

Abstract
Familial hemiplegic migraine type 2 (FHM2) is associated with inherited point-mutations in the Na,K-ATPase α2 isoform, including G301R mutation. We hypothesized that this mutation affects specific aspects of vascular function, and thus compared cerebral and systemic arteries from heterozygote mice bearing the G301R mutation (Atp1a2+/−G301R) with wild type (WT). Middle cerebral (MCA) and mesenteric small artery (MSA) function was compared in an isometric myograph. Cerebral blood flow was assessed with Laser speckle analysis. Intracellular Ca2+ and membrane potential were measured simultaneously. Protein expression was semi-quantified by immunohistochemistry. Protein phosphorylation was analysed by Western blot. MSA from Atp1a2+/−G301R and WT showed similar contractile responses. The Atp1a2+/−G301R MCA constricted stronger to U46619, endothelin and potassium compared to WT. This was associated with an increased depolarization, although the Ca2+ change was smaller than in WT. The enhanced constriction of Atp1a2+/−G301R MCA was associated with increased cSrc activation, stronger sensitization to [Ca2+]i and increased MYPT1 phosphorylation. These differences were abolished by cSrc inhibition. Atp1a2+/−G301R mice had reduced resting blood flow through MCA in comparison with WT mice. FHM2-associated mutation leads to elevated contractility of MCA due to sensitization of the contractile machinery to Ca2+, which is mediated via Na,K-ATPase/Src-kinase/MYPT1 signalling.
Keywords: Familial hemiplegic migraine type 2, hypoperfusion, Na, K-ATPase, smooth muscle sensitization to Ca2+, tyrosine phosphorylation
Introduction
Familial hemiplegic migraine type 2 (FHM2) is an autosomal dominant form of migraine caused by mutations of the Na,K-ATPase α2 isoform.1,2 FHM2 is a ‘classic’ migraine with aura, which is linked to a propagating wave of neuronal depolarization similar to cortical spreading depression3 followed with long-lasting depression.4 This spreading depolarization is often reported to cause long-lasting oligemia.5 Although this response is often biphasic and highly variable,3 recent studies of FHM2 patients with prolonged aura suggested initial multifocal hypoperfusion in the affected hemisphere.6,7 This initial hypoperfusion is followed by hyperperfusion and is in line with the reports on cerebral blood flow changes during migraine attack over the last ∼30 years.8 The mechanism by which spreading depression leads to changes in cerebral blood flow is not fully understood.
A broad spectrum of functional abnormalities of the Na,K-ATPase α2 isoform have been identified for more than 80 FHM2-associated mutations.2 These mutations lead to changes in the enzymes properties, and may cause haploinsufficiency.1,2 In contrast to the ubiquitously expressed α1 isoform that serves a housekeeping role for Na+/K+ homeostasis, the α2 isoform is characterized by a tissue-specific expression and is suggested to be involved in a number of signalling pathways.9 In the brain, the α2 isoform is mostly expressed in glia and the vasculature. Since glia is an important modulator of neuronal activity, attention was previously given to glia in studies of the mechanisms important in FHM2 with limited interest in vascular (dys)function.1,2,10
Importantly, the vascular Na,K-ATPase α2 isoform has been suggested to control [Ca2+]i signalling by forming a signalosome with other Ca2+ transport proteins such as the Na,Ca-exchanger (NCX).11 In accordance with this model, potentiation of [Ca2+]i signalling upon α2 isoform inhibition is suggested to be responsible for the pro-contractile action of ouabain in the vasculature.12 However, another Na,K-ATPase inhibitor, digoxin does not potentiate vascular contraction,13,14 suggesting that the Na,K-ATPase-dependent modulation of arterial contractility cannot be limited to a direct effect on ion homeostasis.9,12 The Na,K-ATPase has previously been shown to initiate several signalling pathways including cSrc kinase signaling.15 This is supported by Y418 phosphorylation-dependent activation of cSrc kinase upon ouabain application,16–22 although the mechanism for this cSrc activation is debated.23–25 Of note, acute application of ouabain can activate a cSrc-dependent pathway in rat mesenteric arteries, and modulate intercellular communication in the vascular wall.26 Chronic ouabain treatment also activates cSrc in rat mesenteric arteries.14 Src kinase family signalling plays an important role in cerebrovascular myogenic contraction27–29 and vasospasm,30 although a detailed mechanistic understanding is lacking.
Mice bearing one of the mutations found in FHM2 patients, i.e. G301R,31 were recently generated and their behavioural and neuronal properties characterized.32 Homozygous knock-in mutant mice die after birth, while heterozygous mice (Atp1a2+/−G301R) demonstrate FHM2-relevant disease traits, including mood depression, stress-induced anhedonia, obsessive-compulsive disorder and increased acoustic startle response.32 Moreover, electrocorticographic recordings in vivo showed that regeneration after induction of cortical spreading depression was significantly reduced in brains from Atp1a2+/−G301R mice, suggesting that the spontaneous activity is more likely affected in these mice than the evoked activity.32
In the current study, it is hypothesized that these Atp1a2+/−G301R mice have abnormal cerebrovascular function, affecting cerebral blood flow. The Atp1a2+/−G301R mice were found to have increased vascular contractility specific for cerebral compared to systemic mesenteric arteries. Accordingly, resting cerebral blood flow was reduced in these mice. This hypercontractility is suggested to be a factor behind the mechanism of aura-associated regional hypoperfusion in the brain, linked to the significance of cSrc-dependent sensitization of vascular smooth muscle cells to Ca2+.
Methods
A detailed ‘Methods’ is available in the Supplementary Information file.
All experiments conformed to guidelines from the European Convention for the Protection of Vertebrate Animals used for Experimental and other Scientific Purposes and were approved by and conducted with permission from the Animal Experiments Inspectorate of the Danish Ministry of Environment and Food. Animal experiments were reported in accordance with the ARRIVE (Animal Research: Reporting in Vivo Experiments) guidelines (www.nc3rs.org.uk/arrive-guidelines).
Atp1a2+/−G301R mice
The Atp1a2+/−G301R mice were generated by introduction of the G301R mutation in Atp1a2 gene, as described previously.32 Homozygote pups died immediately after birth, resembling the lethal phenotype previously reported for other genetic disturbances in the Atp1a2 gene.1,2,33 Heterozygous Atp1a2+/−G301R (n = 82) and wild type (WT) (n = 86) mice ∼12–16 weeks old were used in the current study. A previous study found some sex-coupled differences in behavioural tests.32 However, in the current study, an equal number of males and females were used, as no sex-coupled difference was seen, and data from male and females were pooled in the final analyses. Notably, the lack of sex difference suggests that female sex hormone/s may have a modulatory action on migraine prevalence via neuronal/astrocytic signalling32 rather than via a vascular wall effect. Middle cerebral arteries and mesenteric small arteries from littermate Atp1a2+/−G301R and WT mice were used.
Quantitative polymerase chain reaction
The quantitative polymerase chain reaction (qPCR) was carried out using Taqman probe (FAM) technology. Gene expression (the total Atp1a2 expression or expression of WT Atp1a2 allele only) was normalized to reference gene (Gapdh) and presented as a ΔCt value. Comparison of gene expression was derived from subtraction of averaged WT ΔCt in the experiment from ΔCt value in the analysed probe (either WT or Atp1a2+/−G301R), producing ΔΔCt. Relative gene expression was calculated as 2–ΔΔCt.
Semi-quantification of cSrc and MYPT phosphorylation
Arterial segments were fixed, lysated and used for Western blotting as previously described.26 Membranes for identification of proteins were cut at approximately 100 kDa, 50 kDa and 35 kDa, and the upper segment used for MYPT detection (expected band ∼ 130 kDa), middle segment for cSrc detection (expected band ∼ 65 kDa), and the lower segment for the reference protein thioredoxin 2 detection (expected band ∼ 12 kDa). The membranes were incubated with primary antibodies overnight at +4℃ and then with horseradish-peroxidase-conjugated secondary antibody (1:2000; Dako, Denmark) for 1.5 h. Bound antibodies were detected by an enhanced chemiluminescence kit (ECL, Amersham, UK).
Proteins (MYPT or cSrc) were semi-quantified by measuring band intensities using ImageJ software (NIH, USA). Protein phosphorylation was semi-quantified as a ratio between phospho-protein and total protein. The average measurements from WT group under resting conditions (i.e. in the absence of drugs) were set to 100% to compare the effects of drug interventions detected from the same gel. The total cSrc and MYPT protein expression were individually normalized to the corresponding band intensity of thioredoxin 2 from the same load, and semi-quantified by setting normalized intensity measured for WT group to 100%.
Whole-mount staining of arterial segments
The brain and mesentery were removed from perfusion-fixed mice34 and arteries dissected out. Tissues were incubated at room temperature for 2 h in blocking buffer (1% BSA and 0.2% Triton in PBS), further washed and incubated overnight with primary antibody in blocking buffer at 4℃, washed again and incubated in secondary antibody in 0.1% Triton in PBS for 2 h at room temperature. Tissue was subsequently mounted in anti-fade mounting media; with the media of select preparations also containing 0.002% propidium iodide to clarify cell layer patency. Tissue was imaged with uniform confocal settings. Controls involved peptide block of primary antibody in a 1:10 (v/v) excess of the immunizing peptide; and secondary only was used as a ‘zero’ setting.
Isometric force measurement of isolated arteries
Mice were sacrificed by cervical dislocation, and the brain and mesentery dissected into ice-cold physiological salt solution. Arteries were cleaned of connective tissue and mounted in a wire myograph (Danish Myo Technology A/S, Denmark) for isometric force measurements as previously described.26 The study was performed on endothelium intact arteries, unless otherwise stated.
Simultaneous measurements of isometric force and [Ca2+]i
Ratiometric [Ca2+]i measurements using fura 2-acetoxymethyl ester were obtained simultaneously with force measurements in a myograph as described elswhere.35 It has been previously shown that this loading protocol enables measurements of smooth muscle [Ca2+]i with negligible contribution from endothelial cells.36 [Ca2+]i was expressed as the ratio of fluorescence during excitation at 340 nm and 380 nm.
Simultaneous measurements of isometric force and membrane potential
Smooth muscle membrane potential in the intact vascular wall was measured in arteries mounted in an isometric myograph (Danish Myo Technology A/S, Denmark) as previously described.37 Membrane potential was averaged over a 1 min recording, at least 1 min after U46619 application.
Morphometric measurements
Arteries were mounted in a wire myograph and morphometric measurements performed using a light microscope (40× water immersion lens) as previously described.38,39 Measurements were obtained at three different points on either side of the vessel.
Isobaric tone of isolated cerebral arteries
A segment of middle cerebral artery was dissected and cannulated using glass micro cannulas and mounted in a pressure myograph (111P, DMT), as previously described.35 The pressure-step protocol was repeated for arteries under control conditions in PSS and after 15 min incubation in Ca2+-free PSS in the presence of 30 µM papaverine and 10 µM Y27632. The outer diameters obtained under these conditions were considered active (AD) and passive diameters (PD), respectively. The degree of tone at each pressure level was quantified as [1 – (AD/PD)].
Laser speckle analysis
Laser speckle (LS) imaging experiments were performed in order to assess resting blood flow in cerebral arteries in vivo (Figure 1(a) and Supplemental Figure 1(a)). A segmentation algorithm40,41 was applied to extract diameter and corresponding speckle contrast values for selected arteries (Supplemental Figure 1(b)). These values were then averaged over the vessel segment and used to estimate volumetric blood flow dynamics (blood flow index).
Figure 1.

Atp1a2+/−G301R mice have reduced resting blood flow through branches of middle cerebral artery, in comparison with WT. Whole-brain averaged Laser Speckle intensity image showing cerebral blood vessels (a). White arrows identify branches of middle cerebral artery (Supplemental Figure 1(a)) that were used for Laser Speckle analyses. Analyses did not found any significant changes in arterial diameter (b; n = 6–7), while blood flow index (an estimate volumetric blood flow dynamics) was significantly reduced in Atp1a2+/−G301R mice in comparison with WT (C; *P < 0.05).
pNaKtide peptide
The pNaKtide peptide22 (HD Biosciences, China) is composed of 20 amino acids from the N–domain of the Na,K-ATPase and a leader peptide sequence of 13 amino acids that allows cell membrane penetration. Prior studies have shown that it is effective in blocking the Na,K-ATPase-dependent cSrc activation.20,22,26,42–44
Data analyses
Microsoft Excel and GraphPad Prism software (v.5.02) were used for graphing and statistical analysis. Data are summarized as the mean value ± SEM of the sample group. Concentration-response curves were fitted to experimental data using four-parameter, non-linear regression curve fitting. From these curves, pD2 (-log to the concentration required to produce a half-maximal response), the Hill slope and maximal response were derived and compared using an extra sum-of-squares F test to assess the overall significance for a regression model.45,46 Area under the concentration-response curve (AUC) was calculated based on trapezoidal rule. Significant differences between means were determined by either one-way or two-way ANOVA, where appropriate, followed by Bonferroni correction for multiple comparison or by Student t-test. A probability (P) level of <0.05 was considered significant.
Results
Mice bearing G301R mutation have reduced brain perfusion
Estimation of diameter and blood flow in branches of middle cerebral artery in vivo (Supplemental Figure 1(a)) in anaesthetized mice demonstrated significantly reduced resting blood flow in Atp1a2+/−G301R mice in comparison with WT, although no significant difference in diameter was seen (Figure 1).
G301R mutation modifies expression of the Na,K-ATPase in the vascular wall
The total amount of the α2 isoform Na,K-ATPase mRNA in middle cerebral artery isolated from the Atp1a2+/−G301R mice was similar to that from WT (Figure 2(a)). However, when primers amplifying only the WT allele were used, cerebral artery from the Atp1a2+/−G301R mice had ∼50% of mRNA transcripted from the WT allele (Figure 2(b)).
Figure 2.

The α2 isoform Na,K-ATPase haploinsufficiency of middle cerebral arteries from Atp1a2+/−G301R mice. Quantitative PCR results showed an unchanged expression of total Atp1a2 mRNA (n = 6) in comparison with WT (n = 8) (a). In arteries from the Atp1a2+/−G301R mice, approximately a half of the α2 isoform mRNA is from the WT allele and the rest is from the mutated G301R allele, as it is shown with primers amplifying only the WT allele (b; n = 6–8). Whole-mount staining indicated the presence of α2 isoform protein in both endothelial cells and smooth muscles (c; representative images). Semi-quantitative fluorescence demonstrated a reduction of the α2 isoform in middle cerebral arteries from the Atp1a2+/−G301R mice in comparison with WT (d; n = 5). The α1 isoform Na,K-ATPase is reduced in endothelial cells from the Atp1a2+/−G301R mice in comparison with WT (e, representative images; and f, averaged results; n = 5). In inserts, ‘+peptide’ indicates a control experiment where primary antibody was blocked with an excess of the immunizing peptide. ‘2° only’ shows staining with secondary antibody only that was used as a ‘zero’ setting. * and ***, P < 0.05 and 0.001 vs. WT (one-way ANOVA).
Consistent with previous reports on FHM2-associated α2-isoform haploinsufficiency,2 we found an ∼50% reduction in α2 isoform protein in cerebral arteries from the Atp1a2+/−G301R mice in comparison with WT (Figure 2(c) and (d)). This reduction was seen in both endothelial and smooth muscle cells. The α1 isoform Na,K-ATPase (Figure 2(e) and (f)) and the NCX (Supplemental Figure 2) were also significantly reduced in cerebral arteries from the Atp1a2+/−G301R mice.
Middle cerebral arteries from Atp1a2+/−G301R mice demonstrated stronger agonist-induced constriction and depolarization in comparison with WT
Middle cerebral arteries from Atp1a2+/−G301R mice mounted in isometric myograph had larger lumen diameter (P < 0.026) than arteries from WT; 175 ± 5 µm (n = 33) in comparison 161 ± 4 µm (n = 41), respectively. Media thickness and media-to-lumen ratio were also increased in arteries from Atp1a2+/−G301R mice in comparison to WT (Supplemental Figure 3). Pressurized arteries from Atp1a2+/−G301R mice had increased diameter in Ca2+-free solution (Supplemental Figure 4(a)). In the presence of Ca2+, pressurized cerebral arteries developed myogenic tone, which was similar in arteries from WT and Atp1a2+/−G301R mice (Supplemental Figure 4(a) and (b)).
The thromboxane A2 agonist, U46619, constricted and depolarized mouse middle cerebral arteries (Figure 3). This constriction was significantly potentiated in arteries from Atp1a2+/−G301R mice compared with WT (Figure 3(a)). Since endothelium-dependent relaxation of the two groups were similar (Supplemental Figure 4(c)), and contractile responses of cerebral arteries from the Atp1a2+/−G301R mice remained stronger than WT after removal of the endothelium (Suppl. Fig. 4D), the differences in the contractile responses are unlikely to be due to changes in endothelial function. The rest of the study was performed with endothelium intact arteries.
Figure 3.

Middle cerebral but not small mesenteric arteries from Atp1a2+/−G301R mice had increased agonist-induced constriction and depolarization in comparison with WT. Concentration-response curves to U46619 of middle cerebral arteries (a) from Atp1a2+/−G301R (n = 7) and WT mice (n = 9). F test indicated a significant difference between experimental groups (***, P < 0.05). No difference in U46619-induced constriction was seen between mesenteric small arteries from Atp1a2+/−G301R and WT mice (b; n = 6). A representative simultaneous recording of wall tension and membrane potential in WT middle cerebral artery stimulated with U46619 (c). Wall tension (d) and membrane potentials (e) under resting conditions and after 0.3 µM U46619 stimulation of middle cerebral arteries from Atp1a2+/−G301R (n = 6) and WT mice (n = 8). The experimental protocol is similar to data shown in c. *, P < 0.05, comparison between Atp1a2+/-G301R and WT groups; +++, P < 0.001, the effect of U46619 (two-way ANOVA).
When wall tension and membrane potential were measured simultaneously (Figure 3(c)), no difference between the groups was observed under resting conditions (Figure 3(d) and (e)). However, in the presence of 0.3 µM U46619, a stronger constriction of cerebral arteries from Atp1a2+/−G301R (Figure 3(d)) was associated with larger smooth muscle depolarization (Figure 3(e)).
The enhanced constriction of Atp1a2+/−G301R cerebral arteries is associated with increased sensitization to [Ca2+]i
To test whether the increased depolarization of the arteries from Atp1a2+/−G301R are associated with elevated Ca2+ influx, [Ca2+]i was measured simultaneously with wall tension (Figure 4). No significant difference in the resting [Ca2+]i between the groups was seen. Surprisingly, the increased vasoconstriction of the arteries from Atp1a2+/−G301R (Figure 4(a)) was associated with a smaller increase in [Ca2+]i in comparison with arteries from WT (Figure 4(b)). This relation between wall tension and [Ca2+]i suggests an increased sensitization of contractile machinery to Ca2+. Indeed, when wall tension is plotted as a function of [Ca2+]i (Figure 4(c)), arteries from Atp1a2+/−G301R demonstrated a steeper slope, i.e. an increased sensitization to [Ca2+]i.
Figure 4.

Potentiated constriction to U46619 of middle cerebral arteries from Atp1a2+/−G301R mice is associated with smaller changes in [Ca2+]i suggesting higher sensitivity to Ca2+. U46619 concentration-dependent vasoconstriction (a) of middle cerebral arteries from Atp1a2+/−G301R mice (n = 6) was significantly (***, P < 0.01; F test) potentiated in comparison with WT (n = 5). However, changes of [Ca2+]i (b) were less pronounced (***, P < 0.01; F test) in arteries from Atp1a2+/−G301R (n = 6) compared to WT (n = 5). Arteries from Atp1a2+/−G301R mice had a steeper relation between [Ca2+]i and wall tension (**, P < 0.01 for slopes based on linear regression analysis) in comparison with WT (c; re-plotted data from a and b). The relation between [Ca2+]i and membrane potential (d; re-plotted data from Figures 2(d) and 3(b)) suggested a lesser [Ca2+]i rise in arteries from Atp1a2+/−G301R despite the larger depolarization.
Interestingly, in the presence of 0.3 µM U46619 arteries from Atp1a2+/−G301R mice experienced a reduced [Ca2+]i increase despite a larger membrane depolarization (Figure 4(d)). This suggests that arteries from Atp1a2+/−G301R mice have reduced voltage-dependent Ca2+ influx in compared to WT.
An increased Ca2+ sensitization of middle cerebral arteries from Atp1a2+/−G301R mice is not agonist-specific
To test whether the observed Ca2+ sensitization was specific for U46619 induced constriction, we stimulated middle cerebral arteries with endothelin (Figure 5(a) to (c)). Arteries from Atp1a2+/−G301R mice constricted to endothelin stronger than those from WT (Figure 5(a)). However, endothelin-induced [Ca2+]i increase was not significantly different between arteries from WT and Atp1a2+/−G301R mice (Figure 5(b)). The slope of wall tension as a function of [Ca2+]i was not significantly different between the groups, although there was a tendency for increased Ca2+-sensitivity in cerebral arteries from Atp1a2+/−G301R mice (Figure 5(c); P = 0.13).
Figure 5.

Increased Ca2+ sensitization of middle cerebral arteries from Atp1a2+/−G301R mice is independent of vasoconstrictor type. Constriction of WT (n = 5) and Atp1a2+/−G301R (n = 6) arteries to endothelin (a) and simultaneous recordings of [Ca2+]i expressed as a ratio of Fura-2 fluorescence at 340 and 380 nm (b). The relation between [Ca2+]i and wall tension at different concentrations of endothelin (c; data re-plotted from a and b). Constriction (d) and simultaneous changes in [Ca2+]i (e) in response to K+-induced depolarization. Changes in relation between [Ca2+]i and wall tension in response to K+-induced depolarization (f). ** and *** indicates P < 0.01 and 0.001; n = 5–6.
Arteries from Atp1a2+/−G301R mice constricted also to a greater extent to K+-induced depolarization (in the presence of 1 µM phentolamine) than WT (Figure 5(d)), but this was associated with an attenuated [Ca2+]i increase (Figure 5(e)). This led to a steeper slope of [Ca2+]i – wall tension relation for cerebral arteries from Atp1a2+/−G301R mice in comparison with WT, suggesting increased Ca2+-sensitization (Figure 5(f)).
Contractile responses of cerebral arteries from Atp1a2+/−G301R and WT mice became similar in the presence of ouabain, but not digoxin
Constriction of middle cerebral arteries from the two groups of mice was compared before and after pre-treatment with Na,K-ATPase inhibitors, digoxin and ouabain (Supplemental Figure 5). Digoxin (10 µM) potentiated slightly (P = 0.059) contractile response of arteries from WT, but this was without any effect (P = 0.483) in arteries from Atp1a2+/−G301R mice (Supplemental Figure 5(a)). The difference in contractile responses between the groups (i.e. a stronger constriction in arteries from Atp1a2+/−G301R compared to WT) was not affected by digoxin (Supplemental Figure 5(a)). Ouabain (10 µM) potentiated cerebral artery constriction from both groups (Supplemental Figure 5(b)), while 0.1 µM (n = 6) and 1 µM (n = 10) ouabain was without significant effect (data not shown). In the presence of 10 µM ouabain, no difference in the constriction of cerebral arteries from Atp1a2+/−G301R and WT mice was seen (Supplemental Figure 5(b)).
Inhibition of cSrc kinase abolished the difference in contractile responses of cerebral arteries from the Atp1a2+/−G301R and wild type mice
Pre-incubation of cerebral arteries with 3 µM PP2 abolished the difference in contractile responses between the groups (Figure 6(a)). PP2 suppressed constriction of cerebral arteries and there was a tendency for a larger effect of PP2 inhibition on the arteries from Atp1a2+/−G301R mice in comparison with WT (Figure 6(a)). This was associated with reduced [Ca2+]i changes, which were similar between the groups in the presence of PP2 (Figure 6(b)). Consistent with this, the relationship between wall tension and [Ca2+]i was not different between cerebral arteries from Atp1a2+/−G301R and WT mice in the presence of PP2 (Figure 6(c)).
Figure 6.

Inhibition of cSrc kinase abolished the increased force development, [Ca2+]i, and Ca2+ sensitivity in cerebral arteries from Atp1a2+/−G301R mice. PP2 (3 µM) significantly suppressed constriction of cerebral arteries from Atp1a2+/−G301R (***, P < 0.001; n = 5) and WT (+++, P < 0.001; n = 5), and abolished the difference between the groups seen under control conditions (##, P < 0.01) (a). This was associated with suppression of [Ca2+]i changes in response to U46619 stimulation (b). Under control conditions [Ca2+]i changes were larger (###, P < 0.001; n = 5) in WT than in Atp1a2+/−G301R, but PP2 significantly suppressed responses in Atp1a2+/−G301R (***, P < 0.001; n = 5) and WT (+++, P < 0.001; n = 5) groups and the difference between the groups was abolished. The relation between [Ca2+]i and wall tension (c) was re-plotted from the data obtained in the presence of PP2 and shown in A and B. No difference in the slopes was found. pNaKtide (2 µM) also suppressed contraction of cerebral arteries from Atp1a2+/− G301R (***, P < 0.001; n = 6–7) and WT (+++, P < 0.001; n = 6–7) mice and abolished the difference between the groups seen under control conditions (###, P < 0.001) (d). [Ca2+]i changes in response to U46619 were less in cerebral arteries from Atp1a2+/-G301R mice in comparison with WT (###, P < 0.001; n = 6–7) (e). [Ca2+]i changes were significantly suppressed by pNaKtide in Atp1a2+/-G301R (***, P < 0.001; n = 6–7) and WT (+++, P < 0.001; n = 6–7) groups. The relation between [Ca2+]i and wall tension (f) in the presence of pNaKtide was re-plotted from the data shown in D and E; no difference between the slopes was found. Inserts in A, B, D, E shows differences in area under concentration-response curves (AUC) before and after treatment with PP2 (a, b) and pNaKtide (d, e). & indicates P < 0.05.
pNaKtide (2 µM) also suppressed contraction and abolished the difference in contractile responses between the groups (Figure 6(d)). The effect of pNaKtide on wall tension was significantly larger for cerebral arteries from Atp1a2+/−G301R mice in comparison with arteries from WT, but this was associated with similar suppression of [Ca2+]i changes (Figure 6(e)). Under control conditions [Ca2+]i, changes were larger in cerebral arteries from WT than in Atp1a2+/−G301R (Figure 4(b)), but this difference was abolished in the presence of pNaKtide (Figure 6(e)). The relationship between [Ca2+]i and wall tension in the presence of pNaKtide was similar for cerebral arteries from Atp1a2+/−G301R and WT mice, suggesting a similar level of sensitization for Ca2+ under these conditions (Figure 7(f)).
Figure 7.
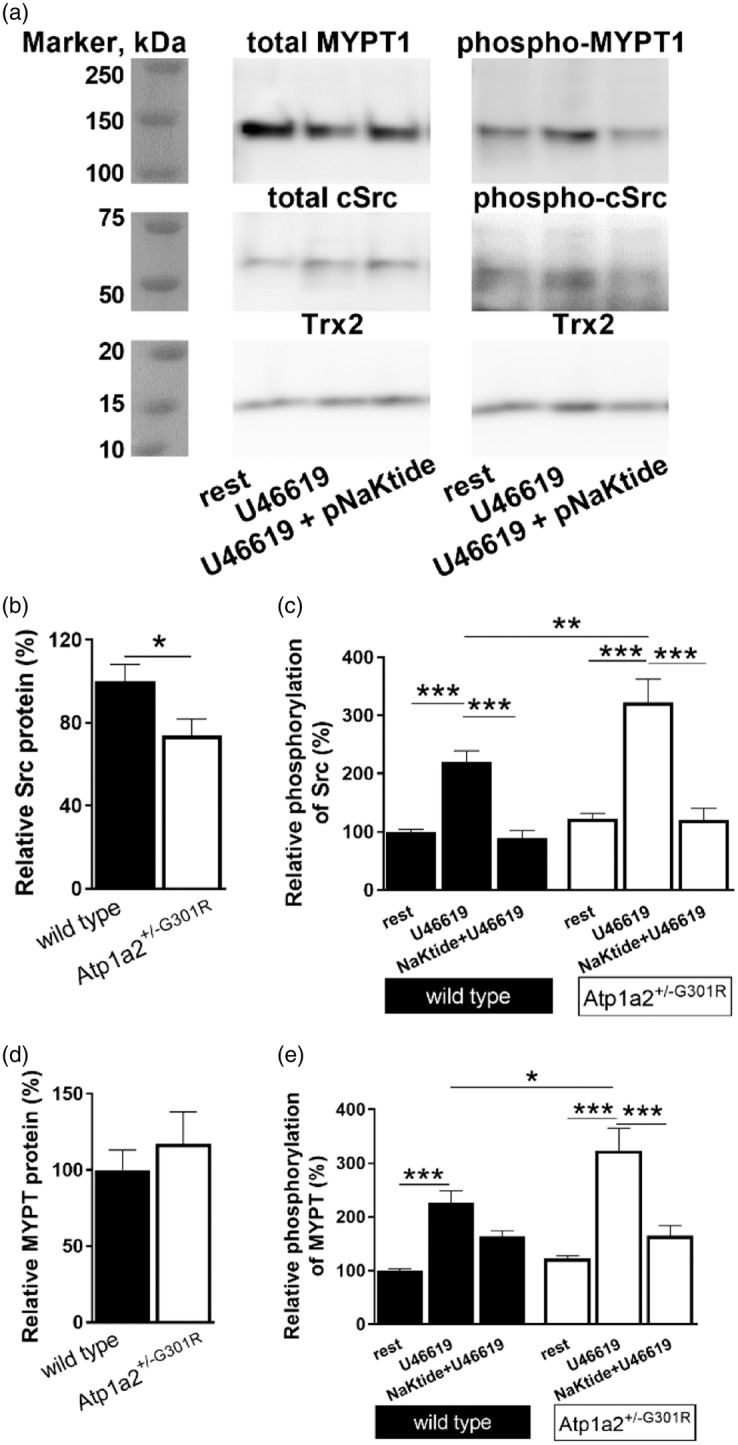
Semi-quantitative analyses of expression and phosphorylation of cSrc kinase and MYPT1 proteins in middle cerebral arteries from Atp1a2+/−G301R and WT mice. Representative WT Western blot where MYPT 1 and cSrc expression and phosphorylation were studied (a). Thioredoxin 2 (Trx2) was used as loading control. Total cSrc kinase expression (normalized to Trx2 band density) was reduced in cerebral arteries from Atp1a2+/−G301R compared to WT mice (b; n = 6–9). Averaged results suggest an increased cSrc phosphorylation (calculated as a ratio between phosphorylated and total cSrc, and normalised to averaged values for WT under resting conditions) in arteries from Atp1a2+/−G301R mice compared to WT after U46619 (10−5 M) stimulation (C; n = 5–11). Incubation with pNaKtide (2 µM) abolished this difference. Expression of total MYPT1 (normalized to Trx2 band density) was not different between the groups (n = 4–12; d, representative Western blot; e, averaged data). U46619 significantly increased MYPT1 phosphorylation at Thr850 (n = 4–12; f) (calculated as a ratio of phosphorylated and total MYPT1, and normalised to averaged values for WT under resting conditions). This potentiation is stronger in cerebral arteries from Atp1a2+/−G301R than WT. In the presence of pNaKtide this potentiation was abolished, and there was no difference between arteries from Atp1a2+/−G301R and WT. *, ** and ***, P < 0.05, < 0.01 and < 0.001 (two-way ANOVA).
Enhanced phosphorylation of cSrc and MYPT in middle cerebral arteries from Atp1a2+/−G301R compared to WT mice
Despite a slightly reduced total expression of cSrc kinase in middle cerebral arteries from Atp1a2+/−G301R mice (Figure 7(b)), no significant difference in phosphorylated cSrc was found between cerebral arteries from Atp1a2+/−G301R and WT mice under resting conditions (Figure 7(c)). When cerebral arteries were stimulated with U46619, the phosphorylation of cSrc was significantly increased and this increase was stronger in arteries from Atp1a2+/−G301R than in WT mice. Moreover, pNaKtide significantly suppressed cSrc phosphorylation induced by U46619 (Figure 7(c)).
The expression of myosin phosphatase targeting protein 1 (MYPT) was similar between cerebral arteries from Atp1a2+/−G301R and WT mice (Figure 7(d)), as well as MYPT1 phosphorylation under resting conditions (Figure 7(d)). U46619 induced MYPT1 phosphorylation, which was significantly larger in arteries from Atp1a2+/−G301R compared to WT mice (Figure 7(d)). This difference in U46619-induced MYPT1 phosphorylation was abolished by pNaKtide.
Contractile responses of mesenteric small arteries did not differ between Atp1a2+/−G301R and WT mice
In contrast to middle cerebral arteries, mesenteric arteries from the Atp1a2+/−G301R and WT mice showed similar agonist-induced constriction (Figure 3(b)). Ouabain significantly potentiated constriction of both groups (Supplemental Figure 6(a) and (b)). In the presence of ouabain, there was no significant difference in constriction of mesenteric arteries from the Atp1a2+/−G301R and WT mice.
Mesenteric arteries from the Atp1a2+/−G301R and WT mice constricted in a similar manner to different contractile agonists, e.g. noradrenaline (NA; Supplemental Figure 7(a)) and U46619 (Supplemental Figure 7(b)). Incubation with pNaKtide was without any significant effect on contractile response of arteries from Atp1a2+/−G301R; however, it suppressed the constriction of arteries from WT mice (Supplemental Figure 7). This difference in the effects of pNaKtide between the groups was not due to endothelial function, since it was similarly affected by pNaKtide in both groups, i.e. pNaKtide was without any effect on relaxation to 3 µM ACh (Supplemental Figure 7(c)) and suppressed relaxation to 0.1 mM ACh (Supplemental Figure 7(d)).
In contrast to cerebral arteries (Figure 2), whole-mount immunostaining of mesenteric arteries demonstrated an elevation of both α1 and α2 isoforms of the Na,K-ATPase in the Atp1a2+/−G301R mice in comparison with WT (Supplemental Figure 8(a) to (d)). However, the expression of NCX in mesenteric arteries from the Atp1a2+/−G301R mice was reduced (Supplemental Figure 8(e) to (f)), similar to cerebral arteries (Supplemental Figure 2).
No difference in total cSrc and its phosphorylation levels was found between the Atp1a2+/−G301R and WT mice (Supplemental Figure 9(a) to (c)). Stimulation with NA increased cSrc phosphorylation in both groups without any difference between them (Supplemental Figure 9(c)). Incubation with pNaKtide suppressed cSrc phosphorylation.
Total MYPT1 expression and baseline MYPT1 phosphorylation were also the same in mesenteric arteries from the Atp1a2+/−G301R and WT mice (Supplemental Figure 9(a), (d) and (e)). NA elevated MYPT1 phosphorylation, and this increase was the same for both Atp1a2+/−G301R and WT mice. Incubation with pNaKtide reduced NA-induced MYPT1 phosphorylation in mesenteric arteries from both groups (Supplemental Figure 9(d) and (e)).
Discussion
Mutation of the Na,K-ATPase α2 isoform causes FHM2 in humans. In this study, increased middle cerebral artery contractility in mice with a mutation in the Na,K-ATPase α2 isoform was found. This was found to be associated with complex changes in excitation-contraction coupling. These changes can be summarized (Supplemental Figure 10) as cerebral artery smooth muscle cells from the Atp1a2+/−G301R mice being depolarized more to agonist stimulation, with this being paradoxically associated with a lesser increase in [Ca2+]i compared to WT. Furthermore, the reduced [Ca2+]i increase was unexpectedly associated with an increased force development. These findings suggest that an activation of cSrc kinase may be the mechanistic link, which can explain the complex changes in the excitation-contraction coupling (Supplemental Figure 10). A second important finding of this study is that the increased contractility may be confined to the cerebral, over systemic mesenteric arteries.
FHM2 is characterized with perfusion abnormalities in the brain
Biphasic changes in cerebral blood flow during a migraine attack in FHM2 patients are established.6,47 Although it is highly variable between reports,47,48 an initial hypoperfusion (i.e. vasoconstriction) in the affected hemisphere is linked to spreading depolarization of neural tissue and followed by hyperperfusion.3,49 Vasoconstriction has been hypothesized to be a consequence of abnormal neuronal excitation,8,50 although it might also be a result of increased concentration of contractile factors in the circulation.51–53 Thus, plasma thromboxane A2 is known to be elevated in migraine patients.51 Moreover, interical plasma concentration of endothelin-1, and especially its ictal concentration at the beginning of attack, is also known to be elevated in migraine with aura.52,53
How spreading depression leads to cerebral artery constriction is unknown. It has been hypothesized that an abnormally high accumulation of [K+]o and/or other metabolites, neuromediators, or hyperactivation of perivascular innervation could contribute to regional vasoconstriction in the brain.3 Data from the present study suggest that cerebral artery smooth muscle cells with the FHM2-associated mutation have increased sensitivity to [Ca2+]i. Thus, stimuli, which are usually subthreshold for a normal subject, might lead to regional cerebral vasoconstriction and hypoperfusion in FHM2 patients, and in this mouse model. This also implies that changes in perfusion might at least be partly independent of altered neuronal activity, to originate within the vascular wall. Consistent with this, we found that the Atp1a2+/−G301R mice have reduced perfusion through middle cerebral arteries under resting condition. We did not find any significant difference in arterial diameter in vivo. This suggests a possible elevation of downstream resistance of microvessels exposed to circulatory and perivascular vasoactive factors in vivo.
Atp1a2+/−G301R mouse model for vascular haploinsufficiency of the Na,K-ATPase α2 isoform
Previously, attention was given to abnormal function of the mutated Na,K-ATPase α2 isoform in glial cells in FHM2.2,54 However, it is also suggested that the FHM2 disease phenotype cannot be explained by impaired astrocytic function alone, as the α2 isoform constitutes a relatively minor component (∼20%) of the total astrocytic Na,K-ATPase,55 and even less (∼10%) in terms of activity.56 An ∼50% reduction in the Na,K-ATPase α2 isoform in all brain regions of the Atp1a2+/−G301R mice has been reported.32 This is consistent with other reports on haploinsufficiency of the FHM2-associated mutations.54,57 The present study also found a reduction of α2 Na,K-ATPase protein expression in middle cerebral arteries from Atp1a2+/−G301R mice of ∼50%. The present and other findings32,54,57 are not in agreement with another report where no significant reduction of the Na,K-ATPase α2 isoform was found in Atp1a2+/−G301R mouse hippocampus.58 The reason for this discrepancy is unknown, but may represent variability within the brain, to reflect the current data comparison of cerebral and systemic (mesenteric) vascular beds.
Surprisingly, an increased α2 Na,K-ATPase expression in mesenteric arteries from the Atp1a2+/−G301R mice was found. This suggests that α2 isoform maturation, transport, insertion and / or degradation are controlled differently in cerebral and mesenteric arteries. Since the present study is unable to discriminate between mutated and WT proteins, we do not know how much of the mutated protein is expressed, and what the functional consequence of this is in these arteries. However, based on similar contractile responses, we suggest that the functional contribution of the α2 isoform may be unchanged in mesenteric arteries from Atp1a2+/−G301R mice. It is unknown which factor(s) are responsible for the differences in the expression profile of these two vascular beds, but notably, the G301R mutant was previously shown to be expressed differently in different cell types, e.g. the G301R mutant was normally expressed in oocytes,59 but not in neurons32 and oocytes58 in other studies, alluding to differences in endothelial and smooth muscle phenotype and function between different arteries and states.60
The G301R mutation is associated with changes in the expression of membrane transporters
The present study found that expression of the α1 isoform was also suppressed in cerebral arteries from Atp1a2+/−G301R mice. This is in contrast to previous reports where the expression of other isoforms of the Na,K-ATPase was not affected by changes in the expression of the α2 isoform.32,54 The reason for this is unclear, but an increase of cSrc activation, discussed below, could potentially contribute to changes in the smooth muscle phenotype and the expression profile.61 The present study also found a reduction of NCX expression, which is consistent with suppression of the NCX after siRNA-induced downregulation of the α2 isoform in rat mesenteric arteries.35 Accordingly, chronic ouabain treatment upregulated Na,K-ATPase α2 isoform expression in rat mesenteric artery smooth muscle cells, and this was associated with increased NCX expression.62 Collectively, this suggests that expression of the Na,K-ATPase α2 isoform and the NCX is closely linked and might be associated with smooth muscle cell phenotype and vascular remodelling of cerebral arteries, per the present data.
It has been suggested that the Na,K-ATPase modulates expression of other proteins via the cSrc kinase pathway.14 In the present study, changes in cSrc kinase expression and activation in cerebral arteries of Atp1a2+/−G301R mice were found. The mechanism for cSrc-dependent transcriptional regulation is unclear. Notably, tyrosine phosphorylation of STATs (signal transducers and activators of transcription) has been shown to trigger their activation and translocation to the nucleus where it can subsequently upregulate nuclear factor of activated T-cells (NFAT) expression.63 The calcineurin/NFAT pathway can modulate expression of Ca2+ transport proteins including the NCX.64 It is however unclear whether changes in NCX expression in the present study are dependent on cSrc kinase activation or mediated via altered Ca2+ homeostasis. Further work with chronic inhibition of NFAT and cSrc kinase is necessary to test these possibilities.
Expression changes in cerebral arteries of Atp1a2+/−G301R mice are associated with an increased arterial contractility due to sensitization to Ca2+
The increased contractility of cerebral arteries cannot simply be explained by reduced pumping activity of the Na,K-ATPase α2 isoform, or by the secondarily reduced NCX. Expression changes and pharmacological manipulation of the Na,K-ATPase α2 isoform can affect vascular tone via modulation of [Ca2+]i through an interaction of the Na,K-ATPase with the NCX.9,12 In this scenario, suppression of Na,K-ATPase ion pumping activity was suggested to suppress the Na+-dependent net Ca2+ extrusion leading to increased [Ca2+]i and potentiation of vasoconstriction.35,65 This is not consistent with the present results, where increased contraction of cerebral arteries of Atp1a2+/−G301R mice is associated with reduced [Ca2+]i. Previous studies reported that reduced expression of the NCX leads to reduced net Ca2+ influx and suppressed arterial contraction.66,67 Consistently, the present work detected a reduction in the NCX to be associated with a smaller increase of [Ca2+]i. It has been suggested that the reduced [Ca2+]i is a result of functional suppression of the voltage-gated Ca2+ influx, consequent to the reduced NCX activity.67 The present observation of reduced net Ca2+ influx in response to K+-induced depolarization and reduced net Ca2+ influx in spite of stronger depolarization supports this proposal.66,67
Reduced [Ca2+]i changes can also be achieved by hyperactivation of the plasma membrane Ca2+ ATPase, which has been shown to be activated by the Src kinase family in neurons.68 The effect of cSrc kinase can also be mediated via membrane potential due to phosphorylation and inhibition of the voltage-gated and Ca2+-activated K+ channels.69 Accordingly, the present data show a stronger agonist-induced depolarization of cerebral artery smooth muscles in Atp1a2+/−G301R than in WT.
The difference in contractility between Atp1a2+/−G301R and WT mice cannot be explained by changes in Ca2+ transport proteins, since in mesenteric arteries, the present data found a reduced NCX expression, which was not associated with any significant change in agonist-induced contraction. This suggests limited significance of the NCX for agonist-induced contraction of smooth muscles, at least in mesenteric arteries, although some importance of cerebral artery [Ca2+]i homeostasis cannot be excluded. Reversal potential for the NCX in vascular smooth muscle cells is approximately −30 mV;70 i.e. near the maximal depolarization level in response to maximal agonist stimulation. This suggests that the NCX cannot be the major contributor for Ca2+ influx during agonist stimulation and therefore the reduction in its expression did not affect contraction of mesenteric small arteries and cannot explain lower [Ca2+]i changes in cerebral arteries from the Atp1a2+/−G301R mice.
Stronger contractile responses of cerebral arteries from Atp1a2+/−G301R mice may result from the increased media-to-lumen ratio found described herein. However, per below, the ‘normalizing’ effect of acute maximal activation of cSrc as well as inhibition of cSrc does not support this suggestion. The mechanism/s underlying cerebral artery remodelling in the present study is unclear, but may be due to changes in hemodynamic parameters, and/or to a cSrc-dependent phenotypic switch in smooth muscle cell signalling.
Elevated cSrc-dependent signalling is responsible for increased Ca2+ sensitivity
The Na,K-ATPase has been suggested to form a membrane microdomain with several proteins involved in intracellular signalling, including Src kinase.15 Ouabain binding to the Na,K-ATPase has been shown to activate cSrc kinase via autophosphorylation at Y418.22–24,26,71 There are two views on how the activity of cSrc kinase is modulated by the Na,K-ATPase. One suggestion is that cSrc kinase interacts physically with the Na,K-ATPase.16–21,72 This view contrasts with the suggestion that changes in ATP consumption by the Na,K-ATPase play a key role for cSrc autophosphorylation.23–25 The current study is consistent with an interaction between cSrc kinase and the Na,K-ATPase, but cannot distinguish between the two possibilities. Previous reports suggested the importance of either α1 or α2 Na,K-ATPase for cSrc-dependent signaling.26,43 In the current study, it is not possible to distinguish between these options since the expression of both isoforms was suppressed in the cerebral arteries of Atp1a2+/−G301R mice.
The present data suggest that potentiation of cerebral vasoconstriction is mediated by elevated cSrc kinase activation via the sensitization of the contractile machinery to [Ca2+]i. The contribution of cSrc kinase to Ca2+-sensitization was suggested to be mediated via Rho kinase translocation and subsequent MYPT1 phosphorylation in studies of rat pulmonary arteries,73 aorta,74 and mouse mesenteric arteries.75 Pharmacological analyses suggest that agonist-stimulated cSrc kinase activation and contribution to the arterial contraction is upstream to Rho kinase activation.74 The mechanism important for this interaction is unknown, but leukaemia-associated RhoGEF, which is expressed in smooth muscle76 and which can enhance RhoA, is known to be activated by tyrosine phosphorylation.77 Moreover, ouabain was suggested to constitutively activate Rho kinase via a caspase-dependent cleavage pathway.78 Accordingly, the present results suggest that elevated cSrc kinase activation in the Atp1a2+/−G301R mice increases the Ca2+-sensitivity of cerebral arteries via MYPT1 phosphorylation. The role of Na,K-ATPase-cSrc-MYPT1 signalling was also supported by the effects of tyrosine kinase inhibition. The present work used two non-related inhibitors, a conventional cSrc kinase inhibitor, PP2 and a peptide that inhibits the Na,K-ATPase-dependent cSrc activation,20,22,43 and both suppressed the contraction of cerebral arteries and partially antagonized MYPT1 phosphorylation. These findings argue against remodelling as a major contributor to elevated cerebrovascular contractility in Atp1a2+/−G301R mice.
The present work found that although the expression of total cSrc protein was slightly reduced in cerebral arteries from the Atp1a2+/−G301R mice, phosphorylated cSrc in the arteries from Atp1a2+/−G301R mice was not significantly different under resting conditions and significantly increased upon agonist stimulation in comparison with WT. This supports the hypothesis that the reduction in Na,K-ATPase permits autophosphorylation of cSrc kinase.17 The observed reduction in total cSrc expression in cerebral arteries of Atp1a2+/−G301R mice could be a result of compensatory downregulation when a significant part of the Na,K-ATPase was lost. Consistent with this, no difference in resting tone, [Ca2+]i and membrane potential was seen in vitro. Agonist stimulation further activated cSrc kinase and this was significantly potentiated in cerebral arteries of the Atp1a2+/−G301R mice; albeit probably due to reduced ‘buffering’ by the Na,K-ATPase. If the level of cSrc activation inversely correlates with the availability of the Na,K-ATPase, two predictions can be made. When the Na,K-ATPase is blocked with ouabain in arteries from the two groups of mice, no difference in contraction should be seen, and when cSrc kinase activity is blocked by PP2 or pNaKtide, no difference in contraction should be seen. Both predictions were confirmed in this study, supporting the hypothesis that elevated contractility of cerebral arteries in Atp1a2+/−G301R mice is mediated by higher activation of cSrc kinase.
In the present study, digoxin did not activate cSrc signalling in smooth muscles14and nor did it abolish the difference in contraction of cerebral arteries. Notably, a tendency for digoxin to potentiate contraction of cerebral arteries from WT, but not Atp1a2+/−G301R mice, suggests that inhibition of the Na,K-ATPase can affect arterial contraction in a mechanism other than that involving the cSrc activation pathway. This is possibly mediated via the NCX-dependent increase in [Ca2+]i,35,65 which cannot explain the difference between contractile responses cerebral arteries of Atp1a2+/−G301R and WT mice. Importantly, the difference in contractile responses between the groups was observed at agonist concentrations where [Ca2+]i reached sub-maximal levels. This suggest that the proposed cSrc-dependent Ca2+ sensitization has a potentiating action on [Ca2+]i initiated constriction, and is of primary importance in association with vasoconstrictor stimulation, such as at the first stage of a migraine attack.
Limitations
Although FHM2 is a relatively rare form of migraine,1 further insight into its pathology will contribute to a broader understanding of migraine in general. The increased Ca2+ sensitivity of cerebral arteries from mutated mice likely contributes to the aura-associated initial hypoperfusion. This cannot be generalized to all forms of migraine since FHM2 is the only migraine subtype known to be associated with mutations in the α2 isoform Na,K-ATPase. However, the principle can also be applied to other types of migraine where cerebrovascular hypercontractility associated with neuronal excitation may be elicited by other pathways.
This study has used an animal model, and it will be important to translate the findings to patients. It is also unclear whether aura-associated hypoperfusion can be elicited in mice, although we have seen some reduction in resting cerebral blood perfusion. Finally, the mechanisms responsible for regional specific translational modulation/s of the Na,K-ATPase α isoform, NCX and cSrc kinase remain to be elicited.
Conclusion
This study suggests a novel mechanism for hypoperfusion of the brain, which is an initial phase in the migraine attack of FHM2. The proposed mechanism suggests that the α2 isoform dependent regulation of cSrc kinase explains the Ca2+-sensitization of the contractile machinery in smooth muscle cells of cerebral arteries. The present work suggests that this signalling pathway has significant importance for cerebral artery contractility, but is of lesser significance for extra-cerebral systemic vessels.
Supplemental Material
Supplemental material for Smooth muscle Ca2+ sensitization causes hypercontractility of middle cerebral arteries in mice bearing the familial hemiplegic migraine type 2 associated mutation by Christian Staehr, Lise Hangaard, Elena V Bouzinova, Sukhan Kim, Rajkumar Rajanathan, Peter Boegh Jessen, Nathan Luque, Zijian Xie, Karin Lykke-Hartmann, Shaun L Sandow, Christian Aalkjaer and Vladimir V Matchkov in Journal of Cerebral Blood Flow & Metabolism
Acknowledgements
We thank Jane Holbæk Rønn, Viola Smed Mose Larsen, Jørgen Andresen and Dan Shelley for excellent technical assistance. We thank Dr. D.D. Postnov for expert help with Laser Speckle technique.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by the Lundbeck Foundation [grant number R183-2014-3618]; the Novo Nordisk Foundation [grant numbers NNF14OC0012731 and NNF17OC0026198]; the Independent Research Fund Denmark – Medical Sciences [grant number 7025-00015B] the Brain Foundation, Australia.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Authors’ contributions
CS, LH, SK, RK and PBJ performed myograph experiments, CS and VVM made perfusion fixation, CS, VVM and EVB performed immunoblotting, NL and SLS made whole-mount staining, and data analysis. CS, LH, EVB, ZX, KLH, SLS, CA and VVM took part in the conception and designed the study. CS, ZX, SLS, CA and VVM completed the manuscript writing.
Supplementary material
Supplementary material for this paper can be found at the journal website: http://journals.sagepub.com/home/jcb
References
- 1.Isaksen TJ, Lykke-Hartmann K. Insights into the pathology of the alpha2-Na(+)/K(+)-ATPase in neurological disorders: lessons from animal models. Front Physiol 2016; 7: 161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Friedrich T, Tavraz NN, Junghans C. ATP1A2 mutations in migraine: seeing through the facets of an ion pump onto the neurobiology of disease. Front Physiol 2016; 7: 239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ayata C, Lauritzen M. Spreading depression, spreading depolarizations, and the cerebral vasculature. Physiol Rev 2015; 95: 953–993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Pietrobon D, Striessnig J. Neurobiology of migraine. Nat Rev Neurosci 2003; 4: 386–398. [DOI] [PubMed] [Google Scholar]
- 5.Olesen J, Larsen B, Lauritzen M. Focal hyperemia followed by spreading oligemia and impaired activation of rCBF in classic migraine. Ann Neurol 1981; 9: 344–352. [DOI] [PubMed] [Google Scholar]
- 6.Iizuka T, Tominaga N, Kaneko J, et al. Biphasic neurovascular changes in prolonged migraine aura in familial hemiplegic migraine type 2. J Neurol Neurosurg Psychiatry 2015; 86: 344–353. [DOI] [PubMed] [Google Scholar]
- 7.Blicher JU, Tietze A, Donahue MJ, et al. Perfusion and pH MRI in familial hemiplegic migraine with prolonged aura. Cephalalgia 2016; 36: 279–283. [DOI] [PubMed] [Google Scholar]
- 8.Andersen AR, Friberg L, Olsen TS, et al. Delayed hyperemia following hypoperfusion in classic migraine. Single photon emission computed tomographic demonstration. Arch Neurol 1988; 45: 154–159. [DOI] [PubMed] [Google Scholar]
- 9.Matchkov VV, Krivoi II. Specialized functional diversity and interactions of the Na,K-ATPase. Front Physiol 2016; 7: 179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Larsen BR, Stoica A, MacAulay N. Managing brain extracellular K(+) during neuronal activity: the physiological role of the Na(+)/K(+)-ATPase subunit isoforms. Front Physiol 2016; 7: 141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Blaustein MP, Wier WG. Local sodium, global reach: filling the gap between salt and hypertension. Circ Res 2007; 101: 959–961. [DOI] [PubMed] [Google Scholar]
- 12.Blaustein MP, Chen L, Hamlyn JM, et al. Pivotal role of alpha2 Na+ pumps and their high affinity ouabain binding site in cardiovascular health and disease. J Physiol 2016; 594: 6079–6103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Song H, Karashima E, Hamlyn JM, et al. Ouabain-digoxin antagonism in rat arteries and neurones. J Physiol 2014; 592: 941–969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zulian A, Linde CI, Pulina MV, et al. Activation of c-SRC underlies the differential effects of ouabain and digoxin on Ca(2+) signaling in arterial smooth muscle cells. Am J Physiol 2013; 304: C324–C333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tian J, Xie ZJ. The Na-K-ATPase and calcium-signaling microdomains. Physiology 2008; 23: 205–211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tian J, Cai T, Yuan Z, et al. Binding of Src to Na+/K+-ATPase forms a functional signaling complex. Mol Biol Cell 2006; 17: 317–326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ye Q, Lai F, Banerjee M, et al. Expression of mutant alpha1 Na/K-ATPase defective in conformational transition attenuates Src-mediated signal transduction. J Biol Chem 2013; 288: 5803–5814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Banerjee M, Duan Q, Xie Z. SH2 ligand-like effects of second cytosolic domain of Na/K-ATPase alpha1 subunit on Src kinase. PLoS One 2015; 10: e0142119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Liang M, Cai T, Tian J, et al. Functional characterization of Src-interacting Na/K-ATPase using RNA interference assay. J Biol Chem 2006; 281: 19709–19719. [DOI] [PubMed] [Google Scholar]
- 20.Lai F, Madan N, Ye Q, et al. Identification of a mutant alpha1 Na/K-ATPase that pumps but is defective in signal transduction. J Biol Chem 2013; 288: 13295–13304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Haas M, Askari A, Xie Z. Involvement of Src and epidermal growth factor receptor in the signal-transducing function of Na+/K+-ATPase. J Biol Chem 2000; 275: 27832–27837. [DOI] [PubMed] [Google Scholar]
- 22.Li Z, Cai T, Tian J, et al. NaKtide, a Na/K-ATPase-derived peptide Src inhibitor, antagonizes ouabain-activated signal transduction in cultured cells. J Biol Chem 2009; 284: 21066–21076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Weigand KM, Swarts HG, Fedosova NU, et al. Na,K-ATPase activity modulates Src activation: a role for ATP/ADP ratio. Biochim Biophys Acta 2012; 1818: 1269–1273. [DOI] [PubMed] [Google Scholar]
- 24.Yosef E, Katz A, Peleg Y, et al. Do Src kinase and caveolin interact directly with Na,K-ATPase? J Biol Chem 2016; 291: 11736–11750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gable ME, Abdallah SL, Najjar SM, et al. Digitalis-induced cell signaling by the sodium pump: on the relation of Src to Na+/K+-ATPase. Biochem Biophys Res Commun 2014; 446: 1151–1154. [DOI] [PubMed] [Google Scholar]
- 26.Hangaard L, Bouzinova EV, Staehr C, et al. Na,K-ATPase regulates intercellular communication in the vascular wall via cSrc kinase dependent connexin43 phosphorylation. Am J Physiol 2017; 312: C385–C397. [DOI] [PubMed] [Google Scholar]
- 27.Zubkov A, Miao L, Zhang J. Signal transduction of ET-1 in contraction of cerebral arteries. J Cardiovasc Pharmacol 2004; 44(Suppl 1): S24–S26. [DOI] [PubMed] [Google Scholar]
- 28.Gonzales AL, Yang Y, Sullivan MN, et al. A PLCgamma1-dependent, force-sensitive signaling network in the myogenic constriction of cerebral arteries. Sci Signal 2014; 7: ra49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Colinas O, Moreno-Dominguez A, Zhu HL, et al. alpha5-Integrin-mediated cellular signaling contributes to the myogenic response of cerebral resistance arteries. Biochem Pharmacol 2015; 97: 281–291. [DOI] [PubMed] [Google Scholar]
- 30.Kusaka G, Kimura H, Kusaka I, et al. Contribution of Src tyrosine kinase to cerebral vasospasm after subarachnoid hemorrhage. J Neurosurg 2003; 99: 383–390. [DOI] [PubMed] [Google Scholar]
- 31.Spadaro M, Ursu S, Lehmann-Horn F, et al. A G301R Na+/K+ -ATPase mutation causes familial hemiplegic migraine type 2 with cerebellar signs. Neurogenetics 2004; 5: 177–185. [DOI] [PubMed] [Google Scholar]
- 32.Bottger P, Glerup S, Gesslein B, et al. Glutamate-system defects behind psychiatric manifestations in a familial hemiplegic migraine type 2 disease-mutation mouse model. Sci Rep 2016; 6: 22047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yan Y, Shapiro JI. The physiological and clinical importance of sodium potassium ATPase in cardiovascular diseases. Curr Opin Pharmacol 2016; 27: 43–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gage GJ, Kipke DR, Shain W. Whole animal perfusion fixation for rodents. J Vis Exp 2012; 65: 3564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Matchkov VV, Moeller-Nielsen N, Secher DV, et al. The alpha2 isoform of the Na,K-pump is important for intercellular communication, agonist-induced contraction and EDHF-like response in rat mesenteric arteries. Am J Physiol 2012; 303: H36–H46. [DOI] [PubMed] [Google Scholar]
- 36.Jensen PE, Mulvany MJ, Aalkjaer C. Endogenous and exogenous agonist-induced changes in the coupling between [Ca2+]i and force in rat resistance arteries. Pflügers Arch 1992; 420: 536–543. [DOI] [PubMed] [Google Scholar]
- 37.Dam VS, Boedtkjer DM, Nyvad J, et al. TMEM16A knockdown abrogates two different Ca2+-activated Cl- currents and contractility of smooth muscle in rat mesenteric small arteries. Pflugers Arch 2014; 466: 1391–1409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Mulvany MJ, Hansen OK, Aalkjaer C. Direct evidence that the greater contractility of resistance vessels in spontaneously hypertensive rats is associated with a narrowed lumen, a thickened media, and an increased number of smooth muscle cell layers. Circ Res 1978; 43: 854–864. [DOI] [PubMed] [Google Scholar]
- 39.Kudryavtseva O, Herum KM, Dam VS, et al. Downregulation of L-type Ca2+ channel in rat mesenteric arteries leads to loss of smooth muscle contractile phenotype and inward hypertrophic remodeling. Am J Physiol Heart Circ Physiol 2014; 306: H1287–H1301. [DOI] [PubMed] [Google Scholar]
- 40.Postnov DD, Tuchin VV, Sosnovtseva O. Estimation of vessel diameter and blood flow dynamics from laser speckle images. Biomed Opt Exp 2016; 7: 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Nyvad J, Mazur A, Postnov DD, et al. Intravital investigation of rat mesenteric small artery tone and blood flow. J Physiol 2017; 595: 5037–5053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sodhi K, Maxwell K, Yan Y, et al. pNaKtide inhibits Na/K-ATPase reactive oxygen species amplification and attenuates adipogenesis. Sci Adv 2015; 1: e1500781. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 43.Xie J, Ye Q, Cui X, et al. Expression of Rat Na/K-ATPase alpha2 enables ion pumping but not ouabain-induced signaling in alpha1-deficient porcine renal epithelial cells. Am J Physiol 2015; 309: C373–C382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Li Z, Zhang Z, Xie JX, et al. Na/K-ATPase mimetic pNaKtide peptide inhibits the growth of human cancer cells. J Biol Chem 2011; 286: 32394–32403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kenakin TP. A pharmacology primer: Theory, application, and methods, 3rd ed Burlington, MA, USA: Elsevier Inc., 2009. [Google Scholar]
- 46.Motulsky H, Christopoulos A. Fitting models to biological data using linear and nonlinear regression. A practical guide to curve fitting, San Diego CA: GraphPad Software, Inc., 2003. [Google Scholar]
- 47.Hansen JM, Schytz HW, Larsen VA, et al. Hemiplegic migraine aura begins with cerebral hypoperfusion: imaging in the acute phase. Headache 2011; 51: 1289–1296. [DOI] [PubMed] [Google Scholar]
- 48.Iizuka T, Takahashi Y, Sato M, et al. Neurovascular changes in prolonged migraine aura in FHM with a novel ATP1A2 gene mutation. J Neurol Neurosurg Psychiatry 2012; 83: 205–212. [DOI] [PubMed] [Google Scholar]
- 49.Jacobs B, Dussor G. Neurovascular contributions to migraine: MOVING beyond vasodilation. Neuroscience 2016; 338: 130–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hadjikhani N, Sanchez Del RM, Wu O, et al. Mechanisms of migraine aura revealed by functional MRI in human visual cortex. Proc Natl Acad Sci USA 2001; 98: 4687–4692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kitano A, Shimomura T, Takeshima T, et al. Increased 11-dehydrothromboxane B2 in migraine: platelet hyperfunction in patients with migraine during headache-free period. Headache 1994; 34: 515–518. [DOI] [PubMed] [Google Scholar]
- 52.Kallela M, Farkkila M, Saijonmaa O, et al. Endothelin in migraine patients. Cephalalgia 1998; 18: 329–332. [DOI] [PubMed] [Google Scholar]
- 53.Gallai V, Sarchielli P, Firenze C, et al. Endothelin 1 in migraine and tension-type headache. Acta Neurol Scand 1994; 89: 47–55. [DOI] [PubMed] [Google Scholar]
- 54.Leo L, Gherardini L, Barone V, et al. Increased susceptibility to cortical spreading depression in the mouse model of familial hemiplegic migraine type 2. PLoS Genet 2011; 7: e1002129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Segall L, Daly SE, Blostein R. Mechanistic basis for kinetic differences between the rat alpha 1, alpha 2, and alpha 3 isoforms of the Na,K-ATPase. J Biol Chem 2001; 276: 31535–31541. [DOI] [PubMed] [Google Scholar]
- 56.Crambert G, Hasler U, Beggah AT, et al. Transport and pharmacological properties of nine different human Na, K-ATPase isozymes. J Biol Chem 2000; 275: 1976–1986. [DOI] [PubMed] [Google Scholar]
- 57.Santoro L, Manganelli F, Fortunato MR, et al. A new Italian FHM2 family: clinical aspects and functional analysis of the disease-associated mutation. Cephalalgia 2011; 31: 808–819. [DOI] [PubMed] [Google Scholar]
- 58.Stoica A, Larsen BR, Assentoft M, et al. The alpha2beta2 isoform combination dominates the astrocytic Na+ /K+ -ATPase activity and is rendered nonfunctional by the alpha2.G301R familial hemiplegic migraine type 2-associated mutation. Glia 2017; 65: 1777–1793. [DOI] [PubMed] [Google Scholar]
- 59.Tavraz NN, Durr KL, Koenderink JB, et al. Impaired plasma membrane targeting or protein stability by certain ATP1A2 mutations identified in sporadic or familial hemiplegic migraine. Channels 2009; 3: 82–87. [DOI] [PubMed] [Google Scholar]
- 60.Sandow SL, Grayson TH. Limits of isolation and culture: intact vascular endothelium and BKCa. Am J Physiol 2009; 297: H1–H7. [DOI] [PubMed] [Google Scholar]
- 61.Zhang L, Zhang Z, Guo H, et al. Na+/K+-ATPase-mediated signal transduction and Na+/K+-ATPase regulation. Fundam Clin Pharmacol 2008; 22: 615–621. [DOI] [PubMed] [Google Scholar]
- 62.Pulina MV, Zulian A, Berra-Romani R, et al. Upregulation of Na+ and Ca2+ transporters in arterial smooth muscle from ouabain-induced hypertensive rats. Am J Physiol 2010; 298: H263–H274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.MacKay CE, Knock GA. Control of vascular smooth muscle function by Src-family kinases and reactive oxygen species in health and disease. J Physiol 2015; 593: 3815–3828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Jordan MC, Quednau BD, Roos KP, et al. Cyclosporin A regulates sodium-calcium exchanger (NCX1) gene expression in vitro and cardiac hypertrophy in NCX1 transgenic mice. Ann N Y Acad Sci 2002; 976: 259–267. [DOI] [PubMed] [Google Scholar]
- 65.Aalkjaer C, Mulvany MJ. Effect of ouabain on tone, membrane potential and sodium efflux compared with [3H]ouabain binding in rat resistance vessels. J Physiol 1985; 362: 215–231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Zhang J, Ren C, Chen L, et al. Knockout of Na+/Ca2+ exchanger in smooth muscle attenuates vasoconstriction and L-type Ca2+ channel current and lowers blood pressure. Am J Physiol 2010; 298: H1472–H1483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Ren C, Zhang J, Philipson KD, et al. Activation of L-type Ca2+ channels by protein kinase C is reduced in smooth muscle-specific Na+/Ca2+ exchanger knockout mice. Am J Physiol 2010; 298: H1484–H1491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Ghosh B, Green MV, Krogh KA, et al. Interleukin-1beta activates an Src family kinase to stimulate the plasma membrane Ca2+ pump in hippocampal neurons. J Neurophysiol 2016; 115: 1875–1885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Alioua A, Mahajan A, Nishimaru K, et al. Coupling of c-Src to large conductance voltage- and Ca2+-activated K+ channels as a new mechanism of agonist-induced vasoconstriction. Proc Natl Acad Sci USA 2002; 99: 14560–14565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Kimura J, Noma A, Irisawa H. Na-Ca exchange current in mammalian heart cells. Nature 1986; 319: 596–597. [DOI] [PubMed] [Google Scholar]
- 71.Wang XQ, Yu SP. Novel regulation of Na, K-ATPase by Src tyrosine kinases in cortical neurons. J Neurochem 2005; 93: 1515–1523. [DOI] [PubMed] [Google Scholar]
- 72.Liang M, Tian J, Liu L, et al. Identification of a pool of non-pumping Na/K-ATPase. J Biol Chem 2007; 282: 10585–10593. [DOI] [PubMed] [Google Scholar]
- 73.Knock GA, Shaifta Y, Snetkov VA, et al. Interaction between src family kinases and rho-kinase in agonist-induced Ca2+-sensitization of rat pulmonary artery. Cardiovasc Res 2008; 77: 570–579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Lu R, Alioua A, Kumar Y, et al. c-Src tyrosine kinase, a critical component for 5-HT2A receptor-mediated contraction in rat aorta. J Physiol 2008; 586: 3855–3869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Matsumoto T, Kobayashi T, Ishida K, et al. Enhancement of mesenteric artery contraction to 5-HT depends on Rho kinase and Src kinase pathways in the ob/ob mouse model of type 2 diabetes. Br J Pharmacol 2010; 160: 1092–1104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Somlyo AP, Somlyo AV. Ca2+ sensitivity of smooth muscle and nonmuscle myosin II: modulated by G proteins, kinases, and myosin phosphatase. Physiol Rev 2003; 83: 1325–1358. [DOI] [PubMed] [Google Scholar]
- 77.Suzuki N, Nakamura S, Mano H, et al. Galpha 12 activates Rho GTPase through tyrosine-phosphorylated leukemia-associated RhoGEF. Proc Natl Acad Sci USA 2003; 100: 733–738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Ark M, Ozdemir A, Polat B. Ouabain-induced apoptosis and Rho kinase: a novel caspase-2 cleavage site and fragment of Rock-2. Apoptosis 2010; 15: 1494–1506. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental material for Smooth muscle Ca2+ sensitization causes hypercontractility of middle cerebral arteries in mice bearing the familial hemiplegic migraine type 2 associated mutation by Christian Staehr, Lise Hangaard, Elena V Bouzinova, Sukhan Kim, Rajkumar Rajanathan, Peter Boegh Jessen, Nathan Luque, Zijian Xie, Karin Lykke-Hartmann, Shaun L Sandow, Christian Aalkjaer and Vladimir V Matchkov in Journal of Cerebral Blood Flow & Metabolism