

Abstract
Multidrug-resistant enterococcal strains emerged in the early 1980s and are now among the leading causes of drug-resistant bacterial infection worldwide. We used functional genomics to study an early bacterial outbreak in patients in a Wisconsin hospital between 1984 and 1988 that was caused by multidrug-resistant Enterococcus faecalis. The goal was to determine how a clonal lineage of E. faecalis became adapted to growth and survival in the human bloodstream. Genome sequence analysis revealed a progression of increasingly fixed mutations and repeated independent occurrences of mutations in a relatively small set of genes. Repeated independent mutations suggested selection within the host during the course of infection in response to pressures such as host immunity and antibiotic treatment. We observed repeated independent mutations in a small number of loci, including a little studied polysaccharide utilization pathway and the cydABDC locus. Functional studies showed that mutating these loci rendered E. faecalis better able to withstand antibiotic pressure and innate immune defenses in the human bloodstream. We also observed a shift in mutation pattern that corresponded to the introduction of carbapenem antibiotics in 1987. This work identifies pathways that allow enterococci to survive the transition from the human gut into the bloodstream, enabling them to cause severe bacteremia associated with high mortality.
INTRODUCTION
Enterococcus faecalis bacteria are ubiquitous members of the gastrointestinal tract consortia of land animals (1, 2). In the antibiotic era, E. faecalis has become a leading hospital pathogen, causing opportunistic infections that are often refractory to antibiotic treatment (3, 4). Whereas E. faecalis is well adapted to life in the intestinal tract, bacteria causing bloodstream infections face substantially different selective pressures. These include altered nutrient availability and restriction, different host defenses including abundant neutrophils, and different concentrations of antibiotics (5). Enterococcal bloodstream infections occur mainly in hospitalized patients and are usually caused by hospital-adapted lineages. Multidrug-resistant hospital-adapted enterococcal strains are believed to be readily acquired into the antibiotic-destabilized gut microbial community by ingestion from contaminated objects and surfaces (6). This is followed by proliferation in the lower gastrointestinal tract (7, 8), increasing the likelihood of active or passive translocation across the gut epithelium and into the bloodstream. Enterococci are known to be resistant to killing by phagocytes (9), allowing them to persist and adapt to conditions in the bloodstream. Patient-to-patient transmission occurs predominantly via the fecal-oral route (10) and rarely, if at all, via bloodstream-to-bloodstream spread. Therefore, beneficial mutations that occur during outgrowth of enterococci in the bloodstream represent an evolutionary cul-de-sac and are unlikely to be transmitted between patients. As a consequence, mutations that confer survival advantages in the bloodstream must evolve de novo each time the bacteria infect the bloodstream of a new patient.
RESULTS
Genome comparisons between outbreak and nonoutbreak E. faecalis strains
One of the earliest outbreaks of multidrug-resistant enterococcal bacteremia associated with a demonstrated epidemiological pattern implicating patient-to-patient transmission was caused by an unusually pathogenic and multidrug-resistant strain of E. faecalis. This outbreak occurred over a 4-year period at the University of Wisconsin Hospital and Clinics in the mid-1980s (11). To understand the E. faecalis bacteremia landscape at the time and how the outbreak strains differed genetically from other lineages that caused sporadic infections, we compared the genomes of 93 strains isolated from patient blood samples between 1984 and 1988 and preserved by one of us (M.M.H.) since that time (table S1). The outbreak was represented by 62 isolates collected between July 1985 and February 1988. The genomes of these strains were compared to those of 27 strains isolated immediately before and during the outbreak to identify genetic features that predisposed the outbreak strains to cause a sustained outbreak. Outbreak isolates belonged to sequence type 6 (ST6), a lineage not present among pre-outbreak bacteremia strains (Fig. 1). Outbreak isolates had 10% larger genomes (mean ungapped assembly length, 3.28 Mb versus 2.97 Mb; P < 0.0001) as the result of abundant mobile elements, and they lacked a functional CRISPR-Cas locus (Fig. 1).
Fig. 1. Genetic diversity of E. faecalis strains before and during a hospital outbreak.
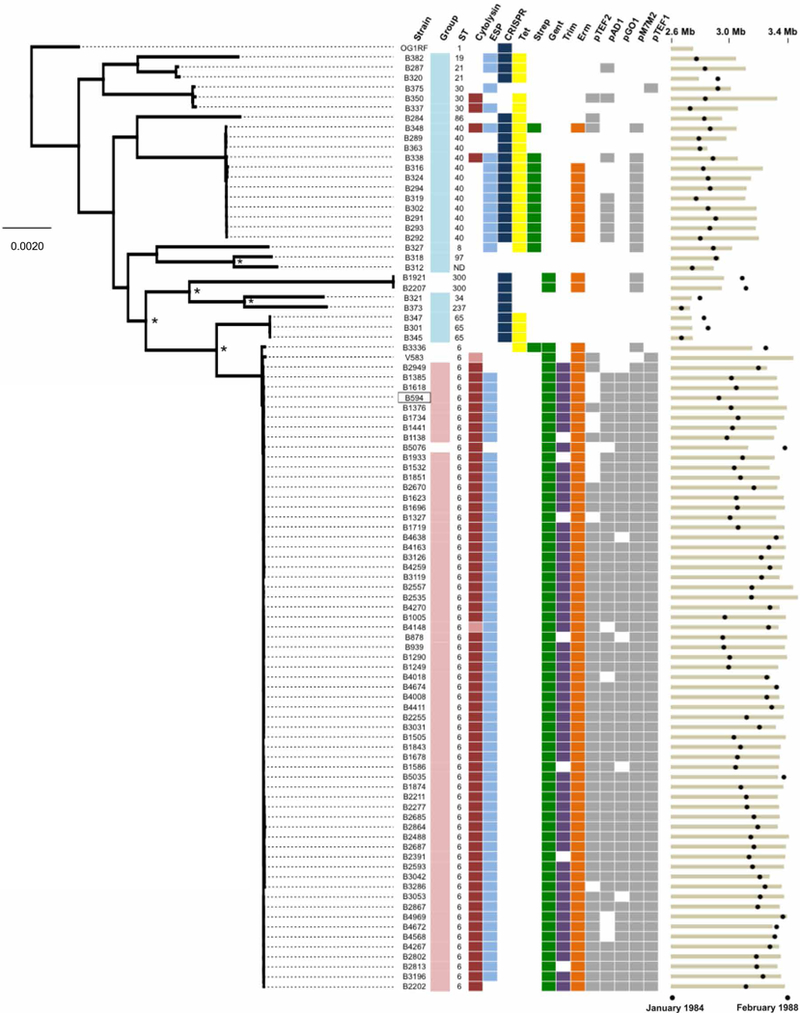
Genetic diversity of E. faecalis strains before (teal) and during (pink) an outbreak from 1984 to 1988 in a Wisconsin hospital is shown. The maximum likelihood phylogeny is shown for the 93 strains sequenced in this study and for the V583 and OG1RF strains (root) that were sequenced previously. The B594 outbreak reference strain is boxed. Orthogroups were clustered with Synergy2, an alignment was generated from single-nucleotide polymorphisms (SNPs) in 1976 single-copy core genes, and a phylogenetic tree was constructed with RAxML. Scale bar shows the number of nucleotide substitutions per site. Asterisks show nodes with bootstrap values of 100; all other nodes have bootstrap values of <90. Colored boxes show the prevalence of virulence (cytolysin and ESP), bacterial immunity (CRISPR-Cas), drug resistance, and plasmid replication genes among pre-outbreak and outbreak E. faecalis strains. Lighter shading in the cytolysin column corresponds to partial, nonfunctional cytolysin operons. Black circles at the far right show the date of isolation of each strain, and tan bars indicate ungapped genome length for each strain. Genome assembly statistics and National Center for Biotechnology Information accession information for all strains are available in table S1.
Among the 324 genes in outbreak isolates that were rare or absent from pre-outbreak genomes were elements associated with virulence, drug resistance, prophages, and mobile element function (table S2). Strains in the outbreak lineage also harbored traits known to confer increased survival in hospital settings, colonization, and virulence. Such traits included production of the cytolysin toxin bacteriocin (12), the E. faecalis pathogenicity island encoding enterococcal surface protein (ESP) (13), and high-level gentamicin resistance (11). Compared to pre-outbreak strains, outbreak strains were more resistant to β-lactam and cephalosporin antibiotics (fig. S1). In addition, they were more resistant to the commonly used hospital disinfectant chlorhexidine and were more tolerant to acidic pH (fig. S1). Comparison of the genome of the prototype outbreak isolate, B594, to the first vancomycin-resistant isolate in the United States, strain V583 isolated in St. Louis, MO in 1987 (14), showed that B594 and V583 were of the same lineage and shared a recent common ancestor (fig. S2).
Analysis of variants among outbreak strains
To serve as a basis for the identification of genetic changes that occurred over the course of the outbreak, we generated a closed genome sequence for the chromosome and two resident plasmids of the earliest outbreak isolate, B594. Derivatives of this isolate have been propagated in the laboratory as MMH594, which has been studied as a representative pathogenic, multidrug-resistant, hospital-adapted strain (15). The closed B594 genome was used as the reference for variant calling in subsequent outbreak isolates (table S3). We identified 15 chromosomal variants that became progressively fixed over time among the outbreak strains. These variants were used to group strains by genotype and infer the genotypes of the transmitted strains over time (Fig. 2, A and B). Multiple bloodstream isolates (two, three, or four) were obtained from six different patients at intervals from 11 to 160 days after the initial E. faecalis-positive blood culture. Analysis of variants in these same-patient isolates suggested that they likely emerged from the diversity harbored within the gastrointestinal tract or a site of localized infection rather than being sustained in the bloodstream (table S4).
Fig. 2. Genetic diversity of E. faecalis outbreak strains.
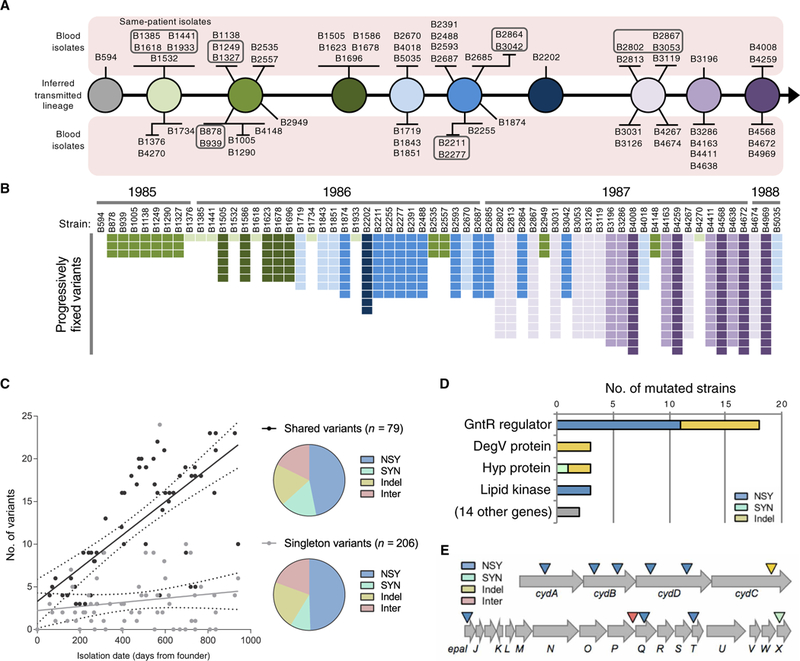
(A) Transmission model of the E. faecalis outbreak lineage. Variants found in 62 outbreak strains were used to infer the genotypes transmitted from patient to patient by the fecal-oral route. The sampled bloodstream isolates are shown as spokes connected to their background genotype and are grouped according to shared variants. Boxed strains were isolated from the same patient. (B) Distribution of progressively fixed variants used to order outbreak strains in (A). Strains are ordered by isolation date, with corresponding years shown on top. The variants are shaded to correspond to the strains in (A). (C) Accumulation of variants over time. Number of shared or singleton variants versus date of isolation is plotted for each strain. NSY, nonsynonymous SNPs; SYN, synonymous SNPs; Indel, insertion/deletion variant; Inter, intergenic variant. (D) Summary of genes that were repeatedly and independently mutated among E. faecalis outbreak strains. Bars show the number of different mutations detected within each gene. (E) Diagram of the cydABDC and epa operons, which were repeatedly and independently mutated among different E. faecalis outbreak strains. Colored triangles indicate the approximate location of mutations.
We next compared all 62 outbreak strains to one another to identify mutations and changes in mobile genetic elements. A phylogenetic tree of SNPs enabled broad clustering of strains by approximate date of isolation (fig. S3). Analysis of mobile gene content revealed additional differences, including a prophage recombination or replacement in five isolates, a recombination within the E. faecalis pathogenicity island (13) in 44 isolates, and six different plasmids that were variably present (fig. S3). One particularly interesting chimeric plasmid contained four distinct predicted rep genes along with four different drug-resistant genes, including a gene encoding high-level aminoglycoside resistance (11).
Point mutations were observed to accumulate over time among outbreak isolates, with shared variants increasing at a rate of one variant every 51 days (Fig. 2C). The number of singleton variants, however, did not change over time, suggesting that, on average, three unique variants arose de novo within each patient, but these variants were not transmitted to the other patients. Approximately half of all variants detected were nonsynonymous SNPs, with the remaining variants equally split among synonymous SNPs, insertion/ deletion variants in coding sequences, and intergenic variants (Fig. 2C). To identify genes and pathways important for E. faecalis survival in the human bloodstream, we next focused on loci that were repeatedly targeted for independent mutation in different isolates (Fig. 2, D and E). Because beneficial mutations that occurred within the bloodstream of a patient were unlikely to become fixed in the population, owing to the lack of bloodstream-to-bloodstream transmission, loci that were repeatedly targeted were examined as possible signatures of bloodstream adaptation.
Exploration of loci independently mutated in different patients
We detected significant enrichment (P < 0.0001) for mutations in the cydABDC operon, with independent mutations occurring in six different outbreak strains (Fig. 2E and table S3). This operon encodes a cytochrome bd–type terminal oxidase that E. faecalis uses to carry out respiration in the presence of heme (16,17). We confirmed that the MMH594 strain exhibited increased growth in vitro in the presence of heme and that this growth increase was due to the ability of the cydABDC operon to use heme (Fig. 3A). A recent study showed that induction of respiration in Staphylococcus aureus and E. faecalis caused increased susceptibility to killing by neutrophils (18). Consistent with this finding, we observed that an E. faecalis strain lacking the cydABDC operon was more resistant to killing in whole human blood (Fig. 3B), suggesting that cydABDC operon mutations were selected for in vivo by pressure from the host immune system.
Fig. 3. cydABDC cytochrome operon mutations confer resistance to bacterial killing in whole human blood.
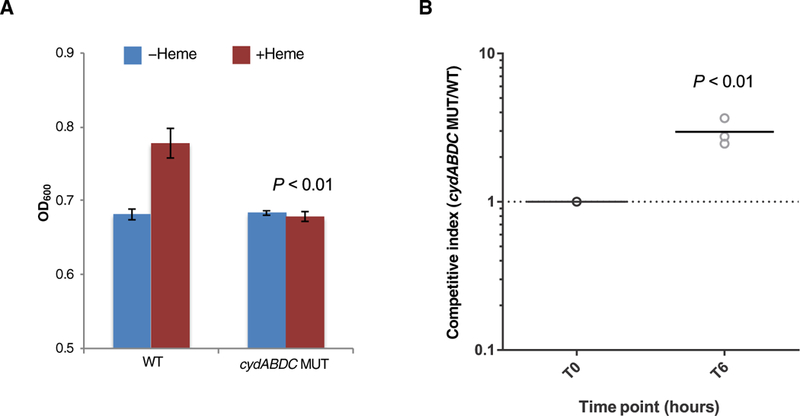
(A) Maximum growth [optical density at 600 nm (OD600)] of MMH594 wild-type (WT) and cydABDC operon knockout (MUT) E. faecalis strains is shown. Bacteria were cultured in brain heart infusion (BHI) medium in the absence or presence of 10 μM heme. Mean values are plotted, and error bars show SDs of triplicate experiments. Growth of WT and MUT strains in the presence of heme was compared. All P values were calculated with a two-tailed t test. (B) A killing assay in whole human blood for MMH594 wild-type (WT) and cydABDC operon knockout (MUT) E. faecalis strains is shown. WT and MUT strains were mixed (1:1) and were incubated with fresh whole human blood from three independent donors for 6 hours. Competitive index was calculated as the ratio of MUT/WT cells and was determined by polymerase chain reaction (PCR) genotyping of at least 70 individual colonies from three biological replicates. A comparison of time zero (T0) with 6 hours of culture (T6) is shown. P value was calculated with a two-tailed t test.
The strongest signal for adaptive evolution within the human bloodstream occurred within a gntR transcriptional regulator (gene ID: SAW_02997 in the B594 strain, EF3156 in the V583 strain, and OG1RF_12423 in the OG1RF strain), which independently acquired 18 distinct mutations in 21 different outbreak isolates (χ2 = 6267, P < 0.0001; Fig. 2D and table S3). Each of these 18 mutations would be predicted to affect the coding sequence of the gene, indicating that loss of function of this regulator conferred enhanced survival within the human bloodstream by some mechanism. Because the gene was predicted to encode a winged helix-turn-helix transcriptional regulator, we identified its regulon by transcriptome comparison of a matched gntR wild-type and mutant E. faecalis strain pair (Fig. 4A and fig. S4). This analysis showed that gntR mutations derepress the adjacent glycoside hydrolase family 65 gene (ghf65), the β-phosphoglucomutase (pgmB) gene, and an unlinked sugar phosphotransferase gene (treB).
Fig. 4. gntR mutants express a glycosyl hydrolase that alters lipoteichoic acid.
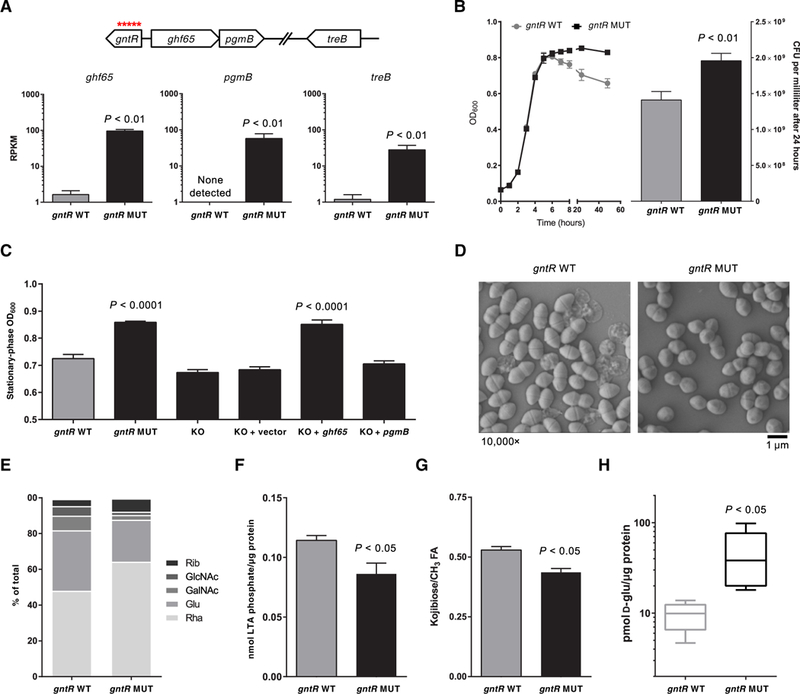
(A) RNA sequencing of isogenic gntR wild-type (WT) and mutant (MUT) E. faecalis strains. Reads per kilobase per million mapped reads (RPKM) are shown for the three genes that constitute the gntR regulon. Genomic context of the genes is shown above. (B) Growth curves for gntR WT and MUT strain are shown on the left. Colony-forming units (CFU) per milliliter for gntR WT and MUT strains, which were enumerated after 24 hours of culture by dilution plating, are shown on the right. (C) Optical densities (OD600) in the stationary phase of growth for gntR WT and MUT strains and for isogenic gntR MUT strain derivatives in culture are shown. KO, knockout strains. (D) Scanning electron micrographs of gntR WT and MUT E. faecalis strains in culture. (E) Monosaccharide composition analysis of cell surface-associated polysaccharides isolated from gntR WT and MUT strains. Rha, rhamnose; Glu, glucose; GalNAc, N-acetylgalactosamine; GlcNAc, N-acetylglucosamine; Rib, ribose. (F) Total lipoteichoic acid (LTA) isolated from gntR WT and MUT stationary-phase cultures. Lipoteichoic acid was quantified by measuring inorganic phosphate and was normalized to total protein in lysate. (G) Quantification of side-chain kojibiose per molecule of lipoteichoic acid. ‘h nuclear magnetic resonance spectroscopy peaks were integrated and normalized to fatty acid (FA) (−CH3) proton peaks. (H) Quantification of intracellular D-glucose in gntR WT and MUT strains, normalized to total protein, as quantified by a Bradford assay. Data in all panels except (D) and (E) show mean values of at least three biological replicates, with error bars showing SDs. All P values were calculated with a two-tailed t test.
To understand how constitutive expression of a carbohydrate metabolism pathway confers a selective advantage to E. faecalis in the bloodstream, we closely examined gntR mutants for distinguishing phenotypes. Culture in BHI medium showed that, whereas gntR wild-type and mutant strains grew at similar rates through the exponential growth phase, gntR mutants consistently grew to a higher optical density (OD600) and were more resistant to subsequent autolysis during the stationary growth phase (Fig. 4B). This could be recapitulated with a Triton X-100 autolysis assay, which confirmed that gntR mutants were indeed more resistant to autolysis (fig. S5). Increased stationary-phase OD600 measurements correlated with resistance to autolysis and could be used to distinguish between gntR wild-type and mutant strains (Fig. 4C). We wondered whether the increased stationary-phase OD600 values observed in gntR mutants stemmed from the up-regulation of ghf65 and pgmB expression. Deletion of both ghf65 and pgmB in a gntR mutant strain restored the OD600 values to that of the wild-type strain (Fig. 4C). Moreover, complementation of the deletion mutant in trans with the reading frame encoding ghf65, but not that encoding pgmB, restored the mutant phenotype (Fig. 4C). These results imply that the constitutive expression of ghf65 conferred a survival advantage to E. faecalis in the human bloodstream by some mechanism.
Electron microscopy was used to visualize ultrastructural differences between gntR mutant and wild-type E. faecalis. In agreement with the autolysis assay (fig. S5), transmission electron micrographs showed markedly decreased cell lysis among stationary-phase gntR mutant cells, as well as lower cellular length-to-width aspect ratios and thinner cell walls compared to wild-type bacteria (fig. S6). Scanning electron micrographs confirmed that gntR mutants were less prone to lysis and were also shorter and less lancet-shaped than wild-type bacteria (Fig. 4D). Electron micrographs of gntR mutants were reminiscent of those for other E. faecalis mutants reported previously that showed altered cell surface-associated polysaccharides (19–21). E. faecalis of the ST6 lineage is known to make four surface-associated polysaccharides: a capsular polysaccharide, the rhamnose-containing enterococcal polysaccharide antigen (Epa), lipoteichoic acid, and wall teichoic acid (22). We extracted cell surface-associated polysaccharides containing all four classes of molecules and analyzed their monosaccharide composition. The largest difference was that surface-associated polysaccharides from the gntR mutants contained less glucose and more rhamnose than did those of wild-type cells (Fig. 4E). This suggested that the gntR mutant bacteria produced more rhamnopolysaccharide and less of other glucose-containing polysaccharides.
The GHF65 enzyme that is constitutively expressed in gntR mutants showed sequence similarity to a trehalose 6-phosphate phosphorylase (TrePP; EC 2.4.1.216) that was previously characterized in Lactococcus lactis (23). We confirmed that an E. faecalis strain lacking GHF65, as well as a strain lacking the TreB phosphotransferase system that is derepressed in gntR mutants, could not grow on the Glc(α1 ↔ α1)Glc disaccharide trehalose as a sole carbon source (fig. S7). Trehalose is largely absent in the human bloodstream; its use as a food additive did not begin until the early 2000s (24). Thus, we wondered whether GHF65 could act on related disaccharide substrates, as has been recently shown for the TrePP enzyme of L. lactis (25). As a potential candidate, we noted that the closely related Glc(α1 → α2)Glc disaccharide kojibiose is used by E. faecalis to build and decorate lipoteichoic acid with glycan chains (26, 27). E. faecalis could not grow on kojibiose as the sole carbon source (fig. S7), probably because the bacteria were unable to transport the disaccharide into the cytoplasm. E. faecalis do, however, synthesize kojibiose through an as yet uncharacterized pathway (27). This disaccharide is incorporated into the lipoteichoic acid anchor diglucosyl diacylglycerol (28) and is also attached to the glycerol phosphate chain of the lipoteichoic acid polymer (27). Either of these might serve as substrates for an enzyme with kojibiose phosphorylase activity, liberating D-glucose in the process. We compared the amount of lipoteichoic acid isolated from gntR wild-type and mutant E. faecalis bacteria and found that the mutant cells contained less lipoteichoic acid overall (Fig. 4F). We also quantified kojibiose in the lipoteichoic acid polymer and found that lipoteichoic acid from gntR mutants had less kojibiose decorating the molecule (Fig. 4G). Last, consistent with increased kojibiose phosphorylase activity, we observed a concomitant increase in cytoplasmic D-glucose in gntR mutant strains compared to wild-type bacteria (Fig. 4H).
To better understand its substrate specificity, we next built a homology model of the E. faecalis GHF65 enzyme (fig. S8). The sequence of the enzyme served as the basis for a homology search against all sequences in the Protein Data Bank (PDB). The strongest hit was the structure of the kojibiose phosphorylase from Caldicellulosiruptor saccharolyticus, which has been solved in a complex with glucose or kojibiose (PDB no. 3wir and 3wiq, respectively; 28% sequence identity across the complete protein sequence) (29). These structures also included a phosphate/sulfate ion in the active site, mimicking the substrate-bound complex. Using the published structures as templates, we built a comparative model of the E. faecalis GHF65 enzyme bound to kojibiose and phosphate using SWISS-MODEL (fig. S8) (30). Relative to the template structure, all key active site side chains were almost identically preserved in both sequence and active site geometries. Mutagenesis studies of the C. saccharolyticus enzyme have shown the positions responsible for recognizing various disaccharides (kojibiose, nigerose, maltose, and trehalose) (29). On the basis of this recognition code, it is likely that the E. faecalis GHF65 enzyme is most active against kojibiose.
What could be the selective advantage gained by inactivating the GntR repressor, constitutively expressing the GHF65 hydrolase, and altering the abundance and composition of lipoteichoic acid on the E. faecalis cell surface? Aside from regulating autolysis, lipoteichoic acid has been implicated in antibiotic susceptibility and killing by immune cells (31). Thus, we hypothesized that gntR mutants could have greater tolerance to antibiotic challenge, an enhanced ability to withstand innate immune killing, or both. Mutant and wild-type cells showed similar minimum inhibitory concentrations (MICs) for the β-lactam antibiotic ampicillin (MIC = 4 mg/ml for both wild-type and mutant strains), suggesting that gntR mutants were not overtly more resistant to the β-lactam antibiotics in common use at the time (32). However, gntR mutants were more tolerant than wild-type strains to killing by cell wall–targeting agents, including ampicillin and lysozyme (Fig. 5A). We confirmed that this resistance to killing was due to the constitutive expression of GHF65 (fig. S9). Strains that had mutated gntR were also found to be more resistant to killing upon incubation with human blood (Fig. 5B). To determine whether these changes were of consequence in an infection model where phagocytic clearance could be measured, we compared competitive survival of gntR mutant and wild-type strains in a subcutaneous abscess mouse model (33). In as little as 24 hours, gntR mutants showed a significant survival advantage in this animal model compared to wild-type bacteria (P < 0.01; Fig. 5C). This indicated that gntR mutants could be readily isolated from the bloodstream of patients because of enhanced resistance to phagocytic killing.
Fig. 5. Resistance of gntR mutants to antibiotic treatment and innate immune stress.
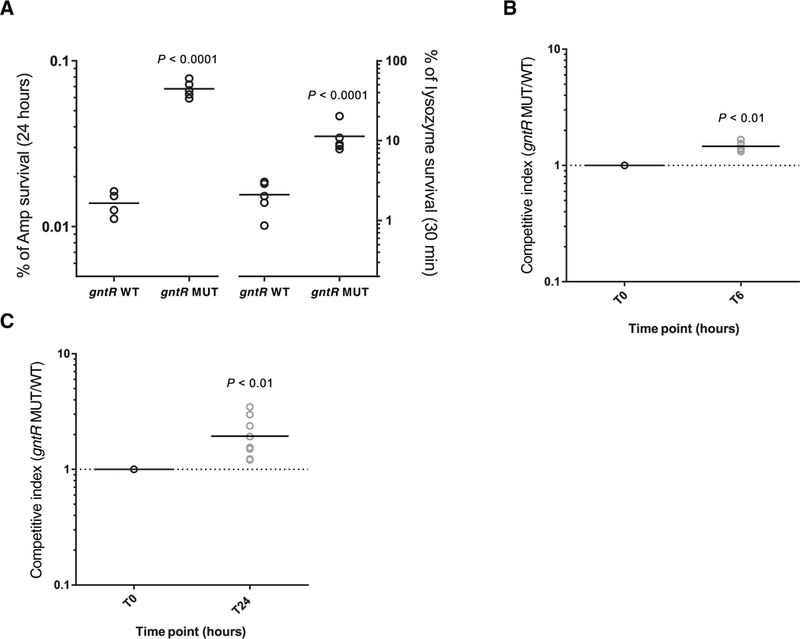
(A) Survival of gntR wild-type (WT) and mutant (MUT) E. faecalis strains in culture after treatment either for 24 hours with a lethal dose (10× WT MIC) of ampicillin (Amp) or for 30 min with lysozyme (10 mg/ml). CFU per milliliter were determined at the beginning and at the end of the assay, and survival was calculated as a percentage of the starting CFU per milliliter. (B) For the killing assay, gntR WT and MUT strains were mixed (1:1) and were incubated with fresh whole human blood from six different donors for 6 hours. Competitive index was calculated as the ratio of MUT/WT cells and was determined by selective plating on media containing tetracycline. (C) Competitive index of gntR MUT/WT strains in a mouse abscess infection model. Bacterial strains were mixed (1:1) and were injected along with cytodex beads into the hind flanks of Swiss Webster mice. After 0 and 24 hours, bacteria were sampled from the injected inoculum or were harvested from hind flank abscesses. Individual colonies were genotyped by high-resolution melting or by PCR and Sanger sequencing. At least 70 colonies were genotyped for each of nine abscesses. Data points in all plots represent independent biological replicates, and horizontal lines indicate mean values. All P values were calculated with a two-tailed t test.
Appearance of a previously unidentified mutation coincident with the introduction of carbapenem antibiotics
Unexpectedly, despite the high frequency of occurrence of gntR mutations and the survival advantages they conferred, gntR mutations were found only in isolates from the first half of the outbreak and not after January 1987 (Fig. 6A). The disappearance of gntR mutants corresponded to the appearance of a previously unidentified mutation upstream of the penicillin-binding protein 4 (pbp4) coding sequence (gene ID: SAW_02336 in the B594 strain, EF2476 in the V583 strain, and 0G1RF_11907 in the OG1RF strain). PBP4 is a high-molecular weight, class B transpeptidase that has been previously implicated in resistance to bacterial cell wall-targeting antibiotics (34, 35). Because the pbp4 mutation that we identified occurred 5’ to the coding sequence in a potential regulatory region, we quantified its effect by real-time PCR. Mutant strains were found to express fivefold more pbp4 mRNA compared to wild-type E. faecalis (Fig. 6B). We next tested whether this increased pbp4 expression affected susceptibility to cell wall-active antibiotics. Mutants were found to be more resistant to ampicillin, ceftriaxone, and imipenem but not vancomycin (Fig. 6C). The greatest increase in resistance was observed for imipenem, the first carbapenem antibiotic, which entered wide-spread use in 1987 (Fig. 6A) (36). Carbapenems exert their antibacterial effects by covalently binding to penicillin-binding proteins, and imipenem has been previously shown to bind strongly to the ortholog of the PBP4 enzyme in E. coli (37).
Fig. 6. A mutation near the pbp4 gene appears in later outbreak strains of E. faecalis.
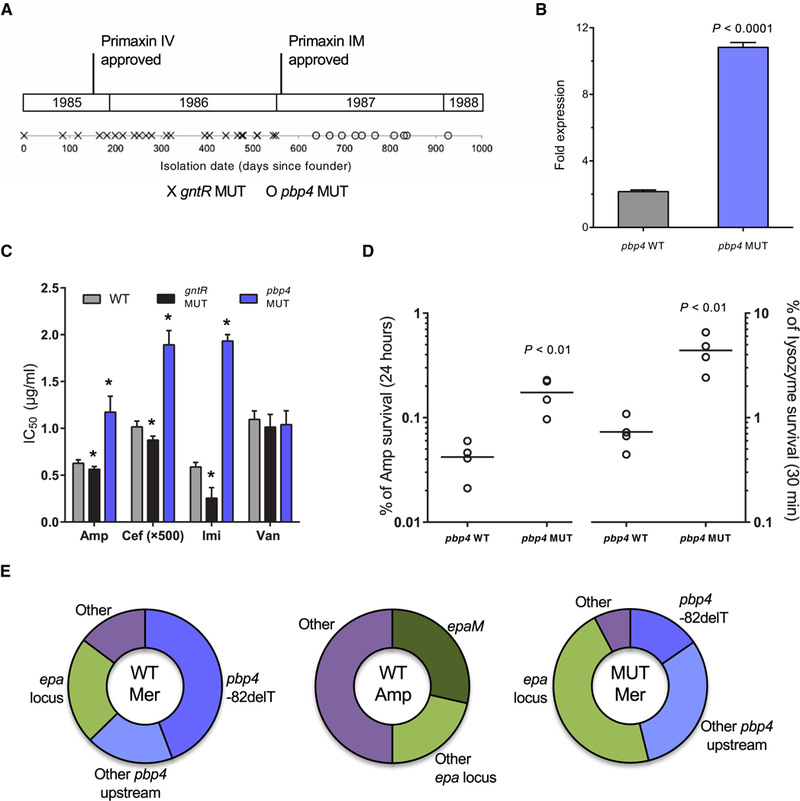
A mutation near the gene encoding pbp4 appeared in later strains most likely in response to pressure from carbapenem treatment. (A) Timeline for outbreak strains containing mutations in gntR (gntR MUT) or a mutation 82 nucleotides upstream of pbp4 (pbp4 MUT). Approval dates for intravenous (IV) and intramuscular (IM) formulations of Primaxin (imipenem/cilastatin), the first carbapenem antibiotic, are shown. (B) Fold mRNA expression of pbp4 in wild-type (WT) and MUT E. faecalis outbreak strains. (C) The 50% inhibitory concentration (IC50) values after treatment of WT, gntR MUT, and pbp4 MUT strains with ampicillin (Amp), ceftriaxone (Cef), imipenem (Imi), and vancomycin (Van). *P < 0.005 calculated with a two-tailed t test of each MUT strain relative to WT. (D) Survival of pbp4 WT and MUT strains after treatment for 24 hours with a lethal dose (10x WT MIC) of Amp or for 30 min with lysozyme (10 mg/ml). CFU per milliliter were determined at the beginning and at the end of the assay, and survival was calculated as a percentage of the starting CFU per milliliter. Data points represent independent biological replicates, and horizontal lines indicate mean values. (E) Distribution of mutations among antibiotic-resistant mutants on WT or gntR MUT strain backgrounds that were selected on meropenem (Mer) or Amp in vitro. After confirmation of resistant phenotypes, mutants were genotyped by PCR and Sanger sequencing or by genome sequencing. Bar charts in (B) and (C) show mean values with error bars indicating the SD of at least three biological replicates. All P values were calculated with a two-tailed t test.
The appearance of pbp4 mutants corresponded to the disappearance of gntR mutants. To test whether pbp4 mutants could pheno-copy gntR mutants, we compared pbp4 and gntR mutants for growth and survival in ampicillin, lysozyme, and whole human blood killing assays. Similar to gntR mutants, pbp4 mutants were more resistant to killing by both ampicillin and lysozyme (Fig. 6D); however, they were not more resistant than wild-type strains to killing in a whole human blood killing assay (fig. S10).
Because treatment records for patients from whom isolates were derived could not be obtained, we tested whether the introduction of imipenem, which occurred in 1987, could have resulted in selection for pbp4 mutants during the second half of the outbreak. We selected for single-step mutants in vitro using either meropenem (a more stable carbapenem) or ampicillin. Genotyping of mutants that were selected for either a two- or fourfold increase in MIC revealed that the dominant mutation conferring meropenem resistance was the same pbp4 mutation observed among outbreak strains (Fig. 6E). An additional 20% of meropenem-resistant mutants had different mutations upstream of the pbp4 gene (fig. S11). Among ampicillin-resistant mutants, no mutations in or near pbp4 were identified, suggesting that carbapenem pressure was specific in selecting for altered expression of pbp4. We also generated meropenem-resistant mutants in a gntR mutant strain background and found that roughly half of all mutants had a mutation upstream of pbp4 (Fig. 6E), suggesting that gntR and pbp4 mutations could coexist in the same strain. Examination of other publicly available E. faecalis genomes identified additional strains with the same mutation upstream of pbp4 but only in clinically derived strains isolated after the mid-1980s (table S5). In contrast, gntR mutants appear to have arisen in a variety of settings and over a broad time span, including six of the unrelated pre-outbreak strains examined in this study.
DISCUSSION
Here, we focused on E. faecalis strains isolated early in the emergence of enterococci as leading nosocomial pathogens. We used comparative genomics and functional approaches to describe the adaptation of E. faecalis from life as a gut commensal to survival in the face of strong selective pressures in the human bloodstream in an early enterococcal outbreak that occurred between 1984 and 1988 in a Wisconsin hospital. Overall, we found multiple pathways that enabled the survival of E. faecalis in the face of antibiotic and host immune pressures. This finding underscores both the adaptability of enterococci and the complexity of this process.
By studying a collection of closely related E. faecalis outbreak strains, we were able to focus on understanding whether any of the relatively few mutations that arose in these strains were biologically or medically relevant. Of the 285 variants we detected, 15 became progressively fixed over the course of the outbreak and an additional 64 were found in more than one E. faecalis strain. These shared variants might affect bacterial survival during transmission from one patient to the next. For example, they could confer greater tolerance to desiccation or starvation, resistance to disinfectants, or an enhanced ability to colonize the antibiotic-perturbed gastrointestinal tract (10). Here, we focused on characterizing de novo adaptation within each new patient; the shared variants we identified, however, could be similarly explored in future studies to investigate their impact on transmission of drug-resistant enterococci in the hospital environment.
Our analysis uncovered multiple mutations in distinct pathways that contributed to enterococcal survival in the face of selective pressures that are relevant to bloodstream infection. Every mutation that we identified in the cydABDC cytochrome locus and the gntR regulator appeared to affect protein function, suggesting that diminishing or abolishing their functions was important for the bacteria to proliferate and persist in the bloodstream. Note that none of the cydABDC mutations and very few of the gntR mutations were found in more than one bacterial strain. This observation suggests that these mutants arose in the human bloodstream or another extraintestinal site and then proliferated because of their survival advantage over wild-type bacteria. Alternatively, mutants could have first occurred in the gastrointestinal tract and then translocated into the bloodstream; however, in this case, the mutants would be expected to be transmitted between patients. Unfortunately, the lack of available gastrointestinal tract isolates from these patients makes it impossible to know exactly where and how these mutations arose. Nonetheless, it seems likely that, after translocation of a small number of bacteria from the gut into the bloodstream (38, 39), the population grew big enough to generate these mutants, which were then driven to high frequency by antibiotic treatment, host innate immune selection, and other possible selective pressures.
In contrast to the cydABDC and gntR mutations that we detected, the mutation uncovered in pbp4 resulted in the deletion of a deoxyadenosine from a run of seven deoxyadenosine residues. This made it impossible to know whether the same base deletion was shared among patients with E. faecalis strains and potentially was transmitted from patient to patient. The pbp4 mutation identified here is not unique to our study, but its appearance among E. faecalis strains worldwide is coincident with the introduction of carbapenems into widespread use (36). We suspected that the use of imipenem as an empirical therapy contributed to the emergence of E. faecalis strains bearing pbp4 mutations. Our conclusion that antibiotic selection likely drove emergence of the pbp4 mutation is supported by other studies (40, 41), including a recent report of the identical mutation in an E. faecalis strain infecting the joint of a patient who was treated long-term with cell wall-targeting antibiotics (41). We observed distinct mutations associated with increased resistance to carbapenems or β-lactam antibiotics in our in vitro resistance selection experiments. Mutations in or near pbp4 did not occur among ampicillin-resistant E. faecalis mutants; instead, mutations in the epa locus were detected in approximately half of these ampicillin-resistant mutants. The Epa surface polysaccharide remains to be fully characterized, but it may serve a role similar to that of cell wall teichoic acids (42). We also detected five independent mutations in the epa locus among the outbreak strains, which might have arisen within patients in response to β-lactam antibiotic treatments or other related pressures. Additional work is needed to understand the role of Epa in enterococcal growth and persistence in the human bloodstream.
A limitation of this study is the lack of available patient data to associate with the outbreak strains that were collected. The bacteria were originally isolated in the mid-1980s and were cryopreserved for nearly three decades. Despite our best efforts, patient information was determined to be no longer available for metadata analysis. In addition, our theory that the GHF65 enzyme works to cleave kojibiose from bacterial lipoteichoic acid remains to be fully validated. A more complete understanding of how E. faecalis synthesizes and modifies its lipoteichoic acid is needed, as is biochemical information regarding the substrate affinities of the enterococcal GHF65 enzyme for different disaccharides. Last, it seems counterintuitive that having less lipoteichoic acid on the bacterial surface would confer a survival advantage in the face of cell envelope-targeting stressors. We suspect that having less lipoteichoic acid on the bacterial surface may allow for more cell wall teichoic acid or Epa polysaccharide to be laid down on the peptidoglycan and that having more of these molecules on the surface confers greater tolerance to cell wall stresses; however, this theory remains to be proven.
Our study shows how an enterococcal outbreak lineage emerged and evolved over an extended hospital outbreak and how outbreak strains appeared to have responded to host immune selection and changing antibiotic regimens. These findings highlight pathways that could be further leveraged in the future for control and management of nosocomial enterococcal infections.
MATERIALS AND METHODS
Study design
This study was a retrospective functional genomics investigation of an outbreak of E. faecalis bacteremia at the University of Wisconsin Hospital and Clinics from 1984 to 1988. The outbreak was described previously (11), and the analyses presented here build upon previous work. The objectives of this study were to (i) use comparative genomics to characterize the outbreak lineage and to contrast the outbreak strains with E. faecalis strains causing bacteremia before the outbreak, (ii) identify bacterial loci undergoing selection during bloodstream infection, and (iii) characterize mechanisms by which E. faecalis can adapt to antibiotic pressure and innate immune pressure within the human host.
Collection of bacterial strains
Bacterial strains were collected from the Clinical Microbiology Laboratory at the University of Wisconsin Hospital and Clinics as part of a retrospective cohort study (11). For this study, 66 putative outbreak E. faecalis bacteremia strains isolated over the 32-month period from July 1985 to February 1988 were included. These strains were selected on the basis of high-level gentamicin resistance and high genetic relatedness, as determined by pulsed-field gel electrophoresis (11). An additional 27 pre-outbreak E. faecalis bacteremia strains were also included and represent genetically diverse strains that were isolated from the same hospital before July 1985. After collection, blood culture isolates were stored in BHI broth with 10% glycerol at −70°C. Bacterial strains were collected as part of routine diagnostic purposes, contained no human genetic material, and were deidentified before the initiation of this study.
Genomic DNA isolation, whole-genome sequencing, and assembly
Strains were first streaked onto BHI agar and grown overnight at 37°C. A single bacterial colony was then inoculated into BHI broth and grown overnight at 37°C. Genomic DNA was isolated with the Qiagen DNeasy Blood and Tissue Kit (catalog no. 69506, Valencia, CA). The earliest isolated outbreak strain, called B594, was sequenced with both Illumina (San Diego, CA) and Pacific Biosciences (PacBio, Menlo Park, CA) technology; the remaining pre-outbreak and outbreak strains were sequenced with Illumina technology. Seven outbreak strains were also sequenced with MinION technology (Oxford Nanopore Technologies, Oxford, UK). Additional details regarding genome sequencing, assembly, annotation, phylogenetic and variant analyses, and other genome comparisons are provided in the Supplementary Materials.
gntR mutant assays
Detailed methods regarding strains and growth conditions for functional assays, construction of deletion mutants, RNA sequencing, and quantitative PCR are provided in the Supplementary Materials. For gntR growth assays, gntR wild-type and mutant strains were inoculated into 200-ml BHI media in a 96-well plate at OD600 = 0.01 and were incubated at 37°C. OD600 was measured every 30 min with a Synergy2 BioTek plate reader (Winooski, VT) with Gen5 software. For CFU per milliliter measurements, serial 10-fold dilutions of each culture were made, and then, 10 ml of each dilution was spotted and tracked onto nutrient agar plates. Individual colonies were counted the next day and multiplied by the dilution factor to determine CFU per milliliter in the original cultures. Between six and nine biological replicates were included for initial growth experiments. For follow-up experiments, strains were grown overnight in BHI broth at 37°C, and the overnight OD600 was measured and recorded. Electron microscopy, monosaccharide composition analysis, and lipoteichoic acid extraction and analysis methods are described in the Supplementary Materials.
In vitro resistance selection and genotyping
MMH594 wild-type bacteria were selected for in vitro resistance to ampicillin or meropenem with a single-step selection approach. Briefly, 50 μl of a stationary-phase culture of bacteria was spread onto a 10-cm petri dish containing BHI agar with ampicillin (16 mg/ml) or meropenem (32 μg/ml). Plates were incubated at 37°C for 2 to 5 days to allow resistant mutants to emerge. Individual colonies were passaged in BHI without drug and were then tested for resistance by determining their MIC values and comparing to the wild-type parental strain. Mutants with two- or fourfold higher MICs as measured after 24 hours were retained and were genotyped either by PCR and Sanger sequencing of the region upstream of pbp4 or by whole-genome sequencing. Variants were identified from genome sequence data with CLC Genomics Workbench v8 and Pilon (43).
Statistical analysis
Changes in the numbers of shared and singleton variants over time were assessed by linear regression. Enrichment of mutations at the gntR, cydABDC, and epa loci was calculated with Fisher’s exact test. Significance of all other differences was assessed with a two-tailed t test.
Supplementary Material
Acknowledgments:
We thank all members of the Gilmore Lab and, particularly, F. Lebreton, A. Gaca, E. Selleck Fiore, J. Schwartzman, J. Saavedra, and J. Wurster. We also thank D. Hooper and C. Chen for assistance with mouse experiments and F. Ausubel and S. Walker for helpful feedback on the manuscript. We acknowledge the contributions made to this project by T. Shea, S. Saif, A. D. Griggs, T. J. Straub, and S. Young. In addition, we thank C. Spiegel for helping to collect and store the bacterial strains from the 1980s.
Funding: This work was supported by PHS grant nos. AI083214 (Harvard-wide Program on Antibiotic Resistance) and AI072360 to M.S.G. and grant no. EY028222 to D.V.T. Additional support for genome sequence analysis was provided by NIAID contract no. HHSN272200900018C and grant no. U19AI110818 to the Broad Institute. M.M.H. was supported by the Francis Duffy Endowment and J.K. was supported by NIH/NCI Cancer Center Support Grant P30 CA006927.
Footnotes
Competing interests: M.S.G. has served as a consultant to Arietis Pharma on unrelated matters, and A.L.M. and A.M.E. served as consultants to Synlogic Therapeutics and Janssen Pharmaceutica on unrelated matters. All other authors declare that they have no competing interests.
Data and materials availability: All data associated with this study are present in the paper or the Supplementary Materials. The genome sequence data for all strains reported in this study are archived in the National Center for Biotechnology Information with accession numbers presented in table S1. Bacterial strains analyzed in this study are available through a material transfer agreement and can be obtained by contacting the corresponding author at michael_gilmore@meei.harvard.edu.
SUPPLEMENTARY MATERIALS
www.sciencetranslationalmedicine.org/cgi/content/full/11/487/eaat8418/DC1
REFERENCES AND NOTES
- 1.Van Tyne D, Gilmore MS, Friend turned foe: Evolution of enterococcal virulence and antibiotic resistance. Annu. Rev. Microbiol. 68, 337–356 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lebreton F, Manson AL, Saavedra JT, Straub TJ, Earl AM, Gilmore MS, Tracing the enterococci from paleozoic origins to the hospital. Cell 169, 849–861.e13 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gilmore MS, Lebreton F, van Schaik W, Genomic transition of enterococci from gut commensals to leading causes of multidrug-resistant hospital infection in the antibiotic era. Curr. Opin. Microbiol. 16, 10–16 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Agudelo Higuita NI, Huycke MM, in Enterococci: From Commensals to Leading Causes of Drug Resistant Infection, Gilmore MS, Clewell DB, Ike Y, Shankar N, Eds. (Boston, 2014). [PubMed] [Google Scholar]
- 5.Van Tyne D, Gilmore MS, Raising the alarmone: Within-host evolution of antibiotictolerant Enterococcus faecium. MBio 8, e00066–17 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Billington EO, Phang SH, Gregson DB, Pitout JDD, Ross T, Church DL, Laupland KB, Parkins MD, Incidence, risk factors, and outcomes for Enterococcus spp. blood stream infections: A population-based study. Int. J. Infect. Dis. 26, 76–82 (2014). [DOI] [PubMed] [Google Scholar]
- 7.Taur Y, Xavier JB, Lipuma L, Ubeda C, Goldberg J, Gobourne A, Lee YJ, Dubin KA, Socci ND, Viale A, Perales M-A, Jenq RR, van den Brink MRM, Pamer EG, Intestinal domination and the risk of bacteremia in patients undergoing allogeneic hematopoietic stem cell transplantation. Clin. Infect. Dis. 55, 905–914 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ubeda C, Taur Y, Jenq RR, Equinda MJ, Son T, Samstein M, Viale A, Socci ND, van den Brink MRM, Kamboj M, Pamer EG, Vancomycin-resistant Enterococcus domination of intestinal microbiota is enabled by antibiotic treatment in mice and precedes bloodstream invasion in humans. J. Clin. Invest. 120, 4332–4341 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gentry-Weeks CR, Karkhoff-Schweizer R, Pikis A, Estay M, Keith JM, Survival of Enterococcus faecalis in mouse peritoneal macrophages. Infect. Immun. 67, 2160–2165 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.C. A. Arias, B. E. Murray, The rise of the Enterococcus: Beyond vancomycin resistance. Nat. Rev. Microbiol. 10, 266–278 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Huycke MM, Spiegel CA, Gilmore MS, Bacteremia caused by hemolytic, high-level gentamicin-resistant Enterococcus faecalis. Antimicrob. Agents Chemother. 35, 1626–1634 (1991). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Van Tyne D, Martin MJ, Gilmore MS, Structure, function, and biology of the Enterococcus faecalis cytolysin. Toxins 5, 895–911 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Shankar N, Baghdayan AS, Gilmore MS, Modulation of virulence within a pathogenicity island in vancomycin-resistant Enterococcus faecalis. Nature 417, 746–750 (2002). [DOI] [PubMed] [Google Scholar]
- 14.Paulsen IT, Banerjei L, Myers GSA, Nelson KE, Seshadri R, Read TD, Fouts DE, Eisen JA, Gill SR, Heidelberg JF, Tettelin H, Dodson RJ, Umayam L, Brinkac L, Beanan M, Daugherty S, DeBoy RT, Durkin S, Kolonay J, Madupu R, Nelson W, Vamathevan J, Tran B, Upton J, Hansen T, Shetty J, Khouri H, Utterback T, Radune D, Ketchum KA, Dougherty BA, Fraser CM, Role of mobile DNA in the evolution of vancomycin-resistant Enterococcus faecalis. Science 299, 2071–2074 (2003). [DOI] [PubMed] [Google Scholar]
- 15.McBride SM, Coburn PS, Baghdayan AS, Willems RJL, Grande MJ, Shankar N, Gilmore MS, Genetic variation and evolution of the pathogenicity island of Enterococcus faecalis. J. Bacteriol. 191, 3392–3402 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Winstedt L, Frankenberg L, Hederstedt L, von Wachenfeldt C, Enterococcus faecalis V583 contains a cytochrome bd-type respiratory oxidase. J. Bacteriol. 182, 3863–3866 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Huycke MM, Moore D, Joyce W, Wise P, Shepard L, Kotake Y, Gilmore MS, Extracellular superoxide production by Enterococcus faecalis requires demethylmenaquinone and is attenuated by functional terminal quinol oxidases. Mol. Microbiol. 42, 729–740 (2001). [DOI] [PubMed] [Google Scholar]
- 18.Painter KL, Hall A, Ha KP, Edwards AM, The electron transport chain sensitizes Staphylococcus aureus and Enterococcus faecalis to the oxidative burst. Infect. Immun. 85, e00659–17 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Teng F, Singh KV, Bourgogne A, Zeng J, Murray BE, Further characterization of the epa gene cluster and Epa polysaccharides of Enterococcus faecalis. Infect. Immun. 77, 3759–3767 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dale JL, Cagnazzo J, Phan CQ, Barnes AMT, Dunny GM, Multiple roles for Enterococcus faecalis glycosyltransferases in biofilm-associated antibiotic resistance, cell envelope integrity, and conjugative transfer. Antimicrob. Agents Chemother. 59, 4094–4105 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rigottier-Gois L, Madec C, Navickas A, Matos RC, Akary-Lepage E, Mistou M-Y, Serror P, The surface rhamnopolysaccharide Epa of Enterococcus faecalis is a key determinant of intestinal colonization. J. Infect. Dis. 211, 62–71 (2015). [DOI] [PubMed] [Google Scholar]
- 22.Hancock LE, Gilmore MS, The capsular polysaccharide of Enterococcus faecalis and its relationship to other polysaccharides in the cell wall. Proc. Natl. Acad. Sci. U.S.A. 99, 1574–1579 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Andersson U, Levander F, Radstrom P, Trehalose-6-phosphate phosphorylase is part of a novel metabolic pathway for trehalose utilization in Lactococcus lactis. J. Biol. Chem. 276, 42707–42713 (2001). [DOI] [PubMed] [Google Scholar]
- 24.Collins J, Robinson C, Danhof H, Knetsch CW, van Leeuwen HC, Lawley TD, Auchtung JM, Britton RA, Dietary trehalose enhances virulence of epidemic Clostridium difficile. Nature 553, 291–294 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Taguchi Y, Saburi W, Imai R, Mori H, Evaluation of acceptor selectivity of Lactococcus lacti sssp. lactis trehalose 6-phosphate phosphorylase in the reverse phosphorolysis and synthesis of a new sugar phosphate. Biosci. Biotechnol. Biochem. 81, 1512–1519 (2017). [DOI] [PubMed] [Google Scholar]
- 26.Kessler RE, Duke J, Goldstein IJ, Interaction of anti-kojibiose antibody with the lipoteichoic acids from Streptococcus faecalis and Streptococcus faecium. Infect. Immun. 46, 279–281 (1984). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Theilacker C, Kaczynski Z, Kropec A, Fabretti F, Sange T, Holst O, Huebner J, Opsonic antibodies to Enterococcus faecalis strain 12030 are directed against lipoteichoic acid. Infect. Immun. 74, 5703–5712 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Theilacker C, Sanchez-Carballo P, Toma I, Fabretti F, Sava I, Kropec A, Holst O, Huebner J, Glycolipids are involved in biofilm accumulation and prolonged bacteraemia in Enterococcus faecalis. Mol. Microbiol. 71, 1055–1069 (2009). [DOI] [PubMed] [Google Scholar]
- 29.Okada S, Yamamoto T, Watanabe H, Nishimoto T, Chaen H, Fukuda S, Wakagi T, Fushinobu S, Structural and mutational analysis of substrate recognition in kojibiose phosphorylase. FEBS J. 281, 778–786 (2014). [DOI] [PubMed] [Google Scholar]
- 30.Biasini M, Bienert S, Waterhouse A, Arnold K, Studer G, Schmidt T, Kiefer F, Gallo Cassarino T, Bertoni M, Bordoli L, Schwede T, SWISS-MODEL: Modelling protein tertiary and quaternary structure using evolutionary information. Nucleic Acids Res. 42, W252–W258 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Pasquina LW, Santa Maria JP, Walker S, Teichoic acid biosynthesis as an antibiotic target. Curr. Opin. Microbiol. 16, 531–537 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gross MH, Barriere SL, Pathogenesis and treatment of enterococcal infections. Clin. Pharm. 3, 161–166 (1984). [PubMed] [Google Scholar]
- 33.Chen C, Hooper DC, Effect of Staphylococcus aureus Tet38 native efflux pump on in vivo response to tetracycline in a murine subcutaneous abscess model. J. Antimicrob. Chemother. 73, 720–723 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ono S, Muratani T, Matsumoto T, Mechanisms of resistance to imipenem and ampicillin in Enterococcus faecalis. Antimicrob. Agents Chemother. 49, 2954–2958 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hiraga N, Muratani T, Naito S, Matsumoto T, Genetic analysis of faropenem-resistant Enterococcus faecalis in urinary isolates. J. Antibiot. 61, 213–221 (2008). [DOI] [PubMed] [Google Scholar]
- 36.Clissold SP, Todd PA, Campoli-Richards DM, Imipenem/cilastatin. A review of its antibacterial activity, pharmacokinetic properties and therapeutic efficacy. Drugs 33, 183–241 (1987). [DOI] [PubMed] [Google Scholar]
- 37.Yang Y, Bhachech N, Bush K, Biochemical comparison of imipenem, meropenem and biapenem: Permeability, binding to penicillin-binding proteins, and stability to hydrolysis by β-lactamases. J. Antimicrob. Chemother. 35, 75–84 (1995). [DOI] [PubMed] [Google Scholar]
- 38.Zeng J, Teng F, Weinstock GM, Murray BE, Translocation of Enterococcus faecalis strains across a monolayer of polarized human enterocyte-like T84 cells. J. Clin. Microbiol. 42, 1149–1154 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.van der Heijden KM, van der Heijden IM, Galvao FH, Lopes CG, Costa SF, Abdala E, D’Albuquerque LA, Levin AS, Intestinal translocation of clinical isolates of vancomycin-resistant Enterococcus faecalis and ESBL-producing Escherichia coli in a rat model of bacterial colonization and liver ischemia/reperfusion injury. PLOS ONE 9, e108453 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Duez C, Zorzi W, Sapunaric F, Amoroso A, Thamm I, Coyette J, The penicillin resistance of Enterococcus faecalis JH2–2r results from an overproduction of the low-affinity penicillin-binding protein PBP4 and does not involve a psr-like gene. Microbiology 147, 2561–2569 (2001). [DOI] [PubMed] [Google Scholar]
- 41.Rice LB, Desbonnet C, Tait-Kamradt A, Garcia-Solache M, Lonks J, Moon TM, D’Andréa ED, Page R, Peti W, Structural and regulatory changes in PBP4 trigger decreased β-lactam susceptibility in Enterococcus faecalis. MBio 9, e00361–18 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Mistou M-Y, Sutcliffe IC, van Sorge NM, Bacterial glycobiology: Rhamnose-containing cell wall polysaccharides in gram-positive bacteria. FEMSMicrobiol. Rev. 40, 464–479 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Walker BJ, Abeel T, Shea T, Priest M, Abouelliel A, Sakthikumar S, Cuomo CA, Zeng Q, Wortman J, Young SK, Earl AM, Pilon: An integrated tool for comprehensive microbial variant detection and genome assembly improvement. PLOS ONE 9, e112963 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ribeiro FJ, Przybylski D, Yin S, Sharpe T, Gnerre S, Abouelleil A, Berlin AM, Montmayeur A, Shea TP, Walker BJ, Young SK, Russ C, Nusbaum C, MacCallum I, Jaffe DB, Finished bacterial genomes from shotgun sequence data. Genome Res. 22, 2270–2277 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Fisher S, Barry A, Abreu J, Minie B, Nolan J, Delorey TM, Young G, Fennell TJ, Allen A, Ambrogio L, Berlin AM, Blumenstiel B, Cibulskis K, Friedrich D, Johnson R, Juhn F, Reilly B, Shammas R, Stalker J, Sykes SM, Thompson J, Walsh J, Zimmer A, Zwirko Z, Gabriel S, Nicol R, Nusbaum C, A scalable, fully automated process for construction of sequence-ready human exome targeted capture libraries. Genome Biol. 12, R1 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Chin C-S, Alexander DH, Marks P, Klammer AA, Drake J, Heiner C, Clum A, Copeland A, Huddleston J, Eichler EE, Turner SW, Korlach J, Nonhybrid, finished microbial genome assemblies from long-read SMRT sequencing data. Nat. Methods 10, 563–569 (2013). [DOI] [PubMed] [Google Scholar]
- 47.Chaisson MJ, Tesler G, Mapping single molecule sequencing reads using basic local alignment with successive refinement (BLASR): Application and theory. BMC Bioinformatics 13, 238 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Li H, Durbin R, Fast and accurate short read alignment with burrows-wheeler transform. Bioinformatics 25, 1754–1760 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R; 1000 Genome Project Data Processing Subgroup, The sequence alignment/ map format and SAMtools. Bioinformatics 25, 2078–2079 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Gordon D, Abajian C, Green P, Consed: A graphical tool for sequence finishing. Genome Res. 8, 195–202 (1998). [DOI] [PubMed] [Google Scholar]
- 51.Bourgogne A, Garsin DA, Qin X, Singh KV, Sillanpaa J, Yerrapragada S, Ding Y, Dugan-Rocha S, Buhay C, Shen H, Chen G, Williams G, Muzny D, Maadani A, Fox KA, Gioia J, Chen L, Shang Y, Arias CA, Nallapareddy SR, Zhao M, Prakash VP, Chowdhury S, Jiang H, Gibbs RA, Murray BE, Highlander SK, Weinstock GM, Large scale variation in Enterococcus faecalis illustrated by the genome analysis of strain OG1RF. Genome Biol. 9, R110 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wick RR, Judd LM, Gorrie CL, Holt KE, Unicycler: Resolving bacterial genome assemblies from short and long sequencing reads. PLOS Comput. Biol. 13, e1005595 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Lebreton F, van Schaik W, McGuire AM, Godfrey P, Griggs A, Mazumdar V, Corander J, Cheng L, Saif S, Young S, Zeng Q, Wortman J, Birren B, Willems RJL, Earl AM, Gilmore MS, Emergence of epidemic multidrug-resistant Enterococcus faecium from animal and commensal strains. MBio 4, e00534–13 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Hyatt D, Chen G-L, LoCascio PF, Land ML, Larimer FW, Hauser LJ, Prodigal: Prokaryotic gene recognition and translation initiation site identification. BMC Bioinformatics 11, 119 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lowe TM, Eddy SR, tRNAscan-SE: A program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res. 25, 955–964 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Lagesen K, Hallin P, Rodland EA, Stærfeldt H-H, Rognes T, Ussery DW, RNAmmer: Consistent and rapid annotation of ribosomal RNA genes. Nucleic Acids Res. 35, 3100–3108 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Finn RD, Tate J, Mistry J, Coggill PC, Sammut SJ, Hotz H-R, Ceric G, Forslund K, Eddy SR, Sonnhammer ELL, Bateman A, The Pfam protein families database. Nucleic Acids Res. 36, D281–D288 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Haft DH, Loftus BJ, Richardson DL, Yang F, Eisen JA, Paulsen IT, White O, TIGRFAMs: A protein family resource for the functional identification of proteins. Nucleic Acids Res. 29, 41–43 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Ogata H, Goto S, Sato K, Fujibuchi W, Bono H, Kanehisa M, KEGG: Kyoto Encyclopedia of Genes and Genomes. Nucleic Acids Res. 27, 29–34 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Tian W, Arakaki AK, Skolnick J, EFICAz: A comprehensive approach for accurate genome-scale enzyme function inference. Nucleic Acids Res. 32, 6226–6239 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Krogh A, Larsson B, von Heijne G, Sonnhammer ELL, Predicting transmembrane protein topology with a hidden Markov model: Application to complete genomes. J. Mol. Biol. 305, 567–580 (2001). [DOI] [PubMed] [Google Scholar]
- 62.Wapinski I, Pfeffer A, Friedman N, Regev A, Automatic genome-wide reconstruction of phylogenetic gene trees. Bioinformatics 23, i549–i558 (2007). [DOI] [PubMed] [Google Scholar]
- 63.Stamatakis A, RAxML-VI-HPC: Maximum likelihood-based phylogenetic analyses with thousands of taxa and mixed models. Bioinformatics 22, 2688–2690 (2006). [DOI] [PubMed] [Google Scholar]
- 64.Salazar A, Earl A, Desjardins C, Abeel T, Normalizing alternate representations of large sequence variants across multiple bacterial genomes. BMC Bioinformatics 16 (suppl. 2), A8 (2015). [Google Scholar]
- 65.Matos RC, Lapaque N, Rigottier-Gois L, Debarbieux L, Meylheuc T, Gonzalez-Zorn B, Repoila F, de Fatima Lopes M, Serror P, Enterococcus faecalis prophage dynamics and contributions to pathogenic traits. PLOS Genet. 9, e1003539 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Darling ACE, Mau B, Blattner FR, Perna NT, Mauve: Multiple alignment of conserved genomic sequence with rearrangements. Genome Res. 14, 1394–1403 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Kurtz S, Phillippy A, Delcher AL, Smoot M, Shumway M, Antonescu C, Salzberg SL, Versatile and open software for comparing large genomes. Genome Biol. 5, R12 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Zhang G, Mills DA, Block DE, Development of chemically defined media supporting high-cell-density growth of lactococci, enterococci, and streptococci. Appl. Environ. Microbiol. 75, 1080–1087 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Thurlow LR, Thomas VC, Hancock LE, Capsular polysaccharide production in Enterococcus faecalis and contribution of CpsF to capsule serospecificity. J. Bacteriol. 191, 6203–6210 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Trieu-Cuot P, Carlier C, Poyart-Salmeron C, Courvalin P, A pair of mobilizable shuttle vectors conferring resistance to spectinomycin for molecular cloning in Escherichia coli and in gram-positive bacteria. Nucleic Acids Res. 18, 4296 (1990). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Santander J, Martin T, Loh A, Pohlenz C, Gatlin III DM, Curtiss III R, Mechanisms of intrinsic resistance to antimicrobial peptides of Edwardsiella ictaluri and its influence on fish gut inflammation and virulence. Microbiology 159, 1471–1486 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Lowry OH, Lopez JA, The determination of inorganic phosphate in the presence of labile phosphate esters. J. Biol. Chem. 162, 421–428 (1946). [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.