

Abstract
The retrotransposon Long Interspersed Element 1 (LINE‐1 or L1) has played a major role in shaping the sequence composition of the mammalian genome. In our recent publication, “Heritable L1 retrotransposition in the mouse primordial germline and early embryo,” we systematically assessed the rate and developmental timing of de novo, heritable endogenous L1 insertions in mice. Such heritable retrotransposition events allow L1 to exert an ongoing influence upon genome evolution. Here, we place our findings in the context of earlier studies, and highlight how our results corroborate, and depart from, previous research based on human patient samples and transgenic mouse models harboring engineered L1 reporter genes. In parallel, we outline outstanding questions regarding the stage‐specificity, regulation, and functional impact of embryonic and germline L1 retrotransposition, and propose avenues for future research in this field.
1. Introduction
1.1. L1 in Mammalian Genomes
L1 retrotransposon sequences account for approximately 17% of human genomic DNA and approximately 18% of mouse genomic DNA.1, 2 While the vast majority of L1 copies are molecular fossils rendered incapable of further retrotransposition by 5′ truncation, internal mutations and structural rearrangements, a subset of L1s retain the ability to mobilize. A typical human genome harbors around 100 of these mobile L1s,3 while mice are estimated to contain approximately 3000 such elements.4 A retrotransposition‐competent L1 is about 6 kb (human) or 7 kb (mouse) in length,5, 6 and contains a 5′ untranslated region (5′UTR) harboring an internal promoter,7 two intact open reading frames (ORF1 and ORF2), a 3′ untranslated region (3′UTR), and terminates in a poly(A) tract.5 In mice, the 5′UTR is variable in length and comprises ≈200 bp repetitive units, or monomers.8 The sequence of these monomer units is the primary distinguishing characteristic between three presently active L1 subfamilies in the mouse genome: L1 TF, GF, and A.4, 9, 10
A round of L1 retrotransposition initiates with transcription of a retrotransposition‐competent donor, or source, L1 in the genome, translation of the L1‐encoded proteins ORF1p and ORF2p, and association of ORF1p and ORF2p with their encoding L1 mRNA in a phenomenon known as cis‐preference.11 The resultant L1 ribonucleoprotein particle (RNP)12, 13 enters the nucleus, where the L1 ORF2p endonuclease activity initiates the formation of a new L1 insertion by catalyzing a single‐strand nick in genomic DNA at the loose consensus sequence 5′‐TTTT/AA‐3′. This activity liberates a free 3′ hydroxyl residue that serves as a primer from which the L1 ORF2p reverse transcriptase activity initiates synthesis of the first‐strand L1 cDNA on the L1 mRNA template, with this process referred to as target‐primed reverse transcription (TPRT).14, 15, 16, 17 Second‐strand genomic DNA cleavage typically occurs downstream of first‐strand cleavage, resulting in variable length target‐site duplications (TSDs) flanking the newly‐integrated L1 insertion.18, 19, 20, 21
Although cis‐preference is the general rule for L1 retrotransposition, the L1‐encoded proteins can occasionally act in trans to mobilize non‐autonomous retrotransposons known as short interspersed elements (SINEs). In humans, active SINEs include the 7SL‐derived element Alu and the SINE‐R‐VNTR‐Alu (SVA) composite retrotransposon.22, 23, 24 In mice, active SINEs include the 7SL‐derived B1 element and the tRNA‐derived B2 element.25 Cellular mRNAs can also be mobilized in trans by the L1 protein machinery, generating processed pseudogenes.11, 26, 27 A uniting feature of the L1 mRNA and other RNAs that can be trans‐mobilized by L1 is a 3′ poly(A) tract and, indeed, recent work has definitively shown that a 3′ poly(A) tract is required for L1 retrotransposition.28
During retrotransposition, structural changes can occur in the L1 sequence, and in the proximal target site genomic DNA. Nascent L1 insertions often undergo 5′ truncation, as well as internal inversion/deletion events, rendering the new L1 copy incapable of subsequent retrotransposition.1, 21, 29, 30 Alterations to target site DNA include deletions of genomic sequence and, occasionally, severe aberrations such as chromosomal translocations.20, 31 During transcription of a retrotransposition‐competent L1, read‐through of the L1 polyadenylation signal in favor of a downstream polyadenylation signal can ultimately result in the retrotransposition of flanking genomic sequence along with the nascent L1 insertion, in a process known as 3′ transduction.32, 33, 34 Less commonly, 5′ transductions arise when transcription of a donor L1 initiates from an upstream genomic promoter rather than the L1 internal promoter, and 5′ flanking genomic sequence is incorporated into the new insertion.1 L1 insertions into exons of genes can ablate gene function,21, 35, 36 and intronic insertions are associated with mis‐splicing, premature polyadenylation, disruption of regulatory elements, provision of new regulatory elements, and reduced RNA polymerase processivity leading to subtle changes in gene expression.37, 38, 39, 40, 41
L1‐mediated retrotransposition events have been found to be responsible for 130 cases of human genetic disease42 and six spontaneous mouse mutants have been attributed to de novo L1 insertions.43, 44 L1 mobilization clearly can have a negative impact on genome integrity and, indeed, L1 activity is curtailed by a variety of host factors (for reviews, see Refs. 45, 46). New L1 insertions also provide a continuing supply of genetic diversity. Based upon the frequency of polymorphic retrotransposon insertions identified in the human population, one new L1 insertion is estimated to arise per 100–200 live births, and one new Alu insertion per 20 live births.47, 48, 49, 50, 51, 52 The rate of new L1 insertions arising in mice was until recently estimated to be 30‐fold higher, at one insertion per 2–3 live births,53, 54 due provisionally to the much larger number of potentially active L1 copies in the mouse genome.4
2. Examining L1 Retrotransposition In Vivo
2.1. Transgenic L1 Reporter Systems Provide Critical Insights
Cultured cell assays55, 56 for L1 retrotransposition have elucidated the mechanism by which L1 mobilizes, as well as potential consequences for the host genome (reviewed in Ref. 57). In these systems, a retrotransposition‐competent L1 is tagged in its 3′UTR with a selectable (e.g., neomycin phosphotransferase55) or screenable (e.g., enhanced green fluorescent protein (EGFP)58) reporter gene, complete with a heterologous promoter and polyadenylation signal, that will only be expressed if the tagged L1 undergoes a successful round of retrotransposition (Figure 1A). Engineered L1s have also been incorporated into transgenic rodent models to study the dynamics of L1 retrotransposition in vivo59, 60, 61, 62, 63, 64, 65, 66 (Table 1). These systems have considerably informed our understanding of L1 activity in the germline and early embryo. The advantages of transgenic models include highly sensitive detection, quantitation, and mapping of integrated L1 retrotransposition events (Figure 1B), readily distinguished from existing genomic copies by the presence of the spliced reporter cassette. Expression of the EGFP reporter gene can also facilitate detection and identification of cells harboring engineered retrotransposition events by flow cytometry and fluorescence microscopy (Figure 1B), although silencing of the integrated reporter cassette in some physiological contexts may hinder detection.67, 68
Figure 1.
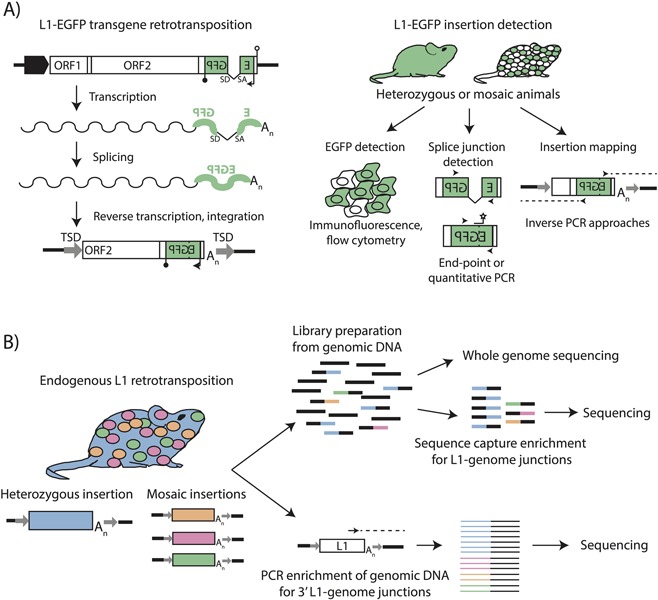
Methods for detecting engineered and endogenous L1 insertions. A) L1‐EGFP transgene retrotransposition. An engineered L1‐EGFP transgene consists of a retrotransposition‐competent L1 element with intact ORFs (white boxes) driven by the native L1 promoter, a heterologous promoter, or both (broad black arrow). The L1 3′UTR contains an enhanced green fluorescent protein (EGFP) reporter gene (green) in antisense orientation to the L1, interrupted by an intron with splice donor (SD) and splice acceptor (SA) sites in sense orientation to the L1. The EGFP reporter is driven by a heterologous promoter (thin black arrow) and terminates in a polyadenylation signal (black lollipop). The EGFP reporter is followed by a polyadenylation signal in sense orientation to the L1 (white lollipop). Upon L1 mRNA transcription (wavy line), the intron in the EGFP gene is spliced. Reverse transcription and integration of the L1 mRNA during target‐primed reverse transcription delivers an intact copy of the EGFP reporter into the genome. B) L1‐EGFP insertion detection. Animals harboring engineered L1‐EGFP insertions can be heterozygous for an insertion (filled green mouse) or mosaic for an insertion (white and green mouse). L1‐EGFP insertions can be detected through expression of the EGFP gene (flow cytometry, fluorescence microscopy), or by PCR‐mediated detection of the spliced EGFP cassette. The genomic locations and structural characteristics of L1‐EGFP insertions can be identified using inverse PCR strategies with primers specific to the EGFP cassette. C) Detection of endogenous L1 retrotransposition. At left, an insertion for which a mouse is consummately heterozygous is represented in blue, and mosaic insertions present at <1 copy per cell are represented in orange, pink, and green. Strategies for detecting endogenous L1 insertions include preparation of Illumina sequencing libraries from genomic DNA (above), which can be directly used for whole genome sequencing (WGS) or enriched for L1‐genome junctions using sequence capture probes, as in RC‐seq. Enrichment for 3′ L1‐genome junctions can also be achieved using PCR‐based approaches (below) in which outward‐facing primers are used to selectively amplify the 3′ termini of active L1 subfamilies, and amplicons subjected to high‐throughput sequencing. In both cases, heterozygous insertions are better represented than mosaic insertions when genomic DNA from bulk tissues is assayed.
Table 1.
Transgenic mouse models used to study heritable L1 retrotransposition events
| Study | L1 element used | Promoter driving L1 transgene | Reporter cassette | Transgene copy number | Developmental origin of identified insertions | Insertions transmitted? |
|---|---|---|---|---|---|---|
| Ostertag et al.59 | Human L1RP | Native L1RP 5′UTR | pAC‐EGFP | n/d | Male germline | n/d |
| pPolII + native L1RP 5′UTR | pAC‐EGFP | n/d | Male germline | Yes | ||
| Prak et al.60 | Human L1RP | pPolII + native L1RP 5′UTR | pCMV‐EGFP | n/d | Early embryonic in founder mouse | Yes |
| Babushok et al.62 | Human LRE3 | pHSP70‐2 + native LRE3 5′UTR | Markerless cassette | n/d | Early embryonic | No |
| An et al.63 | Mouse synthetic ORFeus_Mm | pCAG | pRSV LTR‐EGFP | 31064 | n/d | Yes |
| An et al.64 | Mouse synthetic ORFeus_Mm | pCAG; conditionally activated by Cre‐mediated recombination | pRSV LTR‐EGFP | Single‐copy | n/d | n/d |
| Kano et al.65 | Human LRE3 flanked by chicken β‐globin insulators | Native LRE3 5′UTR | γ‐globin intron in 3′UTR | n/d | Early embryonic | No |
| Human L1RP 61 | Native L1RP 5′UTR | pCMV‐EGFP | n/d | |||
| Human L1RP (tested in rat) | Native L1RP 5′UTR | Markerless cassette | n/d | |||
| Mouse TGF21 | Native TGF21 5′UTR | pCMV‐EGFP | n/d | |||
| Newkirk et al.66 | Mouse synthetic ORFeus_Mm | Native L1spa 5′UTR | pCMV‐EGFP | Single‐copy | Male germline | n/d |
From left to right, columns indicate the study cited, the species of origin and name of the L1 element, the promoter(s) driving L1 transgene expression, the promoter driving reporter cassette expression, the developmental origins of insertions examined by each study, and whether germline transmission of insertions to subsequent generations was observed. pPolII, mouse RNA polymerase II promoter; pHSP70‐2, mouse heat shock protein 70‐2 promoter; pAC, preproacrosin promoter; pCMV, human cytomegalovirus immediate‐early enhancer and promoter; pCAG, CMV immediate‐early enhancer/modified chicken β‐actin promoter; pRSV LTR, Rous sarcoma virus LTR promoter. n/d; not determined. In Kano et al.,65 the L1RP element was used in both transgenic mouse and transgenic rat models; the L1RP transgenic mice were first reported by Muotri et al.61
Other considerations regarding transgenic L1 animal models include the genomic location of the transgene, as randomly integrated reporters may not faithfully recapitulate the activity of endogenous L1s. The choice of L1 element in a transgenic animal model is also important: at least in cultured cells, highly‐active human L1s retrotranspose more efficiently than the mouse L1s that have been studied to date,4, 10, 69, 70 making them an appealing choice to observe high‐frequency L1 mobilization in vivo. However, introducing a human L1 into a rodent genome raises the question of whether it will be regulated similarly to endogenous rodent L1s in vivo. Heterologous promoters have been used in addition to the endogenous L1 promoter,59, 60, 62 or in some cases in its place,64, 71 to drive high levels of L1 transgene expression which may not recapitulate endogenous L1 regulation.
A synthetic, codon‐optimized mouse L1, termed ORFeus_Mm, has provided an intriguing solution to these problems.72 The amino acid sequence of ORFeus_Mm is that of a native mouse L1, but the nucleotide sequence has been altered for optimal codon usage in mammalian cells, resulting in a substantial increase in GC content. ORFeus_Mm is highly expressed compared to native mouse L1s and retrotransposes extremely well in cultured cells.72 This element has been used with heterologous promoters63, 64 as well as a native mouse L1 promoter66 to facilitate efficient L1 retrotransposition in vivo. Whether the synthetic nucleotide sequences of the ORFeus_Mm ORFs affect its regulation in vivo remains to be determined.
2.2. High‐Throughput Sequencing Allows Detection of Endogenous Retrotransposition Events
Endogenous L1 activity in mammalian genomes can be described in unprecedented resolution by high‐throughput DNA sequencing approaches. Such methods typically exploit discordant paired‐end sequencing reads, or split‐reads that span L1‐genome junctions, to identify L1 sequences at loci where an L1 is absent from the reference genome sequence (as reviewed in Refs. 73, 74). L1 insertions can be identified through analysis of whole genome sequencing (WGS) data,48, 75, 76, 77, 78, 79, 80 and targeted enrichment for L1 insertions can be achieved by sequence capture81 or PCR‐based methods in which L1‐genome junctions are selectively amplified using outward‐facing primers specific to an active L1 family (Figure 1C).47, 82, 83, 84, 85 In each case, putative L1 insertions must be validated by PCR and capillary sequencing to exclude chimeric molecules generated during sample preparation and high‐throughput sequencing. One obvious advantage of high‐throughput sequencing strategies is that they allow detection of endogenous L1 insertions, eliminating caveats posed by the somewhat artificial nature of transgenic systems. By contrast, per observed L1 integration event, high‐throughput sequencing is far more expensive at present than tracking engineered L1 insertions. In addition, sequencing‐based approaches more readily detect insertions that are present at a high prevalence in a given sample. When bulk tissue samples are used, insertions that occurred in cells that have undergone extensive clonal expansion have an advantage over insertions that occurred in cells with fewer mitotic descendants, or insertions that are unique to a single cell. Whole genome amplification (WGA) of single cells can facilitate detection of such low prevalence insertions,86, 87 but the WGA process represents an additional source of chimeric molecules that can introduce false positive results,88 making thorough PCR validation especially crucial. Together, transgenic and high‐throughput sequencing based approaches provide an often consistent window into L1 biology in vivo, and both are valuable tools for understanding the dynamics of L1 retrotransposition in vivo.
3. Heritable L1 Retrotransposition Is an Ongoing Process
3.1. How Frequently Do Heritable L1 Insertions Occur in Mice?
The contribution of retrotransposon activity to mouse genetic diversity has been made clear by comparing the genomic sequences of commonly‐used inbred mouse strains. In a 2008 study, Akagi et al. identified structural variants (SVs) present in the C57BL/6J reference genome but absent from WGS reads generated from four additional inbred mouse strains (A/J, DBA/2J, 129S1/SvImJ, and 129 × 1/SvJ).75, 89 L1 polymorphisms indicative of relatively recent retrotransposition events accounted for the majority of these variants, with 6723 putative L1 insertions present in the C57BL/6J reference genome and absent from at least one other inbred strain. Most polymorphic L1s identified were members of the young, presently active TF, GF, and A subfamilies. Similarly, Quinlan et al. carried out a detailed comparison of SV content in the C57BL/6J and DBA/2J genomes78 and found that transposable element sequences accounted for approximately 2/3 of the 7196 SVs detected. Of these SVs, 4412 consisted of a single TE annotation, consistent with retrotransposition rather than another mechanism of structural variation, and L1 sequences were responsible for nearly half of insertions. Subsequent WGS applied to 17 inbred mouse strains confirmed that transposable element sequences accounted for the majority of SVs76, 90 and totaled 103 798 such events, including 40 074 L1 insertions.77 Together, these studies affirmed L1 activity as a major force that continues to shape murine genomes.
The L1 integrants identified in the above studies were clearly the result of evolutionarily recent L1 mobilization, as evidenced by their differential presence/absence among inbred strains. However, they present only a snapshot of L1‐mediated variation that has accumulated during the divergence of inbred strains, rather than a “real‐time” account of ongoing retrotransposition in individual genomes. In a recent publication, we studied the genesis of de novo L1 insertions in mice with unprecedented resolution.91 By analyzing the genomes of individual, related C57BL/6J animals, we directly quantified the frequency of endogenous, de novo L1 retrotransposition across multiple generations of mice, and deduced the developmental timing of many of these events.
Our experimental strategy entailed generating two‐ and three‐generation pedigrees of wild‐type C57BL/6J mice and performing mouse retrotransposon capture sequencing (mRC‐seq) and WGS (Figure 1C) on parental mice and their offspring. Among 85 mouse genomes analyzed, we identified 11 de novo L1 insertions, providing an estimate of one new L1 insertion per 8 births. Of these 11 insertions, we could explicitly demonstrate that 9 were heritable, indicating a frequency of at least one heritable retrotransposition event per 10 births. We regard this figure as a conservative estimate, as the mouse L1 3′ end is refractory to sequencing on Illumina platforms due to the presence of a GC‐rich tract which may form a G‐quadruplex structure92, 93 and this technical hurdle may have prevented the detection of 5′ truncated L1 insertions by mRC‐seq. All 11 insertions belonged to the L1 TF subfamily, which is consistent with reports of spontaneous disease‐causing L1 insertions in mice,43, 44 although examination of de novo retrotransposition events in different inbred mouse strains may reveal strain‐specific activity of GF and A subfamily elements. Indeed, we have very recently uncovered a tumor‐specific L1 GF insertion in a mouse model of hepatocellular carcinoma on a congenic FVB.129 Sv background.94
3.2. What Are the Consequences of Heritable L1 Retrotransposition?
Among 11 de novo mouse L1 insertions uncovered in our study, 10 were intergenic and the 11th intronic insertion had no significant impact upon expression of the disrupted gene, at least in adult tissues from heterozygous animals.91 In addition, insertions were transmitted from heterozygous animals to their offspring at the expected Mendelian ratios. Thus, the vast majority of de novo L1 insertions in live‐born mammals likely produce no immediate phenotype or selective disadvantage, and instead represent new genetic diversity that could ultimately provide the raw material for genome evolution through exaptation of L1 sequences. At the other end of the spectrum, insertions with catastrophic impacts on developmentally essential genes are predicted to result in sterility or embryonic lethality, and hence would not be observed in live‐born individuals. The prevalence and impact of such insertions upon mammalian reproductive outcomes remains an open question. Intriguingly, increased L1 expression and reverse transcriptase activity has been implicated in fetal oocyte attrition in mice, although the role of mutagenic L1 integrants in this process remains to be explored.95 In the future, the contribution of L1 insertions to infertility and embryonic lethality may be resolved, at least in part, by the application of single‐cell genomic approaches to identify mutagenic L1 insertions in early embryonic and germ cells.
3.3. L1 Insertions Are an Ongoing Source of New Genetic Diversity
A distinguishing aspect of L1‐mediated mutagenesis is that retrotransposition is a replicative process. Thus, following a round of retrotransposition, the original donor L1 remains in its genomic position and retains the capacity to generate more L1 insertions. In addition, when a new L1 insertion is retrotransposition‐competent, this daughter element also has the potential to generate retrotransposition events.69 It follows that, in our study, we were able to determine the complete sequence of nine full‐length de novo L1 insertions, and all nine contained intact open reading frames. Two were tested in a cultured cell retrotransposition assay55, 96 and retained the ability to mobilize, highlighting the potential contribution of new L1 insertions to genetic diversity. Furthermore, 3′ transductions carried by three de novo insertions (and two additional insertions that were polymorphic with respect to presence among our mice, indicating relatively recent retrotransposition) allowed the identification of five discrete donor L1s in the mouse reference genome that generated heritable endogenous insertions. As the retrotransposition capacity of a donor L1 depends on its epigenetic regulation in a particular cell, in addition to the enzymatic efficiency of its encoded proteins, it will be interesting to determine whether de novo daughter insertions in vivo are subjected to the same epigenetic and host factor regulation as pre‐existing genomic elements, or whether there is some delay in establishing epigenetic marks upon newly‐integrated L1 copies. Recent studies employing engineered L1 elements in mouse embryonic stem cells and transgenic mice suggest the former scenario is more likely.97, 98
Examination of shared 3′ transductions carried by polymorphic L1 insertions in human genomes has indicated that certain donor L1s tend to produce new heritable insertions, and have propagated “hot” L1 lineages.3, 99, 100 In addition, examination of 3′ transductions and distinguishing internal mutations borne by tumor specific L1 insertions in humans has likewise revealed particular donor L1s that are recurrently active in the context of cancer.36, 101, 102 These findings raise the prospect of specific donor L1s being highly active in mammalian somatic cells.74, 103 Although all five transductions identified in our study were unique, future studies potentially uncovering common transductions in mouse L1s will reveal whether particular donor L1s are presently creating “hot” lineages by being especially active in the early embryo, and thereby recurrently contributing to mouse genomic diversity.
3.4. At What Points During Development Do Heritable L1 Insertions Occur?
To cause genetic disease and contribute to inter‐individual diversity, L1 insertions must occur in cells with the potential to contribute their genetic material to subsequent generations. Previous studies relying on transgenic L1 reporter mice or rare disease‐causing insertions in human patients had indicated that heritable L1 insertions can occur in the pluripotent cells of the early embryo, prior to germline specification, and in cells of the germline proper (Figure 2).59, 60, 63, 65, 70, 104
Figure 2.
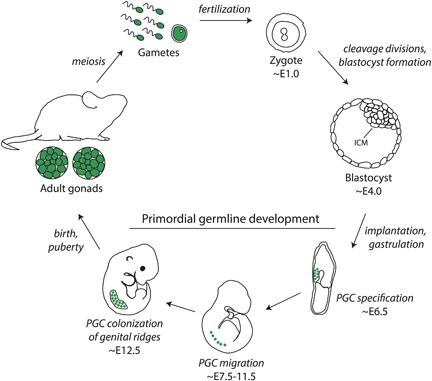
Simplified timeline of mouse embryonic and germline development. Cells of the germ lineage are depicted in green. Clockwise from upper left: fertilization of a mature oocyte by a sperm cell gives rise to the single‐cell zygote (embryonic day 1.0 [E1.0], which progresses through cleavage divisions and the morula stage to form the blastocyst [≈E4.0]). The inner cell mass (ICM) of the blastocyst is composed of pluripotent cells that give rise to the embryo. During the process of gastrulation, these pluripotent embryonic cells differentate to form the three germ layers (endoderm, ectoderm, and mesoderm). At ≈E6.5, a founding population of primordial germ cells (PGCs) are specified, then migrate (≈E7.5–11.5) to colonize the genital ridges by ≈E12.5, followed by formation of the embryonic gonads. Following birth and sexual maturation, completion of meiosis in adult animals generates mature sperm and oocytes.
3.5. Human Patients and Cell Culture Models Provide Insight Into Embryonic Retrotransposition
That new L1 insertions, and retrotransposition intermediates, can arise in pluripotent embryonic cells was established by back‐to‐back studies in 2007.104, 105 Van den Hurk et al. traced the causative mutation in a patient with X‐linked choroideremia to a full‐length L1 insertion in the CHM gene.104 By genotyping the patient's mother and siblings for the insertion and flanking heterozygous single nucleotide polymorphisms (SNPs), Van den Hurk et al. deduced that the patient's mother was both a somatic and germline mosaic for the insertion, consistent with L1 mobilization during her embryonic development prior to the segregation of the germ lineage from the soma.104 In the accompanying study, Garcia‐Perez et al. utilized L1 reporter constructs to demonstrate engineered L1 retrotransposition in human embryonic stem cells (ESCs), which generated new L1 insertions bearing the structural hallmarks of TPRT.105 Engineered L1 retrotransposition was subsequently found to occur in mouse ESCs98 and human induced pluripotent stem cells (iPSCs).106 Endogenous L1, Alu, and SVA retrotransposition has also been identified in human ESCs and iPSCs via retrotransposon capture sequencing (RC‐seq).107 By contrast, evidence for endogenous L1 retrotransposition in mouse iPSCs has been lacking to date108 and the reasons for this apparent discrepancy are unclear.
3.6. Transgenic Animal Models Exhibit Embryonic Retrotransposition
Analysis of transgenic mouse models likewise supports the case for early embryonic L1 retrotransposition (Table 1). Prak et al. generated a transgenic mouse harboring a human L1‐EGFP reporter gene, and identified an engineered insertion arising during early embryonic development of the founder mouse.60 An et al.63 also observed heritable engineered insertions in an L1‐EGFP transgenic mouse model, although individual insertions were not traced to a discrete developmental timeframe.
Two subsequent studies using transgenic mouse models produced results consistent with retrotransposition occurring post‐fertilization. Babushok et al. generated transgenic mice using a human “markerless” reporter cassette, consisting only of an intron rather than an expressed reporter gene.62 While frequent de novo retrotransposition events were detected in F1 and F2 offspring from a transgenic founder animal, these insertions were not transmitted in Mendelian fashion and were present in <1 copy per cell. Together, these results suggested that the L1 insertions frequently contributed to somatic, but not germline, mosaicism.
In a 2009 study, Kano et al. verified this result more robustly by generating multiple transgenic mouse and rat lines harboring human and mouse L1‐EGFP reporter transgenes driven by the corresponding native L1 promoter (Table 1).65 L1 insertions were identified in the offspring of transgenic males and females crossed with wild‐type animals, and in some cases the offspring containing an insertion lacked the transgene. This result is consistent with retrotransposition in the parental germline, prior to the end of meiosis I, and subsequent segregation of the new insertion away from the transgene during meiosis II. In such a scenario, insertion‐positive, transgene‐negative offspring are expected to be heterozygous for the insertion, and transmit it to approximately 50% of their offspring (Figure 3A). Unexpectedly, insertion‐positive, transgene‐negative animals uniformly failed to transmit the insertion to their offspring, and harbored the insertion at <1 copy per cell. This result led to a model in which an L1 mRNA is transcribed in the parental germline and, perhaps complexed in an RNP, can be carried over during fertilization to undergo retrotransposition in a cell of the resultant F1 embryo.65 The failure of such insertions to be inherited by F2 offspring suggests that these insertions contributed to somatic but not germline mosaicism, consistent with retrotransposition after specification of the germ lineage, or in pluripotent cells which did not contribute to the germline (Figure 3A).65
Figure 3.
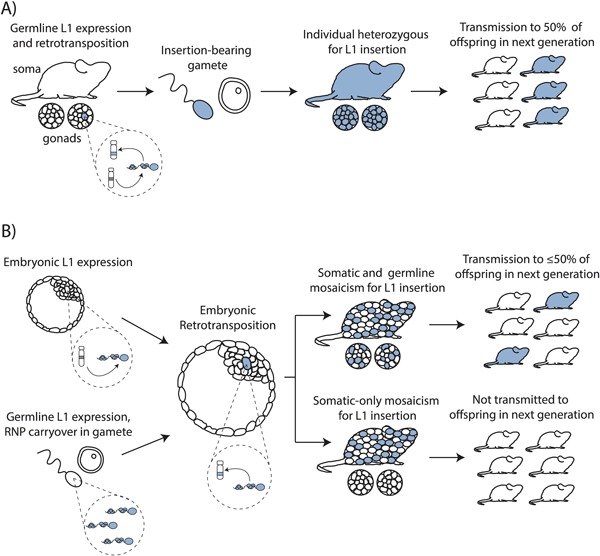
Germline and embryonic L1 retrotransposition. A) Germline L1 retrotransposition. A retrotransposition‐competent L1 (gray) undergoes retrotransposition at some point during germline development (gonads represented by circles under mouse) to generate a new insertion (blue). If a gamete harboring this insertion (blue sperm) undergoes fertilization, the resultant animal will be heterozygous for the insertion (blue mouse) and is expected to transmit the insertion to ≈50% of its offspring. B) Embryonic L1 retrotransposition. From left: L1 expression and RNP formation may occur in a cell of the early embryo (above), or an L1 RNP formed in the parental germline may be carried over in a gamete during fertilization (below). Completion of retrotransposition in a pluripotent embryonic cell will give rise to an insertion for which the embryo is mosaic (initial insertion‐harboring cell shown in blue). Descendants of the insertion‐harboring cell may contribute to the somatic and germ cell lineages (above), rendering the insertion heritable by up to ≈50% of offspring. Alternatively, insertion‐harboring cells may contribute only to somatic cell lineages (below), in which case the insertion is not heritable by offspring.
3.7. Endogenous Heritable Retrotransposition Events Occur in Early Mouse Embryos
Our recent results elucidating endogenous heritable L1 retrotransposition events share several consistencies with the studies described above, but also feature important differences.91 In our study, we were able to trace the developmental timing of multiple endogenous L1 insertions, by using insertion‐specific PCR genotyping to detect each insertion in the various somatic and germ tissues of the mouse of origin and of related mice. Among 11 de novo L1 insertions, we traced 6 to pluripotent embryonic cells, as evidenced by mosaicism for the insertion among the tissues of the originating mouse detected by conventional and quantitative genotyping PCR. Among these six insertions, we demonstrated transmission of four insertions from the mosaic animal of origin to its offspring, consistent with the findings of Van den Hurk et al. in humans and Prak et al. in a transgenic mouse (Figure 3B).60, 104 The remaining two L1 insertions could be detected in multiple tissues from the originating mosaic animal, but were not transmitted to the next generation. One explanation for this result is that the number of offspring produced by the originating mosaic animals was insufficient to observe infrequent transmission. The other possibility is that these two events contributed to somatic but not germline mosaicism during embryonic development of the originating animal, consistent with the results of Babushok et al. and Kano et al. (Figure 3B).62, 65
3.8. What Proportion of Early Embryonic Insertions Are Heritable?
While more than half of the early embryonic insertions we identified contributed to the germline and were demonstrably heritable, the vast majority of engineered L1‐EGFP insertions reported by earlier studies arose in early embryonic cells of transgenic animals and contributed to somatic mosaicism but were not transmitted to subsequent generations.60, 62, 65 The likely explanation for this discrepancy is that our approach favored the detection of transmissible insertions. All four heritable embryonic insertions initially were identified by mRC‐seq in bulk tissues from heterozygous offspring, and were subsequently detected at low prevalence in the corresponding maternal tissues. Thus, many additional somatic‐restricted L1 insertions, such as those occurring in the neuronal lineage,61, 86, 87, 109, 110 may have been present in our animals but fell below the detection capacity of mRC‐seq.74 Indeed, a transgenic mouse model allows much more sensitive detection of such “late” or low‐prevalence insertions a priori, since the spliced reporter cassette is much more readily detected by PCR than a low‐prevalence endogenous insertion is by high‐throughput sequencing. Nevertheless, it is quite striking that Kano et al. never detected transmission of early embryonic insertions, despite using mouse and human L1 transgenes driven by their native promoters, and examining 170 F2 progeny. It is possible that the L1 transgenes preferentially generated insertions slightly later during embryonic development, restricted to cells committed to the somatic lineages, while insertions arising from endogenous donor L1s were more likely to generate earlier insertions in pluripotent cells with the capacity to contribute to the germline. Future studies employing single‐cell genomics have the potential to resolve this discrepancy by elucidating the rate of endogenous embryonic retrotransposition in an unbiased manner, in totipotent, pluripotent, and lineage‐committed cells during early embryonic development.
3.9. Carry‐Over of L1 RNPs: Spatial and Temporal Separation of L1 Expression and Insertion?
Whether endogenous L1 RNPs arising in the maternal or paternal germline can be carried over during fertilization to generate early embryonic insertions, as has been proposed for L1 RNPs arising from engineered transgenes,65 remains an open question. Our study employed inbred C57BL/6J mice, so the vast majority of potential donor L1s responsible for insertions and flanking SNPs were homozygous. Thus, we could not use 3′ transductions to discern whether de novo embryonic L1 insertions arose from a maternal or paternal donor L1, nor whether the mosaic daughter insertion occurred independently of the donor allele, indicative of endogenous L1 RNP carryover during fertilization. Future studies employing a more genetically diverse mouse model, in which the parent of origin and progenitor L1 element responsible for embryonic insertions can potentially be discerned, may allow formal demonstration of endogenous L1 RNP carryover during fertilization.
If L1 expression and TPRT can be spatially and temporally separated in this extreme scenario, it also seems likely that the transcription of a full‐length L1 mRNA, and its translation and subsequent incorporation into an RNP, could occur in a given cell, and the RNP then persist during multiple rounds of cell division, and perhaps differentiation, to undergo retrotransposition in a descendant cell at a later developmental stage (Figure 4). Indeed, endogenous and engineered L1 proteins and mRNA have been demonstrated to form perinuclear aggregates,111, 112, 113 which we hypothesize could facilitate the long‐term protection and persistence of L1 RNPs. In such a scenario, we speculate that L1 expression could occur in pluripotent cells where L1 sequences are demethylated and transcriptionally active, and that L1 RNPs could persist to carry out retrotransposition at a stage where L1 sequences may be highly methylated but intracellular conditions (i.e., the presence or absence of certain host factors) allow TPRT to be completed. The hypothesis of mitotic L1 RNP carryover thus represents an exciting avenue for future research into L1 and host factor dynamics during mammalian development.
Figure 4.
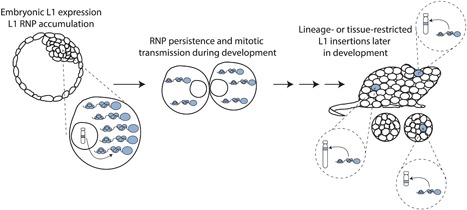
Hypothetical scenario for mitotic transmission of L1 RNPs during development. From left: L1 expression in a permissive cell type (e.g., a pluripotent embryonic cell) could lead to L1 RNP accumulation. If these RNPs persist during subsequent rounds of cell division and differentiation, they may complete retrotransposition much later in development, generating lineage‐, or tissue‐restricted insertions.
4. L1 Retrotransposition Occurs in the Germline
Germ cells represent a clear opportunity for the generation of heritable retrotransposition events, and developmentally regulated L1 expression in the mammalian germline suggests that germ cells may accommodate L1 retrotransposition. Full‐length endogenous L1 mRNAs and ORF1p are expressed in prepuberal mouse testis at postnatal day 10, in leptotene and zygotene spermatocytes during the first wave of meiosis.114 In adult testis, ORF1p is expressed in round and elongating spermatids, and can be faintly and rarely detected in spermatocytes.114, 115 L1 ORF1p can also be detected in fetal male and female germ cells from embryonic day 15.5.94, 116
4.1. Does L1 Retrotransposition Occur in Human Germ Cells?
Evidence for L1 retrotransposition in the human germline is limited. Brouha et al. identified an L1 insertion bearing a 3′ transduction that had occurred into the X‐linked CYBB gene of a male patient, resulting in a case of chronic granulomatous disease.70 The 3′ transduction allowed the donor L1 for the disease‐causing insertion to be identified. Moreover, a polymorphism within this transduced sequence made it possible to determine that the disease causing insertion had been inherited by the patient independently of its donor allele. This result is consistent with retrotransposition in the maternal germline prior to the completion of meiosis I. However, the observation of transgene‐independent early embryonic insertions in mice resulting from L1 RNP carryover during fertilization, as reported by Kano et al.,65 provides an equally likely explanation for this result. Indeed, while human oocytes have been demonstrated to accommodate the retrotransposition of L1‐EGFP reporter constructs,117 analysis of human sperm for endogenous L1 insertions using a PCR‐based strategy uncovered no insertions and resulted in an estimate of fewer than one de novo insertion per 400 sperm.118
4.2. Transgenic Animal Models Undergo Germline L1 Retrotransposition
Results from transgenic animal models have provided some support for germline L1 retrotransposition. Ostertag et al. generated transgenic L1‐EGFP mouse lines in which expression of the L1 was driven by the human L1 5′UTR with and without a heterologous mouse RNA polymerase (pPol II) promoter.59 Two L1‐EGFP insertions were detected among 135 offspring of male pPol II‐L1 EGFP mice mated to wild‐type females. One insertion was inherited along with the transgene, and therefore could have occurred either in the male germline or post‐fertilization in the developing embryo. The second insertion was inherited independently of the transgene, strongly suggesting that retrotransposition had occurred in the male germline prior to the end of meiosis I. Whether this insertion arose during meiosis or at an earlier developmental stage could not be determined.
Very recently, Newkirk et al.66 developed a transgenic mouse model using the synthetic mouse L1 ORFeus_Mm, driven by a native L1 TF promoter10 and bearing an EGFP retrotransposition indicator cassette.58 In contrast to most previous transgenic L1 mouse models (Table 1), the authors performed their study using a mouse line containing a single copy of the transgene, avoiding the complication of repeat‐induced transgene silencing observed for multi‐copy transgene concatamers.119 Indeed, methylation of this transgene recapitulated the dynamics of endogenous L1 TF copies in pre‐ and post‐natal male germ cells and somatic cells. Insertions arising from the L1‐EGFP transgene were detected using a highly sensitive digital droplet PCR (ddPCR) strategy targeting the EGFP splice junction. Newkirk et al. applied this system to mice deficient in Mov10l1, an RNA helicase with an essential role in the piRNA pathway, an adaptive transposon defense mechanism active in male gonadal primordial germ cells.120, 121 Their results clearly demonstrated that deficiency in a piRNA pathway effector results not only in an increase in L1 expression, but also an increase in retrotransposition in germ cells during meiosis.
Although Newkirk et al. primarily focused on the increase in germline L1 retrotransposition in Mov10l1−/− mice, their findings have interesting implications for the dynamics of heritable retrotransposition in wild‐type animals. Based upon a frequency of 0.00024 engineered ORFeus_Mm insertions per cell as detected by ddPCR in adult control (Mov10l1+/−) testes, the estimation of 3000 active L1 copies per diploid mouse genome,4 and the assumption that ORFeus_Mm retrotransposes 200‐fold more efficiently than the average active L1 copy,72 the authors arrive at an estimated frequency of one insertion per 278 cells in the mouse germline. As the authors note, their methodology does not distinguish between an insertion arising de novo specifically in a germ cell, and an insertion that arose in a progenitor cell at an earlier stage of development. Indeed, their data reflect a degree of variability between individual animals in the prevalence of insertions in adult tissues and also in staged spermatogenic cells. This inter‐individual variability could arise from different levels of de novo retrotransposition in individual somatic and germ cells, or from differences in the relative germline prevalence of insertions that occurred earlier during the development of each mouse, either prior to germline specification or during earlier germline development. The developmental timing of engineered insertions in control animals bearing this experimental system therefore remains to be determined. Indeed, development of a transgenic mouse model that allows the distinction of individual engineered L1 insertions could be an invaluable tool for lineage‐tracing experiments that establish the developmental timing of L1 retrotransposition events.
4.3. How Frequently Does L1 Retrotranspose in Mouse Germ Cells?
In our examination of de novo endogenous L1 insertions in mice, we uncovered a single L1 insertion that was uniformly heterozygous in all tissues of the originating mouse, and could not be detected in the somatic or germ tissues of either parental animal (Figure 3A).91 We attributed this insertion to germline development, although the exact timing of this event and the parental origin for the insertion could not be established. Determination of endogenous L1 retrotransposition frequency during later stages of germline development will likely require single‐cell analysis applied to staged spermatogenic cells or individual developing oocytes.
5. L1 Retrotransposition Occurs in Early Primordial Germ Cells
Mouse primordial germline development begins in the posterior primitive streak of the post‐implantation embryo at embryonic day 6.5 (E6.5) with a discrete founding population of around 40 PGCs in the proximal epiblast. PGCs then migrate through the developing hindgut from E7.5–E8.5, and then along the midline of the embryo through the hindgut epithelium and dorsal mesentary at about E9.0–E9.5. The PGCs then form two clusters as they move laterally to colonize the genital ridges from E10.0–E12.5 (Figure 2).122 In our recent study,91 three out of the 11 de novo L1 insertions identified showed a distinctive pattern of inheritance: among the offspring of a given male mouse, 3–5% of siblings were heterozygous for a new L1 insertion that was not present in the somatic tissues of either parent. However, the insertion was present at <1 copy per cell in both testicles of the paternal mouse. Together, these results indicated germline‐restricted mosaicism for the insertion, and that the insertion had originated very early during germline development, most likely in an early PGC prior to colonization of the genital ridges and the formation of two testicles.
L1 retrotransposition at this very early stage of germline development represents a largely unexplored aspect of L1 biology in mammals. Indeed, previous studies have focused on L1 expression and activity in later germ cells, particularly during meiosis. Thus, numerous questions remain to be answered: is L1 expression activated at the onset of PGC specification, or afterwards, as PGCs migrate toward the genital ridges? Genome‐wide demethylation occurs progressively during these processes, including on L1 copies,123 but what other epigenetic mechanisms regulate L1 in early PGCs? Notably, a recent study by Kim et al. demonstrated L1 expression is upregulated in E11.5–E12.5 mouse PGCs with conditional deletion of the histone arginine methyltransferase PRMT5, although a corresponding increase in L1 copy number was not observed.124
The low number of PGCs per mouse, particularly in the PGC founder population, renders PGC specification and early development a particularly difficult process to study in vivo. Ethical considerations limit the extent to which PGC development can be studied in humans. However, recently‐developed protocols for differentiating mouse and human cultured pluripotent cells to PGC‐like cells may be more tractable to gain insight into the dynamics of endogenous L1 regulation during PGC specification, and to high‐throughput sequencing approaches to identify endogenous L1 retrotransposition events occurring during PGC specification in vitro.125, 126 Such cultured cell systems may also be amenable to the use of engineered L1 reporter constructs. Future studies will no doubt elucidate the dynamics of L1 regulation and activity in this important developmental niche.
6. Conclusions and Outlook
The generation of heritable L1 insertions is critically important for the evolutionary success of L1 as a selfish genetic element, and has had a profound effect on the landscape of mammalian genomes. Rare cases where the developmental origins of disease‐causing L1 insertions can be traced, in addition to innovative cultured cell and transgenic mouse model systems allowing the detection of engineered L1 retrotransposition events, have provided invaluable insight regarding L1 dynamics in the mammalian germline and early embryo. Our recent work took advantage of high‐throughput sequencing approaches to identify and track the developmental origins of heritable, endogenous L1 insertions.91 Our results build upon and strengthen the conclusions from previous studies, but also reveal important differences between the results obtained from transgenic model systems and via mapping endogenous L1 retrotransposition events. Going forward, the integration of high‐throughput sequencing approaches, including single‐cell genomics, with engineered reporter systems in cultured cells and transgenic animals, should provide the means to answer remaining questions regarding the rate and timing of L1 retrotransposition during mammalian development, the fate of embryonic cells carrying de novo L1 mutations, and the consequences of this activity on the host genome.
Conflict of Interest
The authors declare no conflict of interest.
Acknowledgments
G.J.F. is supported by a CSL Centenary Fellowship and the Mater Foundation. S.R.R. and G.J.F. are supported by Project Grants funds from the Australian NHMRC (GNT1106206 and GNT1126393).
Contributor Information
Sandra R. Richardson, Email: sandra.richardson@mater.uq.edu.au
Geoffrey J. Faulkner, Email: faulknergj@gmail.com
References
- 1. Lander E. S., Linton L. M., Birren B., Nusbaum C., Zody M. C., Baldwin J., Devon K., Dewar K., Doyle M., FitzHugh W., Funke R., Gage D., Harris K., Heaford A., Howland J., Kann L., Lehoczky J., LeVine R., McEwan P., McKernan K., Meldrim J., Mesirov J. P., Miranda C., Morris W., Naylor J., Raymond C., Rosetti M., Santos R., Sheridan A., Sougnez C., Stange‐Thomann N., Stojanovic N., Subramanian A., Wyman D., Rogers J., Sulston J., Ainscough R., Beck S., Bentley D., Burton J., Clee C., Carter N., Coulson A., Deadman R., Deloukas P., Dunham A., Dunham I., Durbin R., French L., Grafham D., Gregory S., Hubbard T., Humphray S., Hunt A., Jones M., Lloyd C., McMurray A., Matthews L., Mercer S., Milne S., Mullikin J. C., Mungall A., Plumb R., Ross M., Shownkeen R., Sims S., Waterston R. H., Wilson R. K., Hillier L. W., McPherson J. D., Marra M. A., Mardis E. R., Fulton L. A., Chinwalla A. T., Pepin K. H., Gish W. R., Chissoe S. L., Wendl M. C., Delehaunty K. D., Miner T. L., Delehaunty A., Kramer J. B., Cook L. L., Fulton R. S., Johnson D. L., Minx P. J., Clifton S. W., Hawkins T., Branscomb E., Predki P., Richardson P., Wenning S., Slezak T., Doggett N., Cheng J. F., Olsen A., Lucas S., Elkin C., Uberbacher E., Frazier M., Gibbs R. A., Muzny D. M., Scherer S. E., Bouck J. B., Sodergren E. J., Worley K. C., Rives C. M., Gorrell J. H., Metzker M. L., Naylor S. L., Kucherlapati R. S., Nelson D. L., Weinstock G. M., Sakaki Y., Fujiyama A., Hattori M., Yada T., Toyoda A., Itoh T., Kawagoe C., Watanabe H., Totoki Y., Taylor T., Weissenbach J., Heilig R., Saurin W., Artiguenave F., Brottier P., Bruls T., Pelletier E., Robert C., Wincker P., Smith D. R., Doucette‐Stamm L., Rubenfield M., Weinstock K., Lee H. M., Dubois J., Rosenthal A., Platzer M., Nyakatura G., Taudien S., Rump A., Yang H., Yu J., Wang J., Huang G., Gu J., Hood L., Rowen L., Madan A., Qin S., Davis R. W., Federspiel N. A., Abola A. P., Proctor M. J., Myers R. M., Schmutz J., Dickson M., Grimwood J., Cox D. R., Olson M. V., Kaul R., Shimizu N., Kawasaki K., Minoshima S., Evans G. A., Athanasiou M., Schultz R., Roe B. A., Chen F., Pan H., Ramser J., Lehrach H., Reinhardt R., McCombie W. R., de la Bastide M., Dedhia N., Blocker H., Hornischer K., Nordsiek G., Agarwala R., Aravind L., Bailey J. A., Bateman A., Batzoglou S., Birney E., Bork P., Brown D. G., Burge C. B., Cerutti L., Chen H. C., Church D., Clamp M., Copley R. R., Doerks T., Eddy S. R., Eichler E. E., Furey T. S., Galagan J., Gilbert J. G., Harmon C., Hayashizaki Y., Haussler D., Hermjakob H., Hokamp K., Jang W., Johnson L. S., Jones T. A., Kasif S., Kaspryzk A., Kennedy S., Kent W. J., Kitts P., Koonin E. V., Korf I., Kulp D., Lancet D., Lowe T. M., McLysaght A., Mikkelsen T., Moran J. V., Mulder N., Pollara V. J., Ponting C. P., Schuler G., Schultz J., Slater G., Smit A. F., Stupka E., Szustakowski J., Thierry‐Mieg D., Thierry‐Mieg J., Wagner L., Wallis J., Wheeler R., Williams A., Wolf Y. I., Wolfe K. H., Yang S. P., Yeh R. F., Collins F., Guyer M. S., Peterson J., Felsenfeld A., Wetterstrand K. A., Patrinos A., Morgan M. J., de Jong P., Catanese J. J., Osoegawa K., Shizuya H., Choi S., Chen Y. J., Nature 2001, 409, 860. 11237011 [Google Scholar]
- 2. Waterston R. H., Lindblad‐Toh K., Birney E., Rogers J., Abril J. F., Agarwal P., Agarwala R., Ainscough R., Alexandersson M., An P., Antonarakis S. E., Attwood J., Baertsch R., Bailey J., Barlow K., Beck S., Berry E., Birren B., Bloom T., Bork P., Botcherby M., Bray N., Brent M. R., Brown D. G., Brown S. D., Bult C., Burton J., Butler J., Campbell R. D., Carninci P., Cawley S., Chiaromonte F., Chinwalla A. T., Church D. M., Clamp M., Clee C., Collins F. S., Cook L. L., Copley R. R., Coulson A., Couronne O., Cuff J., Curwen V., Cutts T., Daly M., David R., Davies J., Delehaunty K. D., Deri J., Dermitzakis E. T., Dewey C., Dickens N. J., Diekhans M., Dodge S., Dubchak I., Dunn D. M., Eddy S. R., Elnitski L., Emes R. D., Eswara P., Eyras E., Felsenfeld A., Fewell G. A., Flicek P., Foley K., Frankel W. N., Fulton L. A., Fulton R. S., Furey T. S., Gage D., Gibbs R. A., Glusman G., Gnerre S., Goldman N., Goodstadt L., Grafham D., Graves T. A., Green E. D., Gregory S., Guigo R., Guyer M., Hardison R. C., Haussler D., Hayashizaki Y., Hillier L. W., Hinrichs A., Hlavina W., Holzer T., Hsu F., Hua A., Hubbard T., Hunt A., Jackson I., Jaffe D. B., Johnson L. S., Jones M., Jones T. A., Joy A., Kamal M., Karlsson E. K., Karolchik D., Kasprzyk A., Kawai J., Keibler E., Kells C., Kent W. J., Kirby A., Kolbe D. L., Korf I., Kucherlapati R. S., Kulbokas E. J., Kulp D., Landers T., Leger J. P., Leonard S., Letunic I., Levine R., Li J., Li M., Lloyd C., Lucas S., Ma B., Maglott D. R., Mardis E. R., Matthews L., Mauceli E., Mayer J. H., McCarthy M., McCombie W. R., McLaren S., McLay K., McPherson J. D., Meldrim J., Meredith B., Mesirov J. P., Miller W., Miner T. L., Mongin E., Montgomery K. T., Morgan M., Mott R., Mullikin J. C., Muzny D. M., Nash W. E., Nelson J. O., Nhan M. N., Nicol R., Ning Z., Nusbaum C., O'Connor M. J., Okazaki Y., Oliver K., Overton‐Larty E., Pachter L., Parra G., Pepin K. H., Peterson J., Pevzner P., Plumb R., Pohl C. S., Poliakov A., Ponce T. C., Ponting C. P., Potter S., Quail M., Reymond A., Roe B. A., Roskin K. M., Rubin E. M., Rust A. G., Santos R., Sapojnikov V., Schultz B., Schultz J., Schwartz M. S., Schwartz S., Scott C., Seaman S., Searle S., Sharpe T., Sheridan A., Shownkeen R., Sims S., Singer J. B., Slater G., Smit A., Smith D. R., Spencer B., Stabenau A., Stange‐Thomann N., Sugnet C., Suyama M., Tesler G., Thompson J., Torrents D., Trevaskis E., Tromp J., Ucla C., Ureta‐Vidal A., Vinson J. P., Von Niederhausern A. C., Wade C. M., Wall M., Weber R. J., Weiss R. B., Wendl M. C., West A. P., Wetterstrand K., Wheeler R., Whelan S., Wierzbowski J., Willey D., Williams S., Wilson R. K., Winter E., Worley K. C., Wyman D., Yang S., Yang S. P., Zdobnov E. M., Zody M. C., Lander E. S., Nature 2002, 420, 520. 12466850 [Google Scholar]
- 3. Brouha B., Schustak J., Badge R. M., Lutz‐Prigge S., Farley A. H., Moran J. V., H. H. Kazazian, Jr. , Proc. Natl. Acad. Sci. U. S. A. 2003, 100, 5280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Goodier J. L., Ostertag E. M., Du K., H. H. Kazazian, Jr. , Genome Res. 2001, 11, 1677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Dombroski B. A., Mathias S. L., Nanthakumar E., Scott A. F., H. H. Kazazian, Jr. , Science 1991, 254, 1805. [DOI] [PubMed] [Google Scholar]
- 6. Fanning T. G., Nucl. Acids Res. 1983, 11, 5073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Swergold G. D., Mol. Cell Biol. 1990, 10, 6718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Loeb D. D., Padgett R. W., Hardies S. C., Shehee W. R., Comer M. B., Edgell M. H., C. A. Hutchison, 3rd , Mol. Cell Biol. 1986, 6, 168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. DeBerardinis R. J., Goodier J. L., Ostertag E. M., H. H. Kazazian, Jr. , Nat. Genet. 1998, 20, 288. [DOI] [PubMed] [Google Scholar]
- 10. Naas T. P., DeBerardinis R. J., Moran J. V., Ostertag E. M., Kingsmore S. F., Seldin M. F., Hayashizaki Y., Martin S. L., Kazazian H. H., EMBO J. 1998, 17, 590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Wei W., Gilbert N., Ooi S. L., Lawler J. F., Ostertag E. M., Kazazian H. H., Boeke J. D., Moran J. V., Mol Cell Biol 2001, 21, 1429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Hohjoh H., Singer M. F., EMBO J. 1996, 15, 630. [PMC free article] [PubMed] [Google Scholar]
- 13. Martin S. L., Mol. Cell Biol. 1991, 11, 4804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Feng Q., Moran J. V., H. H. Kazazian, Jr. , Boeke J. D., Cell 1996, 87, 905. [DOI] [PubMed] [Google Scholar]
- 15. Cost G. J., Feng Q., Jacquier A., Boeke J. D., EMBO J. 2002, 21, 5899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Luan D. D., Korman M. H., Jakubczak J. L., Eickbush T. H., Cell 1993, 72, 595. [DOI] [PubMed] [Google Scholar]
- 17. Kulpa D. A., Moran J. V., Nat. Struct. Mol. Biol. 2006, 13, 655. [DOI] [PubMed] [Google Scholar]
- 18. Grimaldi G., Skowronski J., Singer M. F., EMBO J. 1984, 3, 1753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Gilbert N., Lutz‐Prigge S., Moran J. V., Cell 2002, 110, 315. [DOI] [PubMed] [Google Scholar]
- 20. Gilbert N., Lutz S., Morrish T. A., Moran J. V., Mol. Cell Biol. 2005, 25, 7780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. H. H. Kazazian, Jr. , Wong C., Youssoufian H., Scott A. F., Phillips D. G., Antonarakis S. E., Nature 1988, 332, 164. [DOI] [PubMed] [Google Scholar]
- 22. Dewannieux M., Esnault C., Heidmann T., Nat. Genet. 2003, 35, 41. [DOI] [PubMed] [Google Scholar]
- 23. Raiz J., Damert A., Chira S., Held U., Klawitter S., Hamdorf M., Lower J., Stratling W. H., Lower R., Schumann G. G., Nucl Acids Res. 2012, 40, 1666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Hancks D. C., Goodier J. L., Mandal P. K., Cheung L. E., H. H. Kazazian, Jr. , Hum. Mol. Genet. 2011, 20, 3386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Dewannieux M., Heidmann T., J. Mol. Biol. 2005, 349, 241. [DOI] [PubMed] [Google Scholar]
- 26. Esnault C., Maestre J., Heidmann T., Nat. Genet. 2000, 24, 363. [DOI] [PubMed] [Google Scholar]
- 27. Ewing A. D., Ballinger T. J., Earl D., Harris C. C., Ding L., Wilson R. K., Haussler D., Genome Biol. 2013, 14, R22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Doucet A. J., Wilusz J. E., Miyoshi T., Liu Y., Moran J. V., Mol. Cell. 2015, 60, 728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Myers J. S., Vincent B. J., Udall H., Watkins W. S., Morrish T. A., Kilroy G. E., Swergold G. D., Henke J., Henke L., Moran J. V., Jorde L. B., Batzer M. A., Am. J. Hum. Genet. 2002, 71, 312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Ostertag E. M., H. H. Kazazian, Jr. , Genome Res. 2001, 11, 2059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Gilbert N., Bomar J. M., Burmeister M., Moran J. V., Hum. Mutat. 2004, 24, 9. [DOI] [PubMed] [Google Scholar]
- 32. Moran J. V., DeBerardinis R. J., H. H. Kazazian, Jr. , Science 1999, 283, 1530. [DOI] [PubMed] [Google Scholar]
- 33. Pickeral O. K., Makalowski W., Boguski M. S., Boeke J. D., Genome Res. 2000, 10, 411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Goodier J. L., Ostertag E. M., H. H. Kazazian, Jr. , Hum. Mol. Genet. 2000, 9, 653. [DOI] [PubMed] [Google Scholar]
- 35. Miki Y., Nishisho I., Horii A., Miyoshi Y., Utsunomiya J., Kinzler K. W., Vogelstein B., Nakamura Y., Cancer Res. 1992, 52, 643. [PubMed] [Google Scholar]
- 36. Scott E. C., Gardner E. J., Masood A., Chuang N. T., Vertino P. M., Devine S. E., Genome Res. 2016, 26, 745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Perepelitsa‐Belancio V., Deininger P., Nat. Genet. 2003, 35, 363. [DOI] [PubMed] [Google Scholar]
- 38. Belancio V. P., Roy‐Engel A. M., Deininger P., Gene 2008, 411, 38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Han J. S., Szak S. T., Boeke J. D., Nature 2004, 429, 268. [DOI] [PubMed] [Google Scholar]
- 40. Shukla R., Upton K. R., Munoz‐Lopez M., Gerhardt D. J., Fisher M. E., Nguyen T., Brennan P. M., Baillie J. K., Collino A., Ghisletti S., Sinha S., Iannelli F., Radaelli E., Dos Santos A., Rapoud D., Guettier C., Samuel D., Natoli G., Carninci P., Ciccarelli F. D., Garcia‐Perez J. L., Faivre J., Faulkner G. J., Cell 2013, 153, 101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Denli A. M., Narvaiza I., Kerman B. E., Pena M., Benner C., Marchetto M. C., Diedrich J. K., Aslanian A., Ma J., Moresco J. J., Moore L., Hunter T., Saghatelian A., Gage F. H., Cell 2015, 163, 583. [DOI] [PubMed] [Google Scholar]
- 42. H. H. Kazazian, Jr. , Moran J. V., N. Eng. J. Med. 2017, 377, 361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Ostertag E. M., H. H. Kazazian, Jr. , Annu. Rev. Genet. 2001, 35, 501. [DOI] [PubMed] [Google Scholar]
- 44. Chen J., Rattner A., Nathans J., Hum. Mol. Genet. 2006, 15, 2146. [DOI] [PubMed] [Google Scholar]
- 45. Crichton J. H., Dunican D. S., Maclennan M., Meehan R. R., Cell. Mol. Life Sci. 2014, 71, 1581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Goodier J. L., Mobile DNA 2016, 7, 16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Ewing A. D., H. H. Kazazian, Jr. , Genome Res. 2010, 20, 1262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Xing J., Zhang Y., Han K., Salem A. H., Sen S. K., Huff C. D., Zhou Q., Kirkness E. F., Levy S., Batzer M. A., Jorde L. B., Genome Res. 2009, 19, 1516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. H. H. Kazazian, Jr. , Nat. Genet. 1999, 22, 130. [DOI] [PubMed] [Google Scholar]
- 50. Cordaux R., Hedges D. J., Herke S. W., Batzer M. A., Gene 2006, 373, 134. [DOI] [PubMed] [Google Scholar]
- 51. Li X., Scaringe W. A., Hill K. A., Roberts S., Mengos A., Careri D., Pinto M. T., Kasper C. K., Sommer S. S., Hum. Mutat. 2001, 17, 511. [DOI] [PubMed] [Google Scholar]
- 52. Hancks D. C., H. H. Kazazian, Jr. , Curr. Opin. Genet. Dev. 2012, 22, 191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. H. H. Kazazian, Jr. , Moran JV., Nat. Genet. 1998, 19, 19. [DOI] [PubMed] [Google Scholar]
- 54. H. H. Kazazian, Jr. , Science 2000, 289, 1152. [DOI] [PubMed] [Google Scholar]
- 55. Moran J. V., Holmes S. E., Naas T. P., DeBerardinis R. J., Boeke J. D., H. H. Kazazian, Jr. , Cell 1996, 87, 917. [DOI] [PubMed] [Google Scholar]
- 56. Rangwala S. H., H. H. Kazazian, Jr. , Methods 2009, 49, 219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Richardson S. R., Doucet A. J., Kopera H. C., Moldovan J. B., Garcia‐Perez J. L., Moran J. V., Microbiol. Spectr. 2015, 3, MDNA3‐0061‐2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Ostertag E. M., Prak E. T., DeBerardinis R. J., Moran J. V., H. H. Kazazian, Jr. , Nucl. Acids Res. 2000, 28, 1418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Ostertag E. M., DeBerardinis R. J., Goodier J. L., Zhang Y., Yang N., Gerton G. L., H. H. Kazazian, Jr. , Nat. Genet. 2002, 32, 655. [DOI] [PubMed] [Google Scholar]
- 60. Prak E. T., Dodson A. W., Farkash E. A., H. H. Kazazian, Jr. , Proc. Natl. Acad. Sci. U. S. A. 2003, 100, 1832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Muotri A. R., Chu V. T., Marchetto M. C., Deng W., Moran J. V., Gage F. H., Nature 2005, 435, 903. [DOI] [PubMed] [Google Scholar]
- 62. Babushok D. V., Ostertag E. M., Courtney C. E., Choi J. M., H. H. Kazazian, Jr. , Genome Res. 2006, 16, 240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. An W., Han J. S., Wheelan S. J., Davis E. S., Coombes C. E., Ye P., Triplett C., Boeke J. D., Proc. Natl. Acad. Sci. U. S. A. 2006, 103, 18662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. An W., Han J. S., Schrum C. M., Maitra A., Koentgen F., Boeke J. D., Genesis 2008, 46, 373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Kano H., Godoy I., Courtney C., Vetter M. R., Gerton G. L., Ostertag E. M., H. H. Kazazian, Jr. , Genes. Dev. 2009, 23, 1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Newkirk S. J., Lee S., Grandi F. C., Gaysinskaya V., Rosser J. M., Vanden Berg N., Hogarth C. A., Marchetto M. C. N., Muotri A. R., Griswold M. D., Ye P., Bortvin A., Gage F. H., Boeke J. D., An W., Proc. Natl. Acad. Sci. U. S. A. 2017, 114, E5635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Garcia‐Perez J. L., Morell M., Scheys J. O., Kulpa D. A., Morell S., Carter C. C., Hammer G. D., Collins K. L., O'Shea K. S., Menendez P., Moran J. V., Nature 2010, 466, 769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Macia A., Widmann T. J., Heras S. R., Ayllon V., Sanchez L., Benkaddour‐Boumzaouad M., Munoz‐Lopez M., Rubio A., Amador‐Cubero S., Blanco‐Jimenez E., Garcia‐Castro J., Menendez P., Ng P., Muotri A. R., Goodier J. L., Garcia‐Perez J. L., Genome Res. 2017, 27, 335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Kimberland M. L., Divoky V., Prchal J., Schwahn U., Berger W., H. H. Kazazian, Jr. , Hum. Mol. Genet. 1999, 8, 1557. [DOI] [PubMed] [Google Scholar]
- 70. Brouha B., Meischl C., Ostertag E., de Boer M., Zhang Y., Neijens H., Roos D., H. H. Kazazian, Jr. , Am. J. Hum. Genet. 2002, 71, 327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. An W., Boeke J. D., Genome Biol. 2005, 6, 361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Han J. S., Boeke J. D., Nature 2004, 429, 314. [DOI] [PubMed] [Google Scholar]
- 73. Ewing A. D., Mobile DNA 2015, 6, 24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Faulkner G.J., Garcia‐Perez J.L., Trends Genet. 2017, 33, 802. [DOI] [PubMed] [Google Scholar]
- 75. Akagi K., Li J., Stephens R. M., Volfovsky N., Symer D. E., Genome Res. 2008, 18, 869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Keane T. M., Goodstadt L., Danecek P., White M. A., Wong K., Yalcin B., Heger A., Agam A., Slater G., Goodson M., Furlotte N. A., Eskin E., Nellaker C., Whitley H., Cleak J., Janowitz D., Hernandez‐Pliego P., Edwards A., Belgard T. G., Oliver P. L., McIntyre R. E., Bhomra A., Nicod J., Gan X., Yuan W., van der Weyden L., Steward C. A., Bala S., Stalker J., Mott R., Durbin R., Jackson I. J., Czechanski A., Guerra‐Assuncao J. A., Donahue L. R., Reinholdt L. G., Payseur B. A., Ponting C. P., Birney E., Flint J., Adams D. J., Nature 2011, 477, 289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Nellaker C., Keane T. M., Yalcin B., Wong K., Agam A., Belgard T. G., Flint J., Adams D. J., Frankel W. N., Ponting C. P., Genome Biol. 2012, 13, R45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Quinlan A. R., Clark R. A., Sokolova S., Leibowitz M. L., Zhang Y., Hurles M. E., Mell J. C., Hall I. M., Genome Res. 2010, 20, 623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Lee E., Iskow R., Yang L., Gokcumen O., Haseley P., Luquette L. J., 3rd, Lohr J. G., Harris C. C., Ding L., Wilson R. K., Wheeler D. A., Gibbs R. A., Kucherlapati R., Lee C., Kharchenko P. V., Park P. J., Science 2012, 337, 967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Hazen J. L., Faust G. G., Rodriguez A. R., Ferguson W. C., Shumilina S., Clark R. A., Boland M. J., Martin G., Chubukov P., Tsunemoto R. K., Torkamani A., Kupriyanov S., Hall I. M., Baldwin K. K., Neuron 2016, 89, 1223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Baillie J. K., Barnett M. W., Upton K. R., Gerhardt D. J., Richmond T. A., De Sapio F., Brennan P. M., Rizzu P., Smith S., Fell M., Talbot R. T., Gustincich S., Freeman T. C., Mattick J. S., Hume D. A., Heutink P., Carninci P., Jeddeloh J. A., Faulkner G. J., Nature 2011, 479, 534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Sheen F. M., Sherry S. T., Risch G. M., Robichaux M., Nasidze I., Stoneking M., Batzer M. A., Swergold G. D., Genome Res. 2000, 10, 1496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Ovchinnikov I., Troxel A. B., Swergold G. D., Genome Res. 2001, 11, 2050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Badge R. M., Alisch R. S., Moran J. V., Am. J. Hum. Genet. 2003, 72, 823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Iskow R. C., McCabe M. T., Mills R. E., Torene S., Pittard W. S., Neuwald A. F., Van Meir E. G., Vertino P. M., Devine S. E., Cell 2010, 141, 1253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Upton K. R., Gerhardt D. J., Jesuadian J. S., Richardson S. R., Sanchez‐Luque F. J., Bodea G. O., Ewing A. D., Salvador‐Palomeque C., van der Knaap M. S., Brennan P. M., Vanderver A., Faulkner G. J., Cell 2015, 161, 228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Evrony G. D., Cai X., Lee E., Hills L. B., Elhosary P. C., Lehmann H. S., Parker J. J., Atabay K. D., Gilmore E. C., Poduri A., Park P. J., Walsh C. A., Cell 2012, 151, 483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Richardson S. R., Morell S., Faulkner G. J., Annu. Rev. Genet. 2014, 48, 1. [DOI] [PubMed] [Google Scholar]
- 89. Mural R. J., Adams M. D., Myers E. W., Smith H. O., Miklos G. L., Wides R., Halpern A., Li P. W., Sutton G. G., Nadeau J., Salzberg S. L., Holt R. A., Kodira C. D., Lu F., Chen L., Deng Z., Evangelista C. C., Gan W., Heiman T. J., Li J., Li Z., Merkulov G. V., Milshina N. V., Naik A. K., Qi R., Shue B. C., Wang A., Wang J., Wang X., Yan X., Ye J., Yooseph S., Zhao Q., Zheng L., Zhu S. C., Biddick K., Bolanos R., Delcher A. L., Dew I. M., Fasulo D., Flanigan M. J., Huson D. H., Kravitz S. A., Miller J. R., Mobarry C. M., Reinert K., Remington K. A., Zhang Q., Zheng X. H., Nusskern D. R., Lai Z., Lei Y., Zhong W., Yao A., Guan P., Ji R. R., Gu Z., Wang Z. Y., Zhong F., Xiao C., Chiang C. C., Yandell M., Wortman J. R., Amanatides P. G., Hladun S. L., Pratts E. C., Johnson J. E., Dodson K. L., Woodford K. J., Evans C. A., Gropman B., Rusch D. B., Venter E., Wang M., Smith T. J., Houck J. T., Tompkins D. E., Haynes C., Jacob D., Chin S. H., Allen D. R., Dahlke C. E., Sanders R., Li K., Liu X., Levitsky A. A., Majoros W. H., Chen Q., Xia A. C., Lopez J. R., Donnelly M. T., Newman M. H., Glodek A., Kraft C. L., Nodell M., Ali F., An H. J., Baldwin‐Pitts D., Beeson K. Y., Cai S., Carnes M., Carver A., Caulk P. M., Center A., Chen Y. H., Cheng M. L., Coyne M. D., Crowder M., Danaher S., Davenport L. B., Desilets R., Dietz S. M., Doup L., Dullaghan P., Ferriera S., Fosler C. R., Gire H. C., Gluecksmann A., Gocayne J. D., Gray J., Hart B., Haynes J., Hoover J., Howland T., Ibegwam C., Jalali M., Johns D., Kline L., Ma D. S., MacCawley S., Magoon A., Mann F., May D., McIntosh T. C., Mehta S., Moy L., Moy M. C., Murphy B. J., Murphy S. D., Nelson K. A., Nuri Z., Parker K. A., Prudhomme A. C., Puri V. N., Qureshi H., Raley J. C., Reardon M. S., Regier M. A., Rogers Y. H., Romblad D. L., Schutz J., Scott J. L., Scott R., Sitter C. D., Smallwood M., Sprague A. C., Stewart E., Strong R. V., Suh E., Sylvester K., Thomas R., Tint N. N., Tsonis C., Wang G., Wang G., Williams M. S., Williams S. M., Windsor S. M., Wolfe K., Wu M. M., Zaveri J., Chaturvedi K., Gabrielian A. E., Ke Z., Sun J., Subramanian G., Venter J. C., Pfannkoch C. M., Barnstead M., Stephenson. Barnstead L. D., Stephenson L. D., Science 2002, 296, 1661. [DOI] [PubMed] [Google Scholar]
- 90. Yalcin B., Wong K., Agam A., Goodson M., Keane T. M., Gan X., Nellaker C., Goodstadt L., Nicod J., Bhomra A., Hernandez‐Pliego P., Whitley H., Cleak J., Dutton R., Janowitz D., Mott R., Adams D. J., Flint J., Nature 2011, 477, 326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Richardson S. R., Gerdes P., Gerhardt D. J., Sanchez‐Luque F. J., Bodea G. O., Munoz‐Lopez M., Jesuadian J. S., Kempen M. H. C., Carreira P. E., Jeddeloh J. A., Garcia‐Perez J. L., H. H. Kazazian, Jr. , Ewing A. D., Faulkner G. J., Genome Res. 2017, 27, 1395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Chambers V. S., Marsico G., Boutell J. M., Di Antonio M., Smith G. P., Balasubramanian S., Nat. Biotechnol. 2015, 33, 877. [DOI] [PubMed] [Google Scholar]
- 93. Sahakyan A. B., Murat P., Mayer C., Balasubramanian S., Nat. Struct. Mol. Biol. 2017, 24, 243. [DOI] [PubMed] [Google Scholar]
- 94. Schauer S. N., Carreira P. E., Shukla R., Gerhardt D. J., Gerdes P., Sanchez‐Luque F. J., Nicoli P., Kindlova M., Ghisletti S., Santos A. D., Rapoud D., Samuel D., Faivre J., Ewing A. D., Richardson S. R., Faulkner G. J., L1 retrotransposition is a common feature of mammalian hepatocarcinogenesis. Genome Res 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Malki S., van der Heijden G. W., O'Donnell K. A., Martin S. L., Bortvin A., Dev. Cell 2014, 29, 521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Wei W., Morrish T. A., Alisch R. S., Moran J. V., Anal. Biochem. 2000, 284, 435. [DOI] [PubMed] [Google Scholar]
- 97. Kannan M., Li J., Fritz S. E., Husarek K. E., Sanford J. C., Sullivan T. L., Tiwary P. K., An W., Boeke J. D., Symer D. E., Mobile DNA 2017, 8, 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. MacLennan M., Garcia‐Canadas M., Reichmann J., Khazina E., Wagner G., Playfoot C. J., Salvador‐Palomeque C., Mann A. R., Peressini P., Sanchez L., Dobie K., Read D., Hung C. C., Eskeland R., Meehan R. R., Weichenrieder O., Garcia‐Perez J. L., Adams I. R., eLife 2017, 6, e26152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Beck C. R., Collier P., Macfarlane C., Malig M., Kidd J. M., Eichler E. E., Badge R. M., Moran J. V., Cell 2010, 141, 1159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Macfarlane C. M., Collier P., Rahbari R., Beck C. R., Wagstaff J. F., Igoe S., Moran J. V., Badge R. M., Hum. Mutat. 2013, 34, 974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Tubio J. M., Li Y., Ju Y. S., Martincorena I., Cooke S. L., Tojo M., Gundem G., Pipinikas C. P., Zamora J., Raine K., Menzies A., Roman‐Garcia P., Fullam A., Gerstung M., Shlien A., Tarpey P. S., Papaemmanuil E., Knappskog S., Van Loo P., Ramakrishna M., Davies H. R., Marshall J., Wedge D. C., Teague J. W., Butler A. P., Nik‐Zainal S., Alexandrov L., Behjati S., Yates L. R., Bolli N., Mudie L., Hardy C., Martin S., McLaren S., O'Meara S., Anderson E., Maddison M., Gamble S., Group I. B. C., Group I. B. C., Group I. P. C., Foster C., Warren A. Y., Whitaker H., Brewer D., Eeles R., Cooper C., Neal D., Lynch A. G., Visakorpi T., Isaacs W. B., van't Veer L., Caldas C., Desmedt C., Sotiriou C., Aparicio S., Foekens J. A., Eyfjord J. E., Lakhani S. R., Thomas G., Myklebost O., Span P. N., Borresen‐Dale A. L., Richardson A. L., Van de Vijver M., Vincent‐Salomon A., Van den Eynden G. G., Flanagan A. M., Futreal P. A., Janes S. M., Bova G. S., Stratton M. R., McDermott U., Campbell P. J., Science 2014, 345, 1251343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Gardner E. J., Lam V. K., Harris D. N., Chuang N. T., Scott E. C., Pittard W. S., Mills R. E., Genomes Project C., Devine S. E., Genome Res. 2017, 27, 1916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Scott E. C., Devine S. E., Viruses 2017, 9, 131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. van den Hurk J. A., Meij I. C., Seleme M. C., Kano H., Nikopoulos K., Hoefsloot L. H., Sistermans E. A., de Wijs I. J., Mukhopadhyay A., Plomp A. S., de Jong P. T., Kazazian H. H., Cremers F. P., Hum. Mol. Genet. 2007, 16, 1587. [DOI] [PubMed] [Google Scholar]
- 105. Garcia‐Perez J. L., Marchetto M. C., Muotri A. R., Coufal N. G., Gage F. H., O'Shea K. S., Moran J. V., Hum. Mol. Genet. 2007, 16, 1569. [DOI] [PubMed] [Google Scholar]
- 106. Wissing S., Munoz‐Lopez M., Macia A., Yang Z., Montano M., Collins W., Garcia‐Perez J. L., Moran J. V., Greene W. C., Hum. Mol. Genet. 2012, 21, 208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Klawitter S., Fuchs N. V., Upton K. R., Munoz‐Lopez M., Shukla R., Wang J., Garcia‐Canadas M., Lopez‐Ruiz C., Gerhardt D. J., Sebe A., Grabundzija I., Merkert S., Gerdes P., Pulgarin J. A., Bock A., Held U., Witthuhn A., Haase A., Sarkadi B., Lower J., Wolvetang E. J., Martin U., Ivics Z., Izsvak Z., Garcia‐Perez J. L., Faulkner G. J., Schumann G. G., Nat. Commun. 2016, 7, 10286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Quinlan A. R., Boland M. J., Leibowitz M. L., Shumilina S., Pehrson S. M., Baldwin K. K., Hall I. M., Cell Stem Cell 2011, 9, 366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Evrony G. D., Lee E., Mehta B. K., Benjamini Y., Johnson R. M., Cai X., Yang L., Haseley P., Lehmann H. S., Park P. J., Walsh C. A., Neuron 2015, 85, 49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Erwin J. A., Paquola A. C., Singer T., Gallina I., Novotny M., Quayle C., Bedrosian T. A., Alves F. I., Butcher C. R., Herdy J. R., Sarkar A., Lasken R. S., Muotri A. R., Gage F. H., Nat. Neurosci. 2016, 19, 1583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Doucet A. J., Hulme A. E., Sahinovic E., Kulpa D. A., Moldovan J. B., Kopera H. C., Athanikar J. N., Hasnaoui M., Bucheton A., Moran J. V., Gilbert N., PLoS Genet. 2010, 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Goodier J. L., Mandal P. K., Zhang L., H. H. Kazazian, Jr. , Hum. Mol. Genet. 2010, 19, 1712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Martin S. L., Branciforte D., Mol. Cell Biol. 1993, 13, 5383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Branciforte D., Martin S. L., Mol. Cell Biol. 1994, 14, 2584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Soper S. F., van der Heijden G. W., Hardiman T. C., Goodheart M., Martin S. L., de Boer P., Bortvin A., Dev. Cell 2008, 15, 285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Trelogan S. A., Martin S. L., Proc. Natl. Acad. Sci. U. S. A. 1995, 92, 1520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Georgiou I., Noutsopoulos D., Dimitriadou E., Markopoulos G., Apergi A., Lazaros L., Vaxevanoglou T., Pantos K., Syrrou M., Tzavaras T., Hum. Mol. Genet. 2009, 18, 1221. [DOI] [PubMed] [Google Scholar]
- 118. Freeman P., Macfarlane C., Collier P., Jeffreys A. J., Badge R. M., Hum. Mutat. 2011, 32, 978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Garrick D., Fiering S., Martin D. I., Whitelaw E., Nat. Genet. 1998, 18, 56. [DOI] [PubMed] [Google Scholar]
- 120. Aravin A. A., Sachidanandam R., Bourc'his D., Schaefer C., Pezic D., Toth K. F., Bestor T., Hannon G. J., Mol. Cell. 2008, 31, 785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. Zheng K., Xiol J., Reuter M., Eckardt S., Leu N. A., McLaughlin K. J., Stark A., Sachidanandam R., Pillai R. S., Wang P. J., Proc. Natl. Acad. Sci. U. S. A. 2010, 107, 11841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Ewen K. A., Koopman P., Mol. Cell. Endocrinol. 2010, 323, 76. [DOI] [PubMed] [Google Scholar]
- 123. Seisenberger S., Andrews S., Krueger F., Arand J., Walter J., Santos F., Popp C., Thienpont B., Dean W., Reik W., Mol. Cell. 2012, 48, 849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124. Kim S., Gunesdogan U., Zylicz J. J., Hackett J. A., Cougot D., Bao S., Lee C., Dietmann S., Allen G. E., Sengupta R., Surani M. A., Mol. Cell. 2014, 56, 564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125. Hayashi K., Ohta H., Kurimoto K., Aramaki S., Saitou M., Cell 2011, 146, 519. [DOI] [PubMed] [Google Scholar]
- 126. Sasaki K., Yokobayashi S., Nakamura T., Okamoto I., Yabuta Y., Kurimoto K., Ohta H., Moritoki Y., Iwatani C., Tsuchiya H., Nakamura S., Sekiguchi K., Sakuma T., Yamamoto T., Mori T., Woltjen K., Nakagawa M., Yamamoto T., Takahashi K., Yamanaka S., Saitou M., Cell Stem Cell 2015, 17, 178. [DOI] [PubMed] [Google Scholar]