

Abstract
Introduction:
PDAC is a lethal malignancy with a clear unmet need; almost all patients fail 1st, 2nd, and 3rd line multi-agent cytotoxic chemotherapy. The mammalian target of rapamycin (mTOR) has been identified as a key signaling node enhancing tumor survival and drug resistance in PDAC, hence it is considered a promising therapeutic target.
Areas covered:
We comprehensively reviewed the evidence from preclinical and phase I and II clinical trials, based on the authors’clinical experience and a pubmed, Chochrane library, Embase, and Google Scholar search everolimus + pancreatic cancer.
Expert opinion:
Everolimus has not demonstrated efficacy in PDAC; however, an mTOR inhibitor in combination with stroma-targeted therapies may be a promising area to explore in clinical trials.
Keywords: Everolimus, pancreatic adenocarcinoma, mTOR
1. Introduction:
PDAC is the seventh most deadly malignancy worldwide, with an estimated 418,000 new cases expected to be diagnosed by 2020[1,2]. The relative mortality of PDAC to other malignancies is higher in the United States than in the rest of the world. PDAC is the second and third leading cause of gastrointestinal malignancies and cancer-related deaths, respectively in the U.S.[3]. Compared to other malignancies, advanced PDAC has the lowest survival rate, with a median overall survival of 2–8 months and a five-year survival of 8.5%[3]. This is due to lack of effective screening tools, advanced stage at presentation (~80%) and limited therapeutic efficacy of current FDA approved drugs[4,5].
For advanced PDAC, single agent activity with fluoropyrimidines, gemcitabine, irinotecan, platinum compounds, and taxanes have a low objective response rates of <10% and dismal survival benefit (2 weeks); multi-agent combination therapies provide slightly higher response rates with modest survival benefit5. Two landmark clinical trials, FOLFIRINOX (ACCORD 11) and nab-paclitaxel plus gemcitabine (GA) (MPACT trial), have prolonged survival compared to best standard of care single agent gemcitabine (11.1 vs. 8.5 vs. 5.6 months, respectively)[6,7,8]. Patients who have progressed on first line FOLFIRINOX and continue to have a good performance status (ECOG 0–1) may be considered for gemcitabine-nab-paclitaxel, and vice versa. Nano-liposomal irinotecan plus 5-FU provides a next-line of treatment for metastatic PDAC patients, after progression on gemcitabine-based therapy[9]. There has been no prospective clinical trial comparing FOLFIRINOX versus GA. A retrospective study did not find a significant difference in the efficacy between the two regimens[10]. Multiple novel targeted therapies (including cetuximab, bevacizumab, axitinib, sorafenib, aflibercept, and pegylated recombinant human hyaluronidase) have failed to significantly extend overall survival (OS) compared to single agent gemcitabine, while erlotinib, an epidermal growth factor receptor (EGFR) inhibitor, in combination with gemcitabine showed a marginal clinical benefit (Hazard Ratio 0.81, CI 95% CI, 0.69 to 0.99)[11]. The search for clinically effective therapies for advanced PDAC continues to be challenging.
Omic technologies have been used successfully to subtype and identify potential molecular targets for PDAC. The mitogen-activated protein kinase (MEK), the EGFR and its downstream signaling intermediates, the extracellular signal-regulated kinase (ERK) and phosphoinositide 3-kinase (PI3K)/Akt signaling pathways play integral roles in the survival, growth, and drug resistance in PDAC[12]. Signal transduction pathways involved in tumor cell proliferation or cell survival often are mutated in solid tumors, and molecularly targeted agents that interdict these pathways may represent a promising approach. Given the poor prognosis for gemcitabine and fluoropyrimidine-refractory advanced PDAC, there is an urgent clinical need for improved therapeutic strategies. Based on the unique molecular biology of PDAC, therapeutic agents that target cell fate and autophagy may hold the key for prolonged remissions. KRAS proto-oncogene is mutated in >90% of PDAC and is key to cell growth and proliferation. Gain of function KRAS mutations in amino acid residues 12, 13, and 61 enhance autonomous interactions with diverse downstream signaling pathways to promote tumor growth. Development of KRAS inhibitors is challenging and clinical trials based on KRAS inhibitors have been unsuccessful. Therefore, it is prudent to concentrate efforts to identify therapeutic targets downstream from KRAS. The few crucial downstream players of KRAS signaling include PI3K and AKT[13]. These two molecules connect upstream membrane growth factor receptors with several activated kinases, including mammalian target of rapamycin (mTOR). The mTOR pathway (Figure 1) is involved in the proliferation of cancer cells via angiogenesis and regulation of autophagy[14,15]. The activation of this pathway leads to the production of pro-angiogenic factors including vascular endothelial growth factor (VEGF) which in turn promote endothelial cell growth. mTOR also activates hypoxia-inducible factor 1 (HIF-1), through activation of its downstream mediators including the 40S ribosomal S6 kinases, thus promoting metastasis via activated transcription of FSCN1 and LASP1 in PDAC cells[16]. mTOR exists as part of 2 different functional complexes, mTORC1 and mTORC2. mTORC1 is rapamycin sensitive and activates downstream effectors via S6 kinase 1, HIF1alpha, 4EBP1, and SREBP, and thereby it is mostly involved in cell survival and angiogenesis. On the other hand, mTORC2 is rapamycin insensitive and activates RICTOR, which in turn phosphorylates PKC and AKT, thereby promoting cell survival and cell migration[17]. Everolimus (RAD 001) is a first generation mTOR inhibitor, proven to be effective in pancreatic neuroendocrine cancers, hormone receptor (HR)-positive breast cancers, and kidney cancers. In this review, we will focus on the preclinical studies and clinical trials highlighting the use of everolimus (Figure 2) in advanced PDAC[16,18].
Figure 1. The mTOR signaling pathway.

The key signaling pathway that regulates mTORC1 and mTORC2 is depicted above. mTOR is activated by signaling through the PI3K pathway, regulating cell growth, and proliferation. PTEN, TSC1, and TSC2 act as negative regulators of the mTOR pathway and inhibition of the aforementioned leads to hyperactivation of mTOR. mTOR1 signaling requires activation of the adaptor protein raptor and mTORC2, which is insensitive to acute rapamycin treatment, requires activation of rictor protein. Multiple signals from growth factors, amino acids, cellular energy status, and stress are integrated into mTORC1. Activated mTORC1 plays a major role in promoting cell growth and proliferation by stimulating various anabolic processes including protein, lipid, nucleotide synthesis, and ribosome biogenesis, and inhibiting catabolic processes such as autophagy. mTORC2 is regulated by growth factors and although mechanisms are poorly defined, it has been linked to cytoskeleton.
Abbv: IRS1:Insulin Receptor substrate 1; mTOR: mammalian target of rapamycin; PDK1: 3-phosphoinositide-dependent protein kinase 1; PTEN: Phosphatase and tensin homolog; PI3K: Phosphoinositide 3-kinase; TSC1: TSC Complex Subunit 1; TSC2 TSC Complex Subunit 2; RHEB: Ras homolog enriched in brain; S6K1: Ribosomal protein S6 kinase beta-1; eIF4: Eukaryotic initiation factor 4F; 4E-BP1: eIF4E-binding protein.
Figure 2: The mechanism of action of rapamycin and rapalogs.

Rapamycin (also known as sirolimus) is a potent immunosuppressive and anti-cancer agent that binds two proteins: FK506-binding protein (FKBP12) and FKBP-rapamycin-associated protein (FRAP). The 2.7°A structure (pdb:1FAP)18of the complex between the FRB domain of mTOR (cyan), rapamycin and FKBP12 (green) is shown. Rapamycin mediates FKBP12 dimerization with mTOR, which then blocks access to the mTOR kinase active site located in a deep cleft and hydrophobic aromatic pocket behind the FKBP12-rapamycin binding domain. Rapamycin binding to FKBP12 and subsequent selective association of the FKBP12-rapamycin complex with mTORC1 conveys highly sensitive and targeted mTORC1 inhibition. Chemical structures of sirolimus and the rapalogs (e.g. everolimus, temsirolimus, and ridaforolimus) all share a ‘central macrolide’ structure and have unique R groups at the C40 position implicating a common mechanism of mTOR inhibition [18]
Choi J, Chen J, Schreiber SL, Clardy J. Structure of the FKBP12-rapamycin complex interacting with the binding domain of human FRAP. Science. 1996;273(5272):239–242.
2. Preclinical and Clinical activity of everolimus in PDAC:
2.1. Preclinical studies:
See Table 1.
Table-1:
Summary of Preclinical Studies:
| Tx | Model | Results | Ref |
| Everolimus + gemcitabine | MIAPaCa / Panc-1 | Combo had an additive anti-proliferative effect. | 19 |
| EGFR and PI3K/AKT/mTOR pathway inhibitors | GS MIAPaCa-E vs GR MIAPaCa-M | GR – insensitive to EGFR, AKT, and PI3K inhibitors. Hypersensitive to everolimus. | 20 |
| Everolimus and sorafenib | MIAPaCa-2 and Panc-1 | Combination of the 2 reduced anti-proliferative versus monotherapy. | 21 |
| AZD8055 vs everolimus | PANC-1, AsPC-1, CFPAC-1, Capan-1, Capan-2, Mia-Paca2 | Neither everolimus nor AZD8055 could arrest the cell cycle at the G0/G1 phase. | 23 |
| Everolimus + sorafenib | PDAC xenografts | Combo demonstrated efficacy at higher dose versus monotherapy. | 24 |
| AZD8055 + radiation | PDAC xenografts | Combo has greater efficacy than single agent. | 29 |
| Trametinib (MEK inhibitor) + everolimus | PDAC xenografts | Combo is synergistic | 30 |
GR: gemcitabine resistant; PDAC: Pancreatic ductal adenocarcinoma.
2.1.1. In vitro Studies:
Preclinical studies of everolimus to treat PDAC have had mixed results. In human PDAC cell lines, everolimus has a sensitizing effect toward gemcitabine[19]. Human cell lines MIAPaCa and Panc-1 were treated with gemcitabine (1–106 nM) in combination with everolimus (0.001–1 μg/ml). The anti-proliferative effect of the treatment was assessed by BrdU test (calorimetric immunoassay proliferation). Treatment with gemcitabine alone had an expected dose dependent anti-proliferative effect. The addition of everolimus to gemcitabine showed an additive anti-proliferative effect (76.8–85.2%) with an increase in the inhibitory effect of collagen type I expression necessary for PDAC tumor microenvironment, thus providing promising novel adjunct to standard chemotherapy.
Another study aimed to compare the effect of everolimus on gemcitabine sensitive versus gemcitabine-resistant human PDAC cell lines[20]. MIAPaCa-E, which is relatively sensitive to gemcitabine (GS), and MIAPaCa-M which is highly resistant to gemcitabine (GR), were treated with various inhibitors of the EGFR and PI3K/AKT/mTOR pathways (everolimus dose up to 50 μM) in combination with gemcitabine (dose up to up to 800 μM). In comparison to GS cells, GR cells were resistant to all the tested inhibitors of EGFR, AKT, and PI3K, except for the mTOR inhibitor everolimus for which GR was almost twice as sensitive as GS (40% versus 25% inhibition detected). This was attributed to the effect of everolimus in decreasing phosphorylated mTOR protein in GR over GS cells, thus leading to greater inhibition of mTOR in GR cells. The ability of everolimus to overcome resistance in GR cells was demonstrated by high levels of Thr389 and Thr371 phosphorylated p70S6 protein (an mTOR substrate) in the GR cells treated with gemcitabine, but not in the cells treated with everolimus. mTOR inhibition by everolimus also led to G2 arrest in GR cells but G1 arrest in GS cells. Additionally, everolimus led to increased caspase 3/7 activation in GR cells relative to GS cells. The aforementioned study suggested that everolimus could enhance efficacy of gemcitabine-based chemotherapy in GR PDAC.
Interestingly, in another in vitro study, everolimus (5–8 μM) and sorafenib (5 nM) had the opposite effect on proliferation of PDAC cell lines MIAPaCa-2 and Panc-1[21]. The study found that the use of each agent had an anti-proliferative effect, while combining both agents showed antagonism with less of an anti-proliferative effect. This was attributed to the site of action of everolimus and sorafenib in the cell cycle, where everolimus causes arrest of cells in the late G1 phase, whereas sorafenib inhibits cells from transitioning into G1 phase. Thus, the arrest of cells from transitioning into G1 by sorafenib would lead to fewer cells being susceptible to the inhibitory effect of everolimus. Another interesting finding was that both drugs used as single agents or in combination were ineffective on a third PDAC cell line BXPC-3, which is KRAS wild type, while MIAPaCa-2 and Panc-1 have additional pathogenic mutations in TP53, CDKN2A, and SMAD4.
The effect of first generation mTOR inhibitors, like everolimus, is curtailed by positive feedback relief of the insulin-like growth factor-1 receptor (IGF-1R)/AKT mTOR complex 2 on PDAC cell growth[22]. In a preclinical study, AZD8055, a second generation mTOR inhibitor, which overcomes activation of the (IGF-1R)/AKT mTOR complex 2 was compared to everolimus in six PDAC cell lines (PANC-1, AsPC-1, CFPAC-1, Capan-1, Capan-2 and Mia-Paca2)[23]. By flow cytometry, neither everolimus nor AZD8055 could arrest the cell cycle at the G0/G1 phase. Additionally, there was no large difference in cell viability with either treatment after 72 hours. The failure of AZD8055 to inhibit cell growth despite its mTORC1/C2 dual inhibition was further investigated by treating three cell lines with AZD8055 and examined by Western blotting and Akt in vitro kinase activity. On Western blotting, phosphorylation of Akt (S473/T308), mTOR substrates S6 (ribosomal protein S6) and 4E-BP1 (the eukaryotic translation inhibition factor 4E-binding protein 1) were synchronous, with a nadir in phosphorylation at 1 hour and a subsequent increase at 4, 8, and 24 hours. Akt kinase activity also showed an initial decrease at 1 hour after treatment with AZD8055, with a gradual increase at 4, 8, and 24 hours. The authors concluded that these data suggest a temporal inhibition of Akt activity by AZD8055 and that Akt re-phosphorlylation can be attributed to PDAC cell resistance to AZD8055. The mechanism behind AZD8055’s induction of re-phosphorylation of Akt was further investigated in the same study. It was found that AZD8055 induces EGFR up-regulation via Akt inhibition. AZD8055, but not everolimus, was found to induce EGFR over-expression and activation in association with the transient inhibition of Akt tested by a phospho-RTK array, Western blotting, and mRNA levels. Thus, combined inhibition of mTOR by AZD8055 and EGFR by erlotinib showed effective transcriptional activity by Western blotting of downstream proteins, synergistically enhanced cell growth inhibition (60% compared to control), increased apoptosis (30% compared to control) and significantly reduced tumor volume in vivo of PDAC xenografts (p <0.05). This was not the case with either erlotinib or AZD8055 alone. Table-1 provides a summary of in vitro and in vivo studies.
2.1.2. In vivo studies:
In an in vivo study of PDAC xenografts in mice, the combination of everolimus plus sorafenib was compared to the use of each drug alone by measuring tumor volume progression[24]. Sorafenib, a multi-kinase inhibitor acts, by inhibiting Raf kinases (CRAF, BRAF and mutated BRAF) and cell surface receptor tyrosine kinases (VEGFR-1, VEGFR-2, VEGFR-3, PDGFR-beta, cKIT, FLT-3, RET, and RET/PTC), thus inhibiting tumor growth and angiogenesis[25]. Treatment for 5 weeks with 0.5 mg/kg of everolimus and 10 mg/kg of sorafenib, had no effect on tumor volume progression whether as single agents or in combination. However, after dose escalation to 1 mg/kg of everolimus and 20 mg/kg of sorafenib for 5 weeks with both agents, it was found that the combination of both drugs had a greater efficacy in decreasing tumor progression than either drug alone. The mean tumor to control ratio of tumor volume was 0.2 for the combination as compared to 0.6 for treatment with everolimus alone and 0.5 for treatment with sorafenib alone. This was attributed to an additive effect of inhibiting both the MAPK and the PI3K-AKT-mTOR pathways and thereby inhibiting both compensatory and collateral signaling pathways thus overcoming resistance to monotherapy. The synergistic effect of everolimus plus sorafenib was also demonstrated in melanoma and hepatocellular carcinoma[26,27,28].
Concurrent chemo-radiation therapy is widely used on PDAC, particularly for locally advanced/borderline resectable disease or for palliative intent in metastatic disease. Wei F et al showed that ionizing radiation promoted mTOR expression, thus inhibition of tumor xenografts with AZD8055 (500 nM) synergistically with radiation (0–10 Gy) significantly increased anti-tumor effect compared to either treatment modality alone[29].
In the era of precision oncology, identifying known gene vulnerabilities and exploiting them has become a promising tool of personalized medicine. Witkiewicz AK et al, using high-throughput screening statistics of patient-derived PDAC cells lines with therapeutic vulnerabilities, uncovered that the mitogen-activated protein kinase (MEK) inhibitor (trametinib) in combination of several agents (e.g. everolimus) could translate disease control in vivo. The combination of everolimus plus trametinib had a synergistic effect on decreasing the tumor volume in a mouse xenograft model (EMC519 PDX; resistant to trametinib + docetaxel and trametinib + dasatinib, but sensitive to trametinib + everolimus)[30]. This has led to clinical studies evaluating the combination of mTOR inhibitors with EGFR and MEK inhibitors as outlines below.
Mechanistic translational studies led to evaluating everolimus in PDAC patients to identify its toxicity profile and efficacy as a single agent or in combination with systemic or targeted therapies on selected targeted biological vulnerabilities.
2.2. Early clinical trials:
Everolimus has been tested in early clinical trials with disappointing results as a single agent, although modest activity has been observed when used in combination with other agents. The most relevant early stage clinical trials are presented in Table 2, and a summary of the basic characteristics of the drugs in these trials are presented in the drug information table. A rationally based dose of everolimus was assessed in a dose escalation phase I pharmacodynamics study (PDAC N=4 [7%], out of a total of 55 patients with other solid tumors)[31]. The recommended dose for development was 10 mg/day or 50 mg/week. Dose limiting toxicities (DLTs) were observed at 10 mg/day in one patient (grade 3 stomatitis), and at 70 mg/week four patients experienced DLTs that included grade 3 stomatitis (2 patients) and grade 3 anemia and hyperglycemia (1 patient each). There was one partial response (PR, colorectal cancer), 3 stable disease (SD, 1 renal cell carcinoma and 2 breast cancer), and no objective responses in PDAC. The study demonstrated an increase in pAkt and mTOR activation due to feedback activation of the upstream signaling through the insulin-like growth factor 1 receptor (IGF-1R). This feedback activation of the PI3K/Akt/mTOR (PAM) pathway plays a key role in the attenuation of cellular response to mTOR inhibition. This was not tumor specific as it was found in the skin biopsies and did not occur in all patients’ tumor tissue, and it was not always sustained between doses. However, targeting tumors with both IGF-1R plus mTOR inhibitors could be a strategy to inhibit the PAM pathway reactivation.
Table-2:
Summary of Early Clinical Studies:
| Tx | Trial | Population | Results | Ref |
| Everolimus | Dose escalation Phase I | Patients with refractory advanced solid tumors including PDAC. | 0% ORR; increased pAkt and mTOR activation. | 31 |
| Everolimus | Phase II | GR metastatic PDAC | 0% ORR and 7% SD. | 32 |
| Everolimus | Phase II | PIK3CA amplified/mutated and/or PTEN loss in advanced refractory solid tumors. | 0% ORR/SD. | 33 |
| Everolimus + erlotinib | Phase II | Advanced PDAC | 0% ORR/SD. | 34 |
| Capecitabine + cetuximab + everolimus | Phase I/II | Advanced PDAC | Everolimus limited to 5 mg daily; 6.5% PR, 16% SD; mOS 5 months. | 35 |
| Capecitabine + everolimus | Followup phase II | Advanced PDAC | 6% ORR; 32% SD; mOS 8.9 months; 45% hyperglycemia. | 37 |
| Everolimus + gemcitabine + cisplatin | Phase I, (3+3) | Advanced solid tumors | Cohort I: 1 PDAC pt had CR, Cohort II: 0%ORR, Cohort III: 0% ORR. Overall 18.5% ORR and 66.6% DCR. | 38 |
| Everolimus + gemcitabine | Phase I | Advanced PDAC | MTD: gemcitabine 400 mg/m2/week and everolimus 5mg daily. 78% clinical benefit rate, 65% SD, and 13% PR. | 39 |
| Everolimus + trametinib | Phase Ib | Advanced refractory solid tumors | 33% serious adverse event; PDAC pt 5%PR, 29%SD. Phase II dose not identified. | 41 |
| Everolimus + ribociclib | Phase I (3+3) | Advanced PDAC refractory to 5-fluorouracil (5-FU) and gemcitabine-based chemotherapy | Ongoing – endpoints: PFS, OS, adverse events, best response. | 42 |
CR: complete response; DCR: disease control rate; GR: gemcitabine resistant; MTD: maximum tolerated dose; OS: overall survival; ORR: overall response rate; PDAC: pancreatic ductal adenocarcinoma; PFS: progression free survival; PR: partial response; SD: stable disease.
A multi-national, single-arm, phase II study with a different dosing schedule of everolimus (10 mg daily) in gemcitabine refractory metastatic PDAC patients did not show clinically relevant anti-tumor effects[32]. Treatment was tolerated well with modest side effects, most commonly thrombocytopenia and mild hyperglycemia. Good compliance was maintained with only 2 patients reporting missing more than one tablet. Of 33 patients, 21% of patients (N=7) had SD at first restaging scans after 2 months with no complete responses (CRs) or PRs. One patient had a biochemical response with >50% decrease in cancer antigen 19–9 (CA19–9). The study concluded that everolimus was not effective as a single agent and recommended pursuing further studies with everolimus in combination with other agents targeting upstream components of the PAM pathway or patients with pathogenic mutations in the aforementioned pathway.
A single-arm, open label phase II trial investigated the role of everolimus (10 mg/daily on 28 day cycle) in PIK3CA amplified/mutated and/or PTEN loss in advanced refractory solid tumors. Of 10 patients (1 PDAC), none had CRs or PRs and 4 patients had SD (2 gastric, 1 colorectal, and 1 cholangiocarcinoma). There were no new safety concerns, but the study did not meet its primary objective[33]. The aforementioned studies showed no significant activity for everolimus as a single agent in patients with PDAC and hence investigators pursued combination treatments.
A phase II study in patients with advanced PDAC investigated the combination of everolimus (30 mg weekly) plus erlotinib (150 mg daily) in 16 adult patients with advanced PDAC[34]. Disease progression was observed after 1 cycle in 15 patients and one patient was non-evaluable. There was one grade 4 toxicity of hyponatremia, six grade 3 toxicities which included cholangitis (n = 3), diarrhea (n = 1), hyperglycemia (n = 1), and fatigue (n = 1). There were no objective responses and/or disease stability. One proposed hypothesis that may explain the negative results is that mTOR inhibitors increase Akt phosphorylation and activation, along with cyclin D expression thus promoting proliferation of cancer cells[21]. This supports the premise of investigating an IGF-1R plus mTOR dual inhibition approaches. Another reason for failure was that everolimus was dosed Q1weekly while daily dosing of everolimus demonstrated a more sustained inhibition of ribosomal S6 kinase when compared to Q1weekly dosing in early clinical trials[30].
A statistically significant survival advantage was previously shown with combination chemotherapy which included FOLFIRINOX, gemcitabine + nab-paclitaxel and gemcitabine + erlotinib versus gemcitabine monotherapy in PDAC[6,7]. This has led to studies investigating the additional benefit that targeted drugs, such as MAPK and EGFR resistance pathway inhibition, may offer when combined with systemic chemotherapy agents to attain higher responses.
A phase I/II multi-center, open label study, enrolled patients with advanced PDAC and investigated the combination of capecitabine (oral fluoropyrimidine; dose 600–800 mg/m2 bid, 2 weeks on and one week off, every 3 weeks), cetuximab (EGFR monoclonal antibody; a fixed dose of 250 mg/m2, with a start-up dose of 400 mg/m2, weekly treatment), and everolimus (5–10 mg/day)[35]. The maximum tolerated dose (MTD) was the standard dose cetuximab in addition to 5 mg/day of everolimus and 600mg/m2 bid, 2 weeks on and one week off, of capecitabine. The observed DLTs were mucositis, rash, and hand-foot syndrome. On the phase II part, 31 patients with advanced PDAC were treated; 2 patients (6.5%) exhibited PRs and 5 patients (16.1%) had SD. A median OS of 5 months was lower than historical controls (MPACT and ACCORD trials). Most notably, 74% of patients experienced grade 3–4 treatment-related toxicities. Hyperglycemia, a known side effect of everolimus, was more pronounced in this study than in other everolimus monotherapy PDAC trials. The combination was found to have a high toxic burden with low clinical efficacy. The poor outcome of the study was attributed by the authors to be related to toxicities limiting the dose of everolimus to 5 mg daily. Furthermore, the authors hypothesized that the desmoplastic and fibrotic nature of PDAC with poor vascularization could lead to suboptimal drug delivery of cetuximab. Moreover, the predominance of KRAS mutations in PDAC is the cause for a poor response to anti-EGFR treatment with cetuximab[36] and everolimus.
On a follow up phase II trial in advanced PDAC, after progression on first-line, a higher dose of capecitabine (dose; 1000 mg/m2 BID day 1–14) and everolimus (10 mg/daily-21 day cycle) eliminating cetuximab[37] was evaluated. The study showed a 6% response rate in 31 enrolled patients and 32% with SD and the median OS of 8.9 months. Grade 3/4 treatment-related toxicities remained high, with 45% of patient having hyperglycemia resulting in dose adjustments. Other commonly reported adverse events include stomatitis, fatigue, and hand-foot syndrome. Hence, everolimus plus capecitabine had minimal efficacy with increased toxicity when indirectly compared to other regimens.
Other trials that investigated a combination regimen included a phase I, 3+3 modified Fibonacci dose escalation study, that investigated a 2-drug combination regimen of everolimus plus gemcitabine (Cohort I, 12 patients) with the addition of cisplatin (cohort II, 15 patients)[38]. In addition, an expansion cohort (cohort III) was planned after determining the MTD in patients with cholangiocarcinoma or gallbladder carcinoma. Everolimus was given on Monday-Wednesday-Friday (M/W/F) or daily depending on the dose level and gemcitabine and cisplatin were administered on days 1 and 8 of a 21-day cycle. The MTD in cohort I was 5 mg of everolimus M/W/F and gemcitabine (800 mg/m2). In cohort II, the everolimus MTD was determined to be 5mg M/W/F when combined with gemcitabine (600 mg/m2) and cisplatin 12.5 mg/m2. All DLTs were hematologic (thrombocytopenia, neutropenia, and leucopenia). Two patients in cohort I had CRs; a patient with refractory metastatic primary peritoneal carcinoma was on the trial for 4 months prior to progression. The second patient had recurrent PDAC and came off the trial after declining further treatment and had CR as a best response for one cycle. In cohort II, two patients had a PR one patient with metastatic ampullary carcinoma who progressed on cycle 7 and a patient with metastatic pheochromocytoma who had best response for one cycle. No patients in cohort III experienced a response and the overall response rate (ORR) and disease control rate (DCR) for all cohorts was 18.5% and 66.6%, respectively[38]. In addition, another phase I study investigated the combination of everolimus plus gemcitabine[39]. Of 27 patients, DLTs were observed in two patients with liver toxicities and one neutropenia leading to discontinuation from trial and the MTD was determined to be 400 mg/m2/week gemcitabine in combination with 5mg everolimus daily. Although 81% of patients had a suspected related adverse event and 41% experienced at least one serious adverse event, the authors reported tolerability of this regimen with no new safety concerns. Clinical benefit rate was observed in 78% (N=18) of patients, with 65% having SD (N=15), 13% PR (N=13) and no CRs were seen. Authors concluded no new safety concerns and that an expansion cohort phase II clinical trial could potentially corroborate efficacy outcomes.
A phase I study combination of sorafenib plus everolimus in patients with solid tumors was reported as an abstract at the 2011 American Society of Clinical Oncology (ASCO)[40]. Although no DLTs were observed, at an everolimus dose of 10mg and sorafenib 400 mg daily, grade 3 toxicities were reported and included upper respiratory tract infection, leukopenia, and thrombocytopenia, in addition to cardiac arrest, potentially due to arrhythmia. Based on these observations the MTD was defined as 7.5 mg of everolimus and 400mg of sorafenib daily. Of 9 evaluable patients, one patient with PDAC had progression of disease and 8 patients with other solid tumors had SD.
Based on preclinical data supporting a role for combining mTOR plus MEK inhibitors, a phase Ib trial studied the combination of everolimus and trametinib (GSK1120212) in patients with advanced refractory solid tumors[41]. Serious adverse events were noted in 33% of patients, of which the most common were mucositis, stomatitis, fatigue, and diarrhea. Of the 67 enrolled heavily pretreated (>3 lines) patients, 5 (7%) achieved PR and 21 (31%) SD. In addition, of the 21 patients with PDAC, PR was observed in 1 patient (5%) and SD in 6 patients (29%). The concern with this trial was the treatment-related adverse events that limited identifying a recommended phase 2 dose. The authors of the trial concluded that the modest activity seen in PDAC patients may be due to inability to combine both drugs at an optimal biologic dose and hence further expansion was aborted[41]. Table-2 provides a summary of early clinical trials.
2.3. Current clinical trials:
There are ongoing clinical trials investigating the role of mTOR inhibitor therapy in PDAC. Ongoing clinical trials on everolimus in PDAC includes a single-arm, open label phase I (3+3 design) to find the recommended dose of a CDK 4/6 inhibitor (ribocilib, LEE011) plus everolimus in 44 patients with metastatic PDAC refractory to 5-fluorouracil (5-FU) and gemcitabine-based chemotherapy []. Treatment regimen includes everolimus 2.5mg once daily continuously plus ribociclib 150, 200, 250, or 300 mg once daily for 21 days of 28-day cycle. The primary outcome is evaluating the progression free survival (PFS) at 8 weeks by imaging. Secondary outcomes include PFS, OS, adverse events, and the best response[42]. This study was conceptualized based on preclinical studies highlighting a potential efficacy for combination of CDK4/6 inhibitors when combined with other compounds including an mTOR inhibitor such as AZD0855 (mTOR inhibitor)[43,44].
3. Conclusion:
PDAC is a lethal disease with poor prognosis. This is due to the advanced stage at presentation, aggressive biology, intense fibrosis, and an immune suppressive tumor microenvironment. All these factors contribute to drug resistance. Current multi-agent cytotoxic systemic therapies modestly improve OS and quality of life in the front-line and second-line setting compared to best supportive care. Due to the modest effectiveness of these combined cytotoxic systemic regimens with toxicities, biology based therapeutic strategies should be encouraged. Although preclinical data supported mTOR inhibition (everolimus) in PDAC, treatment as a single agent and/or with combination with upstream (EGFR) and downstream (CDK4/6) inhibitors or cytotoxic therapies (e.g. gemcitabine) have not resulted in meaningful therapeutic responses. Nonetheless, participation in the many early phase clinical trials with novel investigational agents should be encouraged in patients with PDAC. Perhaps a potential avenue would be to explore mTOR inhibitor combined with stroma-targeted drugs and drugs inhibiting autophagy. It is conceivable that mTOR inhibitors could potentially be combined with immune checkpoint inhibitors, for synergy. Table-3 provides highlights of this review.
Table-3:
Article highlights box:
| • Preclinical: |
| • Preclinical studies demonstrated efficacy of everolimus as a mono-therapy. Combinations with agents targeting the EGFR, MEK, RAF/MAPK, CDK4/6 and chemotherapy have shown promise in mouse stu-dies. |
| • Everolimus plus trametinib demonstrated efficacy in xenograft models (EMC519 PDX). |
| • Gemcitabine resistant pancreatic cancer cell lines were sensitive to everolimus (MIAPaCa-M). |
| • Early Clinical: |
| • Early Clinical Trials investigating the efficacy of everolimus alone or combined with capecitabine, cetuximab, gemcitabine, cisplatin, sora-fenib, and trametinib failed to demonstrate efficacy and was toxic to patients. |
| • Potential Combined Targets: |
| • BET, HGF/c-MET, CTGF, Rho-kinase inhibitors other stroma targeted agents. |
4. Expert opinion:
Despite preclinical mechanistic translational research, which provided a strong rationale for combining mTOR inhibitors with drugs that target treatment-emergent resistance pathways in PDAC, early clinical trials failed to mirror such activity. Everolimus combined with EGFR-targeted drugs such as cetuximab or erlotinib, Raf-MAP kinases inhibitors such as sorafenib, MEK-targeted drugs such as trametinib, or FDA approved chemotherapy such as gemcitabine or capecitabine failed to achieve promising responses in PDAC. In addition, contrary to preclinical data, everolimus failed to achieve meaningful responses in gemcitabine refractory PDAC patients[20,32]. Moreover, the conflicting in vitro and in vivo results of the combination of everolimus and sorafenib is interesting. Many previous preclinical trials demonstrated conflicting results between in vitro and in vivo studies, however, perhaps beyond a certain dose-threshold both drugs can work synergistically as seen when everolimus was increased to 1mg/kg and sorafenib to 20 mg/kg in vivo. This can potentially be due to everolimus inhibiting an escape resistant pathway beyond a specific dose-threshold.
The failure to achieve meaningful responses are multi-factorial due to reactivation of upstream RTK driven pathways, poor vascularization due to the prominent stromal fibrosis, and toxicities limiting optimal biologic dose delivery of therapies. The elephant in the room, the desmoplastic reaction due to tumor-stroma interactions with deposition of collagen, hyaluronic acid, fibronectin, and proteoglycans by pancreatic cancer stellate cells has not been effectively targeted despite trials with molecular targets such as hedgehog inhibitors and hyaluronidase[13,45,46,47]. Pegvorhyaluronidase alfa (PEGPH20) has been studied in a phase II trial and when combined with chemotherapy (gemcitabine plus nab-paclitaxel) in patients with metastatic PDAC and there was an improvement in PFS and ORR in patients with hyaluronan-high tumors[48]. In addition, inhibition of mTOR causes immune suppression which likely amplifies the existing immune suppressive tumor microenviroment (TME) in PDAC[49]. Recently, FG-3019 (pamrevlumab, P), a monoclonal antibody targeting connective tissue growth factor (CTGF), provided promising results when combined with nab-paclitaxel plus gemcitabine in patients with locally advanced unresectable PDAC. This combination showed improvement in the resection rate and median OS of arm A(GN+P) compared to arm B(GN) of 43% vs 25% and 17.6 months vs 13.2 months, respectively[50]. This has led the FDA to grant a fast-track designation to P for the treatment of patients with locally advanced, unresectable PDAC. Moreover, other promising stromal targeted agents include BET, HGF/c-MET, and Rho-kinase13 are under investigation. Combination of an mTOR inhibitor such as everolimus with one of the aforementioned inhibitors may be a strategy to pursue in future clinical studies. Given the marginal efficacy of current standard of care chemotherapy regimens with cumulative toxicities, it would be reasonable to study mTOR inhibition post-chemotherapy in the maintenance setting with/without stroma-directed therapies. An anecdotal case of a PDAC patient with an STK 11 LOH mutation (tumor suppressor gene mutated in Peutz-Jeghers Syndrome) achieved a partial remission for ~9.0 months with everolimus[51] highlighting mTOR as a key regulator of cell signaling. Ongoing studies may identify several genes or mRNA or proteomics signatures, which could potentially identify PDAC subtypes with high mTOR activity. This could be useful as a biomarker in identifying PDAC patients who could benefit from everolimus or other mTOR inhibitors. Another reason for lack of clinical activity in PDAC patients could be escape of mTORC2 and effectors as everolimus inhibits mTORC1 only[52]. In a preclinical study, Driscoll et al, demonstrated that targeting the mTOR pathway, using a dual mTORC1/2 inhibitor AZD2014, increased tumor regression in PDAC. A recent study by Fricke at al, showed activity of TAK-228, a dual mTORC1/2 inhibitor in PI3K mutated colon cancer patient-derived organoid models refractory to everolimus[53]. This response correlated with reduction in the phosphorylation of 4EBP1 and RPS6. They concluded that mTORC1/2 inhibition is sufficient to overcome resistance to everolimus. A further potential avenue includes combining drugs that inhibits autophagy with everolimus, as inhibition of autophagy may sensitize the tumor to metabolic demands of mTOR signaling. Drugs that inhibit mTOR increase autophagy and cancer cell survival and hence combining everolimus with a drug that selectively targets autophagy can potentially be promising. Everolimus can modulate T-lymphocyte homeostasis with an enhanced antitumor T-cell response during disease control whereas immunosuppression develops at the time of progression. Hence, re-introducing an immune checkpoint inhibitor at the time of progression may be a good trial design to pursue. Thus we opinion, scratching the surface in the elucidation of the role of mTOR1/2 inhibition via rapalogs and mTOR Ser/Thr kinase inhibition. Further preclinical and clinical studies are needed to understand the role of this important pathway in targeting PDAC.
Drug Summary
| Drug name | Phase | Indication | Mechanism of action (rsp) | Route of administration (rsp) | Chemical structure | Pivotal trial |
|---|---|---|---|---|---|---|
| Everolimus | Dose escalation Phase I | Patients with refractory advanced solid tumors including PDAC. | mTORC1 inhibitor | Oral |
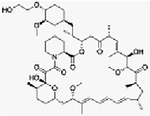 (C53H83NO14) |
31 |
| Everolimus | Phase II | GR metastatic PDAC | mTORC1 inhibitor | Oral | C53H83NO14 | 32 |
| Everolimus | Phase II | PIK3CA amplified/mutated and/or PTEN loss in advanced refractory solid tumors. | mTORC1 inhibitor | Oral | C53H83NO14 | 33 |
| Everolimus + erlotinib | Phase II | Advanced PDAC | mTORC1 inhibitor + EGFR inhibitor | Oral + oral | C53H83NO14 +  (C22H23N3O4) |
34 |
| Capecitabine + cetuximab + everolimus | Phase I/II | Advanced PDAC | Thymidylate synthase inhibitor + EGFR inhibitor + mTORC1 inhibitor | Oral + IV + oral |
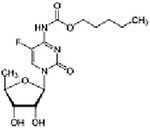 (C15H22FN3O6) + Chimeric antibody C53H83NO14 |
35 |
| Capecitabine + everolimus | Followup phase II | Advanced PDAC | Thymidylate synthase inhibitor + mTORC1 inhibitor | Oral + oral | C15H22FN3O6 C53H83NO14 |
37 |
| Everolimus + gemcitabine + cisplatin | Phase I, (3+3) | Advanced solid tumors | mTORC1 inhibitor + cytotoxic + cytotoxic | Oral + IV + IV | C53H83NO14+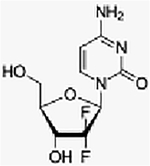 (C9H11F2N3O4) + [Pt(NH3)2Cl2] |
38 |
| Everolimus+ gemcitabine |
Phase I | Advanced PDAC | mTORC1 inhibitor + cytotoxic | Oral + IV | C53H83NO14 C9H11F2N3O4 |
39 |
| Everolimus + trametinib | Phase Ib | Advanced refractory solid tumors | mTORC1 inhibitor + MEK inhibitor | Oral + oral | C53H83NO14+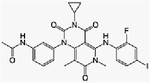 (C26H23FIN5O4) |
41 |
| Everolimus + ribociclib | Phase I (3+3) | Advanced PDAC refractory to 5-fluorouracil (5-FU) and gemcitabine-based chemotherapy | mTORC1 inhibitor + CDK4/6 inhibitor | Oral + oral | C53H83NO14 +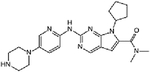 (C23H30N8O) |
42 |
Acknowledgements
The authors would like to thank Dr James Mancuso for reviewing and editing the manuscript.
Funding
The work of the authors is funded by U.S. Department of Health and Human Services, National Institutes of Health, National Cancer Institute Grant 2P30 CA023074
Declaration of interest
The authors have no relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript. This includes employment, consultancies, honoraria, stock ownership or options, expert testimony, grants or patents received or pending, or royalties.
Footnotes
Reviewer disclosures
Peer reviewers on this manuscript have no relevant financial or other relationships to disclose
References
Papers of special note have been highlighted as either of interest (•) or of considerable interest (••) to readers
- 1.Are C, Chowdhury S, Ahmad H, et al. Predictive global trends in the incidence and mortality of pancreatic cancer based on geographic location, socio-economic status, and demographic shift. J Surg Oncol 2016;114(6):736–42. [DOI] [PubMed] [Google Scholar]
- 2.Ilic M, Ilic I. Epidemiology of pancreatic cancer. World J Gastroenterol [Internet] 2016. [cited 2018 Dec 19];22(44):9694–705. Available from: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5124974/ [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Siegel RL, Miller KD, Jemal A. Cancer statistics, 2018. CA Cancer J Clin 2018;68(1):7–30. [DOI] [PubMed] [Google Scholar]
- 4.Kamarajah SK, Burns WR, Frankel TL, et al. Validation of the American Joint Commission on Cancer (AJCC) 8th Edition Staging System for Patients with Pancreatic Adenocarcinoma: A Surveillance, Epidemiology and End Results (SEER) Analysis. Ann Surg Oncol 2017;24(7):2023–30. [DOI] [PubMed] [Google Scholar]
- 5.Tempero MA, Malafa MP, Al-Hawary M, et al. Pancreatic Adenocarcinoma, Version 2.2017, NCCN Clinical Practice Guidelines in Oncology. J Natl Compr Cancer Netw JNCCN 2017;15(8):1028–61. [DOI] [PubMed] [Google Scholar]
- 6.Conroy T, Desseigne F, Ychou M, et al. FOLFIRINOX versus gemcitabine for metastatic pancreatic cancer. N Engl J Med 2011;364(19):1817–25. [DOI] [PubMed] [Google Scholar]; ***combined chemotherapy shows efficacy in PDAC.
- 7.Von Hoff DD, Ervin T, Arena FP, et al. Increased survival in pancreatic cancer with nab-paclitaxel plus gemcitabine. N Engl J Med 2013;369(18):1691–703. [DOI] [PMC free article] [PubMed] [Google Scholar]; ***combined chemotherapy shows efficacy in PDAC.
- 8.Burris HA, Moore MJ, Andersen J, et al. Improvements in survival and clinical benefit with gemcitabine as first-line therapy for patients with advanced pancreas cancer: a randomized trial. J Clin Oncol Off J Am Soc Clin Oncol 1997;15(6):2403–13. [DOI] [PubMed] [Google Scholar]; **1st trial to show improvement of advanced pancreatic cancer with gemcitabine.
- 9.Wang-Gillam A, Li C-P, Bodoky G, et al. Nanoliposomal irinotecan with fluorouracil and folinic acid in metastatic pancreatic cancer after previous gemcitabine-based therapy (NAPOLI-1): a global, randomised, open-label, phase 3 trial. Lancet Lond Engl 2016;387(10018):545–57. [DOI] [PubMed] [Google Scholar]; **Practice changing regimen in 2nd line pancreatic cancer.
- 10.Cartwright TH, Parisi M, Espirito JL, et al. Clinical Outcomes with First-Line Chemotherapy in a Large Retrospective Study of Patients with Metastatic Pancreatic Cancer Treated in a US Community Oncology Setting. Drugs - Real World Outcomes 2018;5(3):149–59. [DOI] [PMC free article] [PubMed] [Google Scholar]; **Only study comparing pancreatic cancer front line regimens retrospectively, no prospective clinical trial.
- 11.Moore MJ, Goldstein D, Hamm J, et al. Erlotinib plus gemcitabine compared with gemcitabine alone in patients with advanced pancreatic cancer: a phase III trial of the National Cancer Institute of Canada Clinical Trials Group. J Clin Oncol Off J Am Soc Clin Oncol 2007;25(15):1960–6. [DOI] [PubMed] [Google Scholar]
- 12.Waddell N, Pajic M, Patch A-M, et al. Whole genomes redefine the mutational landscape of pancreatic cancer. Nature 2015;518(7540):495–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chandana S, Babiker HM, Mahadevan D. Therapeutic trends in pancreatic ductal adenocarcinoma (PDAC). Expert Opin Investig Drugs 2019;28(2):161–77. [DOI] [PubMed] [Google Scholar]
- 14.Rowinsky EK. Targeting the molecular target of rapamycin (mTOR). Curr Opin Oncol 2004;16(6):564–75. [DOI] [PubMed] [Google Scholar]
- 15.Paquette M, El-Houjeiri L, Pause A. mTOR Pathways in Cancer and Autophagy. Cancers [Internet] 2018. [cited 2019 Jan 24];10(1). Available from: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5789368/ [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhao T, Ren H, Li J, et al. LASP1 is a HIF1α target gene critical for metastasis of pancreatic cancer. Cancer Res 2015;75(1):111–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Conciatori F, Ciuffreda L, Bazzichetto C, et al. mTOR Cross-Talk in Cancer and Potential for Combination Therapy. Cancers [Internet] 2018. [cited 2019 Jan 24];10(1). Available from: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5789373/ [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Choi J, Chen J, Schreiber SL, et al. Structure of the FKBP12-rapamycin complex interacting with the binding domain of human FRAP. Science 1996;273(5272):239–42. [DOI] [PubMed] [Google Scholar]
- 19.Tuncyurek P, Mayer JM, Klug F, et al. Everolimus and Mycophenolate Mofetil Sensitize Human Pancreatic Cancer Cells to Gemcitabine in vitro: A Novel Adjunct to Standard Chemotherapy? Eur Surg Res [Internet] 2007. [cited 2018 Dec 20];39(6):380–7. Available from: https://www.karger.com/Article/FullText/107356 [DOI] [PubMed] [Google Scholar]
- 20.Peng T, Dou QP. Everolimus Inhibits Growth of Gemcitabine-Resistant Pancreatic Cancer Cells via Induction of Caspase-Dependent Apoptosis and G2/M Arrest. J Cell Biochem 2017;118(9):2722–30. [DOI] [PubMed] [Google Scholar]
- 21.Pawaskar DK, Straubinger RM, Fetterly GJ, et al. Interactions of Everolimus and Sorafenib in Pancreatic Cancer Cells. AAPS J [Internet] 2012. [cited 2018 Dec 20];15(1):78–84. Available from: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3535103/ [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.O’Reilly KE, Rojo F, She Q-B, et al. mTOR inhibition induces upstream receptor tyrosine kinase signaling and activates Akt. Cancer Res 2006;66(3):1500–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wei F, Zhang Y, Geng L, et al. mTOR inhibition induces EGFR feedback activation in association with its resistance to human pancreatic cancer. Int J Mol Sci 2015;16(2):3267–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pawaskar DK, Straubinger RM, Fetterly GJ, et al. Synergistic interactions between sorafenib and everolimus in pancreatic cancer xenografts in mice. Cancer Chemother Pharmacol [Internet] 2013. [cited 2018 Dec 20];71(5):1231–40. Available from: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3752659/ [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.nexavar-pm-en-pt3.pdf [Internet]. [cited 2018 Dec 20];Available from: https://www.bayer.ca/omr/online/nexavar-pm-en-pt3.pdf
- 26.Lasithiotakis KG, Sinnberg TW, Schittek B, et al. Combined inhibition of MAPK and mTOR signaling inhibits growth, induces cell death, and abrogates invasive growth of melanoma cells. J Invest Dermatol 2008;128(8):2013–23. [DOI] [PubMed] [Google Scholar]
- 27.Molhoek KR, Brautigan DL, Slingluff CL. Synergistic inhibition of human melanoma proliferation by combination treatment with B-Raf inhibitor BAY43–9006 and mTOR inhibitor Rapamycin. J Transl Med [Internet] 2005. [cited 2018 Dec 20];3(1):39 Available from: https://translational-medicine.biomedcentral.com/articles/10.1186/1479-5876-3-39 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Newell P, Toffanin S, Villanueva A, et al. Ras pathway activation in hepatocellular carcinoma and anti-tumoral effect of combined sorafenib and rapamycin in vivo. J Hepatol 2009;51(4):725–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wei F, Liu Y, Guo Y, et al. miR-99b-targeted mTOR induction contributes to irradiation resistance in pancreatic cancer. Mol Cancer 2013;12:81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Witkiewicz AK, Balaji U, Eslinger C, et al. Integrated Patient-Derived Models Delineate Individualized Therapeutic Vulnerabilities of Pancreatic Cancer. Cell Rep 2016;16(7):2017–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tabernero J, Rojo F, Calvo E, et al. Dose- and schedule-dependent inhibition of the mammalian target of rapamycin pathway with everolimus: a phase I tumor pharmacodynamic study in patients with advanced solid tumors. J Clin Oncol Off J Am Soc Clin Oncol 2008;26(10):1603–10. [DOI] [PubMed] [Google Scholar]; *** Phase I trial with everolimus in refractory solid tumors.
- 32.Wolpin BM, Hezel AF, Abrams T, et al. Oral mTOR inhibitor everolimus in patients with gemcitabine-refractory metastatic pancreatic cancer. J Clin Oncol Off J Am Soc Clin Oncol 2009;27(2):193–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kim ST, Lee J, Park SH, et al. Prospective phase II trial of everolimus in PIK3CA amplification/mutation and/or PTEN loss patients with advanced solid tumors refractory to standard therapy. BMC Cancer 2017;17(1):211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Javle MM, Shroff RT, Xiong H, et al. Inhibition of the mammalian target of rapamycin (mTOR) in advanced pancreatic cancer: results of two phase II studies. BMC Cancer 2010;10:368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kordes S, Richel DJ, Klümpen H-J, et al. A phase I/II, non-randomized, feasibility/safety and efficacy study of the combination of everolimus, cetuximab and capecitabine in patients with advanced pancreatic cancer. Invest New Drugs [Internet] 2013. [cited 2018 Dec 20];31(1):85–91. Available from: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3553409/ [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bruns CJ, Solorzano CC, Harbison MT, et al. Blockade of the epidermal growth factor receptor signaling by a novel tyrosine kinase inhibitor leads to apoptosis of endothelial cells and therapy of human pancreatic carcinoma. Cancer Res 2000;60(11):2926–35. [PubMed] [Google Scholar]
- 37.Kordes S, Klümpen HJ, Weterman MJ, et al. Phase II study of capecitabine and the oral mTOR inhibitor everolimus in patients with advanced pancreatic cancer. Cancer Chemother Pharmacol [Internet] 2015. [cited 2018 Dec 20];75(6):1135–41. Available from: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4441736/ [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Costello BA, Borad MJ, Qi Y, et al. Phase I trial of everolimus, gemcitabine and cisplatin in patients with solid tumors. Invest New Drugs 2014;32(4):710–6. [DOI] [PubMed] [Google Scholar]
- 39.Joka M, Boeck S, Zech CJ, et al. Combination of antiangiogenic therapy using the mTOR-inhibitor everolimus and low-dose chemotherapy for locally advanced and/or metastatic pancreatic cancer: a dose-finding study. Anticancer Drugs 2014;25(9):1095–101. [DOI] [PubMed] [Google Scholar]
- 40.Nogova L, Mattonet C, Scheffler M, et al. The combination of sorafenib and everolimus in patients with solid tumors: Results of a phase I study. J Clin Oncol [Internet] 2016. [cited 2018 Dec 20];Available from: http://ascopubs.org/doi/abs/10.1200/jco.2011.29.15_suppl.e13613 [Google Scholar]
- 41.Tolcher AW, Bendell JC, Papadopoulos KP, et al. A phase IB trial of the oral MEK inhibitor trametinib (GSK1120212) in combination with everolimus in patients with advanced solid tumors. Ann Oncol Off J Eur Soc Med Oncol 2015;26(1):58–64. [DOI] [PubMed] [Google Scholar]
- 42.Weinberg BA, Wang H, Witkiewicz AK, et al. A phase I/II study of ribociclib plus everolimus in patients (pts) with metastatic pancreatic adenocarcinoma (mPAC) refractory to chemotherapy. J Clin Oncol [Internet] 2018. [cited 2018 Dec 20];Available from: http://ascopubs.org/doi/abs/10.1200/JCO.2018.36.15_suppl.TPS4150 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Franco J, Witkiewicz AK, Knudsen ES. CDK4/6 inhibitors have potent activity in combination with pathway selective therapeutic agents in models of pancreatic cancer. Oncotarget 2014;5(15):6512–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Franco J, Balaji U, Freinkman E, et al. Metabolic Reprogramming of Pancreatic Cancer Mediated by CDK4/6 Inhibition Elicits Unique Vulnerabilities. Cell Rep 2016;14(5):979–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Feig C, Gopinathan A, Neesse A, et al. The pancreas cancer microenvironment. Clin Cancer Res Off J Am Assoc Cancer Res 2012;18(16):4266–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ko AH, LoConte N, Tempero MA, et al. A Phase I Study of FOLFIRINOX Plus IPI-926, a Hedgehog Pathway Inhibitor, for Advanced Pancreatic Adenocarcinoma. Pancreas 2016;45(3):370–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Catenacci DVT, Junttila MR, Karrison T, et al. Randomized Phase Ib/II Study of Gemcitabine Plus Placebo or Vismodegib, a Hedgehog Pathway Inhibitor, in Patients With Metastatic Pancreatic Cancer. J Clin Oncol Off J Am Soc Clin Oncol 2015;33(36):4284–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hingorani SR, Zheng L, Bullock AJ, et al. HALO 202: Randomized Phase II Study of PEGPH20 Plus Nab-Paclitaxel/Gemcitabine Versus Nab-Paclitaxel/Gemcitabine in Patients With Untreated, Metastatic Pancreatic Ductal Adenocarcinoma. J Clin Oncol Off J Am Soc Clin Oncol 2018;36(4):359–66. [DOI] [PubMed] [Google Scholar]
- 49.Liss AS, Thayer SP. Therapeutic targeting of pancreatic stroma [Internet]. In: Grippo PJ, Munshi HG, editors. Pancreatic Cancer and Tumor Microenvironment. Trivandrum (India): Transworld Research Network; 2012. [cited 2018 Dec 20]. Available from: http://www.ncbi.nlm.nih.gov/books/NBK98931/ [PubMed] [Google Scholar]
- 50.Picozzi VJ, Rocha FG, Helton S, et al. Randomized, open-label trial of gemcitabine/nab-paclitaxel (G/NP) ± pamrevlumab (P) as neoadjuvant chemotherapy in locally advanced, unresectable pancreatic cancer (LAPC). J Clin Oncol [Internet] 2017. [cited 2018 Dec 20];Available from: http://ascopubs.org/doi/abs/10.1200/JCO.2017.35.4_suppl.365 [Google Scholar]
- 51.Klümpen H-J, Queiroz KCS, Spek CA, et al. mTOR inhibitor treatment of pancreatic cancer in a patient With Peutz-Jeghers syndrome. J Clin Oncol Off J Am Soc Clin Oncol 2011;29(6):e150–153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Driscoll DR, Karim SA, Sano M, et al. mTORC2 Signaling Drives the Development and Progression of Pancreatic Cancer. Cancer Res 2016;76(23):6911–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Fricke SL, Payne SN, Favreau PF, et al. MTORC1/2 inhibition as a therapeutic strategy for PIK3CA mutant cancers. Mol Cancer Ther [Internet] 2018. [cited 2019 Jan 26];molcanther.0510.2018. Available from: http://mct.aacrjournals.org/content/early/2018/11/13/1535-7163.MCT-18-0510 [DOI] [PMC free article] [PubMed] [Google Scholar]