

SUMMARY
The interferon γ-inducible protein 16 (IFI16) is known as immune sensor of retroviral DNA intermediates. We show that IFI16 restricts HIV-1 independently of immune sensing by binding and inhibiting host transcription factor Sp1 that drives viral gene expression. This antiretroviral activity and ability to bind Sp1 require the N-terminal pyrin domain and nuclear localization of IFI16, but not the HIN domains involved in DNA binding. Highly prevalent clade C HIV-1 strains are more resistant to IFI16 and less dependent on Sp1 than other HIV-1 subtypes. Furthermore, inhibition of Sp1 by IFI16 or pharmacologically by Mithramycin A suppresses reactivation of latent HIV-1 in CD4+ T cells. Finally, IFI16 also inhibits retrotransposition of LINE-1, known to engage Sp1, and murine IFI16 homologs restrict Friend retrovirus replication in mice. Thus, IFI16 restricts retroviruses and retrotransposons by interfering with Sp1-dependent gene expression and evasion from this restriction may facilitate spread of HIV-1 subtype C.
Graphical Abstract
eTOC BLURB
The interferon γ-inducible protein 16 (IFI16) is an immune sensor of retroviral DNA intermediates. Hotter et al. demonstrate that IFI16 suppresses HIV-1 transcription and latency reactivation by interfering with Sp1-dependent gene expression. However, highly prevalent subtype C HIV-1 strains are less susceptible to IFI16 than other subtypes of HIV-1.
INTRODUCTION
Cell-intrinsic cellular factors, such as TRIM5α, APOBEC3G, Tetherin, SAMHD1, SERINC5 and GBP5, play key roles in the first line of defense against viral pathogens, targeting virtually every step in the viral replication cycle (Kluge et al., 2015). Most of these antiviral factors share certain characteristics, including signatures of positive selection, IFN inducibility, and direct interaction with viral components (Harris et al., 2012). Some of them display broad antiviral activity since they target viral or host factors essential for replication. Finally, several restriction factors not only inhibit viral pathogens directly but also act as pattern recognition receptors inducing immune responses upon sensing of viral infection (Galão et al., 2012; Pertel et al., 2011). We have used these characteristics to search for previously undescribed restriction factors sharing these features (McLaren et al., 2015). This approach identified guanylate-binding protein (GBP) 5 as an IFN-induced antiviral factor (Krapp et al., 2016) and suggested that the γ-IFN-inducible protein 16 (IFI16) may also restrict HIV-1. IFI16 is a member of the pyrin and HIN domain (PYHIN) containing protein family, which includes key mediators of the innate immune response that sense microbial DNAs to induce IFNs and/or inflammasome activation (He et al., 2016; Schattgen and Fitzgerald, 2011). Previous publications suggested that IFI16 acts as a cytosolic immune sensor of HIV-1 DNA species in macrophages (Jakobsen et al., 2013) and promotes IFN induction via the cyclic GMP-AMP synthase (cGAS) and stimulator of IFN genes (STING) pathway (Jønsson et al., 2017). A second study also reported sensing of HIV-1 reverse transcription (RT) intermediates, resulting in a caspase-1 dependent pyroptotic death of abortively HIV infected CD4+ T cells (Monroe et al., 2014).
Although IFI16 is thought to act as cytosolic sensor of viral DNA it has mainly been detected in the nucleus and shown to interact with nuclear herpesviral DNAs (Kerur et al., 2011; Li et al., 2012; Orzalli et al., 2012). PYHIN proteins are also known as transcriptional regulators, and it has been suggested that IFI16 can sense viral DNAs in the cytoplasm and suppress their transcription in the nucleus (Jakobsen and Paludan, 2014). Recent data show that IFI16 inhibits human cytomegalovirus (HCMV) transcription (Gariano et al., 2012) and restricts herpes simplex virus 1 (HSV-1) replication by repressing viral gene expression independently of innate immune sensing (Diner et al., 2016; Johnson et al., 2014). In addition, IFI16 suppresses gene expression of human papillomaviruses (Lo Cigno et al., 2015). Thus, IFI16 restricts transcription of various DNA viruses in the nucleus.
HIV-1 particles contain two copies of a single-stranded RNA genome. Efficient HIV-1 gene expression, however, occurs from integrated proviral DNA in the nucleus. The findings that IFI16 shares properties of known antiretroviral restriction factors (McLaren et al., 2015), while also inhibiting various DNA viruses (Lo Cigno et al., 2015; Diner et al., 2016; Gariano et al., 2012; Johnson et al., 2014), prompted us to investigate whether IFI16 inhibits HIV-1 independently of immune sensing. Indeed, we found that IFI16 inhibits HIV-1 gene expression as well as LINE-1 retrotransposition by interfering with the availability of Sp1. This transcription factor is required for both basal and Tat-mediated HIV-1 gene expression and its inhibition substantially suppressed reactivation of latent HIV-1 proviruses. Interestingly, subtype C strains that account for almost 50% of all HIV-1 infections worldwide are less susceptible to IFI16 inhibition and less dependent on Sp1 than other HIV-1 subtypes. We also show that p204, a mouse homolog of human IFI16, inhibits murine leukemia virus (MLV) in vitro and that replication of Friend virus (FV) is increased in acutely infected PYHIN knockout mice. In summary, our results show that IFI16 is an important effector of intrinsic antiretroviral immunity that might play a role in the spread of HIV/AIDS.
RESULTS
IFI16 inhibits infectious HIV-1 production independently of IFN induction
Coexpression of IFI16 potently inhibited infectious virus yield (determined by infection of TZM-b1 cells) and p24 antigen production by HEK293T cells transfected with the HIV-1 NL4-3 proviral construct (Figures 1A, S1A). Real-time quantitative PCR revealed that IFI16 suppresses the production of initial (R-U5/gag containing mRNA) as well as near (nef) and fully (U3-poly-A) completed HIV-1 RNA transcripts in the transfected HEK293T cells (Figure 1B). Consequently, IFI16 reduced viral protein production and affected processing of the Gag precursor (p55) by the viral protease but not cleavage of the gp160 Env precursor by cellular proteases (Figure 1C). Reduction of infectious virus yield by IFI16 was associated with dose-dependent inhibition of LTR-driven eGFP expression by HIV-1 NL4-3-based proviral constructs containing an IRES-eGFP element downstream of a functional nef gene (Figure S1B). In contrast, IFI16 did not suppress eGFP expression driven by the CMV immediate early (IE) promoter (Figure S1C). The inhibitory effect was confirmed in HEK293T cells infected with HIV-1 IRES-eGFP constructs (Figure S1D) showing that IFI16 reduces expression of the integrated provirus.
Figure 1. IFI16 impairs infectious HIV-1 production independently of type I IFN induction.
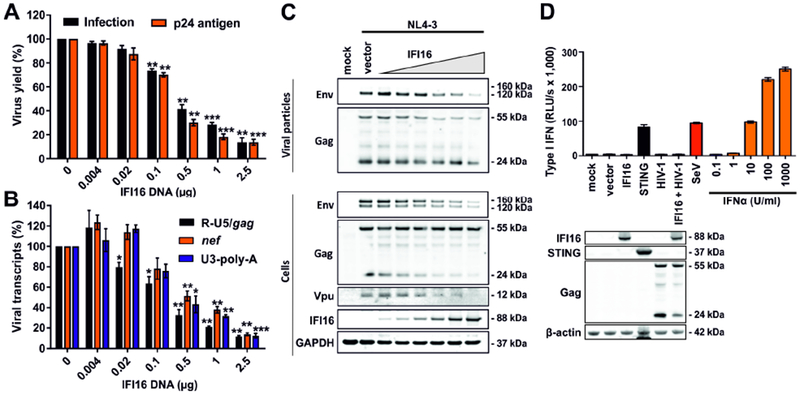
(A-C) HEK293T cells were cotransfected with increasing doses of expression constructs for IFI16 (0.004, 0.02, 0.1, 0.5 and 2.5 μg) and constant quantities (2.5 μg) of the proviral HIV-1 NL4-3 construct. Cells and supernatants were harvested 40 hours post-transfection. Data show mean percentages (±SEM) relative to those detected in the absence of IFI16 (100%) and were derived from three experiments each performed in triplicate. * p < 0.05, ** p < 0.01, *** p < 0.001. (A) Infectious virus yield and p24 antigen levels in the HEK293T cell supernatants determined by TZM-b1 cell infection assay and p24 antigen ELISA, respectively. (B) Levels of different viral transcripts determined by qRT-PCR in multiplex reactions with GAPDH as control. (C) Expression of HIV-1 proteins, IFI16 and GAPDH in viral particles or cellular extracts was determined by Western blot.
(D) IFI16 does not induce IFN in HEK293T cells lacking STING expression. HEK293T cells were transfected with the indicated constructs (1 μg) or infected with Sendai virus (SeV). 40 hours later, cells and supernatants were harvested. Cells were used for Western blot analysis and supernatants were used to stimulate HL116 IFN-reporter cells in triplicates. An IFNα standard curve was used for comparison. After 8 hours, luciferase expression in the HL116 cells was determined. Shown are mean values from triplicates ± SD. See also Figure S1.
It has been reported that IFI16 acts as immune sensor for reverse transcription HIV-1 DNA intermediates, triggering IFN production via cGAS and STING (Jakobsen et al., 2013; Jønsson et al., 2017; Kerur et al., 2011; Unterholzner et al., 2010). However, HEK293T cells lack endogenous STING expression (Burdette et al., 2011) (Figure 1D). Consistent with this, expression of IFI16 alone or together with HIV-1 did not result in type I IFN induction (Figures 1D, S1D). In contrast, IFN expression was induced when STING was transiently expressed or after infection with Sendai virus (SeV) that activates innate immune responses in a STING-independent manner (Figures 1D, S1E). Altogether, the results show that IFI16 restricts HIV-1 in HEK293T cells independently of STING and IFN induction by suppressing the initiation and efficiency of LTR-driven proviral transcription.
Endogenous IFI16 restricts HIV-1 in primary viral target cells
To assess HIV-1 restriction in primary target cells, we examined IFI16 expression in human CD4+ T cells and monocyte-derived macrophages (MDM). We found expression of three IFI16 isoforms, which are generated by alternative splicing (Jakobsen and Paludan, 2014) and further upregulated by type I IFNs in both cell types (Figure 2A). Treatment with TNFα also increased IFI16 levels in CD4+ T cells while activation with anti-CD3/CD28 beads mimicking stimulation by antigen-presenting cells had no enhancing effect (Figure 2A). Restriction factors, such as APOBEC3G, TRIM5α and tetherin, are induced as part of the antiviral immune response but are often unable to inhibit HIV-1 due to viral counteraction mechanisms. Thus, their expression typically correlates directly with HIV-1 RNA loads in vivo (Rotger et al., 2010). A query in the GuavaH (Genomic Utility for Association and Viral Analyses in HIV) database containing a comprehensive dataset of host genes modulated during HIV infection showed that also the levels of IFI16 transcripts correlate (p = 2.8x10−5) with the viral set point RNA loads in HIV-1-infected individuals (Figure S2A).
Figure 2. Endogenous IFI16 is IFN-inducible and restricts HIV-1.
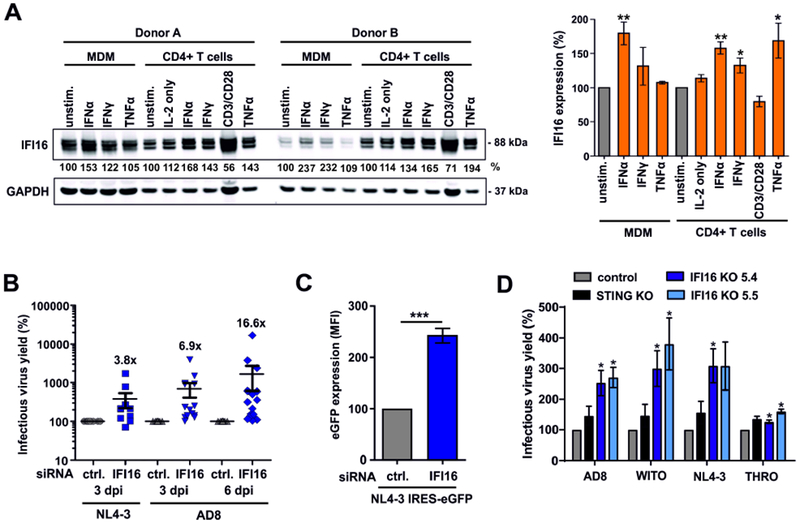
(A) Expression of IFI16 in primary macrophages (MDM) and CD4+ T cells. Cells were either left untreated or stimulated for 3 days with IL-2 [10 ng/ml] alone or together with IFNα2 [500 U/ml], IFNγ [200 U/ml], anti-CD3/CD28 beads or TNFα [20 ng/ml] and analyzed by Western blot. The left panel shows examples of primary data from two donors and the right panel shows mean IFI16 levels of five donors ± SEM. IFI16 expression levels were normalized to the corresponding GAPDH control. * p<0.05, ** p<0.01.
(B) IFI16 restricts infectious HIV-1 production in macrophages. Monocyte-derived macrophages were treated with IFI16-specific or control siRNA and transduced with VSV-G pseudotyped NL4-3 or infected with HIV-1 AD8. Infectious virus production was determined by infection of TZM-b1 cells. Each point represents data obtained from one donor.
(C) LTR-driven eGFP expression in macrophages transduced with VSV-G pseudotyped HIV-1 NL4-3 IRES-eGFP constructs after treatment with control or IFI16-specific siRNA. Shown are mean eGFP fluorescence intensities obtained by flow cytometry at 3 days post-transduction from four donors (±SEM).
(D) PMA-differentiated parental, IFI16 or STING KO THP-1 cells were transduced with the indicated VSV-G pseudotyped viruses. Three days later, supernatants were harvested and infectious virus yield was determined by infection of TZM-b1 cells. Mean values of three independent experiments ± SEM are shown. See also Figure S2.
To determine whether endogenous IFI16 restricts HIV-1 in primary viral target cells, we performed IFI16 siRNA knockdown studies in human macrophages. Despite moderate knockdown efficiencies of ~50% (Figure S2B), reduced IFI16 expression increased infectious virus yield after single-round infection with VSV-G pseudotyped CXCR4-tropic HIV-1 NL4-3 ~3.8-fold (Figure 2B). In the case of CCR5-tropic HIV-1 AD8, which is capable of spreading infection in macrophages, IFI16 knockdown increased infectious virus yield on average 6.9-fold at 3 dpi and 16.6-fold at 6 dpi (Figure 2B). In agreement with an effect on viral transcription, IFI16 silencing enhanced the levels of LTR-driven eGFP expression in macrophages infected with HIV-1 NL4-3 reporter constructs ~2- to 3-fold (Figures 2C, S2C).
To further examine whether endogenous IFI16 inhibits HIV-1 independently of immune sensing, we generated stable knock-outs of the IFI16 and STING genes in THP-1 monocytoid cells (Figure S2D), which adopt a macrophage-like phenotype upon PMA stimulation (Daigneault et al., 2010). Knock-out of IFI16 but not STING significantly increased infectious HIV-1 yield (Figure 2D). As expected from previous data (Jakobsen et al., 2013; Jønsson et al., 2017), lack of STING prevented and knock-out of IFI16 reduced IFN induction upon treatment with herring testis DNA (HT-DNA) or cGAMP (Figure S2E). These data show that endogenous IFI16 restricts HIV-1 in human cells.
The antiretroviral activity of IFI16 is broad and evolutionary conserved
To examine whether the antiviral activity of IFI16 is conserved among primate species, we tested human (hum), chimpanzee (cpz), African green monkey (agm) and rhesus macaque (mac) orthologs against a panel of 22 infectious molecular clones (IMCs) of HIV-1, HIV-2 and SIV. Despite considerable sequence variation (Figure S3A), all four IFI16 orthologs were expressed at similar levels (Figure 3A). To determine their antiviral activity, HEK293T cells were cotransfected with the proviral HIV and SIV constructs and vectors expressing the IFI16 orthologs. Determination of infectious virus yield by TZM-b1 infection assay showed that all primate lentiviruses were inhibited by IFI16 (Figure 3B). However, the magnitude of the effect varied substantially. For example, infectious virus yields of HIV-1 M CH058, SIVcpz EK505, SIVagm and most SIVsmm were reduced by ≥90%, whereas HIV-1 M CH293, HIV-1 N DJO0131 and HIV-2 AB7312 were much less affected. These varying susceptibilities to IFI16 inhibition were not due to differences in absolute infectivity (data not shown) and were independent of the IFI16 species origin (Figures 3B).
Figure 3. Antiretroviral activity of IFI16 is conserved and species independent.

(A) Expression of IFI16 orthologs from different species. HEK293T cells were transfected with IRES-BFP constructs expressing human (hum), chimpanzee (cpz), African green monkey (agm) or rhesus macaque (mac) IFI16 orthologs with or without C-terminal HA-tag and examined by Western blot.
(B) Inhibition of primate lentiviruses by IFI16 orthologs from different species. HEK293T cells were cotransfected with the indicated proviral HIV or SIV constructs (2.5 μg) and a control plasmid or vectors expressing IFI16 from the indicated species (1 μg). Infectious virus yield in the presence of one IFI16 ortholog relative to the vector control (100%) was determined by infection of TZM-b1 cells. Each symbol represents the average values obtained in three independent experiments. See also Figure S3.
IFI16 has been reported to be under positive selection (Cagliani et al., 2014; Cridland et al., 2012; McLaren et al., 2015). However, mutation of seven amino acid residues, showing evidence for positive selection, to alanines had little if any effect on the antiretroviral activity of human IFI16 (Figure S3B). Similarly, the antiviral activity of IFI16 was not impaired by two polymorphisms (S409R and N413Y) found in most individuals of African origin (57%) and a minority of Caucasians (Booiman and Kootstra, 2014) (Figure S3B). All these mutant IFI16 proteins were efficiently expressed (Figure S3C). Altogether, the results showed that the antiretroviral activity of IFI16 is conserved between monkeys, great apes and humans and not species-specific.
The pyrin domain and nuclear localization of IFI16 are critical for antiviral activity
To define the domains involved in antiviral activity, we analyzed a set of twelve IFI16 variants harboring point mutations or deletions in nuclear localization (NLS) signals, oligomerization motifs, DNA-interaction sites or entire domains (Figure 4A, left) for their effect on infectious virus yield and LTR-driven eGFP expression from a proviral HIV-1 reporter construct (Figure S4A). Deletion of the NLS and mutations in its oligomerization sites impaired the antiviral activity of IFI16 (Figures 4A; S4A) without reducing its expression (Figure S4B). Surprisingly, the N-terminal 191 amino acids of IFI16 encompassing the pyrin and NLS-containing linker domains were sufficient for potent anti-HIV-1 activity (Figure 4A). In contrast, the two HIN domains thought to be involved in viral DNA interaction (Ni et al., 2016) and enhancement of cGAS-dependent immune sensing of HIV-1 RT intermediates (Jønsson et al., 2017) were dispensable for the antiviral activity of IFI16 (Figure 4A). Thus, different domains are involved in innate sensing and sensing-independent restriction of retroviruses by IFI16. The effect of the parental and mutant IFI16 proteins on infectious virus yield correlated with their impact on LTR-driven eGFP expression from the proviral HIV-1 constructs (R2= 0.812; p<0.0001), further supporting that IFI16 restricts HIV-1 by suppressing viral transcription.
Figure 4. Role of the pyrin domain and nuclear localization for anti-HIV activity of IFI16.
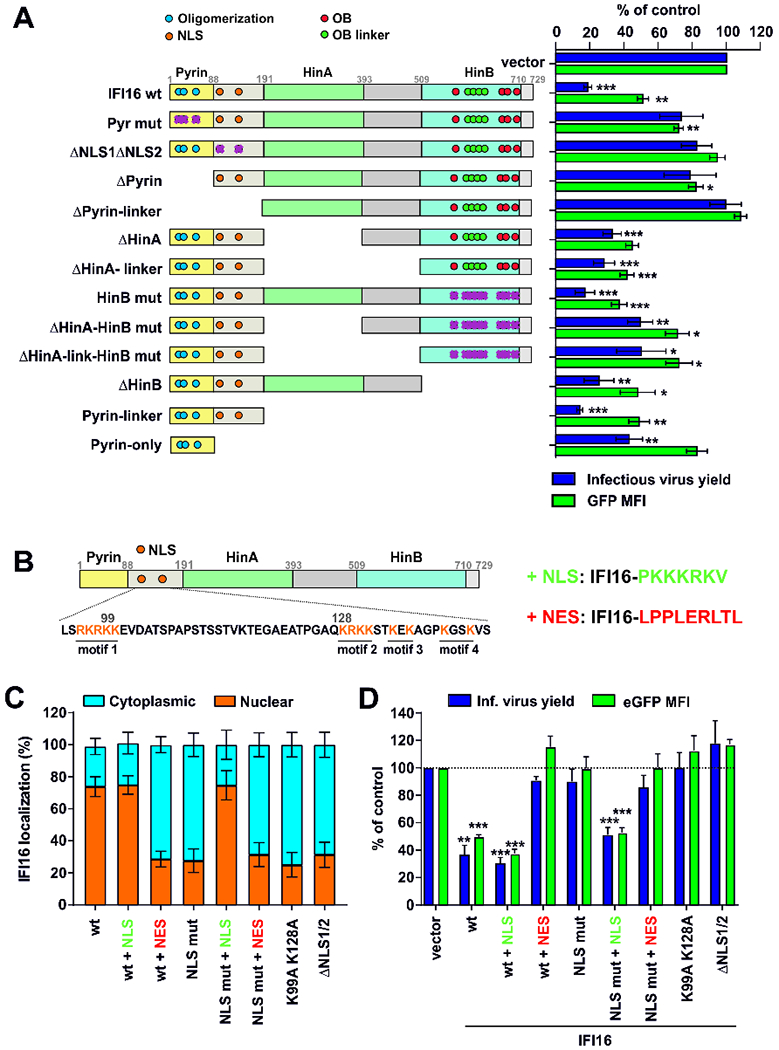
(A) Mutant IFI16 proteins analyzed (left) and infectious virus yield and levels of LTR-dependent eGFP expression (right). HEK293T cells were cotransfected with constructs coexpressing the indicated IFI16 proteins and BFP (1 μg) and proviral HIV-1 NL4-3 IRES-eGFP constructs (2.5 μg). At 40 hours post-transfection, BFP and eGFP expression was analyzed by flow cytometry. NLS, nuclear localization signal; OB, oligonucleotide binding fold. Purple crosses indicate mutated functional sites. Shown are mean values (±SEM) derived from four experiments relative to those obtained in the absence of IFI16 (100%). * p < 0.05, ** p < 0.01, *** p < 0.001.
(B) Schematic presentation of the multipartite NLS in IFI16 and the heterologous SV40 NLS and HIV-1 Rev NES sequences fused to the C-terminus of wt and mutant IFI16 constructs.
(C) Confocal immunofluorescence microscopy analysis of IFI16 localization in HEK293T cells transfected with constructs expressing the indicated IFI16 proteins (1 μg) (Figure S4D). Cytoplasmic and nuclear IFI16 signal intensities were quantified in at least 12 cells using ImageJ software. NLS mut: all positively charged amino acids from the four NLS motifs were mutated to Ala. ΔNLS1/2: NLS motifs 1 and 2 deleted.
(D) Values represent mean levels of infectious virus production and eGFP expression (±SEM) in HEK293T cells cotransfected with constructs coexpressing the indicated IFI16 mutants and BFP (1 μg) and proviral HIV-1 IRES-eGFP constructs (2.5 μg), relative to those obtained in the presence of the vector control (100%). See also Figure S4.
To further elucidate the importance of nuclear localization, we mutated the previously described NLS signals in IFI16 (Li et al., 2012) and/or fused the parental and mutant proteins to the SV40 NLS or an HIV-1 Rev nuclear export signal (NES) (Figure 4B). All IFI16 variants were expressed (Figure S4C) and wt IFI16 was mainly detected in the nucleus (Figures 4C, S4D). Mutations in the NLS motif or addition of a C-terminal NES resulted in relocalization to the cytoplasm, while fusion to the SV40 NLS restored nuclear localization of IFI16 containing mutant NLS (Figures 4C, S4D). The three predominantly nuclear forms of IFI16 suppressed infectious virus yield and proviral eGFP expression, while all five IFI16 derivatives that were mainly found in the cytoplasm lacked inhibitory activity (Figure 4D). Thus, the N-terminal pyrin domain and nuclear localization are essential and sufficient for HIV-1 restriction by IFI16.
IFI16 inhibits HIV-1 gene expression in an Sp1-dependent manner
To determine the specificity of IFI16, we examined its effect on expression of a luciferase reporter gene from various viral promoters. In agreement with the results obtained using IRES-eGFP constructs (Figures S1A, S1B), IFI16 significantly reduced activity of the HIV-1 LTR, as well as the HSV thymidine kinase (TK) promoter but not the CMV IE promoter (Figure 5A). To map elements in the HIV-1 LTR targeted by IFI16, we examined LTR luciferase constructs containing alterations in their Sp1, NF-κB/NFAT, USF, TCF-1α and NF-IL6 transcription factor binding sites (Bosque and Planelles, 2009) (Figure S5A). Mutation of the Sp1 interaction sites impaired LTR activity by about two orders of magnitude (Figure S5B) and significantly reduced the inhibitory effect of IFI16 on basal and Tat-induced LTR-driven gene expression (Figure 5B). Titration experiments confirmed that also at similar activity levels, the wt LTR is inhibited by IFI16, while the Sp1-deficient LTR mutant is not (Figure S5C). In addition, IFI16 suppressed the enhancing effect of Sp1 overexpression on infectious HIV-1 NL4-3 production (Figure 5C).
Figure 5. IFI16 inhibits HIV-1 LTR activity in a Tat and Sp1 dependent manner.
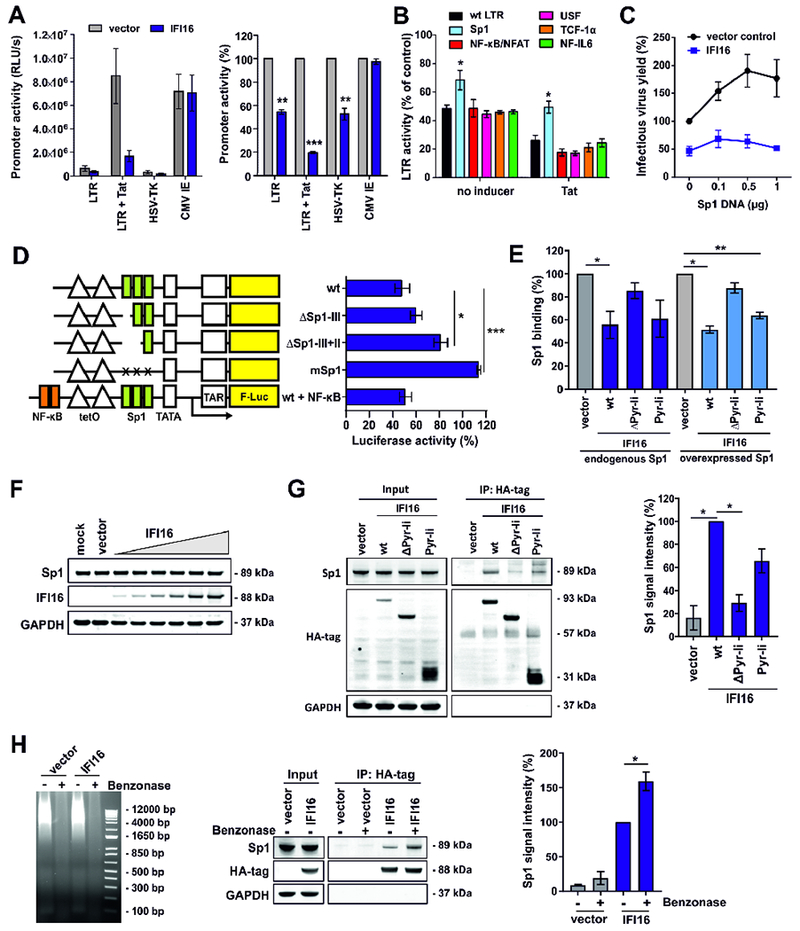
(A) HEK293T cells were cotransfected with luciferase reporter constructs under the control of the indicated promoters and expression constructs for IFI16 or a vector control. A construct expressing NL4-3 Tat under the control of the CMV IE promoter was used to activate the LTR. The left panel shows absolute luciferase values and the right panel values measured in the presence of IFI16 relative to the vector control (100%). All panels show mean values (±SEM) obtained in three to four experiments. * p <0.05, ** p < 0.01, *** p <0.001.
(B) Effect of IFI16 on mutant HIV-1 LTRs. Values represent mean luciferase activities (±SEM, n=4) of LTR reporter constructs with inactivating mutations in the indicated transcription factor binding sites in the presence of IFI16 relative to the vector control (100 %) as described for (A).
(C) HEK293T cells were cotransfected with expression constructs for Sp1, proviral HIV-1 NL4-3 constructs (2.5 μg), and constructs for IFI16 or the vector control (1 μg). 40 h post-transfection infectious virus yield was determined by infection of TZM-b1 cells. Values were normalized to infectious virus yield in the absence of Sp1 and IFI16 (100%).
(D) HEK293T cells were cotransfected with doxycycline-dependent firefly luciferase reporter constructs under the control of minimal wt or mutant LTRs, CMV IE-dependent Gaussia luciferase constructs for normalization, an expression vector for rtTA and expression constructs for IFI16 or a vector control. Doxycycline was added to the cell culture medium to activate transcription. 40 h after transfection, a dual luciferase assay was performed and firefly luciferase activity was normalized to the corresponding Gaussia luciferase activity. The graph shows mean luciferase activity (±SEM, n=3) in the presence of IFI16 relative to the vector control (100%).
(E) HEK293T cells were transfected with expression constructs for the indicated IFI16 variants alone (1 μg) (left) or together with an expression construct for Sp1 (0.2 μg) (right). At 40 h post-transfection, nuclear proteins were isolated and equal quantities were used to determine the levels of active Sp1 using the TransAM Sp1 binding assay (ActiveMotif). Data represent mean values (± SEM) obtained from three experiments. As shown in Figure 4A, ΔPyr-li (ΔPyrin-linker) lacks the N-terminal pyrin and linker regions, while Pyr-li only encompasses these two domains.
(F) HEK293T cells were transfected with increasing doses of IFI16 expression constructs (0.004, 0.02, 0.1, 0.5, 1 and 2.5 μg). At 40 h after transfection, cells were harvested and Sp1, IFI16 and GAPDH levels determined by Western blot.
(G) HEK293T cells were transfected with expression constructs for the indicated HA-tagged IFI16 variants or a vector control. 40 h post-transfection, cells were lysed and IFI16 was immunoprecipitated using anti-HA antibodies and protein A/G magnetic beads. Proteins were blotted and stained using anti-Sp1, anti-HA and anti-GAPDH antibodies. The left panel shows one representative Western blot. The right panel shows mean Sp1 signal intensities (±SEM, n=3) after pull-down.
(H) HEK293T cells were transfected with expression constructs for HA-tagged IFI16 or a vector control. 40 h post-transfection, cells were lysed and nucleic acids were degraded by benzonase treatment before co-immunoprecipitation experiments were performed as described for (G). Left: Protein lysates were run on an agarose gel stained with ethidium bromide to confirm degradation of nucleic acids. Middle: One representative Western blot is shown. Right: Mean Sp1 signal intensities (±SEM, n=3) after pull-down. See also Figure S5.
To further examine the role of Tat and Sp1 in the inhibitory effect of IFI16, we took advantage of HIV-1 constructs harboring a Tet-On gene expression element allowing viral transcription in the absence of Tat and various parts of the LTR (Das et al., 2011). Our analyses confirmed that lack of the Sp1 interaction sites and/or Tat expression abolished the susceptibility of HIV-1 to IFI16 inhibition, while lack of other parts of U3 or the TAR region had only minor effects (Figure S5D). Analysis of the effect of IFI16 on LTR-driven transcription using minimal LTR firefly luciferase reporter constructs containing two tet operator (tetO) sites (Turrini et al., 2015) showed that IFI16 inhibits basal transcription only in the presence of Sp1 binding sites (Figure 5D).
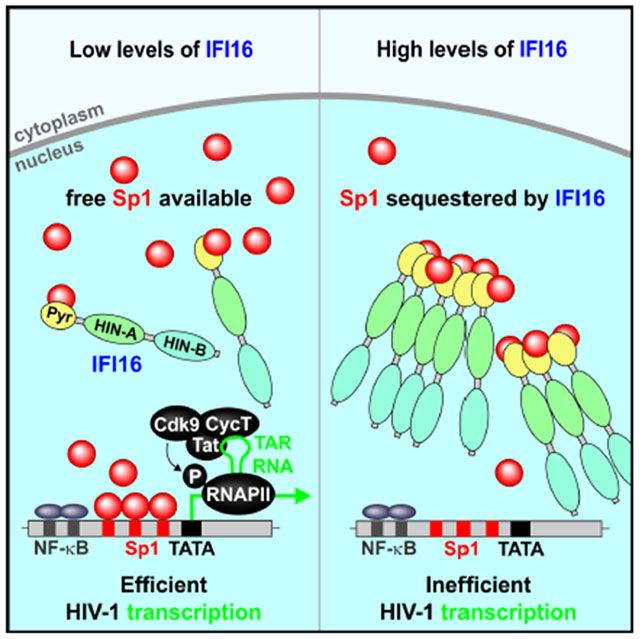
To more directly determine the effect of IFI16 on Sp1 activity, we took advantage of a DNA-binding ELISA quantifying available Sp1 in nuclear extracts. In agreement with the antiviral activity (Figure 4A), the pyrin and linker domains of IFI16 were required and sufficient to reduce the levels of available endogenous or overexpressed Sp1 in nuclear extracts of transfected HEK293T cells (Figure 5E). IFI16 significantly reduced the levels of Sp1 bound to target DNA sequences without affecting its protein levels (Figure 5F). Co-immunoprecipitation experiments with HEK293T cells, transfected with HA-tagged variants of IFI16, revealed that significant quantities of endogenous Sp1 were pulled down together with IFI16 and this effect was dependent on the N-terminal pyrin and linker domain (Figure 5G). To determine if the interaction between Sp1 and IFI16 depends on nucleic acids, we performed co-immunoprecipitation experiments with cell lysates, in which DNA and RNA was removed by benzonase treatment. We found that IFI16 pulled down higher levels of Sp1 in the absence of DNA, possibly because DNA competes with IFI16 for Sp1 binding (Figure 5H). To better assess the relevance of these effects in primary viral target cells, we compared IFI16 and Sp1 expression levels in MDM and CD4+ T cells exposed to various stimuli with the protein levels in HEK293T cells transfected with increasing doses of IFI16. The IFI16/Sp1 expression ratio in macrophages and T cells was generally higher than in transfected HEK293T cells (Figure S5E). Altogether, these results indicate that IFI16 binds Sp1 via its pyrin and/or linker domain and reduces the availability of Sp1 for proviral transcription.
Low IFI16 sensitivity and Sp1 dependency of subtype C HIV-1 strains
To further elucidate the anti-HIV-1 activity of IFI16, we selected five primary HIV-1 IMCs, differing widely in IFI16 sensitivity in transfected HEK293T cells (Figure 6A), for detailed analyses. IFI16 silencing in primary macrophages by siRNA increased infectious yield of IFI16 sensitive HIV-1 strains up to 13-fold but had only ~3-fold effects on IFI16 resistant HIV-1 IMCs (Figure 6B). Our analyses revealed a highly significant correlation between the enhancing effect of IFI16 knock-down on infectious virus yield in primary macrophages and the inhibitory effect of IFI16 overexpression in HEK293T cells (R2=0.936; p=0.007). Sp1 overexpression increased infectious virus yield of IFI16 sensitive HIV-1 strains up to 7-fold but had little effect on IFI16 resistant viruses (Figure 6C). To further determine the Sp1 dependency of the selected HIV-1 IMCs, we took advantage of the chronic myelogenous leukemia cell line HAP1 (Carette et al., 2009) and a derivative thereof lacking Sp1 expression (Figure S6A). We found that lack of Sp1 reduces infectious virus yield of IFI16 sensitive HIV-1 strains more severely than of IFI16 resistant HIV-1 IMCs (Figure 6D). To examine whether the viral accessory vpr, nef, vpu or vif genes affect IFI16 inhibition, we mutated them individually in HIV-1 IMCs showing high (CH058), intermediate (CH198) and low (CH293) susceptibility to inhibition by IFI16. We found that the differential susceptibility of HIV-1 to IFI16 inhibition is independent of viral accessory protein function (Figure S6B).
Figure 6. IFI16 susceptibility and Sp1 dependency of different primate lentiviruses.
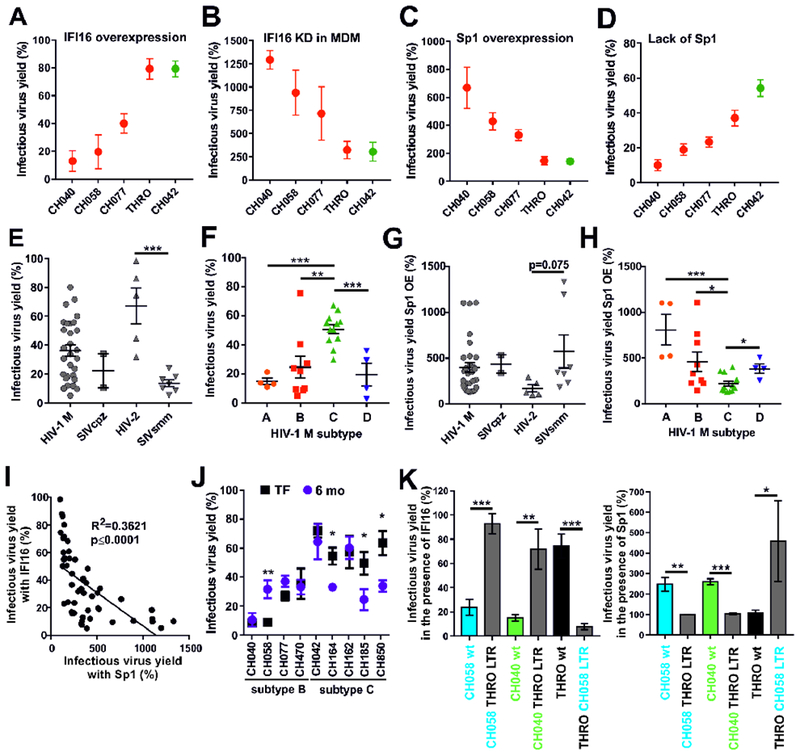
(A) Effect of IFI16 on HIV-1 IMCs. Values represent mean levels of infectious virus production (±SEM) in HEK293T cells in the presence of IFI16 relative to those obtained in its absence (100%). Results in all panels were derived from at least three independent experiments. * p < 0.05, ** p < 0.01, *** p < 0.001.
(B) MDM from five blood donors were treated with ctrl. or IFI16-specific siRNA and transduced with the indicated VSV-G pseudotyped viruses. Three days later, infectious virus yield was determined by TZM-b1 infection assay and normalized to ctrl. siRNA-treated cells (100%).
(C) Effect of Sp1 overexpression on primary HIV-1 IMCs. Values represent mean levels of infectious virus production in HEK293T cells transfected with an Sp1 expression construct (1 μg) relative to those treated with the vector control (100%).
(D) Effect of lack of Sp1 expression on HIV-1 IMCs. Shown are the mean infectious virus yields from Sp1 KO HAP1 cells transfected with the indicated IMCs, compared to those obtained in the parental cell line (100%).
(E, F) Mean infectious virus yields obtained from HEK293T cells cotransfected with 43 different proviral HIV or SIV constructs and expression constructs for IFI16 or a vector control (100%). (E) Comparison between HIVs and their SIV precursors and (F) between different subtypes of HIV-1 group M strains.
(G, H) Effect of Sp1 overexpression (OE) on various primate lentiviruses. Values were determined as described in panel C and grouped based on (G) virus origin and (H) subtype.
(I) Correlation between the effects of IFI16 and Sp1 overexpression on infectious virus yields. Shown are results obtained for all primate lentiviral IMCs analyzed.
(J) IFI16 sensitivity of TF/6-mo HIV-1 pairs. Values represent mean levels (±SEM; n=5) of infectious virus production in HEK293T cells cotransfected with the indicated HIV-1 IMCs and IFI16 or control vector (100%).
(K) Role of the HIV-1 LTRs in IFI16 sensitivity and Sp1 responsiveness. The effects of (left) IFI16 and (right) Sp1 overexpression on the indicated HIV-1 IMCs and otherwise isogenic forms in which both LTRs were replaced are shown. Values represent the mean levels of three experiments (±SEM) relative to the respective vector controls (100%). See also Figure S6.
Our results indicated that the susceptibility of primary HIV-1 strains to IFI16 inhibition correlates with their dependency on Sp1 for efficient virus production. To determine whether this is also the case for other primate lentiviruses, we analyzed a set of 43 different proviral constructs representing pandemic group M of HIV-1, as well as HIV-2 and their simian precursors SIVcpz and SIVsmm, respectively. Remarkably, HIV-2 was substantially less susceptible to IFI16 than SIVsmm, its direct simian counterpart (32.8±12.6% vs 86.3±2.4% inhibition, p=0.0006; mean±SEM) (Figure 6E). In addition, grouping by HIV-1 subtype revealed that subtype C viruses are on average less susceptible to IFI16 (~50% inhibition) than subtypes A, B and D (Figure 6F). All subtype A, B and D HIV-1 strains were inhibited by 60% to 98%, which was similar to the effects of IFI16 on the two SIVcpz strains tested. The only exception was the subtype B HIV-1 THRO IMC, which was largely resistant to IFI16 (Figure 6A) and hardly affected by IFI16 knock-out in THP-1 cells (Figure 2D) or IFI16 knock-down in macrophages (Figure 6B).
On average, HIV-2 strains were less responsive to Sp1 overexpression (172.1±38.64%, n=5) than SIVsmm strains (574.3±180.5%, n=7), although the difference failed to reach statistical significance (Figure 6G). Different subtypes of HIV-1 also showed marked differences in Sp1 responsiveness. For example, Sp1 overexpression increased infectious yield of HIV-1 subtype A strains ~8-fold, while only 2-fold effects were observed for subtype C (Figure 6H). Altogether, we observed a highly significant correlation between the sensitivity of primate lentiviruses to IFI16 and their responsiveness to Sp1 overexpression (Figure 6I). To assess a potential role in virus transmission, we examined IFI16 sensitivity of nine transmitted-founder (TF) HIV-1 IMCs and matched 6-month consensus molecular clones from the same individual (Fenton-May et al., 2013). We found that three of five clade C but none of the four clade B HIV-1 IMCs showed increases in their sensitivity to IFI16 after 6 months of infection (Figure 6J). To map the determinants of IFI16 sensitivity, we swapped both LTRs between susceptible and resistant subtype B HIV-1 IMCs (Figure S6C). The THRO LTR rendered HIV-1 CH058 and CH040 resistant to IFI16, while the CH058 LTR conferred susceptibility to HIV-1 THRO (Figure 6K, left). These LTR sequences also governed the responsiveness of the HIV-1 constructs to Sp1 overexpression (Figure 6K, right). Altogether, these results reveal striking differences in IFI16 susceptibility and Sp1 responsiveness of primate lentiviruses and demonstrate that HIV-1 subtype C is less dependent on Sp1 and less susceptible to IFI16 than subtypes A, B and D.
Inhibition of Sp1 suppresses reactivation of latent HIV-1
Our results show that IFI16 suppresses HIV-1 transcription by reducing the availability of Sp1. To examine the potential relevance of this transcription factor for reactivation of latent HIV, we analyzed the effect of the Sp1 inhibitor Mithramyin A in the Jurkat-based J-Lat 10.6 clone containing a full-length integrated HIV-1 genome that expresses GFP upon reactivation (Jordan et al., 2003). Mithramyin A efficiently suppressed TNFα-induced reactivation of HIV-1 at a concentration of 50 nM (Figure 7A) that did not induce significant cytotoxic effects (Figure S7A). Notably, TNFα treatment did not affect Sp1 expression levels, but moderately increased IFI16 expression (Figure S7B). To further examine the role of Sp1 and IFI16 in reactivation of latent HIV-1, we generated J-Lat clones lacking these factors (Figure 7B, left). We found that induction of LTR-dependent gene expression by TNFα in J-Lat cells is dependent on Sp1 and significantly inhibited by IFI16 (Figure 7B, right).
Figure 7. Effect of Sp1 inhibition and PYHIN proteins on retroviral production in vitro and in vivo.
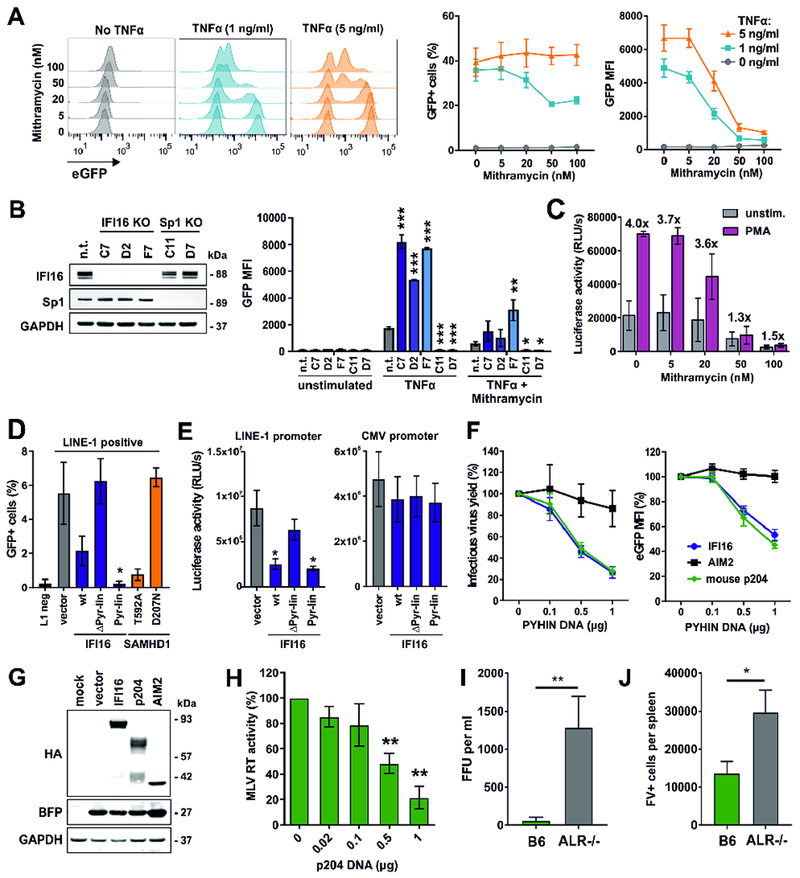
(A) J-Lat cells (clone 10.6) were stimulated with the indicated doses of TNFα in the presence of increasing doses of Mithramycin A. Two days post-stimulation, GFP expression and MFI were determined by flow cytometry. The middle and right panels show mean values (±SEM) derived from three independent experiments.
(B) GFP expression in different clones of J-Lat 10.6 cells lacking IFI16 or Sp1 expression was determined two days post-stimulation with TNFα (1 ng/ml) and Mithramycin A (20 nM) by flow cytometry. Mean GFP fluorescence intensities (±SEM) derived from three independent experiments are shown. Asterisks indicate differences compared to the control cells (n.t.). * p < 0.05, ** p < 0.01, *** p < 0.001.
(C) Luciferase activities in PMA-stimulated or unstimulated CD4+ T cells from two donors latently infected with VSV-G pseudotyped HIV-1 nano-luc reporter constructs in the absence or presence of Mithramycin A.
(D) HEK293T cells were cotransfected with a retrotransposition-competent (L1) or -defective (L1 neg) LINE-1-GFP reporter plasmid and a vector control or expression constructs for IFI16 or SAMHD1. Five days post-transfection, GFP+ cells were quantified by flow cytometry (n=3 ± SEM).
(E) HEK293T cells were cotransfected with LINE-1 or CMV promoter constructs driving luciferase gene expression and expression constructs for IFI16 or a control vector. Luciferase activities (relative light units, RLU) in cell lysates were determined two days later (n=3 ± SEM).
(F) Effects of human IFI16 or AIM2 and murine p204 on (left) infectious virus yield and (right) LTR-driven eGFP expression of HIV-1 in HEK293T cells 40 hours post-transfection. Values represent mean levels of infectious virus production or eGFP expression (±SEM; n=3) in the presence of increasing doses of the indicated PYHIN proteins relative to the vector control (100%).
(G) Expression of HA-tagged PYHIN proteins and co-expressed BFP in cells from (F) was analyzed by Western blot.
(H) HEK293T cells were cotransfected with increasing doses of expression constructs for p204 and a proviral MLV construct. At 40 h post-transfection, MLV RT activity in cell culture supernatants was determined. Shown are mean values (±SEM) derived from three independent experiments.
(I, J) Viral loads in wild type and ALR−/− mice infected with Friend virus. Wild-type C57BL/6 and ALR−/− mice lacking all 13 genes encoding AIM2-like receptors (also name PYHIN proteins; Gray et al., 2016) were infected with 20,000 SFFU (spleen focus-forming units) of Friend virus. At 3 dpi, (D) plasma viremia was determined and (E) spleens were harvested to analyze viral loads in an infectious center (IC) assay. Thirteen mice per group from at least two independent experiments were analyzed. Mean (±SEM) values are indicated by bars. Statistically significant differences between the groups were analyzed using a Mann-Whitney test and are indicated by * for p < 0.05 and ** for p < 0.01.
To verify the relevance of Sp1 in a primary viral target cell latency model, we infected CD4+ T cells with VSV-G pseudotyped NL4.3-deltaEnv-nLuc-2ANef (Figure S7C). This recombinant virus expresses both Nef and Vpu and downregulates CD4 in productively infected cells. Thus, latently infected and uninfected CD4+ cells are purified using anti-CD4 conjugated beads, cultured with or without Mithramycin A, and subsequently treated with PMA or medium (unstimulated) as outlined in Figure S7C. Mithramycin A inhibited HIV-1 gene expression and p24 antigen production in both PMA-stimulated as well as unstimulated CD4+ T cells (Figures 7C, S7D). Notably, Mithramycin A almost entirely abolished PMA-mediated reactivation of HIV-1 at a concentration of 50 nM (Figures 7C; S7D) that did not display marked cytotoxicity (Figure S7E). Thus, inhibition of Sp1 suppresses reactivation of latent HIV-1 in primary CD4+ T cells.
IFI16 restricts LINE-1 retrotransposition
It is estimated that up to 17% of the human genome might be derived from LINE-1, the only autonomously active retrotransposon in humans. Control of retrotransposition is relevant in preventing autoimmunity as well as cancer development and it has been reported that Sp1 and Sp3 interact with the rat LINE-1 promoter (Fedorov et al., 2006). To determine whether IFI16 restricts human LINE-1, we used a well-established reporter assay utilizing GFP expression as surrogate marker for successful retrotransposition (Herrmann et al., 2018). For control, we used active phosphorylation-deficient SAMHD1 (T592A) and an enzymatically inactive mutant (D207N) thereof lacking anti-LINE-1 activity (Herrmann et al., 2018). IFI16 inhibited LINE-1 retrotransposition by 62% (Figure 7D). The N-terminal pyrin and linker domains were required and sufficient for potent inhibition (>95% reduction) of retrotransposition. In agreement with these findings, full-length IFI16 as well as the pyrin-linker fragment significantly inhibited the LINE-1 but not the CMV promoter (Figure 7E). Altogether, these results suggest that IFI16 restricts HIV-1 and LINE-1 by the same mechanism.
PYHIN proteins restrict retroviral replication in vivo
Many previous reports suggested that PYHIN proteins play a key role in DNA sensing and the induction of antiviral IFN responses (reviewed in Bauernfeind and Hornung, 2013; Schattgen and Fitzgerald, 2011). Strikingly, however, a recent study demonstrated that knock-out of all 13 Pyhin genes in mice had no significant effect on their type I IFN response to DNA ligands or various viral pathogens, suggesting that their main function remains unknown (Gray et al., 2016). To determine whether the IFN-independent antiretroviral activity of PYHIN proteins is conserved between mice and men, we cloned and analyzed the IFI16 mouse homolog p204 (Deschamps et al., 2003). IFI16 and p204 reduced infectious HIV-1 yield and LTR-driven proviral eGFP expression with similar efficiency (Figure 7F) and were both efficiently expressed (Figure 7G). For control, we also analyzed human AIM2. This member of the human PYHIN protein family lacks an NLS that is critical for the antiviral activity of IFI16 (Figure 4) and has been detected mainly in the cytoplasm (Lugrin and Martinon, 2018). Indeed, AIM2 did not display significant anti-HIV-1 activity (Figures 7F). To determine restriction of mouse retroviruses, we first examined the effect of p204 on murine leukemia virus (MLV) production in transfected HEK293T cells. p204 efficiently inhibited MLV in a dose-dependent manner (Figure 7H). To analyze whether PYHIN proteins restrict retroviruses in vivo, we infected parental C57BL/6 (B6) and Pyhin KO (ALR−/−; Aim2-like-receptor knock-out) mice (Gray et al., 2016) with equal doses of Friend virus (FV). Early in infection, plasma viremia was ~50-fold reduced in wt mice (Figure 7I) and the number of FV infected cells in the spleen was ~2-fold lower (Figure 7J) compared to mice lacking PYHIN protein expression. Thus, even though they seem to be dispensable for IFN induction (Gray et al., 2016), PYHIN proteins inhibit retroviral replication in vivo.
DISCUSSION
In the present study, we show that the IFN-inducible PYHIN protein IFI16 inhibits retroviral transcription by reducing the availability of the transcription factor Sp1. The N-terminal pyrin domain involved in protein-protein interaction and nuclear localization of IFI16 are critical and sufficient for inhibition of HIV-1 and the LINE-1 retroelement, as well as for Sp1 binding. In contrast, the HIN domains thought to be critical for DNA interaction and sensing are dispensable for this function. Unexpectedly, we found that members of different HIV-1 subtypes exhibit marked variation in their dependency on Sp1 for viral gene expression that correlates with susceptibility to IFI16 inhibition. HIV-1 subtype C strains, which account for almost half of all HIV-1 infections, are less dependent on Sp1 and susceptible to IFI16 inhibition than other subtypes of HIV-1. IFI16 knockdown in primary human macrophages strongly enhanced infectious virus yield of HIV-1 strains that are susceptible to IFI16 inhibition in transient expression assays. Antiretroviral activity is conserved between primate IFI16 proteins and their mouse homolog p204 and knockout of all Pyhin genes in mice was associated with reduced control of Friend virus replication early after infection. Thus, PYHIN proteins restrict retroviruses and presumably endogenous retroelements both in cell culture and in vivo.
Previous studies reported that IFI16 boosts sensing of intracellular DNA and IFN induction via the cGAS-STING pathway in macrophages (Jakobsen et al., 2013; Jønsson et al., 2017). In addition, IFI16 was implicated as cytoplasmic immune sensor mediating pyroptotic death of tissue CD4+ T cells abortively infected with HIV-1 (Monroe et al., 2014) (Figure S7F). A main function of IFI16 as cytoplasmic sensor of viral DNA species, however, is at odds with the observation that this factor is predominantly found in the nucleus (Figure 4C; Li et al., 2012). Indeed, several studies reported that IFI16 acts as nuclear sensor and direct restriction factor of a variety of DNA viruses (Lo Cigno et al., 2015a; Gariano et al., 2012; Johnson et al., 2014). Although retroviruses harbor single-stranded RNA genomes, they use their reverse transcriptase to generate linear double-stranded DNAs that enter the nucleus and integrate into the host cell genome for viral transcription. Our finding that IFI16 inhibits various retroviruses and the LINE-1 retroelement is thus in agreement with a role as a nuclear restriction factor of a diverse array of viral pathogens. Strikingly, it has recently been shown that knock-out of all 13 Pyhin genes in mice does not significantly affect the type I IFN response upon exposure to various DNA ligands or viral pathogens (Gray et al., 2016). Here, we show that lack of PYHIN proteins is associated with about 50-fold increased MLV RNA loads early after infection (Figure 7I). The contribution of individual mouse PYHIN family members to this antiviral effect and their mode of action remains to be determined, but our data clearly support that PYHIN proteins restrict retroviral replication independently of IFN induction.
It has been reported that IFI16 cooperatively binds dsDNA in a length-dependent manner and clusters into protein filaments (Morrone et al., 2014). Assembly into filaments is mediated by conserved residues in the pyrin domain and required for high affinity binding of DNA via the HIN domains (Morrone et al., 2014). Thus, it was unexpected that both HIN domains thought to be critical for DNA interaction and sensing are dispensable for antiretroviral activity of IFI16 suggesting that DNA binding might not be required for the antiviral effect. In contrast, the presence of the pyrin domain in the nucleus was required and sufficient for inhibition of viral transcription (Figures 4, S4). Our results confirm data showing that IFI16 interacts with Sp1 (Gariano et al., 2012) and support the hypothesis that IFI16 limits the availability of this key transcription factor for viral gene expression potentially by competing with DNA for Sp1 binding.
Mutation of Sp1 binding sites abolished IFI16-mediated inhibition of LTR-driven gene expression (Figures 5B, 5D) and the susceptibility of various primate lentiviruses correlated significantly with their dependency on Sp1 for efficient transcription (Figure 6I). Thus, the antiretroviral effects of IFI16 are clearly dependent on Sp1. This result is in agreement with the finding that deletion of a putative Sp1 responsive element reduces inhibition of the HCMV UL44 promoter by IFI16 (Gariano et al., 2012). However, the inhibitory effects of IFI16 were always more pronounced in the presence of the viral transactivator of transcription Tat (Figures 5A, 5B, S5B). Functional interactions between Sp1 and Tat are documented but poorly understood (Das et al., 2011; Jeang et al., 1993; Loregian et al., 2003). It has been shown that even in the absence of a functional TAR element, Tat boosts viral transcription via the Sp1 binding sites in the LTR (Das et al., 2011). Thus, IFI16 might interfere with the functional interactions between Tat and Sp1 that are critical for efficient viral gene expression.
One unexpected finding of our study is that primate lentiviruses differ strongly in their dependency on Sp1. Most notably, we found that subtype C HIV-1 strains are substantially less affected by differences in Sp1 expression levels than HIV-1 subtype A, B and D strains and relatively resistant to IFI16 inhibition (Figure 6). Importantly, these results were obtained using a comprehensive panel of transmitted-founder and chronic HIV-1 IMCs that were generated directly from patient-derived sequences and never passaged in cell culture. Thus, reduced IFI16 susceptibility and Sp1 dependency are characteristic features of primary subtype C HIV-1 strains that might contribute to their effective spread in the human population.
Analyses of recombinant HIV-1 constructs showed that susceptibility to IFI16 is determined by the viral LTR (Figure 6K). HIV-1 IMCs differing in IFI16 susceptibility showed multiple sequence variations in the LTR, although the three potential Sp1 binding sites are usually conserved in the LTRs of all primate lentiviruses (examples shown in Figure S6C). Notably, the only resistant HIV-1 subtype B strain (THRO) differed just by a single nucleotide change across the three Sp1 sites from the susceptible HIV-1 CH058 IMC (Figure S6C). Thus, it is tempting to speculate that the presence of other regulatory elements allowing efficient viral transcription independently of Sp1, rather than differences in Sp1 binding sites themselves, might determine the differential susceptibility of primate lentiviruses to IFI16 inhibition. For example, HIV-1 THRO contains an extra 34 nucleotide stretch compared to other subtype B strains in the LTR (Figure S6C) and HIV-1 subtype C usually contains an additional NF-κB site compared to other subtypes of HIV-1 (Bachu et al., 2012; Montano et al., 2000). Our ongoing studies aim at fully defining the molecular determinants of IFI16 susceptibility and Sp1 dependency.
Although it is long known that Sp1 plays a key role in HIV-1 transcription (Jones et al., 1986; Jeang et al., 1993), its significance for HIV-1 latency and reactivation has hardly been explored because it is commonly thought that Sp1 is constitutively and ubiquitously expressed. However, our results clearly demonstrate that Sp1 is a limiting factor in HIV-1 transcription that is critical for HIV-1 reactivation in J-Lat cells (Figure 7B). In addition, our data show that IFI16 reduces the availability of Sp1, suggesting that this effector of the innate IFN response might promote the establishment of latent viral reservoirs. We also found that pharmacological inhibition of Sp1 by Mithramycin A suppresses reactivation of latent HIV-1 (Figures 7A, 7C). We show that the IFI16/Sp1 expression ratio in primary CD4+ T cells is high (Figure S5E) and it has been reported that IFI16 is expressed at high levels in resting central memory T cells, which harbor a significant part of the latent HIV-1 reservoir (Cerboni et al., 2017). Thus, further studies to elucidate the role of IFI16-mediated suppression of Sp1 activity in the establishment and maintenance of latent HIV reservoirs seem highly warranted and may help to improve “block and lock” approaches aiming to prevent HIV reactivation.
In conclusion, we show that IFI16 is an antiviral factor that is expressed and IFN-inducible in primary CD4+ T cells and macrophages and suppresses viral transcription in an Sp1-dependent manner. Altogether, our results show that IFI16 shares most properties of known antiviral restriction factors (Kluge et al., 2015), although based on more stringent criteria it could also be defined as “negative regulator” of HIV-1 since it targets a virus dependency factor (Sp1) that is also critical for numerous cellular functions (Bieniasz, 2004). Our results further show that HIV-1 subtype C strains are relatively resistant to IFI16 inhibition because they are less dependent on Sp1 for efficient transcription than other subtypes of HIV-1. The antiretroviral effect is conserved between p204 from mice and IFI16 from men. Lack of all PYHIN proteins in mice did not affect type I IFN responses upon exposure to DNA triggers or viruses (Gray et al., 2016) but was associated with increased viral loads early during Friend virus infection. Thus, further studies on the role of PYHIN proteins as effectors of innate antiviral immunity rather than as immune sensors seem highly warranted.
STAR METHODS TEXT
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Frank Kirchhoff (frank.kirchhoff@uni-ulm.de).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Studies in mice
All animal studies were performed under the LANUV NRW-approved animal study number A1536/15 in accordance with the regulations and guidelines of the animal protection law in Germany. The original source of the C57BL/6J controls were the Harlan Laboratories and the ARL−/− mice were from Jackson Laboratories. They were made using C57BL/6N mice, but the N and J strains do not behave differently during FV infection. All mice were females and 8-10 weeks of age at the time point of infection. Prior to the experiment controls and experimental animals were co-housed for at least 3 weeks. The central animal facility in Essen has a species-appropriate animal husbandry under SPF conditions.
Cell lines
All cells were cultured at 37°C in a 5% CO2 atmosphere. HEK293T and TZM-b1 cells were maintained in Dulbecco’s Modified Eagle Medium (DMEM) supplemented with 10% FCS, glutamine (2 mM), streptomycin (100 μg/ml) and penicillin (100 U/ml). HEK293T cells were provided and authenticated by the ATCC. They were originally isolated from a female fetus and first described by DuBridge et al. (DuBridge et al., 1987). TZM-b1 cells were provided and authenticated by the NIH AIDS Reagent Program, Division of AIDS, NIAID, NIH from Dr. John C. Kappes, Dr. Xiaoyun Wu and Tranzyme Inc (Platt et al., 1998). TZM-b1 are derived from HeLa cells, which were isolated from a 30-year-old female. HL116 cells were maintained in DMEM containing 10% FCS, glutamine (2 mM), streptomycin (100 μg/ml) and penicillin (100 U/ml) and HAT supplement (sodium hypoxanthine (50 μM), aminopterin (200 nM) and thymidine (8 μM)). HL116 cells stably express an IFN-inducible firefly luciferase reporter gene and are derivatives of the human fibrosarcoma HT1080 cell line established from a 35-year-old male (Uze et al., 1994). HEK-Blue IFN-α/β cells were cultured in DMEM supplemented with 10% FCS, glutamine (600 μg/ml), penicillin (200 U/ml), streptomycin (100 μg/ml), normocin (100 μg/ml), blasticidin (30 μg/ml) and zeocin (100 μg/ml). HEK-Blue cells were provided and authenticated by InvivoGen. These cells produce SEAP upon stimulation with IFNα/β and were generated by stable transfection of HEK293 cells with the human STAT2 and IRF9 genes as well as a SEAP reporter gene under the control of the IFN-α/β inducible ISG54 promoter. THP-1 and J-Lat cells were maintained in RPMI-1640 medium supplemented with 10% FCS, glutamine (2 mM), streptomycin (100 μg/ml) and penicillin (100 U/ml). THP-1 cells are monocyte-like cells, which adopt a macrophage-like phenotype upon differentiation with PMA and were originally isolated from the peripheral blood of a 1-year-old male with acute monocytic leukaemia. IFI16 and STING knockout cell lines were created using CRISPR-Cas9 technology (Jønsson et al., 2017). J-Lat cells (clone 10.6) were provided and authenticated by the NIH AIDS Reagent Program, Division of AIDS, NIAID, NIH from Dr. Eric Verdin (Jordan et al., 2003). J-Lat cells are latently infected with env- and nef- deficient HIV-1, expressing GFP upon reactivation, and are derived from Jurkat cells which were originally established from a 14-year-old boy with acute lymphoblastic leukemia. IFI16 and Sp1 KO cells were generated by transducing the J-Lat cells with VSV-G pseudotyped lentiviral vectors expressing the Cas9 nuclease and IFI16- (GACCAGCCCTATCAAGAAAG) or Sp1- (CATGGATGAAATGACAGCTG) specific or non-targeting (n.t.) guide RNAs. Successfully transduced cell were selected with Puromycin (0.75 μg/ml) and single cell clones were expanded. HAP1 cells were maintained in Iscove’s Modified Dulbecco’s Medium (IMDM) supplemented with 10% FCS, streptomycin (100 μg/ml) and penicillin (100 U/ml). HAP1 cells were provided and authenticated by Horizon and represent a near-haploid human cell line that was derived from the male chronic myelogenous leukemia (CML) cell line KBM-7 (Carette et al., 2011)
Primary cell cultures
PBMCs from healthy human donors were isolated using lymphocyte separation medium (Biocoll separating solution; Biochrom) or lymphoprep (Stemcell). CD4+ T cells were negatively isolated using the RosetteSep™ Human CD4+ T Cell Enrichment Cocktail (Stem Cell Technologies) or the EasySep™ Human Naïve CD4+ T cell Isolation Kit (Stem Cell Technologies) according to the manufacturer’s instructions. Primary cells were cultured in RPMI-1640 medium containing 10% FCS, glutamine (2 mM), streptomycin (100 μg/ml), penicillin (100 U/ml) and interleukin 2 (IL-2) (10 ng/ml). Monocyte-derived macrophages were obtained by stimulation of PBMC cultures with 15 ng/ml recombinant human M-CSF (R&D systems) and 10 % human serum. The use of human PBMCs was approved by the Ethics Committee of the Ulm University Medical Center. All donors were anonymized and provided informed written consent.
METHOD DETAILS
Expression constructs
Expression vectors for IFI16 orthologs, AIM2, p204, Sp1 and STING were generated by gene amplification via PCR using cDNA as template and subsequent cloning via unique XbaI and MluI restriction sites into a pCG expression vector coexpressing BFP via an IRES. For the generation of the IFI16 Pyrin mutant (S27A, L28A, D50A) and IFI16 HinB mutant (K572A, K607A, R611A, S614A, K618A, N654A, K676A, K678A, K703A) the previously described variants IFI16 mut3 (Morrone et al., 2014) and IFI16 m4 (Jin et al., 2012) were used as templates. To facilitate detection of the expressed proteins a C-terminal HA tag (TACCCATACGATGTTCCAGATTACGCT) was introduced via the reverse PCR primer. Specific mutations or deletions in IFI16 were introduced using overlap-extension PCR. All constructs were sequenced to verify their integrity. PCR primers are listed in Table S1.
Generation of mutant HIV-1 LTR F-luc reporter constructs
Proviral HIV-1 constructs with mutated transcription factor binding sites in the LTR were generated and provided by Vincente Planelles (Bosque and Planelles, 2009). A DNA fragment containing the 3’LTR was excised from the proviral constructs using XhoI and XbaI and used as template to amplify the LTR and insert unique MluI and XhoI restriction sites for cloning into a pGL3 firefly luciferase reporter vector. All constructs were sequenced to verify their integrity. PCR primers are listed in Table S2.
Chimeric HIV-1 proviral constructs
A DNA fragment containing the 3’LTR was excised from the proviral constructs using NotI and BstZ17I for CH058, NotI and AgeI for CH040 and NotI and SbflI for THRO. In parallel, a DNA fragment containing the 5’LTR was excised by digestion with StuI and MluI for CH058, BlpI and MluI for CH040 and MluI and BbvCI for THRO. These DNA fragments were used as template to amplify the 3’ and 5’ LTR of the different proviruses and the regions between the LTR and the next single cutter sites. Using overlap-extension PCR, the different LTRs were added to the proviral parts and religated with the proviral constructs. All primers are listed in Table S3. The 3’and 5’LTR of THRO were inserted into CH040 and CH058 using the respective single cutters. The 3’ and 5’LTR of CH058 were inserted into THRO using SbflI and NotI and MluI and BbvCI. All constructs were sequenced to verify their accuracy.
Transfections and production of virus stocks
HEK293T cells were transiently transfected using the calcium-phosphate precipitation method. One day before transfection, 5x105 HEK293T cells/well were seeded in 6-well plates in 2 ml medium to obtain a confluence of 50-60% at the time of transfection. For transfection, DNA was mixed with 13 μl 2 M CaCl2 and filled up with water to 100 μl Afterwards, 100 μl 2 x HBS was added dropwise to this mixture, which was mixed by pipetting and added dropwise to the cells. To generate virus stocks, cells were transfected with proviral constructs (5 μg). To generate VSV-G pseudotypes, expression plasmids for the vesicular stomatitis virus glycoprotein (1 μg) (Fouchier et al., 1997) were additionally added to the transfection mix. To test the antiviral effect of different proteins, pCG-based expression constructs were cotransfected with proviral constructs. Whenever different amounts of pCG expression vectors were used within an experiment, empty vector control plasmids were used to keep the total DNA amount for all samples constant. The transfected cells were incubated for 8-16 h before the medium was replaced by fresh supplemented DMEM. HAP1 cells were transfected using the TransIT®-LT1 Transfection Reagent (Mirus). One day before transfection, 2.5 x105 HAP1 cells/well were seeded in 12-well plates in 1 ml medium. For transfection, DNA and 4 µl of TransIT®-LT1 Transfection Reagent were mixed with 50 µl Opti-MEM, each. These two solutions were then mixed, incubated at RT for 20 min and added dropwise to the cells. The transfected cells were incubated for 6 h before the medium was replaced by fresh supplemented IMDM. 40 h post transfection, cell culture supernatants were harvested, centrifuged at 1000 x g for 3 min and transferred to fresh tubes to remove cell remnants.
Viral infectivity test
To determine infectious virus yield, 6,000 TZM-b1 reporter cells/well were seeded in 96-well plates and infected with cell culture supernatants in triplicates on the following day. Three days post-infection, cells were lysed and β-galactosidase reporter gene expression was determined using the GalScreen Kit (Applied Bioscience) according to the manufacturer’s instructions with an Orion microplate luminometer (Berthold).
Western blot
To determine expression of cellular and viral proteins, cells were washed in PBS and subsequently lysed in Western blot lysis buffer (150 mM NaCl, 50 mM HEPES, 5 mM EDTA, 0.1% NP40, 500 μM Na3VO4, 500 μM NaF, pH 7.5). Lysates were mixed with Protein Sample Loading Buffer (LI-COR) supplemented with 10% β-mercaptoethanol, heated at 95°C for 5 min, separated on NuPAGE 4±12% Bis-Tris Gels (Invitrogen) and blotted onto Immobilon-FL PVDF membranes (Merck Millipore). Proteins were stained using primary antibodies directed against IFI16 (Santa Cruz #sc-8023), HA-tag (Abcam # ab 18181), STING (Cell Signaling #13647), HIV-1 Env (obtained through the NIH AIDS Reagent Program, Division of AIDS, NIAID, NIH: 16H3 mAb from Drs. Barton F. Haynes and Hua-Xin Liao) (Gao et al., 2009), p24 (Abcam #ab9071), Vpu (kindly provided by S. Bolduan), AU-1 (Novus Biologicals #NB600-453), β-actin (Abcam #ab8226), GAPDH (BioLegend #631401 or Santa Cruz #sc-365062), GFP/BFP (Abcam #ab290), Sp1 (Abcam #157123 or Abcam #227383) and Infrared Dye labeled secondary antibodies (LI-COR IRDye). Proteins were detected using a LI-COR Odyssey scanner and band intensities were quantified using LI-COR Image Studio Lite Version 3.1.
IFN release assay
HL116 cells were used to determine the release of bioactive interferon (IFN) from HEK293T and THP-1 cells. 20,000 HL116 cells/well were seeded in a 96-well plate and stimulated in triplicates with 100 μl cell culture supernatants on the following day. As positive controls, supernatants from HEK293T cells infected with Sendai virus and different concentrations of IFNα were used. 8 h after stimulation, cells were lysed and firefly luciferase activity was determined using the Luciferase Assay Kit (Promega) according to the manufacturer’s instructions with an Orion microplate luminometer (Berthold).
IFNβ-promoter reporter assay
To determine the effect of IFI16 on IFNβ-promoter activity, 22,000 HEK293T cells/well were seeded in poly-L-lysine-coated 96-well plates and transfected in triplicates using the calcium-phosphate transfection method. Cells were cotransfected with a firefly luciferase reporter construct under the control of the IFNβ-promoter (100 ng), a Gaussia luciferase construct under the control of a constitutively active pTAL promoter (5 ng) for normalization, and expression vectors or proviral constructs (50 ng). As a positive control, the IFNβ-promoter was activated by infection of the cells with Sendai virus for 24 h. 40 h after transfection, activity of the secreted Gaussia luciferase was determined in the cell culture supernatants using the Gaussia- Juice Kit (p.j.k.), whereas firefly luciferase activity was determined in cell lysates using a Luciferase Assay Kit (Promega) according to the manufacturer’s instructions with an Orion microplate luminometer (Berthold). Firefly luciferase signals were normalized to the corresponding Gaussia luciferase control values to normalize for transfection efficiency.
qRT-PCR
Viral transcript levels were determined in HEK293T cells cotransfected with proviral constructs of NL4-3 (2.5 μg) and increasing doses of expression constructs for IFI16. 40 h post-transfection cells were washed with PBS and lysed in RLT Plus buffer containing 1% β-mercaptoethanol. Total RNA was isolated using the RNeasy Plus Mini Kit (QIAGEN) according to the manufacturer’s instructions. Residual genomic DNA was removed from the RNA preparations using the DNA-free™ DNA Removal Kit (ThermoFisher). RNA concentrations were determined using the NanoDrop 2000 Spectrophotometer and for each sample equal RNA amounts were subjected to cDNA synthesis using the PrimeScript RT Reagent Kit (TAKARA) with random 6-mers and oligo dT primers. Reactions without reverse transcriptase were included as controls to exclude contaminations with genomic DNA. cDNA was used for qRT-PCR using TaqMan™ Fast Universal PCR Master Mix (ThermoFisher) and viral primer/probe sets (Biomers/TIB Molbiol) in multiplex reactions with GAPDH (ThermoFisher) as control. Viral primers and probes were designed as previously described (Yukl et al., 2018) to measure R-U5/Gag mRNA as indicator of proximal elongation, Nef mRNA as indicator of transcription that has proceeded almost to the 3’LTR and U3-polyA mRNA as indicator for completed transcription. Primers and fluorescent probes are listed in Table S4.
Differentiation, siRNA transfection and infection of macrophages
Monocyte-derived macrophages (MDMs) were obtained by stimulation of PBMC cultures with 15 ng/ml recombinant human M-CSF (R&D systems) and 10% human AB serum (Sigma Aldrich) in DMEM supplemented with glutamine (2 mM), streptomycin (100 μg/ml) and penicillin (100 U/ml) for 6 days. On days 7 and 9 of differentiation, MDMs were transfected with IFI16-sepcific (Invitrogen # IFI16HSS105205, IFI16HSS105206, IFI16HSS105207) or a non-targeting control (Eurofins (UUCUCCGAACGUGUCACGUdTdT)) siRNA. siRNA transfections were performed in 12-well plates with two technical replicates for each sample using the Lipofectamine RNAiMAX transfection reagent (Thermo Fisher). Prior to transfection the medium was replaced by 500 µl fresh supplemented DMEM. For one well, 1.46 μl siRNA (20 μM) and 3 μl Lipofectamine RNAiMAX were mixed with 75 μl Opti-MEM, each. These two solutions were then mixed, incubated at RT for 15 min and added dropwise to the cells. 16 h after transfection on day 7 of differentiation, the medium containing the transfection mix was replaced by fresh supplemented DMEM. 8 h after transfection on day 9 of differentiation, cells were infected. 12 h after infection, the input virus was removed by washing twice with 1 ml PBS and 1 ml fresh DMEM containing 10% FCS, glutamine (2 mM), streptomycin (100 μg/ml), penicillin (100 U/ml) was added to the cells. Infectious virus yield was determined in cell culture supernatants harvested 3 and 6 days after infection. To monitor knockdown efficiencies by Western blot, cells were washed with 1 ml PBS and harvested 6 days after infection.
Flow cytometry
Flow cytometry was used to determine the effect of IFI16 on HIV-1 LTR- or CMV IE-driven eGFP expression. HEK293T cells were cotransfected with expression constructs for IFI16 co-expressing BFP via an IRES and HIV-1 NL4-3 proviral constructs or CMV-IE promoter-driven control plasmids co-expressing eGFP via an IRES. 40 h after transfection cells were harvested, washed in PBS with 1% FCS and fixed in 2% PFA for 30 min at 4°C. Mean fluorescence intensities (MFI) of eGFP in the BFP+/eGFP+ population and percentage of eGFP+ cells in the BFP+ population (P2/(P2+P1)) was determined. Monocyte-derived macrophages treated with IFI16 or non-targeting control siRNA and infected with VSV-G pseudotyped HIV-1 NL4-3 IRES eGFP constructs were harvested 3 days post-infection. Knockdown efficiency was determined by staining with anti-IFI16 (Santa Cruz #sc-8023) and anti-mouse AF647 (ThermoFisher #A31571) antibodies after fixation and permeabilization of the cells using the FIX&PERM kit (Nordic MUbio) according to the manufacturer’s instructions. Flow cytometric measurements were performed using a BD FACS Canto II flow cytometer.
Differentiation, stimulation and infection of THP-1 cells
The replication of HIV-1 in the absence of STING/IFI16 was determined in knockout THP-1 or corresponding control cells. 2x105 cells/well were seeded in 12-well plates in 1 ml RPMI supplemented with with 10% FCS, glutamine (2 mM), streptomycin (100 μg/ml), penicillin (100 U/ml) and PMA (100 nM) to induce differentiation into macrophage-like cells. 24 h later, the medium was replaced by fresh supplemented RPMI without PMA. On the following day, cells were infected with different VSV-G pseudotyped HIV-1 strains. 12 h after infection, the input virus was removed by washing twice with 1 ml PBS and 1 ml fresh supplemented RPMI without PMA was added. Cell culture supernatants were harvested 3 days post-infection to determine infectious virus yield. To determine the ability of the THP-1 cell lines to produce bioactive interferon, the cells were stimulated with 2 μg/ml of HT-DNA or 2′-3′cGAMP using lipofectamine as transfection reagent for 8 hours before harvesting supernatants for analysis.
Microscopy
Confocal immunofluorescence microscopy was used to determine the subcellular localization of various IFI16 mutants. 75,000 HEK293T cells/well were seeded on 13 mm diameter glass cover slips coated with poly-L-lysine in 24-well plates. On the following day, cells were transfected with expression constructs for IFI16 mutants (250 ng) using the calcium phosphate precipitation method. The transfected cells were incubated for 16 h before the medium was replaced by fresh supplemented DMEM. 40 h after transfection, cells were washed with cold PBS, fixed in 4% PFA for 20 min at RT and permeabilized in PBS 0.5% Triton X-100 5% FCS for 20 min at RT. IFI16 was stained using anti-IFI16 (Santa Cruz #sc-8023) and anti-mouse PE (ThermoFisher #P-852) antibodies, whereas the nucleus was stained using NucRed Live 647 ReadyProbes Reagent (ThermoFisher #R37106). Cover slips were mounted on glass slides using Mowiol mounting medium. Confocal microscopy was performed using an LSM710 confocal microscope (Carl Zeiss) and IFI16 signal intensities in at least 12 cells per sample were quantified using Fiji image processing software (ImageJ).
Viral promoter activity
To determine the effect of IFI16 on the activity of different viral promoters, 22,000 HEK293T cells/well were seeded in poly-L-lysine-coated 96-well plates and transfected in triplicates using the calcium-phosphate transfection method. Cells were cotransfected with firefly luciferase reporter constructs (5 ng) under the control of the HIV-1 LTR, the HSV TK promoter or the CMV IE promoter, and expression constructs for IFI16 (50 ng) or a vector control. In some cases, expression constructs for HIV-1 NL4-3 Tat (500 pg) were cotransfected to activate the LTR promoter. 40 h post-transfection, cells were lysed and firefly luciferase activity was determined using the Luciferase Assay Kit (Promega) according to the manufacturer’s instructions with an Orion microplate luminometer (Berthold).
Doxycycline-responsive LTR activity
The effect of IFI16 on LTR reporter constructs lacking Sp1 binding sites were tested as previously described (Turrini et al., 2015). 22,000 HEK293T cells/well were seeded in poly-L-lysine-coated 96-well plates and transfected in triplicates using the calcium-phosphate transfection method. Cells were cotransfected with expression constructs for IFI16 or a vector control (5 ng), CMV IE-dependent Gaussia luciferase constructs for normalization (0.05 ng) and the minimal LTR tetO Luc reporter (wt Sp1), two deletion mutants lacking either one (ΔSp1-III) or two (ΔSp1-III+II) Sp1 binding sites, a mutant with scrambled Sp1 target sequences (mSp1) or a LTR tetO Luc reporter with intact Sp1 binding sites that in contrast to the other constructs still contains the NF-κB binding sites and upstream U3 region (0.5 ng). As these constructs contain two tet operator (tetO) sites for the binding of the doxycycline-inducible transcriptional activator rtTA, rtTA-expressing plasmids (0.01 ng) were cotransfected and transcription was activated adding doxycycline (1 μg/ml) to the culture medium. 40 h after transfection, activity of the secreted Gaussia luciferase was determined in the cell culture supernatants using the Gaussia-Juice Kit (p.j.k.), whereas firefly luciferase activity was determined in cell lysates using a Luciferase Assay Kit (Promega) according to the manufacturer’s instructions with an Orion microplate luminometer (Berthold). Firefly luciferase signals were normalized to the corresponding Gaussia luciferase control values to normalize for transfection efficiency.
Release of doxycycline-responsive HIV-1 variants with mutant LTR
To investigate the influence of the U3 LTR, Sp1 binding sites, TAR and a functional Tat protein on the antiviral effect of IFI16, we took advantage of previously characterized HIV-rtTA variants, which use an incorporated Tet-On gene expression system for activation of transcription (Das et al., 2011). HEK293T cells were seeded in 6-well plates and cotransfected with expression constructs for IFI16 or a vector control (1μg) and different HIV-rtTA mutants (2.5 μg). Transcription was activated adding doxycycline (1 μg/ml) to the culture medium. 40 h post-transfection, HIV-1 p24 levels in cell culture supernatants were determined using an in-house ELISA (Konvalinka et al., 1995). Briefly, 96-well MaxiSorp™ microplates were coated with 500 ng/ml anti-HIV-1 p24 (100 μl/well) and incubated in a wet chamber at RT overnight. The next day, plates were washed 3 times with 350 μl PBS-T (PBS and 0.05% Tween 20) and incubated with 100 μl blocking solution (PBS and 10% (v/v) FCS) for 2 hours at 37 °C. After washing, the plates were loaded with 100 μl serially diluted HIV-1 p24 protein as standard and dilutions of virus stocks lysed with 1% (v/v) Triton X-100. After overnight incubation in a wet chamber at RT, plates were washed to remove unbound capsid proteins. 100 μl/well polyclonal rabbit antiserum against p24 antigen (1:1,000 in PBS-T (PBS, 0.05% Tween 20 and 10% (v/v) FCS)) was added for 1 hour at 37 °C and after another washing step, 100 μl of a goat anti-rabbit HRP-coupled antibody (1:2,000) was loaded on the plate and incubated for another hour at 37°C. Finally, the plate was washed and 100 μl SureBlue TMB 1-Component Microwell Peroxidase Substrate was added. After 20 minutes shaking at 450 rpm and RT, the reaction was stopped with 0.5 M H2SO4 (100 μl/well). The optic density, proportional to the p24 capsid antigen amount, was determined by comparing with a standard curve and measured at 450 nm and 650 nm with the Thermo Max microplate reader (Molecular devices, UK).
Co-IP
The interaction between Sp1 and IFI16 was determined in HEK293T cells transfected with expression constructs for HA-tagged IFI16 mutants or a vector control (1 μg). 40 h after transfection cells were lysed in Western blot lysis buffer and IFI16 was immunoprecipitated using anti-HA antibodies (Abcam #ab18181) and Pierce Protein A/G Magnetic Beads (ThermoFisher). To examine the effect of nucleic acids on the interaction between IFI16 and Sp1, cell lysates were treated with benzonase (750U/ml) in the presence of MgCl2 (1 mM) for 2 hours at room temperature before adding anti-HA antibodies. Beads were washed three times with 1 ml NP40 wash buffer (50 mM HEPES, 300 mM NaCl, 0.5% NP40, pH 7.4) before incubation with 60 μl 1 x Protein Sample Loading Buffer (LI-COR) at 95°C for 10 minutes to remove bound proteins. After addition of 1.75 μl β-mercaptoethanol the eluate was used for Western blotting. Proteins were stained using goat anti-SP1 (Abcam #ab157123), rabbit anti-HA (Cell Signaling #3724) and mouse anti-GAPDH (Santa Cruz #sc-365062) antibodies.
Sp1-DNA binding assay
HEK293T cells were transfected with expression constructs for different IFI16 mutants (1 μg) alone, or cotransfected with expression constructs for Sp1 (0.2 μg). 40 h post transfection, cells were harvested and nuclear proteins were isolated. Cells were washed with PBS and collected in 1 ml ice-cold Phosphatase Inhibitor Buffer (6.25 μΜ NaF, 12.5 mM β-glycerophosphate, 1.25 mM NaVO3, in PBS). Cells were pelleted by centrifugation at 300 x g for 5 minutes at 4°C, resuspended in 1 ml Hypotonic Buffer (20 mM HEPES, 5 mM NaF, 0.1 mM EDTA, pH 7.5) and incubated on ice for 15 min to allow swelling of the cells. After addition of 50 μl 10% NP40, the homogenate was centrifuged at 20,800 x g for 1 min at 4°C. The supernatant was removed and the pellet was resuspended in 50 μl Complete Lysis Buffer AM1 (ActiveMotif). Tubes were incubated for 30 min at 4°C with end-over-end mixing, centrifuged at 20,800 x g for 10 min at 4°C and the supernatants (nuclear cell extract) was transferred to fresh tubes. Protein concentrations were determined using the Pierce Gold BCA Protein Assay Kit (ThermoFisher) according to the manufacturer’s instructions. Volumes corresponding to 30 μg of nuclear extracts from samples with endogenous Sp1 levels and 10 μg of nuclear extracts from cells transfected with Sp1 were used to determine Sp1 binding to consensus target DNA sequences using the TransAM Sp1 Transcription Factor Assay Kit (ActiveMotif) according to the manufacturer’s instructions.
HIV reactivation from J-Lat cells
J-Lat cells (clone 10.6) were seeded in 24-well plates (250,000 cells/well) and treated with increasing doses of TNFα (0; 1; 5 ng/ml) and/or increasing doses of Mithramycin A (0; 5; 20; 50; 100 nM). 48 h later, GFP expression was determined by flow cytometry as a measure of HIV-1 reactivation.
MTT assay
To determine the effect of Mithramycin A on cell viability, J-Lat cells (clone 10.6) were seeded in 96-well plates (50,000 cells/well) and treated with increasing doses of Mithramycin (0; 1; 5; 20; 50; 100; 200; 500 μM). 48 h later, cells were pelleted by centrifugation, the medium was removed and the cells were resuspended in 100 μl MTT solution (0.5 mg/ml). After 2 h incubation at 37°C, 200 μl of a DMSO:ethanol mixture (1:1) was added to dissolve the purple formazan product. The absorbance of the colored solution at 490 nm was determined using a spectrophotometer. The absorbance of a negative control in which cells were treated with DMSO (50 μl/well) was used as background signal and subtracted from all values before normalization to the samples without Mitramycin treatment (=100 %).
Construction of NL4.3-deltaEnv-nLuc-2ANef proviral construct
We constructed NL4.3-deltaEnv-nLuc-2ANef by replacing the 237 bp HpaI-XhoI fragment that contains the env/nef junction in NL4.3-deltaEnv, previously referred to as DHIV (Bosque and Planelles, 2009), with a 930 bp fragment containing 130 bp of the 3’ terminus of the env gene, the nanoluciferase gene (Promega), and the 2A peptide cleavage (Verrier et al. 2011), and a 100 bp fragment containing the 5’ terminus of the nef gene up to the unique XhoI site. The resulting plasmid encodes a bicistronic nanoluciferase and nef gene whose transcription is driven by the HIV-1 LTR. In NL4.3-deltaEnv-nLuc-2ANef, the env gene is not expressed due to deletion of a 600 bp BglII fragment overlapping gp120 coding sequences (Bosque and Planelles, 2009), and the Rev-responsive element is preserved.
HIV reactivation from latently infected CD4+ T cells
Pseudotyped viruses were produced by cotransfection of HEK293T cells with pNL4.3-deltaEnv-nLuc-2ANef and pCMV-VSV-G. Cell supernatant was collected and filtered through a 0.22 μm filter after 2 days. Virus stocks were aliquoted and stored at −80°C until infection of CD4+ T cells. PBMCs were isolated from healthy donors using Lymphoprep and cultured in complete medium (RPMI-1640 with 10% FCS, L-Glutamine and penicillin/streptomycin). Naïve cells were isolated from PBMCs following the instruction of EasySep™ Human Naïve CD4+ T Cell Isolation Kit and then activated by culturing in a 96-well plate with 1 mg/ml anti-IL-4 antibody, 2 µg/ml anti-IL-12 antibody, 10 ng/ml TGF-β1 and anti-CD3/CD28 antibody beads (1 bead/cell). After 3 days, beads were removed using a magnetic column. Cells were cultured in complete medium supplemented with 30 IU/ml IL-2. At day 7, cells were infected with VSV-G pseudotyped NL4.3-deltaEnv-nLuc-2ANef viruses using spinoculation (Morton et al., 2019). At day 17, CD4+ cells were isolated using the Dynabeads™ CD4 Positive Isolation Kit (ThermoFisher). Since the recombinant virus expresses both Nef and Vpu, CD4 is efficiently downregulated in productively infected cells. Only cells that are latently infected or uninfected maintain full levels of CD4 and are purified by this procedure (Martins et al., 2016). At Day 18, CD4+ cells were seeded in a 96-well plate and treated with increasing doses of Mithramycin in the presence or absence of PMA (10 ng/ml). 2 days after stimulation, the luciferase activity in the cell supernatant was measured by Nano-Glo Luciferase Assay System. Cells were stained with cell viability dye, anti-CD4-APC and anti-p24-FITC, and measured by flow cytometry to determine infection rates.
LINE-1 retrotransposition assay
The retrotransposition-competent L1-GFP reporter plasmid (99 PUR L1RP EGFP) and the retrotransposition-defective negative control plasmid (99 PUR JM111 EGFP) have been described previously (Ostertag et al., 2000). Both plasmids contain a CMV-eGFP reporter cassette interrupted by an intron in the opposite transcriptional orientation within the 3’ UTR. eGFP is used as a surrogate marker for successful retrotransposition and is only expressed upon RNA splicing, reverse transcription, and integration. The L1 reporter plasmid and the indicated IFI16 expression vector or empty vector were transfected into HEK293T cells at a molecular ratio of 3:1 using calcium phosphate. Two days post-transfection, 2.5 μg/ml puromycin were added to the medium to boost transfection efficiency. Five days post-transfection, GFP-positive cells were quantified by flow cytometry.
LINE-1 promoter assay
HEK293T cells were cotransfected with luciferase reporter constructs in which expression is driven by the LINE-1 (L1RP) or CMV promoter and expression constructs for IFI16 or a control vector using calcium phosphate. The reporter plasmid L1RP-luc and the L1 promoter activity assay have been described previously (Yu et al., 2001). Two days post-transfection, cells were lysed with Cell Culture Lysis 5x reagent (Promega) and the luciferase activity was quantified using commercially available components (Promega).
MLV RT assay
The inhibitory effect of p204 on murine leukaemia virus (MLV) gene expression was determined by cotransfecting HEK293T cells with env-deficient MLV proviral constructs (1.5 μg) and increasing doses of expression constructs for p204 (0; 0.02; 0.1; 0.5; 1 μg) in 6-well plate format. 40 h post-transfection, MLV reverse transcriptase (RT) activity in the cell culture supernatants was determined using the C-Type RT Activity Kit (CAVIDI) according to the manufacturer’s instructions.
Viral loads in ARL−/− mice infected with Friend virus
C57BL/6J mice (Harlan Laboratories) and homozygously bred ALR−/− mice (Jackson Laboratories) were infected with 20,000 SFFU (spleen focus-forming units) of Friend virus. At 3 dpi, plasma viremia was determined and spleens were harvested to analyze viral loads in an infectious center (IC) assay. 13 mice per group from at least two independent experiments were analyzed. Mean (±SEM) values are indicated by bars. All mice were females and 8-10 weeks of age at the time point of infection. Prior to the experiment controls and experimental animals were co-housed for at least 3 weeks.
Infectious center and viremia assay
FV-infected animals were sacrificed at 3 dpi and plasma and splenocytes were harvested. For the detection of viral loads, serial dilutions of single-cell suspensions or plasma onto Mus dunnis cells were prepared and incubated for 3 days. At approximately 100% confluency, Mus dunnis cells were fixed with ethanol, stained with the F MuLV envelope-specific monoclonal antibody 720 and developed with a peroxidase-conjugated goat anti-mouse antibody and aminoethylcarbazol for the detection of foci.
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistical analyses were performed using GraphPad PRISM 7 (GraphPad Software). P-values were determined using a two-tailed Student’s t test. Correlations were calculated with the linear regression module. In the Friend virus in vivo experiment, statistically significant differences were determined using a Mann-Whitney test Unless otherwise stated, data are shown as the mean of at least three independent experiments ± SEM. Significant differences are indicated as: *, p < 0.05; **, p < 0.01; ***, p < 0.001. Statistical parameters are specified in the figure legends.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Mouse monoclonal anti-IFI16 (1G7) | Santa Cruz | Cat# sc-8023 RRID:AB_627775 |
| Mouse monoclonal anti-HA tag (HA.C5) | Abcam | Cat# ab18181 RRID:AB_444303 |
| Rabbit monoclonal anti-HA tag (C29F4) | Cell Signaling | Cat# 3724S RRID:AB_1549585 |
| Rabbit monoclonal anti-STING (D2P2F) | Cell Signaling | Cat# 13647 RRID:AB_2732796 |
| Mouse monoclonal anti-HIV-1 Env (16H3) | NIH AIDS Reagent Program | Cat# 12559 |
| Mouse monoclonal anti-HIV-1 p24 (39/5.4A) | Abcam | Cat# ab9071 RRID:AB_306981 |
| Rabbit polyclonal anti-Vpu | S. Bolduan | N/A |
| Rabbit polyclonal anti-AU1 tag | Novus Biologicals | Cat# NB600-453 RRID:AB_10003439 |
| Mouse monoclonal anti-β actin | Abcam | Cat# ab8226 RRID:AB_306371 |
| Rabbit polyclonal anti-GAPDH | Biolegend | Cat# 631401 RRID:AB_2247301 |
| Mouse monoclonal anti-GAPDH | Santa Cruz | Cat# sc-365062 RRID:AB_10847862 |
| Rabbit polyclonal anti-GFP | Abcam | Cat# ab290 RRID:AB_303395 |
| Goat polyclonal anti-Sp1 | Abcam | Cat# ab157123 |
| Rabbit polyclonal anti-Sp1 | Abcam | Cat# ab227383 |
| IRDye® 680RD Goat anti-Mouse IgG (H + L) | LI-COR | Cat# 926-68070 RRID:AB_10956588 |
| IRDye® 680RD Goat anti-Rabbit IgG (H + L) | LI-COR | Cat# 926-68071 RRID:AB_10956166 |
| IRDye® 680RD Donkey anti-Goat IgG (H + L) | LI-COR | Cat# 926-68074 RRID:AB_10956736 |
| IRDye® 800CW Goat anti-Mouse IgG (H + L) | LI-COR | Cat# 926-32210 RRID:AB_621842 |
| IRDye® 800CW Goat anti-Rabbit IgG (H + L) | LI-COR | Cat# 926-32211 RRID:AB_621843 |
| Donkey polyclonal anti-mouse, Alexa Fluor 647 | ThermoFisher | Cat# A-31571 RRID:AB_162542 |
| Goat polyclonal anti-mouse, PE | ThermoFisher | Cat# P-852 RRID:AB_2539848 |
| Anti-HIV-1 p24 core antigen (MAK183) | ExBIO | Cat# 11-CM006-BULK |
| Rabbit anti-p24 antiserum obtained by immunization of rabbits | Eurogentec | N/A |
| Peroxidase-AffiniPure goat anti-rabbit IgG, Fc fragment specific antibody | Dianova | Cat#
111-035-008 RRID:AB_2337937 |
| Rabbit polyclonal anti-human IL-4 | PeproTech | Cat# 500-P24 RRID:AB_147568 |
| Goat polyclonal anti-human IL-12 | PeproTech | Cat# 500-P154G RRID:AB_147809 |
| Mouse monoclonal anti-CD4, APC conjugated | Thermo Fisher | Cat# MHCD0405 RRID:AB_10373698 |
| Mouse monoclonal anti-HIV-1 core, FITC conjugated | Beckman Coulter | Cat# 6604665 |
| Bacterial and Virus Strains | ||
| Escherichia coli XL-2 blue™ | Stratagene | Cat# 200150 |
| XL2-Blue MRF’ TM Ultracompetent cells | Agilent Technologies | Cat# 200151 |
| Friend virus | U. Dittmer (University of Essen) | N/A |
| Sendai virus | G. Kochs (University of Freiburg) | N/A |
| Biological Samples | ||
| Human: Peripheral blood mononuclear cells | DRK-Blutspendedienst Baden-Württemberg-Hessen, Ulm, Germany | N/A |
| Chemicals, Peptides, and Recombinant Proteins | ||
| L-Glutamine | Pan Biotech | Cat# P04-80100 |
| Penicillin-Streptomycin | ThermoFisher | Cat# 15140122 |
| HAT supplement | ThermoFisher | Cat# 21060017 |
| Blasticidin | InvivoGen | Cat# ant-bl-1 |
| Zeocin | InvivoGen | Cat# ant-zn-1 |
| Normocin | InvivoGen | Cat# ant-nr-1 |
| QUANTI-Blue | InvivoGen | Cat# rep-qb2 |
| Human IL-2 IS, premium grade | MACS Miltenyi Biotec | Cat# 130-097-745 |
| Recombinant human IL-2 | NIH AIDS Reagent | Cat# 136 |
| Remel™ PHA purified | ThermoFisher | Cat# R30852801 |
| Biocoll separating solution | Biochrom | Cat# L6115 |
| Lymphoprep | Stemcell | Cat# 07851 |
| Human Interferon α 2a | pbl assay science | Cat# 11101-1 |
| Human Interferon γ | Sigma-Aldrich | Cat# I3265 |
| Dynabeads Human T-Activator CD3/CD28 | ThermoFisher | Cat# 111.32D |
| Tumor Necrosis Factor-α human | Sigma-Aldrich | Cat# H8916-10UG |
| Human M-CSF | R&D Systems | Cat# 216-MC-01M |
| Phorbol 12-myristate 13-acetate (PMA) | Sigma-Aldrich | Cat# 79346 |
| Recombinant human TGF-β1 | PeproTech | Cat# 100-21 |
| cGAMP | BioLog | Cat# C161 |
| HT-DNA | Sigma Aldrich | Cat# D6898 |
| Lipofectamine RNAiMAX Transfection Reagent | ThermoFisher | Cat# 13778150 |
| TransIT®-LT1 Transfection Reagent | Mirus | Cat# MIR 2305 |
| β-mercaptoethanol | Sigma Aldrich | Cat# M6250-100ML |
| HIV-1 p24 protein (ELISA standard) | Abcam | Cat# 43037 |
| KPL SureBlue™ TMB Microwell Peroxidase Substrate | Medac | Cat# 52-00-04 |
| Sulfuric acid concentrate for 1l standard solution 0.5 M H2SO4 | Sigma-Aldrich | Cat# 38294-1EA |
| 4X Protein Sample Loading Buffer | LI-COR | Cat# 928-40004 |
| 2-Mercaptoethanol | Sigma-Aldrich | Cat# M6250-100ML |
| NucRed Live 647 ReadyProbes Reagent | ThermoFisher | Cat# R37106 |
| Fixable Viability Dye eFluor™ 450 | eBioscience | Cat# 65-0863-14 |
| Benzonase Nuclease | Sigma-Aldrich | Cat# E1014-25KU |
| Mithramycin A | Cayman Chemicals | Cat# 11434 |
| MTT (Thiazolyl Blue Tetrazolium Bromide) | Sigma-Aldrich | Cat# M5655 |
| Critical Commercial Assays | ||
| RosetteSep™ Human CD4+ T Cell Enrichment Cocktail | Stem Cell Technologies | Cat# 15062 |
| EasySep™ Human Naïve CD4+ T Cell Isolation Kit | Stemcell | Cat# 19555 |
| QuikChange II XL Site-Directed Mutagenesis Kit | Agilent | Cat# 200522 |
| Phusion High-Fidelity PCR Kit | ThermoFisher | Cat# F553L |
| DNA Ligation Kit Ver. 2.1 | TaKaRa | Cat# 6022 |
| GalScreen | Applied Bioscience | Cat# T1027 |
| RNeasy Plus Mini Kit | QIAGEN | Cat# 74136 |
| DNA-free™ DNA Removal Kit | ThermoFisher | Cat# AM1906 |
| PrimeScript RT Reagent Kit | TAKARA | Cat# RR037A |
| TaqMan™ Fast Universal PCR Master Mix | ThermoFisher | Cat# 4352042 |
| GAPDH Endogenous Control (VIC/TAMRA) | ThermoFisher | Cat# 4310884E |
| Luciferase Cell Culture Lysis 5X Reagent | Promega | Cat# E1531 |
| Luciferase Assay System 10-pack | Promega | Cat# E1501 |
| Coelenterazine (native-CTZ) | p.j.k. GmbH | Cat# 102173 |
| FIX & PERM Kit (CE-IVD) | Nordic-MUbio | Cat# GAS-002-1 |
| Pierce Protein A/G Magnetic Beads | ThermoFisher | Cat# 88802 |
| Pierce Gold BCA Protein Assay Kit | ThermoFisher | Cat# A53225 |
| TransAM Sp1 Transcription Factor Assay Kit | ActiveMotif | Cat# 41296 |
| C-Type RT Activity Kit | CAVIDI | Cat# 51020 |
| Nano-Glo Luciferase Assay System | Promega | Cat# N1110 |
| BD Cytofix/Cytoperm™ Fixation and Permeabilization Solution | Becton Dickinson | Cat# 554722 |
| BD Perm/Wash™ Perm/Wash Buffer | Becton Dickinson | Cat# 554723 |
| Experimental Models: Cell Lines | ||
| Human: HEK293T cells | ATCC | Cat# CRL-3216 RRID: CVCL_0063 |
| Human: TZM-b1 cells | NIH AIDS Reagent Program | Cat# 8129 RRID: CVCL_B478 |
| Human: HL116 cells | Christine Goffinet | N/A |
| Human: HEK-Blue | InvivoGen | Cat# hkb-ifnab |
| Human: HAP1 cells | Horizon | Cat#
HZGHC001141c002 RRID:CVCL_TQ04 |
| Human: Sp1 KO HAP1 cells | Horizon | Cat#
HZGHC001141c002 RRID:CVCL_TQ04 |
| Human: THP-1 cells | ATCC | Cat#TIB-202 |
| Human: J-Lat cells (clone 10.6) | NIH AIDS Reagent Program | Cat# 9849 RRID:CVCL_8281 |
| Experimental Models: Organisms/Strains | ||
| B6(129S4)-Del(1Aim2-Ifi205)1Stsn/J Mice | The Jackson Laboratory | Stock No: 029472 |ALR− |
| Oligonucleotides | ||
| Primers used for generation of pCG-based expression constructs | see Table S1 of this paper | N/A |
| Primers used for generation of HIV-1 LTR F-luc reporter constructs | see Table S2 of this paper | N/A |
| Primers used for generation of chimeric HIV-1 proviral constructs | see Table S3 of this paper | N/A |
| Primers and fluorescent probes for qRT-PCR of viral transcripts | see Table S4 of this paper | N/A |
| siRNA targeting IFI16 | Invitrogen | Cat# IFI16HSS105205/IFI16HSS105206/IFI16HSS105207 |
| Non-targeting control siRNA: UUCUCCGAACGUGUCACGUdTdT | Eurofins MWG | N/A |
| Recombinant DNA | ||
| Plasmid: pLKO.1 puro | Stewart et al., 2003 | Addgene Cat# 8453 |
| Plasmid: pCG_IFI16 | This paper | N/A |
| Plasmid: pCG_IFI16 (SV40 NLS) | This paper | N/A |
| Plasmid: pCG_IFI16 (HIV-1 Rev NES) | This paper | N/A |
| Plasmid: pCG_IFI16 NLS mut (R96A, K97A, R98A, K99A, K100A, K128A, R129A, K130A, K131A, K134A, K136A, K140A) | This paper | N/A |
| Plasmid: pCG_IFI16 (K99A, K128A) | This paper | N/A |
| Plasmid: pCG_IFI16 ΔNLS1/2 (ΔR96-100K/K128-131K) | This paper | N/A |
| Plasmid: pCG_IFI16 IRES BFP | This paper | N/A |
| Plasmid: pCG_STING IRES BFP | This paper | N/A |
| Plasmid: pCG_IFI16 C-HA IRES BFP | This paper | N/A |
| Plasmid: pCG_cpz IFI16 C-HA IRES BFP | This paper | N/A |
| Plasmid: pCG_agm IFI16 C-HA IRES BFP | This paper | N/A |
| Plasmid: pCG_mac IFI16 C-HA IRES BFP | This paper | N/A |
| Plasmid: pCG_IFI16 Pyrin mut (S27A, L28A, D50A) C-HA IRES BFP | This paper | N/A |
| Plasmid: pCG_IFI16 ΔNLS1/2 (ΔR96-100K/K128-131K) C-HA IRES BFP | This paper | N/A |
| Plasmid: pCG_IFI16 ΔPyrin (Δ1-87) C-HA IRES BFP | This paper | N/A |
| Plasmid: pCG_IFI16 ΔPyrin-linker (Δ1-190) C-HA IRES BFP | This paper | N/A |
| Plasmid: pCG_IFI16 ΔHinA (Δ191-393) C-HA IRES BFP | This paper | N/A |
| Plasmid: pCG_IFI16 ΔHinA-linker (Δ191-509) C-HA IRES BFP | This paper | N/A |
| Plasmid: pCG_IFI16 HinB mut (K572A, K607A, R611A, S614A, K618A, N654A, K676A, K678A, K703A) C-HA IRES BFP | This paper | N/A |
| Plasmid: pCG_IFI16 ΔHin HinB mut C-HA IRES BFP | This paper | N/A |
| Plasmid: pCG_IFI16 ΔHinA-linker HinB mut C-HA IRES BFP | This paper | N/A |
| Plasmid: pCG_IFI16 ΔHinB (Δ509-729) C-HA IRES BFP | This paper | N/A |
| Plasmid: pCG_IFI16 Pyrin-linker (Δ191-729) C-HA IRES BFP | This paper | N/A |
| Plasmid: pCG_IFI16 Pyrin-only (Δ88-729) C-HA IRES BFP | This paper | N/A |
| Plasmid: pCG_IFI16 (R427A) C-HA IRES BFP | This paper | N/A |
| Plasmid: pCG_IFI16 (E463A) C-HA IRES BFP | This paper | N/A |
| Plasmid: pCG_IFI16 (G464A) C-HA IRES BFP | This paper | N/A |
| Plasmid: pCG_IFI16 (H466A) C-HA IRES BFP | This paper | N/A |
| Plasmid: pCG_IFI16 (G469A) C-HA IRES BFP | This paper | N/A |
| Plasmid: pCG_IFI16 (M472A) C-HA IRES BFP | This paper | N/A |
| Plasmid: pCG_IFI16 (V615A) C-HA IRES BFP | This paper | N/A |
| Plasmid: pCG_IFI16 (S409R, N413Y) C-HA IRES BFP | This paper | N/A |
| Plasmid: pCG_IFI16 (S409R) C-HA IRES BFP | This paper | N/A |
| Plasmid: pCG_IFI16 (N413Y) C-HA IRES BFP | This paper | N/A |
| Plasmid: pCG_AIM2 C-HA IRES BFP | This paper | N/A |
| Plasmid: pCG_mouse p204 C-HA IRES BFP | This paper | N/A |
| Plasmid: pCG_STING IRES BFP | This paper | N/A |
| Plasmid: pCG_Sp1 C-HA IRES BFP | This paper | N/A |
| Plasmid: pBR322_NL4-3 IRES eGFP | Frank Kirchhoff (Ulm University) | N/A |
| Plasmid: pCR-XL-TOPO_HIV-1 M subtype A 191845 (transmitted founder virus) | B. H. Hahn (Baalwa et al., 2013) | N/A |
| Plasmid: pCR-XL-TOPO_HIV-1 M subtype A 9004SDM (transmitted founder virus) | B. H. Hahn (Baalwa et al., 2013) | N/A |
| Plasmid: pCR-XL-TOPO_HIV-1 M subtype A R880F (transmitted founder virus) | B. H. Hahn (Baalwa et al., 2013) | N/A |
| Plasmid: pBR322_HIV-1 M subtype A 191084 (transmitted founder virus) | B. H. Hahn (Baalwa et al., 2013) | N/A |
| Plasmid: pCR-XL-TOPO_HIV-1 M subtype B CH058.c (transmitted founder virus) | B. H. Hahn (Ochsenbauer et al., 2012) | N/A |
| Plasmid: pCR-XL-TOPO HIV-1 M subtype B CH040.c (transmitted founder virus) | B. H. Hahn (Ochsenbauer et al., 2012) | N/A |
| Plasmid: pCR-XL-TOPO HIV-1 M subtype B CH077.t (transmitted founder virus) | B. H. Hahn (Ochsenbauer et al., 2012) | N/A |
| Plasmid: pBR332_HIV-1 M subtype B CH470 (transmitted founder) | B. H. Hahn (Parrish et al., 2013) | N/A |
| Plasmid: pCR-XL-TOPO HIV-1 M subtype B pWITO.c (transmitted founder virus) | B. H. Hahn (Ochsenbauer et al., 2012) | N/A |
| Plasmid: pCR-XL-TOPO_HIV-1 M subtype B CH106.c (transmitted founder virus) | B. H. Hahn (Ochsenbauer et al., 2012) | N/A |
| Plasmid: pCR-XL-TOPO HIV-1 M subtype B pTHRO.c (transmitted founder virus) | B. H. Hahn (Ochsenbauer et al., 2012) | N/A |
| Plasmid: pHIV-1 AD8 | V. Planelles (Zimmerman et al., 2006) | N/A |
| Plasmid: pBR322_HIV-1 M subtype B REJO.c (transmitted founder virus) | B. H. Hahn (Ochsenbauer et al., 2012) | N/A |
| Plasmid: pCR-XL TOPO_HIV-1 M subtype C CH042 (transmitted founder) | B. H. Hahn (Freel et al., 2012) | N/A |
| Plasmid: pCR-XL-TOPO_HIV-1 M subtype C CH162 (transmitted founder) | B. H. Hahn (Parrish et al., 2013) | N/A |
| Plasmid: pCR-XL TOPO_HIV-1 M subtype C CH185 (transmitted founder) | B. H. Hahn (Parrish et al., 2013) | N/A |
| Plasmid: pCR-XL-TOPO_HIV-1 M subtype C CH198 (transmitted founder) | B. H. Hahn (Parrish et al., 2013) | N/A |
| Plasmid: pUC57 HIV-1 M subtype C CH167 (chronic virus) | B. H. Hahn (Parrish et al., 2013) | N/A |
| Plasmid: pBR332_HIV-1 M subtype C CH850 (transmitted founder) | B. H. Hahn (Fenton-May et al., 2013) | N/A |
| Plasmid: pUC57rev_HIV-1 M subtype C CH293 (chronic virus) | B. H. Hahn (Parrish et al., 2013) | N/A |
| Plasmid: pCR-XL-TOPO_HIV-1 M subtype C CH164 (transmitted founder) | B. H. Hahn (Parrish et al., 2013) | N/A |
| Plasmid: pCR-XL-TOPO_HIV-1 M subtype C ZM247Fv-1 (transmitted founder virus) | B. H. Hahn (Salazar-Gonzalez et al., 2009) | N/A |
| Plasmid: pBR332_HIV-1 M subtype C CH236 (transmitted founder) | B. H. Hahn (Fenton-May et al., 2013) | N/A |
| Plasmid: pUC57_HIV-1 M subtype C CH200a (transmitted founder) | B. H. Hahn (Parrish et al., 2013) | N/A |
| Plasmid: pBR322_HIV-1 M subtype C CH264 (transmitted founder) | B. H. Hahn (Salazar-Gonzalez et al., 2009) | N/A |
| Plasmid: pCR-XL-TOPO_HIV-1 M subtype D 191647 (transmitted founder) | B. H. Hahn (Baalwa et al., 2013) | N/A |
| Plasmid: pCR-XL-TOPO_HIV-1 M subtype D 191859 (transmitted founder) | B. H. Hahn (Baalwa et al., 2013) | N/A |
| Plasmid: pCR-XL-TOPO_HIV-1 M subtype D 191882 (transmitted founder) | B. H. Hahn (Baalwa et al., 2013) | N/A |
| Plasmid: pCR-XL-TOPO_HIV-1 M subtype D 190049 (transmitted founder) | B. H. Hahn (Baalwa et al., 2013) | N/A |
| Plasmid: pCR-XL-TOPO_HIV-1 M CH058 with 3’ and 5’ LTR of THRO | This paper | N/A |
| Plasmid: pCR-XL-TOPO_HIV-1 M CH040 with 3’ and 5’ LTR of THRO | This paper | N/A |
| Plasmid: pCR-XL-TOPO_HIV-1 M THRO with 3’ and 5’ LTR of CH058 | This paper | N/A |
| Plasmid: pCR-XL-TOPO SIVcpz Ptt EK505 | B. H. Hahn (Keele et al., 2006) | N/A |
| Plasmid: pCR-XL-TOPO SIVcpz Ptt MB897.2 | F. Kirchhoff (Bibollet-Ruche et al., 2012) | N/A |
| Plasmid: pBluescript II KS+_HIV-2 AB 7312A | NIH AIDS Reagent Program | Cat# 3551 |
| Plasmid: pBR322_HIV-2 ROD9 eGFP | J. Münch(Ulm University) | N/A |
| Plasmid: pBR322_HIV-2 ROD10 | J. Münch (Ulm University) | N/A |
| Plasmid: pSP65 HIV-2 A ST | NIH AIDS Reagent Program (Kumar et al., 1990) | Cat# 12444 |
| Plasmid: pHIV-2 A KR | B. H. Hahn (Talbott et al., 1993) | N/A |
| Plasmid: pGEM_SIVsmm pg | F. Kirchhoff (Ulm University) | N/A |
| Plasmid: pBR322hl_SIVsmm L1.RM174.V1.tf | B. H. Hahn (Mason et al., 2016) | N/A |
| Plasmid: pBR322hl_SIVsmm L1.RM174.V2.tf | B. H. Hahn (Mason et al., 2016) | N/A |
| Plasmid: pBR322hl_SIVsmm L2.RM136.tf | B. H. Hahn (Mason et al., 2016) | N/A |
| Plasmid: pBR322hl_SIVsmm L3.RM175.tf | B. H. Hahn (Mason et al., 2016) | N/A |
| Plasmid: pBR322hl_SIVsmm L4.RM57.tf | B. H. Hahn (Mason et al., 2016) | N/A |
| Plasmid: pBR322MCS_SIVsmm Outlier.DT17.tf | B. H. Hahn (Mason et al., 2016) | N/A |
| Plasmid: pCR-XL-TOPO_HIV-1 N DJO0131 | F. Bibollet-Ruche (Bodelle et al., 2004) | N/A |
| Plasmid: pBluescript_HIV-1 O pCMO2.5 | H.G. Kräusslich (Tebit et al., 2004) | N/A |
| Plasmid: pSMART-topo_HIV-1 O RBF206 | F. Bibollet-Ruche (Mack et al., 2017) | N/A |
| Plasmid: pBR322_HIV-1 O BCF183 | B. Hahn | N/A |
| Plasmid: pBR322_HIV-1 P RBF168 | B. H. Hahn (Plantier et al., 2009) | N/A |
| Plasmid: pBR322_SIVmac 239 | F. Kirchhoff (Ulm University) | N/A |
| Plasmid: pCR-XL-TOPO_SIVagm Sab92018ivTF | F. Kirchhoff (Gnanadurai et al., 2010) | N/A |
| Plasmid: pGL3-enhancer HIV-1 M NL4-3 LTR (wild type) firefly luciferase reporter | This paper | N/A |
| Plasmid: pGL3-enhancer HIV-1 M NL4-3 LTR (Sp1 site mutated) firefly luciferase reporter | This paper | N/A |
| Plasmid: pGL3-enhancer_HIV-1 M NL4-3 LTR (NF-κB/NFAT site mutated) firefly luciferase reporter | This paper | N/A |
| Plasmid: pGL3-enhancer_HIV-1 M NL4-3 LTR (USF site mutated) firefly luciferase reporter | This paper | N/A |
| Plasmid: pGL3-enhancer_HIV-1 M NL4-3 LTR (TCF-1α site mutated) firefly luciferase reporter | This paper | N/A |
| Plasmid: pGL3-enhancer_HIV-1 M NL4-3 LTR (NF-IL6 site mutated) firefly luciferase reporter | This paper | N/A |
| Plasmid: pCG_NL4-3 Tat IRES eGFP | F. Kirchhoff (Ulm University) | N/A |
| Plasmid: pTK-Red_HSV-TK firefly luciferase reporter | ThermoFisher | Cat# 16157 |
| Plasmid: pCMV-Red_CMV-IE firefly luciferase reporter | ThermoFisher | Cat# 16156 |
| Plasmid: pTAL_Gaussia luciferase reporter | D. Sauter (Hotter et al., 2013) | N/A |
| Plasmid: p_HIV-1 M LAI1 wild type | A.T. Das (Das et al., 2011) | N/A |
| Plasmid: p_HIV-1 M LAI1 rtTA-2d15tetO (ΔTAR loop) (tat stop) | A.T. Das (Das et al., 2011) | N/A |
| Plasmid: p_HIV-1 M LAI1 rtTA-2d15tetO (ΔTAR loop) (Tat Y26A) | A.T. Das (Das et al., 2011) | N/A |
| Plasmid: p_HIV-1 M LAI1 rtTA-tetOcmv (tat stop) (lacks all HIV-1 U3 and TAR sequences) | A.T. Das (Das et al., 2011) | N/A |
| Plasmid: p_HIV-1 M LAI1 rtTA-tetOcmv (lacks all HIV-1 U3 and TAR sequences) | A.T. Das (Das et al., 2011) | N/A |
| Plasmid: p_HIV-1 M LAI1 rtTA-tetOcmv (lacks all HIV-1 U3 except for Sp1 binding sites; lacks TAR sequences) | A.T. Das (Das et al., 2011) | N/A |
| Plasmid: p_HIV-1 M LAI1 rtTA-tetOcmv (tat stop) (lacks all HIV-1 U3 except for Sp1 binding sites; lacks TAR sequences) | A.T. Das (Das et al., 2011) | N/A |
| Plasmid: p_HIV-LTR-2d15tetO-dNF-luc | A.T. Das (Turrini et al., 2015) | N/A |
| Plasmid: p_HIV-LTR-2d15tetO-dNF-mSp1-luc (deletion of 1 Sp1 site) | A.T. Das (Turrini et al., 2015) | N/A |
| Plasmid: p_HIV-LTR-2d15tetO-dNF-mSp2-luc (deletion of 2 Sp1 sites) | A.T. Das (Turrini et al., 2015) | N/A |
| Plasmid: p_HIV-LTR-2d15tetO-dNF-mSP-luc (mutation of all 3 Sp1 sites) | A.T. Das (Turrini et al., 2015) | N/A |
| Plasmid: p_HIV-LTR-2d15tetO-X/H-luc | A.T. Das (Turrini et al., 2015) | N/A |
| Plasmid: pHIV-1 NL4-3-deltaEnv-nLuc-2ANef | This paper (generated by Laura Martins in Planelles lab) | N/A |
| Plasmid: pCMV-VSV-G | Stewart et al., 2003 | Addgene Cat# 8454 |
| Plasmid: p99 PUR RPS EGFP | John L. Goodier | N/A |
| Plasmid: p99 JM111 EGFP | John L. Goodier | N/A |
| Plasmid: pL1RP-luc | Gerald Schumann | N/A |
| Software and Algorithms | ||
| BD FACSDiva™ Version 8.0 | BD Biosciences |
https://www.bdbiosciences.com RRID: SCR_001456 |
| Corel DRAW 2017 | Corel Corporation | https://www.coreldraw.com/ |
| GraphPad Prism Version 7 | GraphPad Software, Inc. |
https://www.graphpad.com RRID: SCR_002798 |
| ImageJ | Open source | http://imagej.nih.gov/ij/ |
| LI-COR Image Studio Lite Version 5.0 | LI-COR |
www.licor.com/ RRID: SCR_013715 |
| Other | ||
| Genomic Utility for Association and Viral Analyses in HIV | Open source | http://www.guavah.org/ |
HIGHLIGHTS.
IFI16 targets Sp1 to restrict HIV-1 transcription and LINE-1 retrotransposition
The N-terminal pyrin and NLS domains of IFI16 are sufficient for restriction
Sp1 inhibition by IFI16 or Mithramycin A suppresses reactivation of latent HIV-1
Murine homologs of IFI16 restrict retroviral replication in vitro and in vivo
ACKNOWLEDGEMENTS
We thank Daniela Krnavek, Martha Mayer, Regina Linsenmeyer and Susanne Engelhart for excellent technical assistance, Frederic Bibollet-Ruche and Ronnie Russell for critical reading of the manuscript and Daniel B. Stetson for providing ALR−/− mice. TZM-b1 cells were obtained through the NIH AIDS Reagent Program. LINE-1 GFP reporter constructs 99 PUR RPS EGFP and 99 PUR JM111 EGFP were a gift of John L. Goodier (John Hopkins University, Baltimore). L1RP-luc reporter plasmid was provided by Gerald Schumann (Paul Ehrlich Institute, Langen). This work was supported by funds from the European Research Council (ERC advanced grant “Anti-Virome to F.K.), the German Research Foundation (DFG SPP1923 to F.K., D.S. and U.D.) and the National Institute of Health (R01 AI 114266 and UM1 AI 126620). M.R.J. was funded by the Lundbeck foundation and Danish Research Council (4004-00237). S.W. and T.G. were supported by the German Research Foundation (DFG) grant GR3355/3-1 and IZKF A67 University clinic Erlangen. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
DECLARATION OF INTERESTS
The authors declare no competing interests.
REFERENCES
- Baalwa J, Wang S, Parrish NF, Decker JM, Keele BF, Learn GH, Yue L, Ruzagira E, Ssemwanga D, Kamali A, et al. (2013). Molecular identification, cloning and characterization of transmitted/founder HIV-1 subtype A, D and A/D infectious molecular clones. Virology 436, 33–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bachu M, Yalla S, Asokan M, Verma A, Neogi U, Sharma S, Murali RV, Mukthey AB, Bhatt R, Chatterjee S, et al. (2012). Multiple NF-κB sites in HIV-1 subtype C long terminal repeat confer superior magnitude of transcription and thereby the enhanced viral predominance. J. Biol. Chem 287, 44714–44735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bauernfeind F, and Hornung V (2013). Of inflammasomes and pathogens - sensing of microbes by the inflammasome. EMBO Mol. Med 5, 814–826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bibollet-Ruche F, Heigele A, Keele BF, Easlick JL, Decker JM, Takehisa J, Learn G, Sharp PM, Hahn BH, and Kirchhoff F (2012). Efficient SIVcpz replication in human lymphoid tissue requires viral matrix protein adaptation. J. Clin. Invest 122, 1644–1652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bieniasz PD (2004). Intrinsic immunity: a front-line defense against viral attack. Nat. Immunol 5, 1109–1115. [DOI] [PubMed] [Google Scholar]
- Bodelle P, Vallari A, Coffey R, McArthur CP, Beyeme M, Devare SG, Schochetman G, and Brennan CA (2004). Identification and Genomic Sequence of an HIV Type 1 Group N Isolate from Cameroon. AIDS Res. Hum. Retroviruses 20, 902–908. [DOI] [PubMed] [Google Scholar]
- Booiman T, and Kootstra NA (2014). Polymorphism in IFI16 affects CD4 + T-cell counts in HIV-1 infection. Int. J. Immunogenet 41, 518–520. [DOI] [PubMed] [Google Scholar]
- Bosque A, and Planelles V (2009). Induction of HIV-1 latency and reactivation in primary memory CD4+ T cells. Blood 113, 58–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burdette DL, Monroe KM, Sotelo-Troha K, Iwig JS, Eckert B, Hyodo M, Hayakawa Y, and Vance RE (2011). STING is a direct innate immune sensor of cyclic di-GMP. Nature 478, 515–518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cagliani R, Forni D, Biasin M, Comabella M, Guerini FR, Riva S, Pozzoli U, Agliardi C, Caputo D, Malhotra S, et al. (2014). Ancient and Recent Selective Pressures Shaped Genetic Diversity at AIM2-Like Nucleic Acid Sensors. Genome Biol. Evol 6, 830–845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carette JE, Guimaraes CP, Varadarajan M, Park AS, Wuethrich I, Godarova A, Kotecki M, Cochran BH, Spooner E, Ploegh HL, et al. (2009). Haploid Genetic Screens in Human Cells Identify Host Factors Used by Pathogens. Science (80-. ). 326, 1231–1235. [DOI] [PubMed] [Google Scholar]
- Carette JE, Raaben M, Wong AC, Herbert AS, Obernosterer G, Mulherkar N, Kuehne AI, Kranzusch PJ, Griffin AM, Ruthel G, et al. (2011). Ebola virus entry requires the cholesterol transporter Niemann—Pick C1. Nature 477, 340–343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cridland JA, Curley EZ, Wykes MN, Schroder K, Sweet MJ, Roberts TL, Ragan MA, Kassahn KS, and Stacey KJ (2012). The mammalian PYHIN gene family: phylogeny, evolution and expression. BMC Evol. Biol 12, 140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daigneault M, Preston JA, Marriott HM, Whyte MKB, and Dockrell DH (2010). The identification of markers of macrophage differentiation in PMA-stimulated THP-1 cells and monocyte-derived macrophages. PLoS One 5, e8668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das AT, Harwig A, and Berkhout B (2011). The HIV-1 Tat protein has a versatile role in activating viral transcription. J. Virol 85, 9506–9516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deschamps S, Meyer J, Chatterjee G, Wang H, Lengyel P, and Roe BA (2003). The mouse Ifi200 gene cluster: genomic sequence, analysis, and comparison with the human HIN-200 gene cluster. Genomics 82, 34–46. [DOI] [PubMed] [Google Scholar]
- Diner BA, Lum KK, Toettcher JE, and Cristea IM (2016). Viral DNA Sensors IFI16 and Cyclic GMP-AMP Synthase Possess Distinct Functions in Regulating Viral Gene Expression, Immune Defenses, and Apoptotic Responses during Herpesvirus Infection. MBio 7, e01553–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DuBridge RB, Tang P, Hsia HC, Leong PM, Miller JH, and Calos MP (1987). Analysis of mutation in human cells by using an Epstein-Barr virus shuttle system. Mol. Cell. Biol 7, 379–387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fenton-May AE, Dibben O, Emmerich T, Ding H, Pfafferott K, Aasa-Chapman MM, Pellegrino P, Williams I, Cohen MS, Gao F, et al. (2013). Relative resistance of HIV-1 founder viruses to control by interferon-alpha. Retrovirology 10, 146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fouchier RA, Meyer BE, Simon JH, Fischer U, and Malim MH (1997). HIV-1 infection of non-dividing cells: evidence that the amino-terminal basic region of the viral matrix protein is important for Gag processing but not for post-entry nuclear import. EMBO J 16, 4531–4539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freel SA, Picking RA, Ferrari G, Ding H, Ochsenbauer C, Kappes JC, Kirchherr JL, Soderberg KA, Weinhold KJ, Cunningham CK, et al. (2012). Initial HIV-1 Antigen-Specific CD8+ T Cells in Acute HIV-1 Infection Inhibit Transmitted/Founder Virus Replication. J. Virol 86, 6835–6846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galão RP, Le Tortorec A, Pickering S, Kueck T, and Neil SJD (2012). Innate sensing of HIV-1 assembly by Tetherin induces NFκB-dependent proinflammatory responses. Cell Host Microbe 12, 633–644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao F, Scearce RM, Alam SM, Hora B, Xia S, Hohm JE, Parks RJ, Ogburn DF, Tomaras GD, Park E, et al. (2009). Cross-reactive monoclonal antibodies to multiple HIV-1 subtype and SIVcpz envelope glycoproteins. Virology 394, 91–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gariano GR, Dell’Oste V, Bronzini M, Gatti D, Luganini A, De Andrea M, Gribaudo G, Gariglio M, and Landolfo S (2012). The Intracellular DNA Sensor IFI16 Gene Acts as Restriction Factor for Human Cytomegalovirus Replication. PLoS Pathog 8, e1002498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gnanadurai CW, Pandrea I, Parrish NF, Kraus MH, Learn GH, Salazar MG, Sauermann U, Töpfer K, Gautam R, Münch J, et al. (2010). Genetic Identity and Biological Phenotype of a Transmitted/Founder Virus Representative of Nonpathogenic Simian Immunodeficiency Virus Infection in African Green Monkeys. J. Virol 84, 12245–12254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gray EE, Winship D, Snyder JM, Child SJ, Geballe AP, and Stetson DB (2016). The AIM2-like Receptors Are Dispensable for the Interferon Response to Intracellular DNA. Immunity 45, 255–266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris RS, Hultquist JF, and Evans DT (2012). The Restriction Factors of Human Immunodeficiency Virus. J. Biol. Chem 287, 40875–40883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He Y, Hara H, and Núñez G (2016). Mechanism and Regulation of NLRP3 Inflammasome Activation. Trends Biochem. Sci 41, 1012–1021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hotter D, Kirchhoff F, and Sauter D (2013). HIV-1 Vpu does not degrade interferon regulatory factor 3. J. Virol 87, 7160–7165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jakobsen MR, and Paludan SR (2014). IFI16: At the interphase between innate DNA sensing and genome regulation. Cytokine Growth Factor Rev 25, 649–655. [DOI] [PubMed] [Google Scholar]
- Jakobsen MR, Bak RO, Andersen A, Berg RK, Jensen SB, Tengchuan J, Jin T, Laustsen A, Hansen K, Ostergaard L, et al. (2013). IFI16 senses DNA forms of the lentiviral replication cycle and controls HIV-1 replication. Proc. Natl. Acad. Sci. U. S. A 110, E4571–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeang KT, Chun R, Lin NH, Gatignol A, Glabe CG, and Fan H (1993). In vitro and in vivo binding of human immunodeficiency virus type 1 Tat protein and Sp1 transcription factor. J. Virol 67, 6224–6233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin T, Perry A, Jiang J, Smith P, Curry JA, Unterholzner L, Jiang Z, Horvath G, Rathinam V, Johnstone RW, et al. (2012). Structures of The HIN Domain:DNA Complexes Reveal Ligand Binding and Activation Mechanisms of The AIM2 Inflammasome and IFI16 Receptor. Immunity 36, 561–571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson KE, Bottero V, Flaherty S, Dutta S, Singh VV, and Chandran B (2014). IFI16 restricts HSV-1 replication by accumulating on the hsv-1 genome, repressing HSV-1 gene expression, and directly or indirectly modulating histone modifications. PLoS Pathog 10, e1004503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones KA, Kadonaga JT, Luciw PA, and Tjian R (1986). Activation of the AIDS retrovirus promoter by the cellular transcription factor, Sp1. Science 232, 755–759. [DOI] [PubMed] [Google Scholar]
- Jønsson KL, Laustsen A, Krapp C, Skipper KA, Thavachelvam K, Hotter D, Egedal JH, Kjolby M, Mohammadi P, Prabakaran T, et al. (2017). IFI16 is required for DNA sensing in human macrophages by promoting production and function of cGAMP. Nat. Commun 8, 14391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jordan A, Bisgrove D, and Verdin E (2003). HIV reproducibly establishes a latent infection after acute infection of T cells in vitro. EMBO J, 22(8), 1868–1877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keele BF, Van Heuverswyn F, Li Y, Bailes E, Takehisa J, Santiago ML, Bibollet-Ruche F, Chen Y, Wain LV, Liegeois F, et al. (2006). Chimpanzee Reservoirs of Pandemic and Nonpandemic HIV-1. Science 313, 523–526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerur N, Veettil MV, Sharma-Walia N, Bottero V, Sadagopan S, Otageri P, and Chandran B (2011). IFI16 acts as a nuclear pathogen sensor to induce the inflammasome in response to Kaposi Sarcoma-associated herpesvirus infection. Cell Host Microbe 9, 363–375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kluge SF, Sauter D, and Kirchhoff F (2015). SnapShot: antiviral restriction factors. Cell 163, 774–774.e1. [DOI] [PubMed] [Google Scholar]
- Konvalinka J, Litterst MA, Welker R, Kottler H, Rippmann F, Heuser AM, and Kräusslich HG (1995). An active-site mutation in the human immunodeficiency virus type 1 proteinase (PR) causes reduced PR activity and loss of PR-mediated cytotoxicity without apparent effect on virus maturation and infectivity. J. Virol 69, 7180–7186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krapp C, Hotter D, Gawanbacht A, McLaren PJ, Kluge SF, Stürzel CM, Mack K, Reith E, Engelhart S, Ciuffi A, et al. (2016). Guanylate Binding Protein (GBP) 5 Is an Interferon-Inducible Inhibitor of HIV-1 Infectivity. Cell Host Microbe 19, 504–514. [DOI] [PubMed] [Google Scholar]
- Kumar P, Hui HX, Kappes JC, Haggarty BS, Hoxie JA, Arya SK, Shaw GM, and Hahn BH (1990). Molecular characterization of an attenuated human immunodeficiency virus type 2 isolate. J. Virol 64, 890–901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li T, Diner BA, Chen J, and Cristea IM (2012). Acetylation modulates cellular distribution and DNA sensing ability of interferon-inducible protein IFI16. Proc. Natl. Acad. Sci. U. S. A 109, 10558–10563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lo Cigno I, De Andrea M, Borgogna C, Albertini S, Landini MM, Peretti A, Johnson KE, Chandran B, Landolfo S, and Gariglio M (2015a). The Nuclear DNA Sensor IFI16 Acts as a Restriction Factor for Human Papillomavirus Replication through Epigenetic Modifications of the Viral Promoters. J. Virol 89, 7506–7520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loregian A, Bortolozzo K, Boso S, Sapino B, Betti M, Biasolo MA, Caputo A, and Pald G (2003). The Sp1 transcription factor does not directly interact with the HIV-1 Tat protein. J. Cell. Physiol 196, 251–257. [DOI] [PubMed] [Google Scholar]
- Mack K, Starz K, Sauter D, Langer S, Bibollet-Ruche F, Learn GH, Stürzel CM, Leoz M, Plantier J-C, Geyer M, et al. (2017). Efficient Vpu-Mediated Tetherin Antagonism by an HIV-1 Group O Strain. J. Virol 91, e02177–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martins LJ, Bonczkowski P, Spivak AM, De Spiegelaere W, Novis CL, DePaula-Silva AB, Malatinkova E, Trypsteen W, Bosque A, Vanderkerckhove L, and Planelles V (2016). Modeling HIV-1 Latency in Primary T Cells Using a Replication-Competent Virus. AIDS Res Hum Retroviruses. 32, 187–193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mason RD, Welles HC, Adams C, Chakrabarti BK, Gorman J, Zhou T, Nguyen R, O’Dell S, Lusvarghi S, Bewley CA, et al. (2016). Targeted Isolation of Antibodies Directed against Major Sites of SIV Env Vulnerability. PLoS Pathog 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLaren PJ, Gawanbacht A, Pyndiah N, Krapp C, Hotter D, Kluge SF, Gätz N, Heilmann J, Mack K, Sauter D, et al. (2015). Identification of potential HIV restriction factors by combining evolutionary genomic signatures with functional analyses. Retrovirology 12, 41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monroe KM, Yang Z, Johnson JR, Geng X, Doitsh G, Krogan NJ, and Greene WC (2014). IFI16 DNA sensor is required for death of lymphoid CD4 T cells abortively infected with HIV. Science 343, 428–432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montano MA, Nixon CP, Ndung’u T, Bussmann H, Novitsky VA, Dickman D, and Essex M (2000). Elevated Tumor Necrosis Factor—α Activation of Human Immunodeficiency Virus Type 1 Subtype C in Southern Africa Is Associated with an NF-äB Enhancer Gain-of-Function. J. Infect. Dis 181, 76–81. [DOI] [PubMed] [Google Scholar]
- Morrone SR, Wang T, Constantoulakis LM, Hooy RM, Delannoy MJ, and Sohn J (2014). Cooperative assembly of IFI16 filaments on dsDNA provides insights into host defense strategy. Proc. Natl. Acad. Sci. U. S. A 111, E62–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morton EL, Forst CF, Zheng Y, DePaula-Silva AB, Ramirez GP, Planelles V, and D’Orso I (2019). Transcriptional Circuit Fragility Influences HIV Proviral Fate. Cell Reports (in press). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ni X, Ru H, Ma F, Zhao L, Shaw N, Feng Y, Ding W, Gong W, Wang Q, Ouyang S, et al. (2016). New insights into the structural basis of DNA recognition by HINa and HINb domains of IFI16. J. Mol. Cell Biol 8, 51–61. [DOI] [PubMed] [Google Scholar]
- Ochsenbauer C, Edmonds TG, Ding H, Keele BF, Decker J, Salazar MG, Salazar-Gonzalez JF, Shattock R, Haynes BF, Shaw GM, et al. (2012). Generation of transmitted/founder HIV-1 infectious molecular clones and characterization of their replication capacity in CD4 T lymphocytes and monocyte-derived macrophages. J. Virol 86, 2715–2728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orzalli MH, DeLuca NA, and Knipe DM (2012). Nuclear IFI16 induction of IRF-3 signaling during herpesviral infection and degradation of IFI16 by the viral ICP0 protein. Proc. Natl. Acad. Sci. U. S. A 109, E3008–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ostertag EM, Prak ET, DeBerardinis RJ, Moran JV, and Kazazian HH Jr. (2000). Determination of L1 retrotransposition kinetics in cultured cells. Nucleic Acids Res, 28, 1418–1423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parrish NF, Gao F, Li H, Giorgi EE, Barbian HJ, Parrish EH, Zajic L, Iyer SS, Decker JM, Kumar A, et al. (2013). Phenotypic properties of transmitted founder HIV-1. Proc. Natl. Acad. Sci. U. S. A 110, 6626–6633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pertel T, Hausmann S, Morger D, Züger S, Guerra J, Lascano J, Reinhard C, Santoni FA, Uchil PD, Chatel L, et al. (2011). TRIM5 is an innate immune sensor for the retrovirus capsid lattice. Nature 472, 361–365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plantier J-C, Leoz M, Dickerson JE, Oliveira FD, Cordonnier F, Lemée V, Damond F, Robertson DL, and Simon F (2009). A new human immunodeficiency virus derived from gorillas. Nat. Med 15, 871–872. [DOI] [PubMed] [Google Scholar]
- Platt EJ, Wehrly K, Kuhmann SE, Chesebro B, and Kabat D (1998). Effects of CCR5 and CD4 Cell Surface Concentrations on Infections by Macrophagetropic Isolates of Human Immunodeficiency Virus Type 1. J. Virol 72, 2855–2864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rotger M, Dang KK, Fellay J, Heinzen EL, Feng S, Descombes P, Shianna KV, Ge D, Günthard HF, Goldstein DB, et al. (2010). Genome-wide mRNA expression correlates of viral control in CD4+ T-cells from HIV-1-infected individuals. PLoS Pathog 6, e1000781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salazar-Gonzalez JF, Salazar MG, Keele BF, Learn GH, Giorgi EE, Li H, Decker JM, Wang S, Baalwa J, Kraus MH, et al. (2009). Genetic identity, biological phenotype, and evolutionary pathways of transmitted/founder viruses in acute and early HIV-1 infection. J. Exp. Med 206, 1273–1289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schattgen SA, and Fitzgerald KA (2011). The PYHIN protein family as mediators of host defenses. Immunol. Rev 243, 109–118. [DOI] [PubMed] [Google Scholar]
- Suñé C, and García-Blanco MA (1995). Sp1 transcription factor is required for in vitro basal and Tat-activated transcription from the human immunodeficiency virus type 1 long terminal repeat. J. Virol 69, 6572–6576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Talbott R, Kraus G, Looney D, and Wong-Staal F (1993). Mapping the determinants of human immunodeficiency virus 2 for infectivity, replication efficiency, and cytopathicity. Proc. Natl. Acad. Sci. U. S. A 90, 4226–4230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tebit DM, Zekeng L, Kaptué L, Görtler L, Fackler OT, Keppler OT, Herchenröder O, and Kräusslich H-G (2004). Construction and characterization of an HIV-1 group O infectious molecular clone and analysis of vpr- and nef-negative derivatives. Virology 326, 329–339. [DOI] [PubMed] [Google Scholar]
- Turrini F, Marelli S, Kajaste-Rudnitski A, Lusic M, Van Lint C, Das AT, Harwig A, Berkhout B, and Vicenzi E (2015). HIV-1 transcriptional silencing caused by TRIM22 inhibition of Sp1 binding to the viral promoter. Retrovirology 12, 104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Unterholzner L, Keating SE, Baran M, Horan KA, Jensen SB, Sharma S, Sirois CM, Jin T, Latz E, Xiao TS, et al. (2010). IFI16 is an innate immune sensor for intracellular DNA. Nat. Immunol 11, 997–1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uzé G, Di Marco S, Mouchel-Vielh E, Monneron D, Bandu MT, Horisberger MA, Dorques A, Lutfalla G, and Mogensen KE (1994). Domains of interaction between alpha interferon and its receptor components. J. Mol. Biol 243, 245–257. [DOI] [PubMed] [Google Scholar]
- Verrier JD, Madorsky I, Coggin WE, Geesey M, Hochman M, Walling E, Daroszewski D, Eccles KS, Ludlow R, and Semple-Rowland SL (2011). Bicistronic lentiviruses containing a viral 2A cleavage sequence reliably co-express two proteins and restore vision to an animal model of LCA1. PLoS One 6, e20553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu F, Zingler N, Schumann G, and Strätling WH (2001). Methyl-CpG-binding protein 2 represses LINE-1 expression and retrotransposition but not Alu transcription. Nucleic Acids Res 29, 4493–4501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yukl SA, Kaiser P, Kim P, Telwatte S, Joshi SK, Vu M, Lampiris H, and Wong JK (2018). HIV latency in isolated patient CD4+ T cells may be due to blocks in HIV transcriptional elongation, completion, and splicing. Sci. Transl. Med 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zimmerman ES, Sherman MP, Blackett JL, Neidleman JA, Kreis C, Mundt P, Williams SA, Warmerdam M, Kahn J, Hecht FM, et al. (2006). Human Immunodeficiency Virus Type 1 Vpr Induces DNA Replication Stress In Vitro and In Vivo. J. Virol 80, 10407–10418. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.