

Abstract
Epimerization of cholic and chenodeoxycholic acid (CA and CDCA, respectively) is a notable conversion for the production of ursodeoxycholic acid (UDCA). Two enantiocomplementary hydroxysteroid dehydrogenases (7α‐ and 7β‐HSDHs) can carry out this transformation fully selectively by specific oxidation of the 7α‐OH group of the substrate and subsequent reduction of the keto intermediate to the final product (7β‐OH). With a view to developing robust and active biocatalysts, novel NADH‐active 7β‐HSDH species are necessary to enable a solely NAD+‐dependent redox‐neutral cascade for UDCA production. A wild‐type NADH‐dependent 7β‐HSDH from Lactobacillus spicheri (Ls7β‐HSDH) was identified, recombinantly expressed, purified, and biochemically characterized. Using this novel NAD+‐dependent 7β‐HSDH enzyme in combination with 7α‐HSDH from Stenotrophomonas maltophilia permitted the biotransformations of CA and CDCA in the presence of catalytic amounts of NAD+, resulting in high yields (>90 %) of UCA and UDCA.
Keywords: carboxylic acids, biotransformation, enzymes, cofactors, steroids
Introduction
Ursodeoxycholic acid (UDCA) is an active pharmaceutical ingredient (API) used in clinical therapy for the treatment of cholestatic diseases.1 It is a secondary bile acid that, as reported in several papers and reviews, shows several pharmacological effects.2, 3 Particularly, UDCA dissolves cholesterol gallstones, acting as a surfactant.4, 5 In addition, other functions and influences on human physiology have been discovered; for example, UDCA improves the liver function in cholestatic diseases through secretion of glutathione6, 7 and interaction with several receptors,8, 9 and significantly decreases cholesterol saturation in the bile.10, 11 Recently, other studies demonstrated antitumorigenic effects and a lower risk for some cancers.12 In comparison to chenodeoxycholic acid (CDCA), which is also used in therapies, UDCA provides total absence of side effects maintaining a high efficacy, in terms of pharmacological effects.13, 14 This API has been known in Chinese traditional medicine for many years, but natural UDCA can only be obtained by isolation from bear bile, making it very expensive.15 The current viable route for this API consists of chemical transformation of cholic acid (CA) or CDCA. These bile acids are obtained from the cheaper and more available bovine bile that represents, in terms of industrial feasibility, the only source of hydroxysteroids. Currently, CA is chemically transformed into UDCA by a seven‐step chemical synthesis.16, 17 Unfortunately this route, first proposed by Hofmann,16 requires the employment of toxic and dangerous reagents (e.g., hydrazine, Cr2O, and pyridine) and results in large amounts of waste. In addition, it only has an overall yield of about 30 %. To improve the efficiency of UDCA synthesis, alternative chemical routes have been proposed. Dangate et al.,18 for example, achieved the transformation of CA into UDCA (53 % yield using o‐iodobenzoic acid, hydrazine, and metallic sodium in n‐propanol) without the protection and deprotection steps used in the original route. However, this process is still not optimized in terms of costs and environmental impact. UDCA can also be obtained by epimerization of CDCA (intermediate in the synthesis of CA). In the latter case, a viable production route would need to be complemented by a novel synthetic route for CDCA, because of the low natural availability of this compound.19
An emerging technology for the modification of hydroxysteroids is the use of enzymatic transformations.20, 21 The regiospecificity of enzymes offers the possibility to avoid protection steps of the hydroxyl groups not employed in the transformation. This approach was followed by several research groups: attention was focused on the identification of enzymes able to play a role in this synthesis. In an enzymatic process for the production of UDCA from its precursors, several steps have to be performed. These are hydrolysis/deconjugation of glycine and taurine CA derivatives (used as raw starting material), the stereoinversion of the hydroxyl functions (by oxidation and subsequent reduction) and the specific hydroxylation and dehydroxylation of desirable positions in the steroid rings.20 The epimerization reaction (stereoinversion of 7α‐OH group) can be performed by two enzymes (7α‐ and 7β‐hydroxysteroid dehydrogenase (HSDH)): the first enzyme specifically oxidizes the 7α‐OH group to the ketone, which is further reduced by the second enzyme to the corresponding alcohol group with inverted stereochemistry (7β‐OH). HSDHs are enzymes of the class of alcohol dehydrogenases (ADH) that use NAD(P)+ and NAD(P)H as the electron acceptor and donor, respectively. In the literature, several cascades that use these enzymes are reported;22, 23, 24, 25, 26, 27 however, since known enzymes have different cofactor specificities, that is, 7α‐HSDHs are usually NAD+‐dependent and 7β‐HSDHs NADPH‐dependent, the oxidative and reductive reaction steps are decoupled in currently reported procedures (Figure 1—state of art). The cofactors, in this case, are regenerated by the addition of two other enzymes and two sacrificial substrates.21, 22, 23 In this specific case, the reaction is driven towards completion by the addition of sacrificial substrate (in great surplus): Although good yields were achieved (>80 %), large amounts of side products and waste were formed that have to be removed during the downstream processing steps, increasing the overall cost and the environmental burden of UDCA production.
Figure 1.
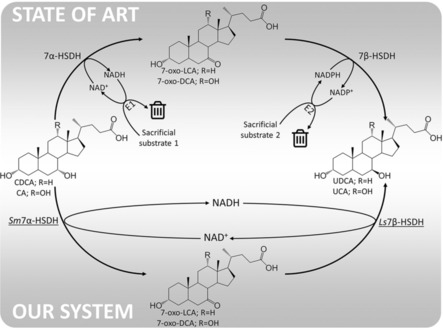
Epimerization of the 7α‐OH group of CA and CDCA. In our work, the NADH produced in the first reaction step (oxidation of 7α‐OH) is reused by a NADH‐dependent 7β‐HSDH giving the 7β‐OH epimer. E1 and E2 are two additional enzymes (i.e., alcohol dehydrogenases) used to regenerate the cofactors converting the sacrificial substrates 1 and 2, respectively, into waste products.
Pedrini et al., already in 2006,28 reported the successful epimerization of CDCA to UDCA by using a catalytic NAD+‐dependent cascade reaction, with two NAD+‐dependent dehydrogenases isolated from Xanthomonas maltophilia CBS 897.97 (recently reclassified as Stenotrophomonas maltophilia). In this way the requirement of external systems for cofactor regeneration was circumvented and UDCA was obtained with a final yield of 75 %. However, the cultivation of this class 2 safety‐level microorganism is complex29 (microaerophilic conditions have to be used) and the 7α‐HSDH and 7β‐HSDH enzymes were only produced in low amounts (4 and 0.2 mg Lculture −1, respectively). In addition, the amino acid or DNA sequences of these enzymes were not reported, making the recombinant expression of these proteins not possible. Since the activity and cofactor specificity is not clear from sequence alone, the univocal identification of the target corresponding gene in genomes databases is not possible. Furthermore, other works23, 24, 30, 31 have been reported on the employment of a NADP+‐dependent redox neutral cascade for CDCA epimerization; however, in comparison to NAD+, the use of NADP+ is nonoptimal because of the higher price, lower stability,32 and lower natural availability. Generally, by the employment of a redox‐neutral cascade, 80 % of UDCA is produced.
In this paper, we report the development of an efficient enzymatic cascade, using well defined and easy to produce enzymes, for the production of UDCA and ursocholic acid (UCA) from CDCA and CA, respectively. A full NAD+‐mediated cascade, that circumvents the need for cofactor regeneration was setup resulting in conversion above 90 %. The key discovery in this paper is the protein engineering of the NADP+‐dependent 7β‐HSDH from Clostridium sardiniense that led to the identification of key residues responsible for NAD/NADP cofactor recognition. With this new sequence at hand, the natural NAD+‐dependent enzyme from Lactobacillus spicheri, was disclosed for the first time as being highly active in the reduction of the 7‐oxo‐derivatives of CA or CDCA to either UCA or UDCA.
Results and Discussion
Protein expression and purification
The genes coding for the Sm7α‐HSDH, Cs7β‐HSDH, and Ls7β‐HSDH were cloned into pET24d(+) plasmid, yielding enzymes containing a C‐terminal 6× His‐tag. The recombinant enzymes were produced in E. coli BL21(DE3) host cells grown at 37 °C in LB medium. Isopropyl β‐D‐1‐thiogalactopyranoside (IPTG) was added at the late exponential phase of growth and the cells were collected after another 18 h of incubation at 25 °C while shaking. The expression level under these conditions of the different proteins is reported in Table 1 and Table S2, Supporting Information. The His‐tagged enzymes were purified by HiTrap chelating chromatography; all of the enzymes were isolated with ≥95 % purity, as judged by SDS‐PAGE analysis (Figure S2, Supporting Information).
Table 1.
Purification of different recombinant His‐tagged HSDHs from E. coli BL21(DE3) cells.
| Protein | Specific activity | Purification | Yield | |
|---|---|---|---|---|
| [mg] | [U mg−1] | [fold] | [%] | |
| Sm7α‐HSDH from 7 g corresponding to 1.0 L of fermentation broth | ||||
| Cell extract | 1050 | 233.2[a] | – | 100.0 |
| HiTrap chelating | 232.3 | 430.4[a] | 1.8 | 40.8 |
| Cs7β‐HSDH from 3.5 g corresponding to 0.5 L of fermentation broth | ||||
| Cell extract | 177 | 0.05[b] | – | 100.0 |
| HiTrap chelating | 10 | 0.74[b] | 14.8 | 83.3 |
| Ls7β‐HSDH from 2.2 g corresponding to 0.3 L of fermentation broth | ||||
| Cell extract | 122 | 1.28[c] | – | 100.0 |
| HiTrap chelating | 29 | 3.10[c] | 2.4 | 57.9 |
[a] Activity was assayed on 1.25 mm CDCA and 2.5 mm NAD+ as a substrate in 50 mm KPi buffer, pH 8.0. [b] Activity was assayed on 1.0 mm UDCA and 2.0 mm NADP+ as a substrate in 50 mm KPi buffer, pH 8.0. [c] Activity was assayed on 1.0 mm UDCA and 2.0 mm NAD+ as substrate in 50 mm KPi buffer, pH 8.0.
Mutagenesis of Cs7β‐HSDH
Since the 3D structure of 7β‐HSDH from C. sardiniense is unknown, a model of the protein was built by using the comparative homology‐modeling server SWISS‐MODEL.33 7β‐HSDH from Collinsella aerofaciens (PDB code 5GT9) was chosen as a template because of the high sequence identity (42 %) with Cs7β‐HSDH.34 The binding mode of the co‐substrate NADP+ in the model of the Cs7β‐HSDH active site was analyzed; the ribose‐bound phosphate group interacts with two arginine residues (R40 and R41) (Figure 2 a).
Figure 2.
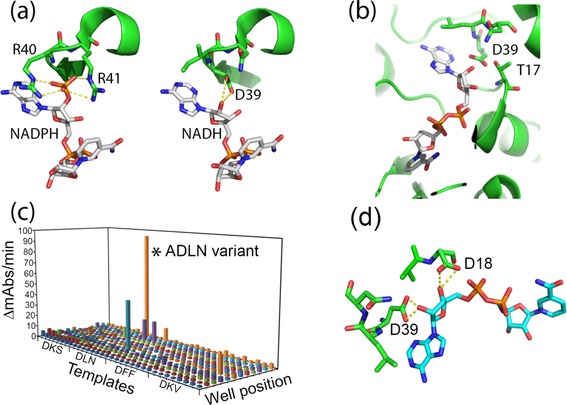
a) Hypothetical binding mode of the cofactor inside the Cs7β‐HSDH active site: on the left) the two arginines R40 and R41 are the main amino acids responsible for recognition and binding of NADPH. The hypothetical NADH binding mode of the ADLN variant (on the right) shows the putative formation of a hydrogen bond between the aspartate side chain (D39) and the 2′‐OH group of ribose (distance 1.8–2.1 Å). b) Interference of T17 with the D39 side chain in the binding of the 2′‐OH group (distance 1.5–1.7 Å). c) Screening results of the second round of SSM on Cs7β‐HSDH libraries. Here, four different templates, isolated during the first round were all subjected to SSM at position 17. The activity of the isolated ADLN variant is indicated with an asterisk. d) Hypothetical binding mode of the ADDLN variant to the NADH: the aspartate in position 18 can form a supplementary hydrogen bond with the 3′‐OH group of the ribose.
On the basis of the in silico analysis, site‐saturation mutagenesis (SSM)35 was performed at positions 39, 40, and 41 by using the QuikChange kit and the wild‐type Cs7β‐HSDH cDNA as a template. In the first round of mutagenesis, an aspartate was specifically introduced at position 39, whereas positions 40 and 41 were more randomly changed. The use of a DHK codon in these positions allows the introduction of 13 different amino acids (A, D, E, F, I, K, L, M, N, S, T, V, and Y) with low repetition, representing different chemical functionalities and steric encumbrances.36 The use of this codon drastically reduces the number of clones that have to be screened. The activity of Cs7β‐HSDH variants on NAD+ as a co‐substrate was screened in a microtiter plate by using a spectrophotometric method (production of NADH, measured at 340 nm) and an automated liquid‐handling system. For the first round of mutagenesis, (G39D, R40X, and R41X), 769 clones were screened, giving a probability of 91 % that every combination of amino acids is measured. The clones most active on NAD+ as identified through the screening procedure were isolated and the substitutions were identified by DNA sequencing.
After the first round of mutagenesis and screening most of the enzyme variants obtained were inactive or had an activity lower than the control activity of wild‐type Cs7β‐HSDH on NAD+ (ca. 1 mU mL−1 under these conditions). However, four Cs7β‐HSDH variants were isolated that showed a clear activity on NAD+: G39D, R40L, R41N (DLN variant); G39D, R40F, R41F (DFF variant); G39D, R40K, R41S (DKS variant); and G39D, R40K, R41V (DKV) variant. From the molecular modeling of these variants it could be observed that the hydroxyl group of threonine at position 17 could interfere with aspartate 39 in the hydrogen bonding of the free OH‐group of ribose (Figure 2 b). A second pair of degenerated primers were designed to mutate that threonine (T17X). For this second round of mutagenesis, all four variants were used as template for the mutagenesis and 380 clones (96 clones for each variant) were screened (99 % coverage). Out of all screened mutants, one variant (T17A, G39D, R40L, R41N—ADLN variant) showed a very high activity towards NAD+ as a cosubstrate in the microtiter plate assay employing cell extract (Figure 2 c).
The ADLN variant was expressed in E. coli BL21(DE3) cells and purified by HiTrap chelating chromatography (>90 % purity). This variant shows an expression yield similar to that of the wild‐type Cs7β‐HSDH (in terms of purified protein/liter of fermentation broth, see Table S2, Supporting Information). The Michaelis–Menten kinetics of all selected enzyme variants were measured using NAD+ or NADP+ as a cofactor.
As shown in Table 2, among the Cs7β‐HSDH variants obtained by SSM, the ADLN was identified because of a thirty‐fold increase in specific activity compared to the wild‐type enzyme.
Table 2.
Kinetic parameters of purified recombinant 7β‐HSDHs on NADP+ and NAD+. The kinetic parameters were determined in the presence of 1.0 mm UDCA. The preference is calculated as the ratio between the catalytic efficiency on the two cofactors.
| NADP+ | NAD+ | |||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| k cat [a] | K m | K i | k cat/K m | Preference | k cat [a] | K m | k cat/K m | Preference | ||||||||||||
| [s−1] | [mmol L−1] | [mmol L−1] | [s−1] | [mmol L−1] | ||||||||||||||||
| wt Cs7β‐HSDH | 2.40 | ± | 0.16 | 0.033 | ± | 0.007 | 10 | ± | 3 | 72.600 | 8354 | 0.023 | ± | 0.001 | 2.62 | ± | 0.21 | 0.009 | 0.0001 | |
| ADLN variant | 0.010 | ± | 0.002 | 2.600 | ± | 0.300 | / | 0.004 | 0.01 | 0.75 | ± | 0.03 | 2.83 | ± | 0.27 | 0.266 | 70 | |||
| ADDLN variant | below detection | 0.08 | ± | 0.01 | 0.33 | ± | 0.02 | 0.234 | / | |||||||||||
| ADDAA variant | below detection | 0.32 | ± | 0.01 | 4.64 | ± | 0.27 | 0.068 | / | |||||||||||
| Ls7β‐HSDH | 0.025 | ± | 0.004 | 10 | ± | 1 | / | 0.002 | 0.0001 | 3.05 | ± | 0.08 | 0.08 | ± | 0.01 | 40.637 | 16 587 | |||
[a] k cat values were calculated considering a MW of 59.4 and 58.8 kDa for Cs7β‐HSDH variants and Ls7β‐HSDH, respectively.
To further increase the activity on NAD+, the T17A, E18D, G39D, R40L, R41N (ADDLN) variant was designed and recombinantly expressed; the substitution of the glutamate with the smaller aspartate at position 18 may result in the formation of a second hydrogen bond between the cofactor and the protein (Figure 2 d). The comparison of the kinetic parameters of the ADLN and ADDLN variants of Cs7β‐HSDH indicates that the addition of a second hydrogen bond increases the affinity of the ADDLN variant for the NAD+ (eight‐fold decrease in K m), see Table 2. However, the specific activity on NAD+ of this variant decreases (10‐fold lower in comparison to the previous isolated one). To restore the specific activity on NAD+ a fourth round of SSM was carried out employing the same primers used in the first round and the ADDLN variant as a template. The T17A, E18D, G39D, R40A, R41A variant (ADDAA) was isolated. The specific activity under standard conditions of this variant is 0.3 U mg−1 (Table 2). Interestingly the affinity for the cofactor is lower than the one observed in the previous variants. However, the expression level of this protein is 15‐fold higher than the wild‐type Cs7β‐HSDH (486 vs. 20 mg Lculture −1), resulting in the higher activity under the screening conditions. Overall, we conclude that ADLN is the best‐obtained variant for our purpose due to its highest catalytic efficiency.
7β‐HSDH from Lactobacillus spicheri
With the new sequence of NADH‐dependent 7β‐HSDH (ADLN and ADDLN variants) in hand, a new alignment search was carried out to find potentially better, previously nonidentified enzymes, able to convert CA and its derivatives. The Ls7β‐HSDH sequence was identified using the Basic Local Alignment Search Tool (BLASTp); the predicted sequence analysis showed a 792 bp ORF corresponding to a protein of 264 amino acid residues.
This enzyme was identified as a putative NADH‐dependent 7β‐HSDH based on the fact that the amino acids relevant to the binding and recognition of NADH are present; specifically, the alanine and aspartate at positions 18 and 19 and the stretch DYS at positions 40–42 are conserved,37 analogous to the situation in analogue Cs7β‐HSDH (Figure 3 b). The predicted M w of 29 kDa and the predicted homodimeric quaternary structure (Figure 3 a) put Ls7β‐HSDH in the short chain dehydrogenase/reductase superfamily.
Figure 3.

3D model of Ls7β‐HSDH built employing the entire structure of 7β‐HSDH from Collinsella aerofaciens (PDB code 5GT9) as a template: the homodimeric quaternary structure (a) is conserved. The hypothetical binding mode of NADH (b) is primary due to the side chains of D19 and D40 to the 3′ and 2′‐OH groups of ribose, respectively.
Biochemical characterization of HSDHs
Sm7α‐HSDH showed a strict NAD+ activity on both CDCA and CA, although the activity on CA is considerably lower (halved). No activity was detected when NADP+ was used as an electron acceptor. Sm7α‐HSDH displayed a K m of 0.22 and 0.96 mm for CDCA and CA, respectively. The Sm7α‐HSDH did not show any substrate inhibition on CA, but a K i of 11 mm on CDCA was measured. The K m value for NAD+ is 0.55 mm (Table 3), which is similar to earlier reported enzymes.
Table 3.
Kinetic parameters of purified recombinant Sm7α‐HSDH.
| k cat [c] | K m | K i | k cat/K m | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| [s−1] | [mmol L−1] | [mmol L−1] | |||||||||||||
| CA[a] | 497.1 | ± | 17.2 | 0.960 | ± | 0.110 | / | 517.8 | ± | 77.2 | |||||
| CDCA[a] | 873.1 | ± | 34.1 | 0.218 | ± | 0.024 | 11 | ± | 1.6 | 4005.2 | ± | 597.5 | |||
| NAD+[b] | 944.1 | ± | 9.1 | 0.560 | ± | 0.017 | / | 1685.8 | ± | 67.4 | |||||
[a] The kinetic parameters were determined in the presence of 2.5 mm NAD+. [b] The kinetic parameters were determined in the presence of 2.0 mm CDCA. [c] k cat values were calculated considering a MW of 107.2 kDa.
The pH and temperature dependence of Sm7α‐HSDH activity was investigated; both the maximum activity and stability occurred at slightly alkaline pH values (Figure 4 a). The enzyme is quite thermophilic, showing an optimum at around 70 °C (Figure S3 a, Supporting Information), and is quite stable; after 24 h incubation at 25 and 37 °C, the enzyme maintained approximately 100 and 70 % of its initial activity, respectively. Incubations at higher temperatures (60 °C) resulted in a complete loss of enzymatic activity. The enzymatic activity of Sm7α‐HSDH was also investigated in the presence of different concentrations of methanol that could be used as a potential co‐solvent in order to increase the solubility of hydroxysteroids in water. The enzyme shows no loss of activity in 10 % methanol and it conserves 90 % of its activity in 20 % methanol (Figure 5 a).
Figure 4.
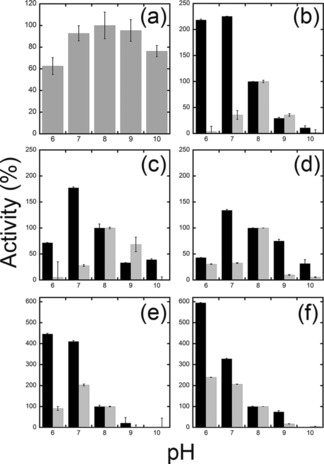
Effect of pH on the oxidative (grey bars) and reductive (black bars) activities of purified a) Sm7α‐HSDH, b) wt Cs7β‐HSDH, c) ADLN, d) ADDLN, and e) ADDAA variants of Cs7β‐HSDH and f) wt Ls7β‐HSDH. The activity at pH 8.0 was taken as 100 %. Enzymatic activities were determined by measuring NAD(P)+ reduction and NAD(P)H oxidation under the same conditions described in materials and methods. Values represent the mean of three independent experiments (mean±standard error).
Figure 5.
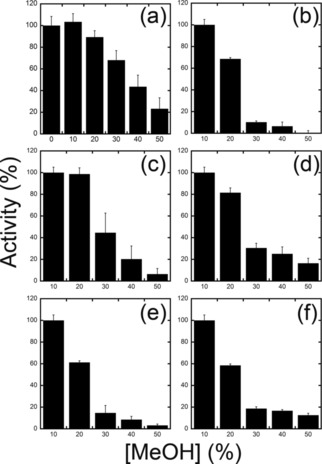
Effect of MeOH concentration on the activities of purified a) Sm7α‐HSDH, b) wt Cs7β‐HSDH, c) ADLN, d) ADDLN, and e) ADDAA variants of Cs7β‐HSDH and f) wt Ls7β‐HSDH. In panel a, the activity in the absence of MeOH was taken as 100 %. Due to the lower solubility of UDCA in water, in all the other panels the activity in the presence of 10 % of MeOH was taken as 100 %. Values represent the mean of three independent experiments (mean±standard error).
On the other hand, wild‐type Cs7β‐HSDH showed a strict NADP+ activity on UDCA (0.74 U mg−1 under standard conditions). The activity on NAD+ is roughly 100‐fold lower showing a K m of 2.6 mm and a specific activity of 0.023 U mg−1. Cs7β‐HSDH displayed a K m of 0.16 mm for UDCA (Table 4). The pH and temperature dependence of Cs7β‐HSDH activity was investigated: both the maximum activity and stability occurred at slightly alkaline pH values (Figure 4 b). Notably, the pH optimum for the reduction reactions was detected at pH 6–7. In the presence of NADPH, this enzyme is able to reduce both 7‐oxo‐DCA and 7‐oxo‐LCA (LCA: lithocholic acid; see Table 4).
Table 4.
Kinetic parameters of purified recombinant 7β‐HSDHs on different substrates.
| UDCA | 7‐oxo‐LCA | 7‐oxo‐DCA | ||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| k cat [a] | K m | K i | k cat/K m | k cat [b] | K m | K i | k cat/K m | k cat [b] | K m | K i | k cat/K m | |||||||||||||||||||||
| [s−1] | [mmol L−1] | [mmol L−1] | [s−1] | [mmol L−1] | [mmol L−1] | [s−1] | [mmol L−1] | [mmol L−1] | ||||||||||||||||||||||||
| wt Cs7β‐HSDH | 3.61 | ± | 0.34 | 0.160 | ± | 0.030 | 1.4 | ± | 0.3 | 22.6 | 1.37 | ± | 0.07 | 0.128 | ± | 0.027 | / | 10.7 | 2.27 | ± | 0.03 | 0.106 | ± | 0.005 | / | 21.3 | ||||||
| ADLN variant | 1.35 | ± | 0.35 | 0.420 | ± | 0.150 | 0.6 | ± | 0.2 | 3.2 | 0.24 | ± | 0.05 | 0.175 | ± | 0.080 | 2.0 | ± | 1.0 | 1.4 | 0.59 | ± | 0.09 | 0.630 | ± | 0.189 | 8.6 | ± | 3.6 | 0.9 | ||
| ADDLN variant | 0.07 | ± | 0.01 | 0.075 | ± | 0.030 | 2.4 | ± | 1.0 | 1.0 | 0.05 | ± | 0.01 | 0.055 | ± | 0.034 | 2.4 | ± | 1.3 | 0.9 | 0.08 | ± | 0.01 | 0.203 | ± | 0.034 | 4.2 | ± | 0.9 | 0.4 | ||
| ADDAA variant | 0.30 | ± | 0.04 | 0.207 | ± | 0.040 | 0.9 | ± | 0.2 | 1.4 | 0.15 | ± | 0.03 | 0.246 | ± | 0.041 | / | 0.6 | 0.18 | ± | 0.01 | 0.323 | ± | 0.070 | / | 0.6 | ||||||
| Ls7β‐HSDH | 11.91 | ± | 2.28 | 0.156 | ± | 0.051 | 0.8 | ± | 0.3 | 76.1 | 7.55 | ± | 0.62 | 0.038 | ± | 0.014 | / | 200.8 | 8.02 | ± | 0.41 | 0.131 | ± | 0.028 | / | 61.0 | ||||||
[a] The kinetic parameters were determined in the presence of 2.0 mm NAD+. [b] The kinetic parameters were determined in the presence of 0.5 mm NADH. The wt Cs7β‐HSDH was characterized by adding NADP instead of the NAD. In all the cases, k cat values were calculated considering a MW of 59.4 and 58.8 kDa for Cs7β‐HSDH variants and Ls7β‐HSDH, respectively.
In general, the enzyme is less thermophilic than the Sm7α‐HSDH, showing an optimum at around 60 °C (Figure S3 b, Supporting Information), and less stable: after 24 h incubation at 25 and 37 °C, Cs7β‐HSDH maintained approximately 85 and 62 % of its initial activity, respectively. Cs7β‐HSDH shows a good tolerance to concentrations of methanol above 10 %, and 70 % of its activity is conserved in the presence of 20 % methanol.
The ADLN, ADDLN, and ADDAA variants showed an increase in activity towards NAD+ and NADH as a cosubstrate, with little change in specificity for the different substrates (Table 4).
Interestingly, the isolated variants showed higher stability than the wild‐type Cs7β‐HSDH. From this comparison, it can be observed that the ADLN variant maintains 95 % of its activity after incubation for 24 h at 25 °C (compared to the 85 % for the wild‐type under the same conditions).
As predicted by in silico analysis, Ls7β‐HSDH showed a strict NAD+ activity (3.10 U mg−1 in standard condition). A weak activity was detected when NADP+ was used as an electron acceptor (Table 2). Ls7β‐HSDH showed a 0.15, 0.04, and 0.13 mm K m for UDCA, 7‐oxo‐LCA, and 7‐oxo‐DCA, respectively. Ls7β‐HSDH is inhibited by UDCA showing a K i of 0.8 mm for this substrate. The K m value for NAD+ is 0.08 mm. From all the 7β‐HSDH‐variants in this paper, this enzyme shows the highest NAD+/NADH activities for both the reduction and the oxidation of 7β‐OH hydroxysteroids and its derivatives. All kinetic data are presented in Tables 2 and 4.
From the biochemical characterization it can be observed that this enzyme showed similar features to the other isolated enzymes, with the exception of the pH dependence; the maximum activity for both oxidation and reduction reactions was observed at lower pH (6–7) (Figure 4 f).
Ls7β‐HSDH is quite thermophilic, showing an optimum at around 70 °C (Figure S3 C, Supporting Information), and it is stable at 25 and 37 °C, maintaining, after 24 h of incubation, approximately 98 and 72 % of its initial activity, respectively. Ls7β‐HSDH conserves 60 % of its activity in the presence of 20 % methanol; thus, for follow‐up bioconversion studies 10 % methanol was used, which limits the amount of substrate that can be loaded in a biotransformation (Figure 5 f).
Bioconversion of CDCA and CA
To assay the applicability of the enzymes described above as a biocatalyst for the production of UDCA and UCA in a batch bioreactor, lab‐scale bioconversions were carried out: 10 μmol of CDCA in the presence of 1 μmol of NAD+ and 10 % MeOH was converted into UDCA (92 % yield) in 200 min by 1 U of Sm7α‐HSDH (0.23 μg) and 0.6 U of Ls7β‐HSDH (190 μg) (Figure 6 a). HPLC analysis showed no formation of undesired products, except for the 7‐oxo‐LCA (0.5 μmol), intermediate of the enzymatic cascade (Figure S4 a, Supporting Information). Under the same conditions, 10 μmol of CA was converted into UCA (91 %) in 120 min (Figure 6 b).
Figure 6.
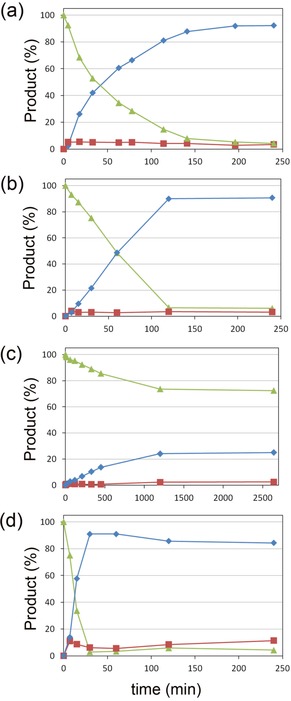
Bioconversion time‐courses of: a) 10 mm CDCA with 1.0 mm NAD+, b) 10 mm CA with 1.0 mm NAD+, c) 10 mm CDCA with 0.2 mm NAD+, employing 1 U of Sm7α‐HSDH (0.23 μg) and 0.6 U of Ls7β‐HSDH (190 μg) in 50 mm KPi, pH 8.0 and 10 % MeOH at 25 °C. d) Bioconversion time‐course of 10 mm CA with 1.0 mm NAD+, employing 1 U of Sm7α‐HSDH (0.23 μg) and 0.6 U of ADLN variant of Cs7β‐HSDH (790 μg) in 50 mm KPi, pH 8.0 and 10 % MeOH at 25 °C. In all cases, the substrates, products, and the corresponding 7‐oxo derivatives are represented by green triangles, blue diamonds, and red squares, respectively.
Surprisingly, the addition of MeOH (10–20 %) resulted in an increase (10 %) of conversion (Table 5): without cosolvent only 80 % of UDCA can be obtained by this biotransformation. The effect of cosolvents on the equilibrium of the epimerization reaction was already discussed in the literature38 but never observed for this specific biotransformation.
Table 5.
Bioconversion results employing different initial concentration of substrate, cofactor, and MeOH. Incubation (1 mL volume) was carried out by employing 1 U of Sm7α‐HSDH (0.23 μg) and 0.6 U of Ls7β‐HSDH (190 μg) in 50 mm KPi, pH 8.0 at 25 °C for 6 h.
| Bioconversion conditions | Conversion [%] | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| [CDCA] [mmol L−1] | [NAD+] [mmol L−1] | [MeOH] [%] | [UDCA] | [7‐oxo‐LCA] | |||||
| 10 | 1 | 0 | 80.4 | ± | 1.9 | 3.5 | ± | 0.7 | |
| 10 | 1 | 10 | 91.3 | ± | 0.5 | 4.7 | ± | 0.6 | |
| 10 | 1 | 20 | 90.2 | ± | 0.2 | 2.5 | ± | 1.0 | |
| 10 | 0.5 | 10 | 92.0 | ± | 0.1 | 1.3 | ± | 0.6 | |
| 10 | 0.2 | 10 | 14.6 (21.1[a]) | ± | 2.9 | 3.0 | ± | 1.3 | |
| 20 | 1 | 10 | 14.0 | ± | 0.0 | 1.1 | ± | 0.1 | |
| 20 | 0.5 | 10 | 19.1 | ± | 2.5 | 1.6 | ± | 0.8 | |
| 50 | 1 | 20 | 0.1 | ± | 0.0 | 0.4 | ± | 0.2 | |
[a] 12 h of incubation.
Additionally, to optimize the reaction conditions, a set of bioconversions were carried out: as reported in Table 5, >90 % of UDCA was observed when employing 1 or 0.5 μmol of NAD+. Biocatalytic conversions were also carried out in the presence of 0.2 μmol of NAD+ by using CDCA as a substrate; in this case, the conversion is slower, yielding 24 % of UDCA in 20 h (Figure 6 c). Notably, here the detected amount of keto intermediate was lower than 1 % (0.1 μmol). Bioconversions were also tested at different pH values (6 and 7) but no improvements were observed (after 150 min in the presence of 1 mm NAD+, 77, 81, and 87 % conversion was observed at pH 6, 7, and 8, respectively). Unfortunately, the increase of substrate loading to 20 mm negatively affects the bioconversion, yielding 20 % of product after 12 h of incubation.
The ADLN variant of Cs7β‐HSDH was also tested in the cascade reaction in the presence of 1 mm NAD+ for the epimerization of CDCA and CA; although, no conversion was observed when CDCA was used as a substrate, 10 μmol of CA was converted into UCA (91 % yield) by 790 μg of enzyme after 30 min (Figure 6 d and Figure S4 b, Supporting Information). This behavior can partially be explained by the kinetic parameters of this variant: K i for the intermediate 7‐oxo‐LCA is four times lower than the one for 7‐oxo‐DCA.
Notably, in the different bioconversions the amount of keto intermediate is limited by the amount of cofactor used. Thus in a typical reaction in which 10 % cofactor was used, 5 % keto intermediate was formed as a byproduct.
Conclusions
Epimerization of CA and its derivatives is a pivotal step in the synthesis of UDCA (see Figure 1). Enzymatic epimerization circumvents the need for protection/deprotection steps and can be carried out with a high yield. A solely NAD+‐dependent cascade for CDCA and CA epimerization, in which NAD+ is present in catalytic amounts, represents a promising method for a more efficient process in terms of atom economy and number of reagents required. To achieve this goal it is fundamental to employ robust, active, and selective enzymes: Sm7α‐HSDH shows kinetic parameters better or comparable to its E. coli or Bacteroides homologues, widely used by researchers in these kinds of processes.22, 27, 39 This enzyme selectively oxidizes the 7α‐OH group of CA and CDCA. In terms of stability it shows promising tolerance to the presence of methanol and elevated temperatures.
Until now, all sequences of 7β‐HSDHs reported in the literature are NADP+‐dependent enzymes.20 However, protein engineering provides a straightforward method to change cofactor specificity; this has been the subject of many pieces of research in both academia and industry over the past few decades.40, 41, 42 Unfortunately, the resulting enzymes usually have a perfect cofactor switch, but in several cases the enzyme activity and/or substrate specificity decreases tremendously.43 A software apparatus for the prediction of the specificity‐determining residues (CSR‐SALAD44, 45) has been developed and is now widely used,46 but there is still no comprehensive solution for cofactor switching.
Following this approach, an iterative mutagenesis of Cs7β‐HSDH was performed. A semirational approach was pursued, based on the identification of the residues responsible for the cofactor recognition discovered in the homology model of the enzyme.
In this way, residues G39, R40, and R41 were identified to be responsible for binding of the phosphate group at the 2′‐position of ribose. Interestingly, the importance of arginine in the binding of NAD(P)+ has been shown previously on the enantiocomplementary 7α‐HSDHs47. The amino acid at position 39 was mutated to an aspartate. Notably, a similar attempt of cofactor switching, performed by Bakonyi and Hummel on the Clostridium difficile 7α‐HSDH,48 has led to the identification of similar variants (A37D). In our case, the free hydroxyl group at 2′‐of ribose forms a hydrogen bond with the aspartate (length 2.2 Å). R40 and R41 residues were randomly mutagenized in order to identify a couple of amino acids that 1) do not interfere with aspartate in NAD+ binding and 2) create a binding pocket for NAD+. SSM with limited codons helped to introduce a certain degree of variability in the protein while generating smart libraries containing <1000 clones for each round.
In the same way, T17 was subjected to SSM. From the 3D structure of the model it was observed that the OH group of the side chain is 2.5 Å from the aspartate. The obtained ADLN variant shows a reasonable activity on NAD+ (in comparison with the wild‐type enzyme on NADP+). The K m of this variant for NAD+ is 2 mm, which is actually close to the value reported by Pedrini et al., for their 7β‐HSDH from S. maltophilia.28 Additionally, the mutation E18D improved the affinity for NAD+ about 10‐fold; however, unfortunately, the specific activity of the protein decreases. We hypothesize that the E18 residue in the Cs7β‐HSDH also has a structural role. From this optimization round we thus concluded that the ADLN variant was the best mutant.
These data suggest that the efficiency and the selectivity in cofactor binding depends, for a large part, on the molecular dynamics of the Cs7β‐HSDH. Although this feature can be reasonably simulated, the engineering of the target enzyme is still complex.49 For this reason, attention was focused on the identification of a wild‐type enzyme that conserved both 1) the structure of Cs7β‐HSDH and 2) the residues identified in Cs7β‐HSDH to be responsible for cofactor recognition. The new analysis led us to identify a sequence of a putative NAD+‐dependent 7β‐HSDH from Lactobacillus spicheri. Ls7β‐HSDH shows indeed a NAD+‐dependent activity (K m 80 μm) and an activity higher than the Cs7β‐HSDH on the respective cofactor. As shown, both the ADLN variant of Cs7β‐HSDH and the Ls7β‐HSDH are suitable for the epimerization of CA (to UCA); however, only the Ls7β‐HSDH is able to epimerize CDCA (to UDCA). This can partially be explained by the comparison of the kinetic parameters of the two enzymes; firstly, the ADLN variant, although it was engineered, shows a K m for the NADH 35‐fold higher than the Lactobacillus one. Secondly, the ADLN variant is inhibited by 7‐oxo‐LCA (K i=2.0 mm). In general, Ls7β‐HSDH is a more interesting biocatalyst because of the high activity on both 7‐oxo derivatives.
Bioconversions were carried out at the substrate concentration used in previous studies (10 mm). This choice was taken in order to 1) have a straight comparison between the reported systems and the current redox neutral cascade and 2) ensure solubility, because solubility of CDCA in a water environment (pH 8.0) is low (around 10 mm as specified by Zheng23).
The developed cascade shows an epimerization yield >90 % for both CA and CDCA under nonoptimized conditions. This shows that the currently developed set of enzymes and the epimerization cascade belonging to it provides a highly promising alternative for both chemical and enzymatic synthetic procedures applied currently.20, 21 This could provide a basis for a much more elegant and atom‐efficient synthesis of UDCA from CDCA.
Since the epimerization of CA and CDCA is carried out with catalytic amounts of NAD+, the shift of the equilibrium depends on the difference in physiochemical properties between reagents and products. Particularly, in a preliminary study using a molecular mechanics approach, we have identified a difference in the ΔG 0 between the two epimers of −1.23 kJ mol−1 (calculated at standard conditions). In addition, the differences in solubility and in critical micelle concentration (CMC) of the two isomers can play a role in the shift of the equilibrium to the products. Similar results were shown by the Dangate et al.18 and Giovannini et al.50 in chemical and chemoenzymatic routes, respectively. Dangate et al. reported that 7‐oxo‐DCA is preferably reduced to the 7β epimer (UCA/CA=80:10 % yield) in the presence of sodium in anhydrous n‐propanol. Under the same conditions, Giovannini et al. showed similar results for the reduction of 7‐oxo‐LCA (UDCA/CDCA 82:15 % yield). In addition, in our case we observed that the reduction towards the 7β epimer is thermodynamically favored; however, in contrast to the chemical cascade, the reversibility of the enzymatic catalyzed reactions allows the thermodynamic equilibrium between the two epimers to be reached. In comparison to the previously reported NADP+‐dependent redox neutral systems we observed a higher conversion yield (>90 vs. 80 %. reported by Zheng et al.23). This increase can be explained by the use of the cosolvent (10 % MeOH) that influences the reaction environment (Table 5) without affecting the activity of the employed enzymes. Another advantage of our system is given by the use of NAD+ instead of NADP+, which lowers the cost of the required cofactor and shows higher stability in solution.
As previously observed by Zheng et al.,23 in all the epimerization reactions, 5 % of keto intermediate (7‐oxo‐DCA or 7‐oxo‐LCA) was obtained: for future studies, since the amount of intermediate is related to the amount of cofactor in solution, lower NAD+ concentrations should be used to decrease this byproduct at the expense of a longer reaction time (e.g., if 0.2 mm NAD+ is used, the amount of intermediate cannot exceed the 2 %). To evaluate the performance of our system, process metrics were calculated: under the conditions described in this study (1 and 0.6 U mL−1 of Sm7α‐HSDH and Ls7β‐HSDH, respectively), the system shows a TTN of 2.9 million and a space‐time yield of 26 g L−1 d−1. Despite a good TTN, the space‐time yield is not yet high enough for industrial application. Further studies, including the use of flow reactors, will be carried out to address the downstream processing aspects of this reaction. This, in combination with solvent engineering and the employment of biphasic systems, both targeted to the increase of substrate loading (and decrease the cofactor loading), could optimize the conditions for the industrial preparation of UDCA.51 In conclusion, we have shown that the epimerization reaction of CA and CDCA can be achieved by a two‐enzyme–one‐cofactor cascade that benefits from favorable thermodynamics for the 7β‐OH isomer. In particular, Sm7α‐HSDH and Ls7β‐HSDH are a compatible, stable set of enzymes and promising candidates as biocatalysts in the synthesis of hydroxysteroid derivatives. Further investigations will be carried out to develop a flow system or a membrane reactor similar to the ones described in literature22, 52 for our newly developed biocatalytic UDCA synthesis procedure and to increase the substrate loading in the process.
Experimental Section
Bacterial strains and materials
Escherichia coli TOP 10 (F− mcrA Δ(mrr‐hsdRMS‐mcrBC) ϕ80lacZΔM15 ΔlacX74 nupG recA1 araD139 Δ(ara‐leu)7697 galE15 galK16 rpsL(StrR) endA1 λ−) and BL21(DE3) (F− ompT gal dcm lon hsdSB(rB − mB −) λ(DE3 [lacI lacUV5‐T7p07 ind1 sam7 nin5]) [malB +]K‐12(λS)) were purchased from Invitrogen (Darmstadt, Germany). Tryptone, yeast extract, cholic acid, ursodeoxycholic acid, and nicotinamide adenine dinucleotide cofactors (NAD(P)+/H) were from Sigma–Aldrich (St. Louis, US). Ursodeoxycholic acid was from Tokio Chemical Industry (TCI) (Tokyo, Japan). Restriction enzymes and Phusion Q‐5 DNA polymerase mastermix were from New England Biolabs (NEB, Ipswich, US). All other reagents were of analytical grade and are commercially available.
Identifying and homology models of 7α‐hydroxysteroid dehydrogenases
The 7α‐hydroxysteroid dehydrogenase from Stenotrophomonas maltophilia (Sm7α‐HSDH) and 7β‐hydroxysteroid dehydrogenase from Lactobacillus spicheri (Ls7β‐HSDH) gene sequences were identified by multiple sequence analysis using BLASTp (NCBI, https://blast.ncbi.nlm.nih.gov): for Sm7α‐HSDH, protein sequences with known activity from Clostridium sardiniense (GenBank: AET80685.1), Clostridium difficile (GenBank: CAJ66880.1, putative), Clostridium sordelli (GenBank: L12058.1), Eubacterium sp. VPI 12708 (GenBank: M58473.1), Bacteroides fragilis (GenBank: AF173833.2), and Escherichia coli (Gene ID: ACI83195.1) were used as queries, restricting the organisms list to Stenotrophomonas maltophilia strains (taxid:40324). A multiple sequence analysis of all 7α‐HSDH is shown in the Supporting Information, Figure S1 a.
A 3D structure model of this enzyme was obtained using SWISS‐MODEL (https://swissmodel.expasy.org/interactive), employing the crystal structure of the 7α‐HSDH from Escherichia coli (PDB ID: 1AHI.1) as a template.
For Ls7β‐HSDH, protein sequences from Clostridium sardiniense (GenBank: AET80684.1), Ruminococcus gnavus (Genbank: WP004843516.1), Ruminococcus torques (GenBank: WP015528793.1) and Collinsella aerofaciens (GenBank: WP006236005.1) together with the variant of the Cs7β‐HSDH obtained (see below) were used as queries. A multiple sequence analysis of all 7β‐HSDH is shown in the Supporting Information Figure S1 b.
The 3D models of Cs7β‐HSDH (GenBank: AET80684.1) and Ls7β‐HSDH were built using the same server employing the entire structure of 7β‐HSDH from Collinsella aerofaciens (PDB code 5GT9) as template.
Cloning and expression of Sm7α‐HSDH, Cs7β‐HSDH, and Ls7β‐HSDH
The synthetic cDNAs encoding the Sm7α‐HSDH, Cs7β‐HSDH, and Ls7β‐HSDH were designed by in silico back translation of the amino acid sequences (GenBank: KRG42928.1, AET80684.1, and WP045806907, respectively). To subclone into the pET24d(+) plasmid (Merck Millipore, Burlington, US), sequences corresponding to NcoI (CCATGG) and XhoI (CTCGAG) restriction sites were added at the 5′‐ and 3′‐ends of the cDNAs, respectively. The codon usage of the synthetic genes was optimized for expression in E. coli and produced by BaseClear B.V. (Leiden, The Netherlands). Sm7α‐HSDH, Cs7β‐HSDH, and Ls7β‐HSDH cDNAs were cloned into the pET24d(+) vector using the NcoI and XhoI sites, resulting in a 6.0‐, 6.0‐, and 6.1‐kb construct (pET24‐Sm7α‐HSDH, pET24‐Cs7β‐HSDH and pET24‐Ls7β‐HSDH). The genes were cloned in frame with the C‐terminal 6‐Histidines tag of the vector. The obtained expression plasmids were then used to transform BL21(DE3) E. coli cells.
Starting cultures (100 mL) were prepared from a single recombinant BL21(DE3) E. coli colony grown in LB medium containing kanamycin (30 μg mL−1), under vigorous shaking (200 rpm) at 37 °C. These cultures were diluted to a starting OD600nm of 0.1 in 1 L of LB medium (LB, 10 g L−1 bactotryptone, 10 g L−1 NaCl and 5 g L−1 yeast extract) and then incubated at 37 °C on a rotatory shaker at 200 rpm until an OD600nm of 1.0 was reached. Protein expression was induced by adding 0.25 mm IPTG: cultures were grown for another 12 h at 25 °C with shaking (200 rpm). Cells were harvested by centrifugation at 10 000 g for 10 min at 4 °C, washed with 50 mm KPi buffer pH 8.0 and stored at −20 °C for at least 1 day before purification.
Protein purification
E. coli cell pellets were resuspended in lysis buffer (50 mm KPi buffer, 1 m NaCl, 5 % glycerol (v/v) and 10 μg mL−1 DNAse, pH 8.0) and disrupted by French press (Constant Systems Limited, Low March, UK) (2 cycles, 180 psi). The insoluble fraction of the lysates was removed by centrifugation at 39 000 g for 30 min at 4 °C. Crude extract was loaded onto a HiTrap chelating affinity column (GE Healthcare, Little Chalfont, UK), previously loaded with Ni2+ metal ions and equilibrated with 50 mm KPi buffer, 1 m NaCl and 5 % glycerol (v/v), pH 8.0. The columns were washed with this buffer until the absorbance value at 280 nm was that of the buffer and the bound proteins were eluted with 50 mm KPi buffer, 250 mm imidazole, and 5 % glycerol (v/v), pH 8.0. The fractions containing the desired activity were dialyzed overnight against 50 mm KPi buffer and 5 % glycerol (v/v), pH 8.0, using a 3‐kDa dialysis tube. During the purification procedure, 7α‐HSDH and 7β‐HSDH activities were assayed by using the standard activity assay (see below).
Activity and kinetic measurements
7α‐HSDH′s enzymatic activity in the crude extract and of the purified enzyme was determined at 25 °C using 1.0 mm CDCA, 2.0 mm NAD+, in 50 mm KPi buffer and 10 % methanol (v/v), pH 8.0. 7β‐HSDH′s enzymatic activity was determined at room temperature (25 °C) using 1.0 mm UDCA, 2.0 mm NAD(P)+, in 50 mm KPi buffer and 10 % methanol (v/v), pH 8.0. The production or the consumption of NAD(P)H was followed at 340 nm (extinction coefficient of NAD(P)H is 6220 L mol−1 cm−1). One unit (U) was defined as the amount of enzyme producing 1 μmol of product per minute at 25 °C and at pH 8.0. Blank measurements were performed in absence of CDCA or UDCA, NAD+, and enzyme.
The kinetic parameters of the purified samples were determined at room temperature in the presence of different concentrations of substrates (5–10 000 μm), 2.0 mm NAD(P)+ in 50 mm KPi buffer and 10 % methanol (v/v), pH 8.0, at 25 °C; different concentrations of NAD(P)+ (1–5000 μm), 2.0 mm CDCA (for 7α‐HSDH) or UDCA (for 7β‐HSDH) in 50 mm KPi buffer and 10 % methanol (v/v), pH 8.0, at 25 °C. The specific activity was expressed as unit per mg of protein (determined by spectrophotometric analysis at 280 nm). The kinetic data were fitted to the Michaelis–Menten equation, or to the one modified to account for substrate inhibition when necessary.
The effect of pH on the enzymatic activities was determined by using 1.0 mm CDCA (for 7α‐HSDH) or UDCA (for 7β‐HSDH), 2.0 mm NAD(P)+, in 100 mm citrate‐phosphate buffer (66 mm citrate, 34 mm Na2HPO4) and 10 % methanol (v/v), in the 3.0–10.0 pH range.
The effect of methanol concentration on the enzymes activity toward CDCA and UDCA was determined using 1.0 mm CDCA (for 7α‐HSDH) or UDCA (for 7β‐HSDH), 2.0 mm NAD(P)+ in 50 mm KPi buffer and different concentrations of methanol (0–50 % (v/v)), pH 8.0, at 25 °C.
Temperature dependence of the enzymatic activities was determined using 1.0 mm CDCA (for 7α‐HSDH) or UDCA (for 7β‐HSDH), 2.0 mm NAD+ in 50 KPi buffer and 10.0 % methanol (v/v), pH 8.0 in the 18–95 °C temperature range.
Enzymatic stability was measured by incubating the enzyme solution in 100 mm citrate–phosphate buffer (66 mm citrate, 34 mm Na2HPO4) in the 3.0–9.0 pH range at 25 °C,53 in 50 mm KPi buffer with different concentrations of methanol (0–50 % (v/v)) at pH 8.0 at 25 °C and in 50 mm KPi buffer, at pH 8.0 at different temperatures: samples were withdrawn at different times and residual activity was determined using the enzymatic activity assay.
SDS‐PAGE
Proteins from crude extract and the purified enzyme fractions were separated by SDS‐PAGE on a 12 % polyacrylamide resolving gel (BioRAD, Hercules, US). Samples were re‐suspended in an appropriate volume of Laemmli sample buffer and boiled. Proteins were visualized by staining with SimplyBlue safe stain (Novex, Carlsbed, US).
Site‐saturation mutagenesis (SSM) and screening NAD+‐dependent enzyme variants
SSM was carried out at different amino acid positions of Cs7β‐HSDH by using whole plasmid PCR;54 pET24d‐Cs7β‐HSDH vector as a template and a set of degenerated synthetic oligonucleotides was employed to prepare the mutant libraries (Table S1, Supporting Information). PCRs were carried out in a final volume of 20 μL. In all cases, 1 μL of template DNA (ca. 25 ng μL−1), 0.5 μL of each primer (final concentration 0.25 μm), 10 μL of Phusion Q‐5 DNA polymerase mastermix and 8 μL of MilliQ water were added in sterile PCR tubes. After an initial denaturation step (98 °C for 1 min), the reaction was carried out for 30 cycles (denaturation at 98 °C for 30 s, annealing at 58 °C for 30 s and elongation at 72 °C for 7 min). A final elongation step (72 °C for 7 min.) was added. The template DNA was eliminated by enzymatic digestion with 1 μL DpnI restriction enzyme at 37 °C for 2 h; the PCR products were used to transform E. coli TOP10 cells. Subsequently, the recombinant plasmids were transferred to E. coli BL21(DE3) cells, and these clones were used for the screening procedure. The introduction of the mutations was confirmed by automated DNA sequencing. The mutant libraries obtained from SSM were screened by means of a rapid colorimetric assay based on the reduction on NAD+ (as described before) and by means of an automated JANUS Liquid Handler Workstations (Perking Elmer, Waltham, US). To a saturated E. coli culture (1 mL, growth in 2 mL DeepWell plate) 0.250 mm IPTG were added and the culture was then incubated at 25 °C for 18 h. The culture was centrifuged at 5000 g for 2 min, and the cell pellet was resuspended in 50 mm KPi buffer, pH 8.0 (200 μL) containing 1 mg mL−1 of lysozyme. Cell lysis was performed incubating the plate for 30 min at 37 °C, 200 rpm. The crude extracts were centrifuged at 5000 g for 30 min and then 50 μL of the supernatant was transferred to a 96‐well flat‐bottom plate. The activity was assayed on the crude extract by adding 150 μL of 1.33X substrate solution (1.33 mm UDCA, 2.66 mm NAD+, 13.3 % MeOH in 50 mm KPi buffer, pH 8.0).
The initial activity was determined by measuring the increase of the absorbance at 340 nm for 5 min at 25 °C in a microtiter plate reader (Synergy2, Biotek, Winoosky, US) and compared with cultures expressing the wild‐type Cs7β‐HSDH and untransformed cells as controls. The selected variants were sequenced and biochemically characterized.
Preparation of 7‐oxo‐DCA, 7‐oxo‐LCA, UCA, and UDCA
In a 2 L round‐bottomed flask CA (2.1 g), NAD+ (133 mg), and oxalacetate (1.98 g) were dissolved in 50 mm KPi buffer, pH 8.0 with 10 % MeOH (final volume: 1 L). The reaction was initiated by adding Sm7α‐HSDH (5100 Utot; 15 mg) and malate dehydrogenase (200 Utot; Sigma–Aldrich, St. Louis, US).
The reaction was gently stirred at 25 °C. Conversion was checked with HPLC. At completion of the reaction (ca. 1 h) the solution was acidified with HCl till pH 2.0 was reached, leading to the formation of a white suspension. To increase the precipitation, NaCl (50 g) was added and dissolved. The product was filtered with a porous glass filter and washed with 0.01 m HCl solution (20 mL). The powder was then dried and crystallized in MeOH, giving 7‐oxo‐DCA. Yield: 1.9 g (95 %); purity: 98 % (assayed by HPLC analysis); 13C NMR (100 MHz, DMSO): δ=211.75 (C7=O), 175.15 (COOH), 70.26 (C12α‐OH), 69.33 ppm (C3α‐OH); full 1H and 13C NMR spectra are reported in Supporting Information.
The same procedure was used for the preparation of 7‐oxo‐LCA from CDCA (1.9 g). Yield: 1.6 g (84 %); purity: 98 % (assayed by HPLC analysis); 13C NMR (100 MHz, DMSO): δ=211.40 (C7=O), 174.86 (COOH), 69.09 ppm (C3α‐OH); full 1H and 13C NMR spectra are reported in the Supporting Information.
UCA and UDCA were obtained by using the enzymatic cascade reported herein starting from CA and CDCA, respectively: CA (0.55 g) or CDCA (0.51 g) and NAD+ (86 mg) were dissolved in 50 mm KPi buffer, pH 8.0 with 10 % MeOH (final volume: 130 mL). The reaction was initiated by adding Sm7α‐HSDH (63 Utot; 0.18 mg) and Ls7β‐HSDH (39 Utot; 12.54 mg). The reaction was gently stirred at 25 °C. After 24 h, the reaction was complete (92 and 91 % conversion of CA into UCA and of CDCA into UDCA, respectively). The reaction was acidified with HCl (final pH 3.0) and extracted twice with 50 mL of diethyl ether. UDCA and UCA were purified by Reveleris X2 apparatus (GRACE, Columbia, US) equipped with C18 cartridge (40 μm, 12 g) employing water (TFA 0.1 %)/acetonitrile gradient. UCA: Yield: 478 mg (87 %); purity: >99 % (assayed by HPLC analysis); 13C NMR (100 MHz, DMSO): δ=174.98 (COOH), 70.66 (C7β‐OH), 69.83 (C12α‐OH), 69.56 ppm (C3α‐OH). UDCA: yield: 438 mg (86 %); purity: >99 % (assayed by HPLC analysis); 13C NMR (100 MHz, DMSO): δ=174.90 (COOH), 69.74 (C7β‐OH), 69.46 ppm (C3α‐OH). Full 1H and 13C NMR spectra are reported in the Supporting Information.
1H and 13C NMR spectra, in agreement with previous reports,18, 29, 50 were acquired with Agilent ProPulse 400 MHz (Santa Clara, US) and are reported in the Supporting Information.
Molecular mass of 7‐oxo‐DCA, 7‐oxo‐LCA, UCA, and UDCA were confirmed using an RP‐ESI‐Q‐TOF‐MS system (M‐Class and Q‐TOF Premier, Waters, UK), which was operated in positive ionization mode (ES+). Mass calibration was performed using [Glu 1]‐Fibrinopeptide B. Data were analyzed using the MassLynx 4.1 tool box. The MS analyses are reported in the Supporting Information.
Bioconversion of CDCA to UDCA
All bioconversions were carried out employing 1 Utot of purified Sm7α‐HSDS and 0.6 Utot of purified 7β‐HSDS on 10 mm (if not differently specified) of CDCA or CA, NAD+ (0.2, 0.5, or 1.0 mm). As a general procedure, 1 mL of reaction mixture containing 10 % MeOH and 50 mm KPi buffer, pH 8.0, was incubated at 25 °C: at fixed times, 50 μL of reaction mixture were withdrawn, diluted with 250 μL of MeOH and centrifuged at 14 000 g for 2 min. 10 μL of the obtained samples were analyzed by HPLC. HPLC analyses were performed on a Shimadzu (Kyoto, Japan) apparatus equipped with a LC20AT pump and an ELSD‐LTII detector and fitted with a XTerra RP C18 column (Waters, Milford, US) (length/internal diameter 150/4.6 mm, pore size 5 μm) under the following conditions: for analyses of CA, 7‐oxo‐DCA and UCA, eluent: H2O/CH3CN/TFA (70:30:0.1), flow 1.0 mL min−1. Retention times CA=6.75 min, 7‐oxo‐DCA=4.82 min, UCA=4.03 min. For analyses of CDCA, 7‐oxo‐LCA and UDCA, eluent: H2O/CH3CN/TFA (65:35:0.1), flow 1.0 mL min−1. Retention times CDCA=4.11 min, 7‐oxo‐LCA=3.49 min, UDCA=3.21 min.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
This work was carried out as part of the ONE‐FLOW project (https://one‐flow.org) that has received funding from the European Union's FET‐Open research and innovation actions (proposal 737266—ONE‐FLOW). We thank Dr. M. Pabst and P. van Dam for LCMS measurements and Prof. U. Hanefeld, Dr. F. Hollmann, Dr. D. McMillan, and all the members of the biocatalysis section of TU Delft for the scientific discussions.
F. Tonin, L. G. Otten, I. W. C. E. Arends, ChemSusChem 2019, 12, 3192.
References
- 1. Ikegami T., Matsuzaki Y., Hepatol. Res. 2008, 38, 123–131. [DOI] [PubMed] [Google Scholar]
- 2. Chen T., Duncan S., Solanki S., Haq K. F., Abdul M. K. M., Soufleris S., Shrestha S., Khan Z., Kamal F., Khan M. A., Gastroenterology 2018, 154, S-782. [Google Scholar]
- 3. Reardon J., Hussaini T., Alsahafi M., Azalgara V. M., Erb S. R., Partovi N., Yoshida E. M., J. Clin. Transl. Hepatol. 2016, 4, 192–205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Crosignani A., Battezzati P. M., Setchell K. D., Invernizzi P., Covini G., Zuin M., Podda M., Dig. Dis. Sci. 1996, 41, 809–815. [DOI] [PubMed] [Google Scholar]
- 5. Salen G., Colalillo A., Verga D., Bagan E., Tint G., Shefer S., Gastroenterology 1980, 78, 1412–1418. [PubMed] [Google Scholar]
- 6. Arisawa S., Ishida K., Kameyama N., Ueyama J., Hattori A., Tatsumi Y., Hayashi H., Yano M., Hayashi K., Katano Y., Biochem. Pharmacol. 2009, 77, 858–866. [DOI] [PubMed] [Google Scholar]
- 7. Pemberton P. W., Aboutwerat A., Smith A., Warnes T. W., Redox Rep. 2006, 11, 117–123. [DOI] [PubMed] [Google Scholar]
- 8. Amaral J. D., Viana R. J., Ramalho R. M., Steer C. J., Rodrigues C. M., J. Lipid Res. 2009, 50, 1721–1734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Amaral J. D., Castro R. E., Solá S., Steer C. J., Rodrigues C. M., J. Biol. Chem. 2007, 282, 34250–34259. [DOI] [PubMed] [Google Scholar]
- 10. Stiehl A., Raedsch R., Czygan P., Götz R., Männer C., Walker S., Kommerell B., Gastroenterology 1980, 79, 1192–1198. [PubMed] [Google Scholar]
- 11. Stiehl A., Czygan P., Kommerell B., Weis H., Holtermüller K., Gastroenterology 1978, 75, 1016–1020. [PubMed] [Google Scholar]
- 12. Akare S., Jean-Louis S., Chen W., Wood D. J., Powell A. A., Martinez J. D., Int. J. Cancer 2006, 119, 2958–2969. [DOI] [PubMed] [Google Scholar]
- 13. Roda E., Bazzoli F., Labate A. M. M., Mazzella G., Roda A., Sama C., Festi D., Aldini R., Taroni F., Barbara L., Hepatology 1982, 2, 804–810. [DOI] [PubMed] [Google Scholar]
- 14. Nilsell K., Angelin B., Leijd B., Einarsson K., Gastroenterology 1983, 85, 1248–1256. [PubMed] [Google Scholar]
- 15. Feng Y., Siu K., Wang N., Ng K.-M., Tsao S.-W., Nagamatsu T., Tong Y., J. Ethnopharmacol. 2009, 5, 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Hofmann A. F., Acta Chem. Scand. 1963, 17, 173–186. [Google Scholar]
- 17. Kanazawa T., Shimazaki A., Sato T., Hoshino T., Nippon Kagaku Zasshi. 1955, 76, 297–301. [Google Scholar]
- 18. Dangate P. S., Salunke C. L., Akamanchi K. G., Steroids 2011, 76, 1397–1399. [DOI] [PubMed] [Google Scholar]
- 19. Washizu T., Tomoda I., Kaneko J. J., J. Vet. Med. Sci. 1991, 53, 81–86. [DOI] [PubMed] [Google Scholar]
- 20. Tonin F., Arends I. W. C. E., Beilstein J. Org. Chem. 2018, 14, 470–483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Eggert T., Bakonyi D., Hummel W., J. Biotechnol. 2014, 191, 11–21. [DOI] [PubMed] [Google Scholar]
- 22. Zheng M. M., Chen F. F., Li H., Li C. X., Xu J. H., ChemBioChem 2018, 19, 347–353. [DOI] [PubMed] [Google Scholar]
- 23. Zheng M.-M., Wang R.-F., Li C.-X., Xu J.-H., Process Biochem. 2015, 50, 598–604. [Google Scholar]
- 24. Ji Q., Tan J., Zhu L., Lou D., Wang B., Biochem. Eng. J. 2016, 105, 1–9. [Google Scholar]
- 25. Bovara R., Carrea G., Riva S., Secundo F., Biotechnol. Lett. 1996, 18, 305–308. [Google Scholar]
- 26. Bovara R., Canzi E., Carrea G., Pilotti A., Riva S., J. Org. Chem. 1993, 58, 499–501. [Google Scholar]
- 27. Monti D., Ferrandi E. E., Zanellato I., Hua L., Polentini F., Carrea G., Riva S., Adv. Synth. Catal. 2009, 351, 1303–1311. [Google Scholar]
- 28. Pedrini P., Andreotti E., Guerrini A., Dean M., Fantin G., Giovannini P. P., Steroids 2006, 71, 189–198. [DOI] [PubMed] [Google Scholar]
- 29. Medici A., Pedrini P., Bianchini E., Fantin G., Guerrini A., Natalini B., Pellicciari R., Steroids 2002, 67, 51–56. [DOI] [PubMed] [Google Scholar]
- 30. Shi J., Wang J., Yu L., Yang L., Zhao S., Wang Z., J. Ind. Microbiol. Biotechnol. 2017, 44, 1073–1082. [DOI] [PubMed] [Google Scholar]
- 31. Yang Q., Wang B., Zhang Z., Lou D., Tan J., Zhu L., RSC Adv. 2017, 7, 38028–38036. [Google Scholar]
- 32. Wu J. T., Wu L. H., Knight J. A., Clin. Chem. 1986, 32, 314–319. [PubMed] [Google Scholar]
- 33. Guex N., Peitsch M. C., Electrophoresis 1997, 18, 2714–2723. [DOI] [PubMed] [Google Scholar]
- 34. Wang R., Wu J., Jin D. K., Chen Y., Lv Z., Chen Q., Miao Q., Huo X., Wang F., Acta Crystallogr. Sect. F 2017, 73, 246–252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Reetz M. T., Carballeira J. D., Nat. Protoc. 2007, 2, 891–903. [DOI] [PubMed] [Google Scholar]
- 36. Kille S., Acevedo-Rocha C. G., Parra L. P., Zhang Z.-G., Opperman D. J., Reetz M. T., Acevedo J. P., ACS Synth. Biol. 2013, 2, 83–92. [DOI] [PubMed] [Google Scholar]
- 37. Savino S., Ferrandi E. E., Forneris F., Rovida S., Riva S., Monti D., Mattevi A., Proteins Struct. Funct. Bioinf. 2016, 84, 859–865. [DOI] [PubMed] [Google Scholar]
- 38. Held C., Sadowski G., Annu. Rev. Chem. Biomol. Eng. 2016, 7, 395–414. [DOI] [PubMed] [Google Scholar]
- 39. Macdonald I., Rochon Y., Hutchison D., Holdeman L., Appl. Environ. Microbiol. 1982, 44, 1187–1195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Machielsen R., Looger L. L., Raedts J., Dijkhuizen S., Hummel W., Hennemann H. G., Daussmann T., Van der Oost J., Eng. Life Sci. 2009, 9, 38–44. [Google Scholar]
- 41. Richter N., Zienert A., Hummel W., Eng. Life Sci. 2011, 11, 26–36. [Google Scholar]
- 42. Xu X., Chen J., Wang Q., Duan C., Li Y., Wang R., Yang S., ChemBioChem 2016, 17, 56–64. [DOI] [PubMed] [Google Scholar]
- 43. Chánique A. M., Parra L. P., Front. Microbiol. 2018, 9, 194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Cahn J. K., Werlang C. A., Baumschlager A., Brinkmann-Chen S., Mayo S. L., Arnold F. H., ACS Synth. Biol. 2017, 6, 326–333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Cahn J. K., Brinkmann-Chen S., Arnold F. H. in Synthetic Methabolic Pathways: Methods and Protocols (Eds.: M. K. Jensen, J. D. Keasling), Springer New York, New York, NY, 2018, pp. 15–26. [Google Scholar]
- 46. Borlinghaus N., Nestl B. M., ChemCatChem 2018, 10, 183–187. [Google Scholar]
- 47. Lou D., Wang Y., Tan J., Zhu L., Ji S., Wang B., Comput. Biol. Chem. 2017, 70, 89–95. [DOI] [PubMed] [Google Scholar]
- 48. Bakonyi D., Hummel W., Enzyme Microb. Technol. 2017, 99, 16–24. [DOI] [PubMed] [Google Scholar]
- 49. Pabis A., Risso V. A., Sanchez-Ruiz J. M., Kamerlin S. C., Curr. Opin. Struct. Biol. 2018, 48, 83–92. [DOI] [PubMed] [Google Scholar]
- 50. Giovannini P. P., Grandini A., Perrone D., Pedrini P., Fantin G., Fogagnolo M., Steroids 2008, 73, 1385–1390. [DOI] [PubMed] [Google Scholar]
- 51. Hessel V., Chem. Int. 2018, 40, 12–16. [Google Scholar]
- 52. Carrea G., Pilotti A., Riva S., Canzi E., Ferrari A., Biotechnol. Lett. 1992, 14, 1131–1134. [Google Scholar]
- 53. Tonin F., Melis R., Cordes A., Sanchez-Amat A., Pollegioni L., Rosini E., New Biotechnol. 2016, 33, 387–398. [DOI] [PubMed] [Google Scholar]
- 54. Siloto R. M., Weselake R. J., Biocatal. Agric. Biotechnol. 2012, 1, 181–189. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary