

Abstract
The study aims to investigate the performance characteristics of the enantiomers of 11C-Me-NB1, a recently reported PET imaging probe that targets the GluN2B subunit of N-methyl-d-aspartate (NMDA) receptors. Methods: Reference compound Me-NB1 (inhibition constant for hGluN1/GluN2B, 5.4 nM) and the phenolic precursor were prepared via multistep synthesis. Following chiral resolution by high-performance liquid chromatography, enantiopure precursor compounds, (R)-NB1 and (S)-NB1, were labeled with 11C and validated in rodents using in vitro/ex vivo autoradiography, PET experiments, and dose–response studies. To illustrate the translational relevance, (R)-11C-Me-NB1 was validated in autoradiographic studies using postmortem human GluN2B-rich cortical and GluN2B-deficient cerebellar brain slices. To determine target engagement, receptor occupancy was assessed at different plasma concentrations of CP101,606, a GluN2B receptor antagonist. Results: The radiosynthesis of (R)- and (S)-11C-Me-NB1 was accomplished in 42% ± 9% (decay-corrected) radiochemical yields. Molar activity ranged from 40 to 336 GBq/μmol, and an excellent radiochemical purity of greater than 99% was achieved. Although (R)-11C-Me-NB1 displayed heterogeneous accumulation with high selectivity for the GluN2B-rich forebrain, (S)-11C-Me-NB1 revealed a homogeneous distribution across all brain regions in rodent brain autoradiograms and predominantly exhibited σ1-receptor binding. Similar to rodent brain, (R)-11C-Me-NB1 showed in postmortem human brain tissues higher binding in the cortex than in the cerebellum. Coincubation of the GluN2B-antagonist CERC-301 (1 μM) reduced cortical but not cerebellar binding, demonstrating the specificity of (R)-11C-Me-NB1 binding to the human GluN2B-containing NMDA receptor. In vivo specificity of (R)-11C-Me-NB1 in the GluN2B-expressing cortex, striatum, thalamus, and hippocampus was demonstrated by PET imaging in rodents. Applying GluN2B-antagonist eliprodil, an evident dose–response behavior was observed with (R)-11C-Me-NB1 but not with (S)-11C-Me-NB1. Our findings further underline the tightrope walk between GluN2B- and σ1-receptor–targeted imaging, illustrated by the entirely different receptor binding behavior of the 2 radioligand enantiomers. Conclusion: (R)-11C-Me-NB1 is a highly selective and specific PET radioligand for imaging the GluN2B subunit of the NMDA receptor. The entirely different receptor binding behavior of (R)-11C-Me-NB1 and (S)-11C-Me-NB1 raises awareness of a delicate balance that is underlying the selective targeting of either GluN2B-carrying NMDA or σ1-receptors.
Keywords: PET imaging, GluN2B, σ1-receptor, receptor occupancy, neurodegeneration
N-methyl-d-aspartate (NMDA) receptors are heterotetrameric assemblies of 3 distinct glutamate subunits—GluN1, GluN2A-D, and GluN3A/B—and regulate numerous vital processes, including neuronal development, synaptic plasticity, learning, and memory functions in the mammalian central nervous system (1). On engagement by the most abundant excitatory neurotransmitter l-glutamate and a coagonist (glycine or d-serine), the ligand-gated ion channel undergoes a conformational change leading to the influx of sodium and calcium ions into the intracellular space (2). Because of its high calcium ion permeability, overactivation of the NMDA receptor by excessive glutamatergic stimulation is associated with elevated intracellular calcium levels, ultimately leading to neuronal apoptosis (3). This insulting excitotoxic state has been described as one of the leading causes in the pathogenesis of various neurodegenerative and psychiatric disorders (4).
Initial attempts to target the NMDA receptor resulted in the development of antagonists that act as channel blockers (5). Despite remarkable antiexcitotoxic properties, this class of compounds largely failed in clinical trials because of adverse psychotomimetic events (1,6). The poor safety profile was attributed to the mechanism of action, as channel blockers suppress virtually all NMDA receptor activity and therefore inhibit physiologic processes that would require NMDA receptor signaling (4). Identification of the GluN2B subunit as a key contributor to excitotoxicity-induced apoptosis, as well as the discovery of the N-terminal domain binding site located at the interface between GluN1 and GluN2B, triggered a wave of enthusiasm and provided the opportunity for the design of subunit-selective NMDA receptor antagonists (4). In particular, GluN2B-selective drug candidates preserve the activity of non-GluN2B containing NMDA receptors, thereby exhibiting a favorable safety profile (7,8).
Despite initial reports on GluN2B antagonists with high affinity and selectivity over other GluN2 subunits, successful clinical translation proved to be challenging because of off-target activity toward nonglutamatergic receptors such as the σ1-receptor (σ1R), a unique ligand-regulated molecular chaperone residing in the endoplasmatic reticulum (9,10). Indeed, improved measures assisting the in vivo evaluation of target engagement are crucial to ensure drug efficacy and to guide successful antagonist development (11). Consequently, validation tools to assess the compound selectivity and in vivo receptor occupancy of GluN2B drug candidates are urgently needed.
Molecular imaging by noninvasive technologies such as PET provides the possibility of studying target engagement both preclinically and clinically (12). Despite nearly 2 decades of preclinical research, no useful PET ligands exist for imaging GluN2B. Imaging probes reported so far have been plagued with 4 major limitations, namely considerable off-target binding, metabolic instability, low brain uptake, and brain uptake not consistent with reported GluN2B expression patterns (13). In the mature brain, GluN2B subunits are predominantly found in forebrain regions such as the striatum, hippocampus, cortex, and thalamus, whereas the cerebellum exhibits only poor receptor subunit density (1).
We recently reported on (rac)-11C-Me-NB1, a GluN2B-specific PET probe that exhibits high target affinity (inhibition constant of 5.4 nM to human GluN1/GluN2B) and excellent pharmacokinetic properties (14). In the present study, we describe remarkable differences in the in vitro and in vivo properties of the (S)- and (R)-enantiomers of 11C-Me-NB1, confirmed by distinct enantiomeric target engagement. An autoradiographic blocker screening assay exploiting the distinct properties of (R)-11C-Me-NB1 and (S)-11C-Me-NB1 was developed, allowing us to account for off-target activity toward σ1R. In sharp contrast to (S)-11C-Me-NB1, (R)-11C-Me-NB1 exhibited excellent GluN2B imaging properties as evidenced by autoradiographic studies on rodent brain tissues and PET imaging of Wistar rats. To illustrate the translational relevance, (R)-11C-Me-NB1 was validated in autoradiographic studies using postmortem human GluN2B-rich cortical and GluN2B-deficient cerebellar brain slices.
MATERIALS AND METHODS
In Vitro Autoradiography
Postmortem human brain samples from healthy volunteers were provided with written consent of the donors by the University Hospital of Zurich, Switzerland. Wistar rat and CD1 mouse brains and human brain tissue were embedded in Tissue-Tek (Sakura Finetek, Optimal Cutting Temperature, O.C.T.) and prepared as 10-μm-thick sections on a cryostat (CryoStar HM 560 MV; Microm International). Slices were adsorbed on SuperFrost Plus (Menzel) and stored at −80°C until further use.
For the autoradiography, slices were thawed on ice (15 min) and preconditioned for 10 min in an aqueous buffer containing 30 mM 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES), 0.56 mM MgCl2, 110 mM NaCl, 5 mM KCl, 3.3 mM CaCl2, and 0.1% fatty-acid–free bovine serum albumin (pH 7.4, 0°C). The tissue samples were air-dried for 3 min at ambient temperature and subsequently incubated with either (R)- or (S)-11C-Me-NB1 (3 nM, calculated on the basis of the molar activity) for 20 min at 21°C in a humidified chamber (complete tissue coverage, with 1–2 mL per tissue slice required).
For the blocker screening, 1 μM of commercially available GluN2B-selective ligands CERC-301, EVT-101, CP101,606, or σ1R-selective ligands SA4503, fluspidine, and (+)-pentazocine was applied. On completion of the incubation time, slices were decanted and washed 5 min in an aqueous buffer containing 30 mM HEPES, 0.56 mM MgCl2, 110 mM NaCl, 5 mM KCl, 3.3 mM CaCl2, and 0.1% fatty-acid–free bovine serum albumin (pH 7.4, 0°C). The samples were further washed (2 × 3 min) in an aqueous buffer containing 30 mM HEPES, 0.56 mM MgCl2, 110 mM NaCl, 5 mM KCl, 3.3 mM CaCl2 (pH 7.4), and dipped twice in distilled water. Finally, the slices were dried and exposed to a phosphor imager plate (Fuji) for 30 min. The films were scanned by a BAS5000 reader (Fuji), and the data were analyzed using AIDA software, version 4.50.010 (Raytest Isotopenmessgeräte GmbH).
In Vivo PET, Dose–Response, and Receptor Occupancy Studies
All animal studies were conducted in accordance with the ARRIVE guidelines and the local animal protection law, with the further approval of the cantonal veterinary office in Zurich, Switzerland.
Wistar rats were purchased from Charles River and kept in a room with controlled temperature (21°C) under a 12-h light/12-h dark cycle, with ad libitum access to food and water. The animals were allowed to acclimatize for 1 wk before the start of the experiments. The animals were anesthetized with isoflurane and scanned in a PET/CT scanner (Super Argus; Sedecal) for 60 min on tail-vein injection of either (R)- or (S)-11C-Me-NB1 (28–37 MBq, 0.5–2.3 nmol/kg). PET scans were followed by CT for anatomic orientation.
For dose–response and receptor occupancy studies, eliprodil (0.002, 0.006, 0.02, and 2 mg/kg) and CP101,606 (0.3, 1, 3, and 10 mg/kg) in an aqueous vehicle (pH 7.0) consisting of glucose (5%), NaCl (0.45%), and citric acid (1 mM) for eliprodil and saline for CP101,606, respectively, were injected shortly before tracer administration. For the CP101,606 scans, the blocker was further infused during the 60-min PET scan with the protocol (0.4, 1.3, 4, and 13.3 mg/kg/h) illustrated in Supplemental Figure 8 (supplemental materials are available at http://jnm.snmjournals.org). For baseline scans, the vehicle was injected shortly before tracer administration and further infused in the control animals of the CP101,606 study.
Data were reconstructed in user-defined time frames with a voxel size of 0.3875 × 0.3875 × 0.775 mm as previously described by our group (14). Time–activity curves were calculated by PMOD, version 3.7 (PMOD Technologies), with predefined regions of interest as previously reported (14). Results are presented as SUVs, indicating the decay-corrected radioactivity per cm3 divided by the injected dose per gram of body weight. Receptor occupancy was calculated as indicated in Supplemental Figure 8B.
Ex Vivo Autoradiography and Subsequent In Vitro Displacement
(R)-11C-Me-NB1 (522 MBq, 20.3 nmol/kg) was injected into a male Wistar rat, and the animal was sacrificed under isoflurane anesthesia by decapitation at 15 min after injection. The brain was dissected, embedded in Tissue-Tek (O.C.T.), and subsequently prepared as 10-μm-thick coronal sections on a cryostat (CryoStar HM 560 MV).
For the displacement screening, tissue sections were incubated with 1 μM CERC-301, EVT-101, CP101,606, fluspidine, (+)-pentazocine, (S)-Me-NB1, or vehicle (aqueous buffer containing 30 mM HEPES, 0.56 mM MgCl2, 110 mM NaCl, 5 mM KCl, 3.3 mM CaCl2, and 0.1% fatty acid free bovine serum albumin, pH 7.4). Untreated sections were included for acquisition and analysis. The sections were exposed to a phosphor imager plate (Fuji) for 60 min, and the film was scanned by a BAS5000 reader (Fuji). Data analysis was performed by AIDA 4.50.010 software (Raytest Isotopenmessgeräte GmbH).
Statistical Analysis
Statistical probability values were based on an independent 2-tailed Student test assuming normal distribution of the data set (n = 4).
RESULTS
The organic syntheses of the desmethyl phenolic precursor NB1 and reference compound Me-NB1, the chiral separation of (rac)-Me-NB1 and the phenolic precursor (rac)-NB1, and the radiolabeling procedures are described in Supplemental Figures 1–3. Absolute configurations of (R)- and (S)-Me-NB1 were determined by x-ray crystallography (Fig. 1A), and circular dichroism spectroscopy was used to confirm the stereochemistry of precursors (R)- and (S)-NB1 (Supplemental Fig. 3). The experimental procedure for the x-ray crystallography can be found in Supplemental Figure 2.
FIGURE 1.

Absolute configuration and binding behavior of (R)- and (S)-Me-NB1. (A) ORTEP view of (R)- and (S)-Me-NB1 in solid state. Me-NB1 exhibits one chiral center depicted at position C13. Thermal ellipsoids are set to 30% probability. For all nonhydrogen atoms, anisotropic displacement parameters were used. Hydrogen atoms were refined in idealized positions using riding model. (B) Representative in vitro autoradiograms of (R)/(S)-11C-Me-NB1 incubated with Wistar rat brain sections. Although (S)-enantiomer binds to virtually all brain regions, (R)-enantiomer exhibits selectivity for GluN2B-rich forebrain. Cx = cortex; CPu = corpus striatum; Hp = hippocampus; Th = thalamus; Cb = cerebellum.
11C labeling of the (R)- and (S)-enantiomers of Me-NB1 was accomplished using 11C-iodomethane in 42% ± 9% radiochemical yield (decay-corrected) and a molar activity of 204 ± 80 GBq/μmol (n = 66). The radiochemical purity was greater than 99%. Autoradiographic experiments revealed significant differences between the binding patterns of (R)-11C-Me-NB1 and (S)-11C-Me-NB1 on rat brain tissue sections as shown in Figure 1B. Although (S)-11C-Me-NB1 displayed a homogeneous distribution across the whole brain, (R)-11C-Me-NB1 revealed a heterogeneous distribution pattern with high binding to GluN2B-rich regions such as the cortex, striatum, thalamus, and hippocampus. The cerebellum, a region with a low density of the GluN2B receptor subunit, showed the lowest (R)-11C-Me-NB1 binding. Similar results were obtained with mouse brain sections (Supplemental Fig. 4).
To assess the specificity and off-target binding toward σ1R of both radiolabeled enantiomers, further in vitro autoradiographic experiments using a series of clinically tested ligands were undertaken. Although (R)-11C-Me-NB1 competed with GluN2B antagonists CERC-301, EVT-101, and CP101,606, the binding of (S)-11C-Me-NB1 was abolished on the addition of σ1R ligands SA4503, fluspidine, and (+)-pentazocine (Fig. 2). In accordance with in vitro autoradiography findings, competitive binding assays revealed inhibition constants of 511 nM for (R)-Me-NB1 and 74 nM for (S)-Me-NB1 toward the σ1R. Additionally, molecular dynamics simulations of the most stable docking poses showed that the (R)-enantiomer exhibits a favored binding mode to the N-terminal domain binding site (Supplemental Figs. 5 and 6). It is common knowledge that enantiomers can exhibit different binding properties; however, our findings remarkably emphasize the tightrope walk between GluN2B- and σ1R-targeted imaging.
FIGURE 2.

Autoradiographic screening of clinically tested GluN2B ligands CERC-301, EVT-101, and CP101,606, as well as σ1R ligands SA4503, fluspidine, and (+)-pentazocine, as blocking agents for (R)-11C-Me-NB1 and (S)-11C-Me-NB1. (R)-11C-Me-NB1 showed heterogeneous binding behavior that was GluN2B-specific whereas (S)-11C-Me-NB1 exhibited considerable σ1R binding.
Spurred by the in vitro and docking results, we investigated both enantiomerically pure radioligands in PET studies using male Wistar rats, and time–activity curves were generated. Brain uptake was generally higher for the GluN2B-specific (R)-form and displayed a slower washout than the (S)-form. Consistently, and as expected, (rac)-11C-Me-NB1 exhibited a time–activity curve that reflected the average of the characteristics of the 2 enantiomers (Fig. 3). An efficient blockade was observed on injection of GluN2B-antagonist eliprodil (2 mg/kg). To assess the utility of (R)-11C-Me-NB1 and (S)-11C-Me-NB1 for dose-dependent target engagement, we performed dose–response experiments applying stepwise dose escalation of eliprodil (0.002, 0.006, 0.02, and 2 mg/kg). Figure 4A shows a concentration-dependent blockade of the (R)-enantiomer, which is reflected by the gradual decrease in time–activity curves with increasing eliprodil doses. In contrast, the (S)-enantiomer did not show a dose-dependent blockade, suggesting that (S)-11C-Me-NB1 is not suitable for studying GluN2B target engagement by PET imaging (Fig. 4B). (S)-11C-Me-NB1 was therefore not considered for further animal studies. An in vitro screening assay with nonradioactive (R)-Me-NB1 revealed no significant binding of the ligand to a series of prominent biologic targets including, but not limited to, the muscarinic M2, dopamine D1 and D3, adrenergic α1A and α2A, histamine H3, serotonin, gamma-aminobutyric acid A, μ-opioid, noradrenaline transporter, monoamine oxidase A, vesicular monoamine transporter 2, vascular endothelial growth factor 2, acetylcholinesterase, cyclooxygenase 1 and 2, androgen receptors, and progesterone receptors.
FIGURE 3.

Whole-brain time–activity curves of racemic and enantiomerically pure 11C-Me-NB1 in Wistar rat brain are depicted as SUVs. GluN2B-specific (R)-form exhibited higher SUVs than (S)-form, whereas racemic mixture displayed time–activity curve that lies between the two enantiomers. For blockade experiment, 2 mg/kg dose of GluN2B antagonist eliprodil was used.
FIGURE 4.

Dose–response curves of (R)-11C-Me-NB1 and (S)-11C-Me-NB1 with eliprodil in Wistar rat brain. Results are reported as SUVs calculated from PET experiments (0–60 min). (A) (R)-11C-Me-NB1 revealed consistent dose–response profile on stepwise dose escalation of eliprodil. (B) (S)-11C-Me-NB1 did not show dose-dependent blockade.
One of the major challenges in the development of GluN2B-targeted PET radioligands is the lack of in vivo selectivity for GluN2B-rich brain regions (15). To address this question for (R)-11C-Me-NB1, we performed ex vivo autoradiography on a male Wistar rat brain. Tracer accumulation was highest in the GluN2B-expressing cortex, striatum, thalamus, and hippocampus, whereas the GluN2B-deficient cerebellum revealed the lowest tracer binding of all brain regions (Fig. 5).
FIGURE 5.
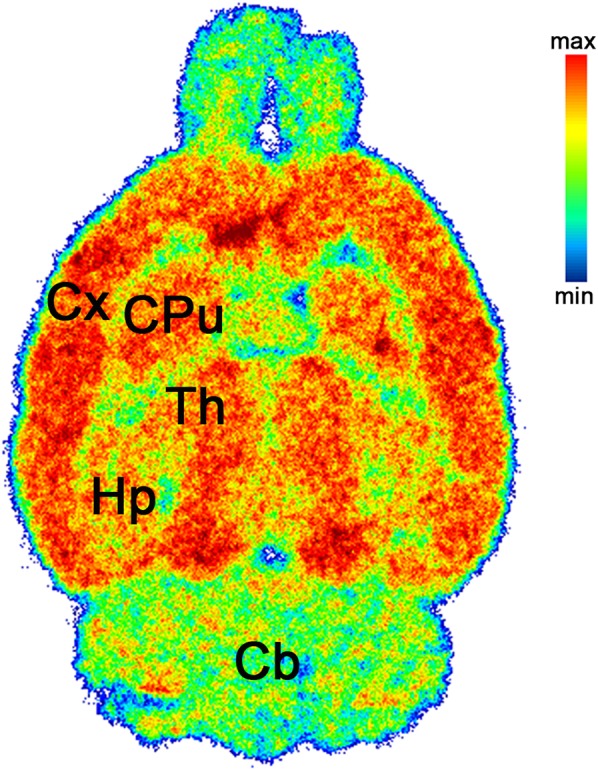
Representative ex vivo autoradiogram of Wistar rat brain at 15 min after injection of (R)-11C-Me-NB1. Selectivity for cortex, striatum, thalamus, and hippocampus over cerebellum was observed. Cb = cerebellum; CPu = corpus striatum; Cx = cortex; Hp = hippocampus; Th = thalamus.
To date, CP101,606 is the only GluN2B-selective antagonist with well-documented clinical efficacy (16). Therapeutic doses applied in clinical trials aim to reach a target plasma concentration of 200 ng/mL (16). We used (R)-11C-Me-NB1 to assess the preclinical dose-dependent receptor occupancy of CP101,606 in male Wistar rats by PET imaging. To maintain the plasma concentration of CP101,606 at steady-state during the scan, the GluN2B antagonist was first injected as a bolus, followed by continuous infusion. Blood samples were harvested after the PET scans with the aim of determining the steady-state plasma concentration of CP101,606 for each dose. CP101,606 plasma concentrations in combination with the time–activity curves (Fig. 6A) were used to calculate the receptor occupancy (Fig. 6B) at different CP101,606 plasma concentrations as indicated in Supplemental Figure 8B. A CP101,606 plasma concentration required for 50% receptor occupancy of 158 nmol/L (52 ng/mL) was deduced. Respective PET images are presented in Figure 6C. On the basis of these results, the therapeutic CP101,606 plasma concentration of 611 nmol/L (200 ng/mL) occupies 80% of the GluN2B binding sites in our rat model. These results highlight the importance of high receptor occupancy for efficacy.
FIGURE 6.

Receptor occupancy by clinically tested GluN2B antagonist CP101,606 and GluN2B PET radioligand (R)-11C-Me-NB in Wistar rats (B), calculated from experimental SUV0–60 min (A), revealed plasma concentration of 158 nmol/L, or 52 ng/mL, required for 50% receptor occupancy. Clinically applied target plasma concentration of 200 ng/mL occupies 80% of GluN2B binding sites in our rat model. Rat brain PET images were superimposed on PMOD MRI template (C). Bs = brain stem; Cb = cerebellum; CPu = corpus striatum; Cx = cortex; Hp = hippocampus; Th = thalamus.
We further conducted a receptor occupancy study with CP101,606 and (R)-11C-Me-NB1 using only bolus injections without any further infusions, with the aim of challenging the need for a labor-intensive infusion-based experimental setup. The study revealed that receptor occupancy results at identical bolus doses with and without infusions are comparable (Supplemental Fig. 8). The in vivo specificity and selectivity of radioligands are conventionally evaluated by the injection of nonradioactive, high-affinity blockers in a severalfold excess; however, this approach is confounded by the metabolism and pharmacokinetics of the blockers. In the absence of an efficient blockade, it remains unclear whether the blocker reached the biologic target. To exclude confounding factors such as blocker metabolism and pharmacokinetics, we performed ex vivo autoradiography on rat brain slices, followed by coincubation of the dissected brain slices with different GluN2B and σ1R ligands. In this autoradiographic displacement assay, CERC-301, EVT-101, CP101,606, fluspidine, (+)-pentazocine, and (S)-Me-NB1 were used as displacing agents. The binding of (R)-11C-Me-NB1 was displaced with the GluN2B antagonists (Fig. 7A), underlining the suitability of (R)-11C-Me-NB1 as a GluN2B imaging agent. Biodistribution experiments conducted at 5, 15, 30, 45, and 60 min after injection (n = 4) confirmed that radioligand accumulation in the cortex was significantly higher than in the cerebellum at every measured time point. At the last measurement, 60 min after injection, the uptake for each GluN2B-expressing brain region was significantly higher than for the cerebellum. The ratios to the cerebellum were as follows: cortex to cerebellum (1.27 ± 0.07, P < 0.01), striatum to cerebellum (1.20 ± 0.06, P < 0.05), hippocampus to cerebellum (1.35 ± 0.05, P < 0.01), and thalamus to cerebellum (1.34 ± 0.05, P < 0.01). Biodistribution data are summarized in Supplemental Tables 2 and 3.
FIGURE 7.

Representative autoradiograms on rodent and human brain tissue. (A) Ex vivo autoradiography at 15 min after injection of (R)-11C-Me-NB1 into Wistar rat and subsequent in vitro displacer screening with GluN2B ligands CERC-301, EVT-101, CP101,606, and σ1R ligands fluspidine, (+)-pentazocine, and (S)-Me-NB1. Displacement of (R)-11C-Me-NB1 binding is observed only for GluN2B antagonists. (B) Representative autoradiograms of (R)-11C-Me-NB1 incubated with human postmortem brain sections. GluN2B-specific binding was observed in temporal cortex (indicated by blockade experiment with GluN2B antagonist CERC-301), whereas cerebellum did not reveal any specific binding of radioligand.
Surprisingly, the Wistar rat brain stem showed considerable tracer uptake, which was blocked by eliprodil and CP101,606. Whether this uptake is related to GluN2B receptor binding or potential interactions with other GluN2 subtypes remains to be investigated. To pave the way for clinical translation, (R)-11C-Me-NB1 was tested on postmortem human brain sections by comparing healthy cortical with cerebellar brain tissue. In accordance with results obtained from the rodent brain, (R)-11C-Me-NB1 displayed higher binding in the cortex than in the cerebellum (Fig. 7B). Coincubation of the GluN2B antagonist CERC-301 (1 μM) reduced cortical but not cerebellar binding, thus demonstrating the specificity of (R)-11C-Me-NB1 binding to the human GluN2B-containing NMDA receptor.
DISCUSSION
The present study revealed a remarkable enantioselectivity of (R)-11C-Me-NB1 over (S)-11C-Me-NB1 for GluN2B-containing NMDA receptors as demonstrated by distinct autoradiographic binding patterns on rat and mouse brain sections, molecular dynamics studies, and in vivo dose–response experiments. Blocker screening assays with GluN2B- and σ1R-selective ligands showed the 2 radiolabeled enantiomers to have entirely different binding properties, thereby raising awareness of the tightrope walk between GluN2B- and σ1R-targeted PET imaging.
Although (R)-11C-Me-NB1 is a GluN2B ligand, (S)-11C-Me-NB1 binds predominantly to the σ1R. There is precedence for showing distinct pharmacologic activities by 2 enantiomers of a given compound, which in part could be related to differences in brain uptake kinetics (17,18). However, the extent to which (R)- and (S)-11C-Me-NB1 reveal different preferences for GluN2B subunits and σ1Rs in the brain is rather remarkable. To our knowledge, there is no PET radioligand reported in the literature that shows such distinct enantiomer-related characteristics for 2 pharmacologically and closely related receptors.
GluN2B antagonists are generally well tolerated preclinically and clinically; however, representative drug candidates such as CERC-301 (MK-0657) failed to demonstrate efficacy in clinical trials (19–22). A phase II clinical trial of EVT-101 was terminated because of difficulties in patient recruitment (ClinicalTrials.gov identifier NCT01128452). No data on clinical efficacy have yet been disclosed. Although CERC-301 and EVT-101 competed with (R)-11C-Me-NB1 during in vitro autoradiography, we did not observe any competition for doses of up to 2 mg/kg in preliminary PET experiments with male Wistar rats (Supplemental Fig. 9). To understand the differences between in vitro and in vivo findings, we performed an ex vivo metabolite study and confirmed the presence of intact (R)-11C-Me-NB1 and the absence of radiometabolites in Wistar rat brain. Ex vivo autoradiography with subsequent displacement of (R)-11C-Me-NB1 by CERC-301 and EVT-101 further confirmed that the radiotracer remains intact and that both CERC-301 and EVT-101 bind to the same binding site. We therefore hypothesize that CERC-301 and EVT-101 exhibit unfavorable in vivo pharmacokinetic or metabolic properties in rodents, which led to the absence of target engagement in vivo. Confirmation of these findings in a clinical setup would shed more light on why these orally available GluN2B antagonists are not living up to their expectations with respect to clinical efficacy.
In sharp contrast to CERC-301 and EVT-101, the GluN2B antagonists CP101,606 and eliprodil efficiently blocked (R)-11C-Me-NB1 in the rodent brain not only in vitro but also in vivo. Receptor occupancy studies revealed that the clinically applied target plasma concentration of CP101,606 occupies most N-terminal domain binding sites, as is in accordance with the clinical efficacy reported on patient treatment with CP101,606 (16,21,23). Finally, to demonstrate the translational relevance of our new radioligand, we performed autoradiographic studies using postmortem human GluN2B-rich cortical and GluN2B-deficient cerebellar brain sections. The results further showed that (R)-11C-Me-NB1 binds specifically, and is sensitive in detecting the GluN2B subunits of the NMDA receptor in postmortem human brain tissue samples. To the best of our knowledge, this is the first report on a GluN2B PET radioligand that exhibits a combination of specificity and selectivity in vitro and in vivo, with appropriate pharmacokinetic properties characterized by high brain uptake and no brain radiometabolites (Supplemental Fig. 7).
Successful clinical validation of (R)-11C-Me-NB1 would enable the assessment of emerging GluN2B antagonists by means of target engagement studies and ultimately lead to accurate patient dosing. Moreover, the availability of a selective GluN2B PET radioligand will allow us to elucidate the role of GluN2B-carrying NMDA receptors in patients with neurodegenerative and psychiatric disorders.
CONCLUSION
After nearly 2 decades of preclinical research, we here report on a successful NMDA PET radioligand that exhibits a combination of GluN2B specificity and selectivity in vitro and in vivo. (R)-11C-Me-NB1 revealed outstanding tracer characteristics and outperformed (S)-11C-Me-NB1. (R)-11C-Me-NB1 has great potential for imaging numerous psychiatric and brain diseases in which the GluN2B subunit of the NMDA receptor is implicated. This new radioligand holds promise both preclinically and clinically for the selection of appropriate doses of candidate drugs that bind to the GluN2B subunit of the NMDA receptor.
DISCLOSURE
This project was supported by Swiss National Science Foundation grant 310030E-160403/1, the Clinical Research Priority Program on Multiple Sclerosis (CRPPMS) of the University of Zurich, and the Stavros Niarchos Foundation. The present work is related to a recently filed patent application (US20170224852A1). No other potential conflict of interest relevant to this article was reported.
Supplementary Material
Acknowledgments
Bruno Mancosu is acknowledged for technical support with the 11C module. We thank Dr. Silvan Boss for assistance with chiral purification, Hui Yuan Oliver for support during biodistribution studies, and Dr. Tao Sun for support with precursor synthesis. The technical assistance of Irina Abakumova and Magdalena Foege is kindly acknowledged. We thank Dr. Ina Dix for the x-ray crystallography, and we acknowledge Prof. Roland Martin for organizing tissue samples.
REFERENCES
- 1.Paoletti P, Bellone C, Zhou Q. NMDA receptor subunit diversity: impact on receptor properties, synaptic plasticity and disease. Nat Rev Neurosci. 2013;14:383–400. [DOI] [PubMed] [Google Scholar]
- 2.Traynelis SF, Wollmuth LP, McBain CJ, et al. Glutamate receptor ion channels: structure, regulation, and function. Pharmacol Rev. 2010;62:405–496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sattler R, Tymianski M. Molecular mechanisms of calcium-dependent excitotoxicity. J Mol Med. 2000;78:3–13. [DOI] [PubMed] [Google Scholar]
- 4.Hardingham GE. Coupling of the NMDA receptor to neuroprotective and neurodestructive events. Biochem Soc Trans. 2009;37:1147–1160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Anis NA, Berry SC, Burton NR, Lodge D. The dissociative anaesthetics, ketamine and phencyclidine, selectively reduce excitation of central mammalian neurones by N-methyl-aspartate. Br J Pharmacol. 1983;79:565–575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hardingham GE, Bading H. Synaptic versus extrasynaptic NMDA receptor signalling: implications for neurodegenerative disorders. Nat Rev Neurosci. 2010;11:682–696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mony L, Kew JN, Gunthorpe MJ, Paoletti P. Allosteric modulators of NR2B-containing NMDA receptors: molecular mechanisms and therapeutic potential. Br J Pharmacol. 2009;157:1301–1317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Karakas E, Simorowski N, Furukawa H. Subunit arrangement and phenylethanolamine binding in GluN1/GluN2B NMDA receptors. Nature. 2011;475:249–253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Contreras PC, Bremer ME, Gray NM. Ifenprodil and SL 82.0715 potently inhibit binding of [3H](+)-3-PPP to sigma binding sites in rat brain. Neurosci Lett. 1990;116:190–193. [DOI] [PubMed] [Google Scholar]
- 10.Maurice T, Su TP. The pharmacology of sigma-1 receptors. Pharmacol Ther. 2009;124:195–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kemp JA, McKernan RM. NMDA receptor pathways as drug targets. Nat Neurosci. 2002;5(suppl):1039–1042. [DOI] [PubMed] [Google Scholar]
- 12.Slough C, Masters SC, Hurley RA, Taber KH. Clinical positron emission tomography (PET) neuroimaging: advantages and limitations as a diagnostic tool. J Neuropsychiatry Clin Neurosci. 2016;28(A4):67–71. [DOI] [PubMed] [Google Scholar]
- 13.Fuchigami T, Nakayama M, Yoshida S. Development of PET and SPECT probes for glutamate receptors. ScientificWorldJournal. 2015;2015:716514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Krämer SD, Betzel T, Mu L, et al. Evaluation of 11C-Me-NB1 as a potential PET radioligand for measuring GluN2B-containing NMDA receptors, drug occupancy, and receptor cross talk. J Nucl Med. 2018;59:698–703. [DOI] [PubMed] [Google Scholar]
- 15.Roger G, Dolle F, De Bruin B, et al. Radiosynthesis and pharmacological evaluation of [11C]EMD-95885: a high affinity ligand for NR2B-containing NMDA receptors. Bioorg Med Chem. 2004;12:3229–3237. [DOI] [PubMed] [Google Scholar]
- 16.Nutt JG, Gunzler SA, Kirchhoff T, et al. Effects of a NR2B selective NMDA glutamate antagonist, CP-101,606, on dyskinesia and Parkinsonism. Mov Disord. 2008;23:1860–1866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Brust P, Deuther-Conrad W, Becker G, et al. Distinctive in vivo kinetics of the new sigma1 receptor ligands (R)-(+)- and (S)-(-)-18F-fluspidine in porcine brain. J Nucl Med. 2014;55:1730–1736. [DOI] [PubMed] [Google Scholar]
- 18.Kranz M, Sattler B, Tiepolt S, et al. Radiation dosimetry of the alpha4beta2 nicotinic receptor ligand (+)-[18F]flubatine, comparing preclinical PET/MRI and PET/CT to first-in-human PET/CT results. EJNMMI Phys. 2016;3:25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Amico-Ruvio SA, Paganelli MA, Myers JM, Popescu GK. Ifenprodil effects on GluN2B-containing glutamate receptors. Mol Pharmacol. 2012;82:1074–1081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Garner R, Gopalakrishnan S, McCauley JA, et al. Preclinical pharmacology and pharmacokinetics of CERC-301, a GluN2B-selective N-methyl-D-aspartate receptor antagonist. Pharmacol Res Perspect. 2015;3:198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Preskorn SH, Baker B, Kolluri S, Menniti FS, Krams M, Landen JW. An innovative design to establish proof of concept of the antidepressant effects of the NR2B subunit selective N-methyl-D-aspartate antagonist, CP-101,606, in patients with treatment-refractory major depressive disorder. J Clin Psychopharmacol. 2008;28:631–637. [DOI] [PubMed] [Google Scholar]
- 22.Ibrahim L, Diaz Granados N, Jolkovsky L, et al. A randomized, placebo-controlled, crossover pilot trial of the oral selective NR2B antagonist MK-0657 in patients with treatment-resistant major depressive disorder. J Clin Psychopharmacol. 2012;32:551–557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bullock MR, Merchant RE, Carmack CA, et al. An open-label study of CP-101,606 in subjects with a severe traumatic head injury or spontaneous intracerebral hemorrhage. Ann N Y Acad Sci. 1999;890:51–58. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.