. Jung SK, Lee KH, Jeon JW, Lee JW, Kwon BO, Kim YJ, Bae JS, Kim D, Lee SY, Chang SJ. Physicochemical characterization of Remsima®. mAbs. 2014;6:1163–1177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Visser J, Feuerstein I, Stangler T, Schmiederer T, Fritsch C, Schiestl M. Physicochemical and functional comparability between the Proposed Biosimilar Rituximab GP2013 and originator Rituximab. BioDrugs. 2013;27:495–507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Colbert A, Umble‐Romero A, Prokopa S, Chow VF, Wonga T, DeSimone D, Zhou L, Pederson S. Bioanalytical strategy used in development of pharmacokinetic (PK) methods that support biosimilar programs. mAbs. 2014;6:1178–1189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Abbas R, Jacobs IA, Li EC, Yin D. Considerations in the early development of biosimilar products [published online ahead of print January 6, 2015]. Drug Discov Today. doi: 10.1016/j.drudis.2014.12.017. In press. [DOI] [PubMed] [Google Scholar]
- 78. van Meer PJ, Ebbers HC, Kooijman M, Wied CC, Silva‐Lima B, Moors EH, Schellekens H. Contribution of animal studies to evaluate the similarity of biosimilars to reference products [published online ahead of print November 20, 2014]. Drug Discov Today. doi: 10.1016/j.drudis.2014.11.009. In press. [DOI] [PubMed] [Google Scholar]
- 79. Brinks V, Jiskoot W, Schellekens H. Immunogenicity of therapeutic proteins: the use of animal models. Pharm Res. 2011;28:2379–2385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Shivhare M, McCreath G. Practical considerations for DoE implementation in quality by design. BioProcess Int. 2010;8:22–30. [Google Scholar]
- 81.FDA. Pharmaceutical cGMPs for the 21st century—a risk‐based approach. Available at:http://www.fda.gov/Drugs/DevelopmentApprovalProcess/Manufacturing/QuestionsandAnswersonCurrentGoodManufacturingPracticescGMPforDrugs/ucm137175.htm. Accessed December 23, 2014.
- 82.FDA/Center for Drug Evaluation and Research (CDER)/Center for Veterinary Medicine (CVM)/Office of Regulatory Affairs (ORA). Guidance for Industry. PAT—A Framework for Innovative Pharmaceutical Manufacturing and Quality Assurance; 2004.
- 83. ICH . ICH Harmonised Tripartite Guideline. Pharmaceutical Development Q8 (R2); 2009. [Google Scholar]
- 84. ICH . ICH Harmonised Tripartite Guideline. Development and Manufacture of Drug Substances (Chemical Entities and Biotechnological/Biological Entities) Q11; 2012. [Google Scholar]
- 85. ICH . ICH Harmonised Tripartite Guideline. Quality Risk Management Q9; 2005. [Google Scholar]
- 86. ICH . ICH Harmonised Tripartite Guideline. Pharmaceutical Quality Systems Q10; 2008. [Google Scholar]
- 87. Calcott P. Implementation of quality by design in vaccine development. BioProcess Int. 2013;11:12–17. [Google Scholar]
- 88. Girard FC. Process optimization of biosimilars production using NMR profiling. BioProcess Int. 2013;11:52–56. [Google Scholar]
- 89.FDA/Department of Health and Human Services. Submission of Chemistry, Manufacturing, and Controls Information in a New Drug Application Under the New Pharmaceutical Quality Assessment System (Docket No. 2005N–0262); 2005.
- 90.FDA/Department of Health and Human Services. Notice of Pilot Program for Submission of Quality Information for Biotechnology Products in the Office of Biotechnological Products (Docket number FDA‐2008‐N‐03551); 2008.
- 91.Krummen L. Lessons Learned from Two Case Studies in the FDA QbD Biotech Pilot. Available at: http://c.ymcdn.com/sites/www.casss.org/resource/resmgr/CMC_Euro_Speaker_Slides/2013_CMCE_KrummenLynne.pdf. 2013. Accessed November 21, 2014.
- 92.FDA. FDA, EMA Announce Pilot for Parallel Assessment of Quality by Design Applications. Available at: http://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm247332.htm. March, 2011. Accessed December 4, 2014.
- 93.FDA. FDA‐EMA Extends Pilot Program of the QbD Parallel‐Assessment. Available at: http://www.fda.gov/Drugs/DrugSafety/ucm388009.htm. 2014. Accessed December 4, 2014.
- 94.Markets and Markets. Biosimilars Market Product [Recombinant Non‐Glycosylated Proteins (Insulin, Filgrastim, Somatropin), Glycosylated (Monoclonal Antibodies, Erythropoietin), Peptides (Glucagon, Calcitonin)] & Application (Oncology, Blood Disorders)—Global Forecast to 2018. Available at: www.marketsandmarkets.com/Market‐Reports/biosimilars‐40.html?gclid=Cj0KEQiA8MSkBRCP5LaRlcOAusMBEiQAiqldkv9Iax6iWLafXqEs9ACqZuy8HkT3210UMyvzTA0zv2waAk9‐8P8HAQ. November, 2013. Accessed December 18, 2014.
- 95. Cornes P. The economic pressures for biosimilar drug use in cancer medicine. Target Oncol. 2012;7:S57–S67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Henry D, Taylor C. Pharmacoeconomics of cancer therapies: considerations with the introduction of biosimilars. Semin Oncol. 2014;41:S13–S20. [DOI] [PubMed] [Google Scholar]
- 97.Mukherjee, S. RAND study: biosimilars could save US $44 billion in 10 years. BioPharma Dive. Available at:http://www.biopharmadive.com/news/rand‐study‐biosimilars‐could‐save‐us‐44‐billion‐in‐10‐years/328924/. November, 2014. Accessed December 18, 2014.
- 98.Lane EJ. Competition to J&J's Remicade looms in U.S. as biosimilar conquers new markets. Fierce Biotech. Available at: http://www.fiercebiotech.com/story/competition‐jjs‐remicade‐looms‐us‐biosimilar‐conquers‐new‐markets/2015‐02‐10?utm_campaign=+SocialMedia. February, 2015. Accessed February 11, 2015.
- 99.Warner JH. Arthritis Advisory Committee; Notice of Meeting. Federal Register. Available at: https://www.federalregister.gov/articles/2015/02/10/2015–02670/arthritis‐advisory‐committee‐notice‐of‐meeting. February, 2015. Accessed February 11, 2015.
- 100.Mintz C. Biosimilars in emerging markets. BioProcess Online Available at: http://www.bioprocessonline.com/doc/biosimilars‐in‐emerging‐markets‐0001. July, 2013. Accessed December 18, 2014.
- 101.Ministério da Saúde. Available at: http://bvsms.saude.gov.br/bvs/saudelegis/gm/2014/prt2531_12_11_2014.html. November, 2014. Accessed January 19, 2015.
- 102.Ministério da Saúde. Available at: http://pt.slideshare.net/MinSaude/medicamentos‐biotecnolgicos‐para‐o‐sus. June, 2013. Accessed December 23, 2014.
- 103. Guillon‐Munos A, Daguet A, Watier H. Antibody biosimilars: fears or opportunities?: First LabEx MAb Improve industrial workshop, May 28, 2013; Tours, France. MAbs. 2014;6:805–809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Reymond E. Dr Reddy's bets high on biosimilars. Indian drug maker Dr Reddy's is planning to launch one biosimilar every year in the next few years, according to Indian media reports. In‐Pharma Technologist.com. Available at: http://www.in‐pharmatechnologist.com/Drug‐Delivery/Dr‐Reddy‐s‐bets‐high‐on‐biosimilars. August, 2007. Accessed December 18, 2014.
- 105.First World Pharma. Big Pharma's march on the biosimilars market gathers pace. First World Pharma. Available at: http://www.firstwordpharma.com/node/1056866#axzz3QzYbRX9j. February, 2013. Accessed February 6, 2015.