

Abstract
Efficient delivery of therapeutic nanoparticles (TNPs) to tumors is critical in improving efficacy, yet strategies that universally maximize tumoral targeting by TNP modification have been difficult to achieve in the clinic. Instead of focusing on TNP optimization, here we show that the tumor microenvironment itself can be therapeutically primed to facilitate accumulation of multiple clinically relevant TNPs. Building on the recent finding that tumor associated macrophages (TAM) can serve as nanoparticle drug depots, we demonstrate that local tumor irradiation substantially increases TAM relative to tumor cells, and thus TNP delivery. High-resolution intravital imaging reveals that following radiation, TAM primarily accumulate adjacent to microvasculature, elicit dynamic bursts of extravasation, and subsequently enhance drug uptake in neighboring tumor cells. TAM depletion eliminates otherwise beneficial radiation effects on TNP accumulation and efficacy, and controls with un-encapsulated drug show radiation effects are more pronounced with TNPs. Priming with combined radiation and cyclophosphamide enhances vascular bursting and tumoral TNP concentration, in some cases leading to a 6-fold increase of TNP accumulation in the tumor, reaching 6% of the injected dose per gram tissue (ID/g). Radiation therapy alters tumors for enhanced TNP delivery in a TAM-dependent fashion, and these observations have implications for the design of next-generation tumor targeted nanomaterials and clinical trials for adjuvant strategies.
One sentence summary:
Radiation therapy enhances nanotherapeutic drug delivery in a tumor associated macrophage-dependent fashion.
Introduction
Therapeutic nanoparticles (TNPs) have been developed to improve the safety and efficacy of encapsulated anti-neoplastic payloads, including clinically-approved nanoformulations that encapsulate doxorubicin (DOXIL, Caelyx, Myocet), irinotecan (Onivyde), paclitaxel (Abraxane), and vincristine (Marqibo). Many other TNPs are undergoing clinical development for cancer indications, using related cytotoxic payloads such as daunorubicin, cyterabine, platinum derivatives, and more recently, molecularly targeted inhibitors (1). Extensive work has demonstrated the potential of TNPs to i) extend drug release and systemic pharmacokinetics, ii) reduce the need for toxic drug solvents and prolonged intravenous infusions, iii) facilitate combination treatments, and iv) increase tumoral drug accumulation. This last feature is thought to occur through “passive targeting” by enhanced permeability and retention (EPR) effects including irregular tumor vasculature, dysfunctional lymphatics, and increased cellular uptake (2). Unfortunately, clinical trials have shown mixed efficacy of TNPs relative to solvent-based drug formulations, presumably due to heterogeneous EPR effects (3) and poor TNP delivery to the tumor. In most cases, >95% of TNPs fail to reach the tumor target, and by some metrics, this poor efficiency has failed to substantially improve over the past decade (4). One strategy to overcome this issue has been active molecular targeting of TNP to cells via affinity ligands and antibodies. However, a recent meta-analysis revealed active targeting typically increases the percent injected dose (%ID) by only 50% compared to passively-targeted TNPs (4), and clinical trials often show equivocal improvement (5, 6).
These results underscore how cellular and anatomical features of the tumor microenvironment represent major limiting factors to delivery of both passive and actively targeted TNPs alike. For instance, vascular permeability is known to be higher in many solid tumors, which theoretically should enable enhanced TNP uptake; nonetheless, recent reports highlight dynamic and heterogeneous macromolecular transport within tumors, characterized by long periods of poor vascular permeability punctuated by brief “bursts” of extravasation into the tumor tissue (7,8). Thus, therapeutically enhancing hyper-permeable “bursts” represents a clear opportunity for improving TNP delivery to tumors. However, the mechanisms to do so are relatively unknown.
Several studies have shown that local tumor radiation therapy (RT) is a clinically ubiquitous tool with the potential to improve TNP delivery via effects on endothelial architecture, permeability, and decreased interstitial fluid pressure in the tumor (9). Yet, the integration of the many RT-mediated microenvironmental and vascular changes with cellular-level mechanisms has been poorly understood, leading to unclear strategies for the timing and fractionation of RT. For instance, although tumor associated macrophages (TAM) have been implicated in RT response (9, 10) and TNP delivery (11), it is still unclear if TAM are a requirement for RT-mediated EPR effects and by what mechanism they may influence TNP uptake post-RT. Consequently, results for clinical trials that combine RT with TNP administration have often been mixed and underwhelming (9).
To address these issues, we combined computational modeling tools (12) with multi-color intravital microscopy (IVM) at single-cell resolution (13) to study how RT may improve TNP delivery. We bridge concepts and mechanisms from disparate studies that show the importance of TAM in modulating RT response via angiogenic and pro-survival signaling (10, 14, 15), along with the prominent behavior TAM display in functioning as drug reservoirs that accumulate substantial TNP and redistribute their cytotoxic payload to neighboring tumor cells (11). Complementary cell depletion experiments ultimately demonstrate that TAM are required for RT-mediated effects. We show that a single dose of RT enhances vascular bursting via a cascade of changes to the tumor vasculature and microenvironment, improving TNP delivery.
Results
Quantitative IVM reveals coordinated changes in the tumor microenvironment after RT.
Multiple studies now document the effect of RT on various disparate features of the tumor vasculature, myeloid populations, and tumor cellularity; nonetheless, little has been done to quantitatively integrate such measurements at a systems-level to discern how various features interact and collectively shift upon therapeutic intervention. To address this need, we combined single-cell multi-color imaging, automated image segmentation, and multivariate statistical analysis to distill key RT effects (Fig. 1). Briefly, the tumor vasculature of HT1080 human fibrosarcoma-bearing mice was imaged using an intravenously (i.v.)-administered fluorescent 70 kDa dextran conjugate; tumor associated phagocytes including TAM were imaged using a recently developed protocol with dextran-coated 20 nm nanoparticles (NPs) administered 24 hours prior to imaging (3); and tumor cells were imaged by the transgenic nuclear-localized 53BP1-mApple reporter protein. Live-animal time-lapse IVM was performed using a dorsal window chamber over the tumor, which enabled acquisition of multiple z-stack images of typical dimension 318 μm x 318 μm x 100 μm with sub-micron resolution (Fig. 1a). Fluorescent dextran revealed vascular structure immediately upon tumor perfusion. Semi-automated image segmentation of IVM data quantified various features of cellular distribution and endothelium geometry within the tumor (Fig. 1b). This analysis pipeline was validated by manual measurements and alternative labeling strategies, showing roughly 95% average accuracy (fig. S1a–d). Over time, effective macromolecular permeability of the tumor vasculature was calculated from the gradual extravasation of dextran into the tumor tissue (16). Altogether, 16 features were derived from a cohort of tumors as a baseline description of the tumor microenvironment (Fig. 1c; see fig. S1e for absolute quantification).
Figure 1. Integrated intravital analysis of RT-treated tumors reveals coordinated changes in the tumor microenvironment in mice.
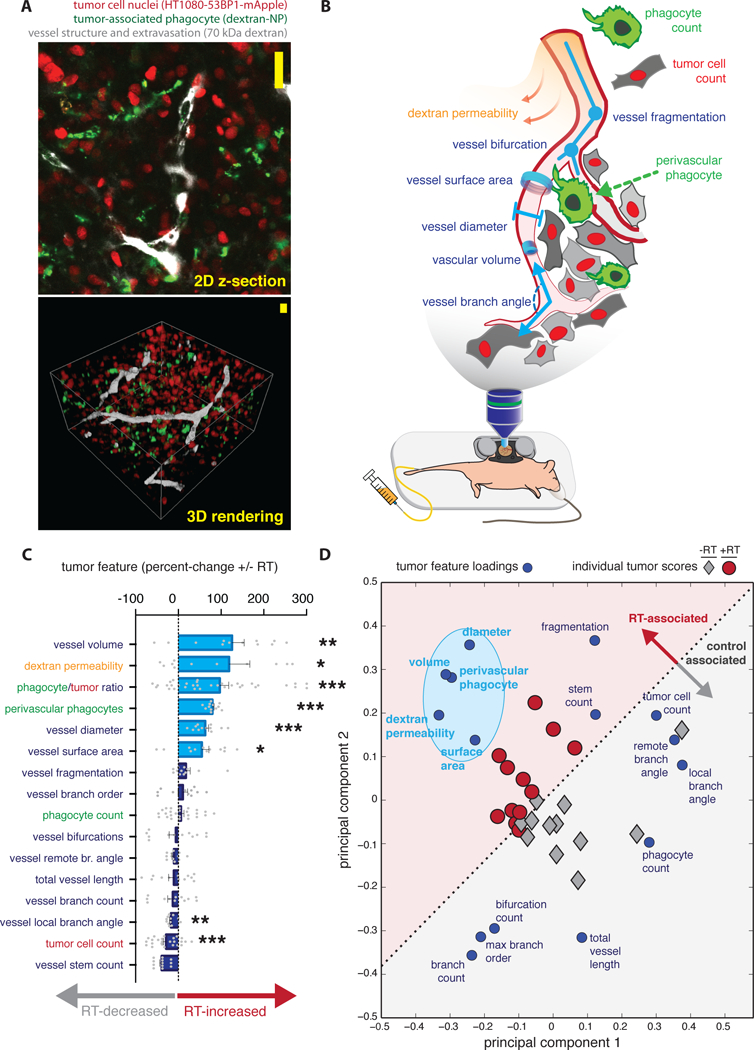
a) Z-stacks comprised of individual image slices of live tumors (top) were compiled and rendered into 3D reconstructions (bottom) for quantitative analysis, either without RT or 3 days post-RT (scale bars = 20 μm). b) 16 distinct image features were semi-automatically calculated from data as in a, 10 of which are depicted here as properties of vascular geometry (blue-purple), phagocyte distribution (green), tumor cell density (red), and rates of dextran extravasation as measured over the course of roughly 2 hours (yellow-orange). c) Image features were ranked by how they changed with RT (n ≥ 12 tumor regions across n ≥ 4 animals; *P<0.05; **P<0.01; ***P<0.001; student’s two-tailed t-test; data are means ± standard error). d) Principal components analysis decomposed 16 image features into two key axes of variation (principal components 1 and 2); principal component loadings of the image features (blue) and scores of the tumor images themselves (black/red) are plotted together, with the light-blue oval highlighting image features that correlatively increase with RT.
Given previous evidence that TNP delivery to tumors occurs indirectly through TAM (11), we hypothesized that RT could maximally improve TNP accumulation under conditions eliciting TAM increase. Therefore, we developed a custom platform for conformal window-chamber RT (fig. S2) and followed a previously optimized treatment scheme showing that 5 Gy local irradiation enhances TAM after 3–4 days (10). We used IVM to examine what impact RT had on not only numbers of TAM, but also their spatial relationship to vascular structure and tumor cell features.
Analysis of individual tumor features revealed that multiple properties of vessel geometry, permeability, and phagocyte localization all change with RT (Fig. 1c; see fig. S3a for representative images). Most substantially, RT-treated tumor vessels were larger, more permeable, and exhibited a higher fraction of TAM located in close proximity to vessels (within 5 μm). Transmission electron microscopy (TEM) of RT-treated tumors provides examples of porous and partially denuded tumor endothelium (fig. S3b), which supports the IVM evidence of enhanced permeability. To understand how such changes in the microenvironment related to each other, we performed principal components analysis (PCA) to decompose the different IVM measurements into two main regulatory axes, or principal components. The PCA scores and loadings plot offers a statistical interpretation of how individual measurements correlate with each other (by their loadings), and how aggregate differences in the ensemble of measurements collectively shift for a given tumor (by their scores). In this unsupervised computational approach, no a priori information is provided to segregate data from the different treatment groups; nonetheless, PCA scores signaled a natural partitioning between tumors that had been irradiated and those that had not (Fig. 1d). Furthermore, PCA loadings indicated close correlation between features related to larger vessel size (surface area, diameter, and volume), dextran permeability, along with the perivascular localization of phagocytes. These features (Fig. 1d, highlighted in light blue) all increased with RT, and their proximity in PCA-loadings suggests that perivascular macrophages are directly influencing vascular permeability, and consequently, drug delivery.
Relative TAM increase in tumor xenografts and patient tumor biopsies after RT corresponds with enhanced TNP uptake.
We next studied the dynamics and generalizability of tumor associated phagocyte enrichment after RT. Using a fluorescent genetic reporter mouse model expressing green fluorescent protein (GFP)+ TAM via the MerTK promoter (Fig. 2a–c), along with 20 nm dextran-NPs as above (Fig. 2d–e, fig. S2), we performed non-invasive longitudinal imaging of tumors to track cellular changes over time. With both of these phagocyte-labeling strategies, the ratio of phagocytes to tumor cells in HT1080 xenografts remained unchanged for two days following RT: phagocytes were outnumbered 3–4 fold by tumor cells, which were found at roughly 20 cells per 104 μm2. However, by 3–4 days post-RT, relative phagocyte numbers increased by >50%, and diminished again by 11 days post-RT (Fig. 2a–e). Similar phagocyte increases did not occur without RT, indicating the changes were not merely a coincidental feature of tumor growth.
Figure 2. Tumor irradiation enhances relative numbers of tumor-associated macrophages.
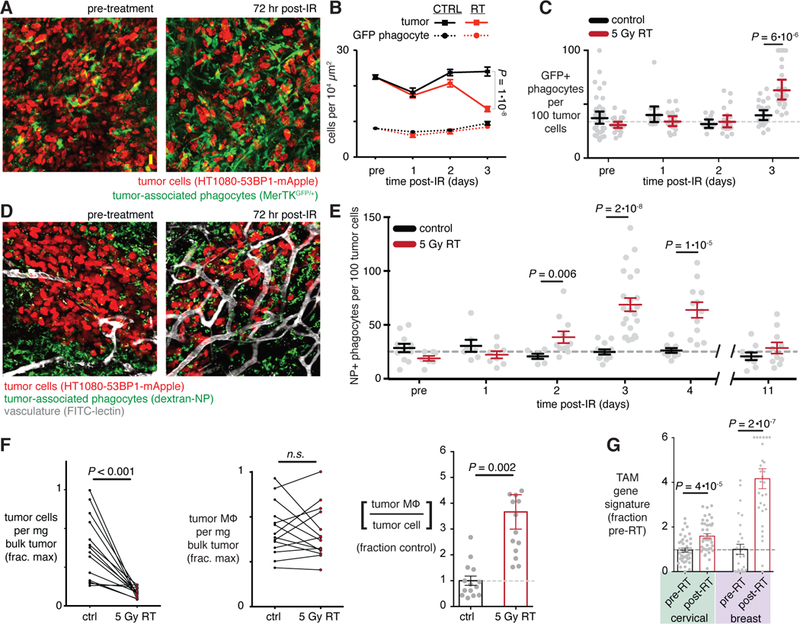
a-e) Tumors were longitudinally imaged immediately before and after 5 Gy irradiation in either MerTKGFP/+ fluorescent reporter mice (a-c) or nu/nu mice with NP-labeled tumor phagocytes (d-e). Representative images (a,d) were quantified according to phagocyte cell density (c,e) before and after RT (n ≥ 4; two-tailed student’s t-test). f) Flow cytometry quantified TAM (CD45+CD11b+CD11c+F4/80+) relative to tumor cells (CD45-hCD29+), after HT1080 tumors were treated 72 hrs post-RT and then excised (n=13; student’s two-tailed t-test). g) Tumor biopsies from cervical and breast cancer patients were collected before and within 1 week of local RT, and were analyzed by microarray to quantify TAM gene expression signature enrichment (n ≥ 26; student’s t-test). For all, data are means ± standard error. Scale bar, 20 μm.
TAM enrichment was driven by a corresponding decrease in tumor cell density, as determined by IVM (Fig. 2b) and evidence of radiation sensitivity in an in vitro clonogenic assay (fig. S4a). In contrast, imaging showed that phagocytes did not substantially decrease (Fig. 2b). As further evidence, we monitored cell proliferation in vivo by measuring how a cell-labeling dye decreases in brightness as cells divide. After intravenous dye injection, one of two bilateral tumors was irradiated. Three days after RT, phagocytes were labeled with dextran-NP, and the following day tumors were excised and analyzed by flow cytometry for dye brightness. Irradiated tumor cells exhibited higher dye concentrations compared to their matched non-irradiated counterparts, indicating RT-induced growth arrest (fig. S4b). In contrast, phagocytes and other stromal cells showed no enhanced growth arrest during this time frame — likely because they minimally proliferate in situ, and in the case of TAM may arise from non-irradiated hematopoietic tissues and monocyte reservoirs (fig. S4b). To study an immunologically defined TAM population, we again performed flow cytometry on matched irradiated and un-irradiated bilateral tumors. Three days after RT, animals were treated with a model polymeric TNP encapsulating a cytotoxic cisplatin prodrug (Pt-NP)(11), and the following day tumors were excised and analyzed for CD45+CD11b+CD11c+F4/80+ TAM amounts in relation to tumor cells. Consistent with the imaging results, flow cytometry indicated a decrease in tumor cell density but no effect on TAM density (defined by cells per mg of tissue). Consequently, RT caused a 3.5-fold increase in the number of TAM relative to tumor cells (Fig. 2f).
To investigate whether similar changes occur in patients, we analyzed TAM amounts in breast and cervical cancer biopsies obtained before and within 10 days following localized RT. Cervical cancer patients were biopsied one week after 9 Gy whole pelvic RT (fractionated 1.8 Gy QDx5) (17), and breast cancer patients received single fraction high-dose radiosurgery (15–21 Gy) 10 days prior to lumpectomy (18). For an unbiased automated analysis, we inferred TAM amounts using a validated computational platform for assessing immune cell abundance(19) by mining available RNA microarray datasets(17, 18). Results show that TAM were the most up-regulated among over 20 cell-types measured when analyzing biopsies pre- and post- RT (Fig. 2g). The greatest TAM enrichment occurred in those patients receiving the highest RT dose, 21 Gy (fig. S4c), although 9 Gy (cervical cancer; Fig. 2g) and 15 Gy (fig. S4c) both showed effects. This may be the result of increased tumor cell killing at higher doses, as we observed using HT1080 in the in vitro clonogenic assay (fig. S4a). However, in the xenograft model we found that higher irradiation (15 Gy) failed to dramatically enhance phagocytic NP uptake in tumors compared to 5 Gy, although both doses showed greater effects than with 2 Gy (fig. S4d).
We next tested whether RT-induced TAM enrichment corresponded with higher TNP accumulation in the tumor. As a syngeneic immunocompetent model of breast cancer, we used 4T1 mammary carcinoma cells implanted into mammary fat pads. Two fluorescently labeled model TNPs — a liposome and a polymeric micelle (Pt-NP) – were injected i.v. 1, 2, and 3 days after local tumor irradiation. The day after liposome and Pt-NP injection, tumors were excised and analyzed by fluorescence for TNP uptake. Just as with the TAM analysis, RT effects on TNP uptake occurred 3 days post-RT, roughly doubling tumor TNP numbers (fig. S5a). We next asked if RT exerted a similar impact on solvent-based drug formulations, or whether the delivery enhancement was specific to TNP. Doxorubicin is clinically administered both as an un-encapsulated drug or encapsulated within PEGylated liposome (DOXIL). Using the same 4T1 model, either un-encapsulated or encapsulated doxorubicin was administered 3 days post-RT. The following day, tumors were excised, digested, and analyzed by HPLC for doxorubicin content. Although RT failed to substantially enhance un-encapsulated drug accumulation, the liposomal-encapsulated doxorubicin (DOXIL) was detected at 300% higher concentrations (to 4 μM) in the irradiated tumors (fig. S5b).
We next used flow cytometry to parse the degree to which enhanced TAM and TNP amounts in the bulk tumor corresponded with drug exposure in tumor cells. Using the HT1080 tumor model, we i.v. injected Pt-NP labeled with two distinct fluorescent dyes that enabled measurement of both the TNP vehicle (PLGA-BODIPY630) and its cytotoxic Pt payload (C16-Pt(IV)-BODIPY) 3 days post-RT (11). 24 hrs post-injection, tumors were excised and analyzed by flow cytometry for drug uptake in tumor, using a human-selective CD29 antibody. Consistent with the bulk tumor analysis, we found that RT increased uptake of both TNP vehicle and its cytotoxic payload in tumor cells (fig. S5c). Interestingly, TNP payload delivery was roughly 40% greater in tumor cells compared to increases in TNP vehicle (fig. S5d), and previous research shows this is caused by redistribution of TNP drug from TAM to neighboring cells (11). Less than half (44%) of increased drug exposure to tumor cells from RT occurred through direct uptake of TNP vehicle (fig. S5e), determined by fitting flow cytometry data to a model of TNP delivery and drug release (11). The other half is attributable to payload redistribution, where the TNP vehicle releases payload either in the tumor interstitium or after having been taken up by neighboring TAM, as demonstrated previously (11). Consistent with this model, Pt-NP causes greater DNA damage in tumor cells’ neighboring phagocytes that have accumulated high amounts of the NP vehicle (11). In this work, we tested this model using a fluorescently labeled liposomal irinotecan and found similar results: tumor cells’ neighboring phagocytes that had accumulated high amounts of liposome exhibited greater amounts of DNA damage response (fig. S5f).
Although distinct TNP formulations exhibit different physicochemical properties, rates of systemic clearance, and rates of uptake by macrophage (fig. S2), using confocal imaging we found that the nanoformulations distribute to phagocytes in irradiated tumors with >90% overlap. In other words, >90% of phagocytes that accumulate any nanoformulation will also accumulate another co-administered nanoformulation (n ≥ 3; fig. S6). Patterns of payload redistribution, metabolism, and activity after being taken up by TAM may be different across TNPs, and analysis of these patterns extends beyond the scope of this work. Nonetheless, co-localization across nanoformulations and phagocyte labeling strategies, including use of dextran-NP (fig. S6a–c) and the MerTKGFP/+ fluorescent reporter model (fig. S6d), suggest that TNPs tend to accumulate in similar TAM populations, even if at different rates as measured in vitro (fig. S2).
TAM co-localize with dynamic bursts of permeability in irradiated tumor vasculature.
Based on the above evidence, we hypothesized that RT could enhance TNP delivery to tumor cells i) by increasing the TAM amounts as already noted, and ii) by influencing macromolecular extravasation efficiency in a TAM-dependent manner, allowing more TNP to directly reach tumor cells. Analysis of the initial IVM dataset already suggested a close relationship between perivascular TAM and vascular permeability (Fig. 1d). Interestingly, vessel permeability was the most heterogeneous among all 16 tumor features (fig. S7a; see video S1 for representative data) and correlated most closely with perivascular phagocyte content (fig. S7b). To better understand these observations, we closely examined time-lapse images of dextran extravasation. Surprisingly, we found not just spatial heterogeneity, but also highly dynamic extravasation behavior. As shown in several examples (Fig. 3), periods of minimal dextran extravasation were interrupted by large bursts in vascular effusion, especially on thicker vessels with phagocytes nearby; similar heterogeneous burst effects were observed using fluorescently labeled TNP. Although some bursts were observed in the absence of irradiation, occurrences increased considerably 3 days post-RT (Fig. 4a). Statistics from multiple bursts across multiple tumors indicated that they happened at random times following dextran administration, with some bursts occurring immediately and others occurring only towards the end of the time-lapse imaging session (fig. S7c). Interestingly, bursts often happened in groups such that multiple bursts on multiple vessels in the same tumor region would occur at similar times (fig. S7d), suggesting bursts may be caused by local microenvironmental cues. Burst dynamics were generally observed within a single image frame of 5 min (Fig. 4b) and produced a transient 6.4-fold increase in effective vessel permeability within that time, as averaged over all bursts. Consistent with the PCA analysis (Fig. 1), we found that hyper-permeable bursts were more likely to occur on larger vessels that are closely neighbored by perivascular phagocytes (Fig. 4c). Averaging the permeability measurements for a burst region over the full two hour time-lapse largely erased the dramatic differences in local effective permeability between neighboring (within 300 μm) tumor regions that contained no bursts (Fig. 4c). This effect may be attributed to the transient nature of these bursts, combined with the potential of other effects such as interstitial diffusion and lymphatic clearance. Nonetheless, effective average permeability, even as averaged over the entire time-course, was still 2.2-fold higher in RT-treated tumors when compared to untreated controls (Fig. 4c).
Figure 3. Tumor associated phagocytes co-localize with heterogeneous extravasation bursting.
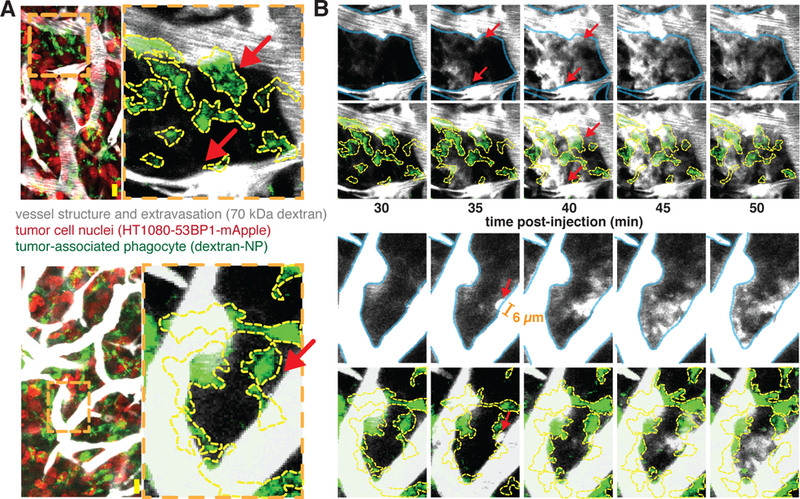
3 days post-RT, tumors were imaged every 5 min after injection of fluorescent 70 kDa dextran (greyscale). a) Zoomed-in regions where bursting will occur (red arrows) are highlighted in two example images collected immediately after dextran injection to show vascular geometry and phagocyte co-localization. Orange dashed lines denote zoomed-in regions where bursts occur, and yellow lines outline phagocytes. b) Images of dextran extravasation in the example regions where bursting will occur shown over 50 minutes post-injection. Vessels (blue, top rows) and phagocytes (yellow, bottom rows) are outlined for context. In the bottom example, the orange bar marks the diameter of a pre-burst protrusion (6 μm). Scale bars, 20 μm.
Figure 4. RT-induced vascular bursts are rapid and associated with thicker phagocyte-associated vessels.
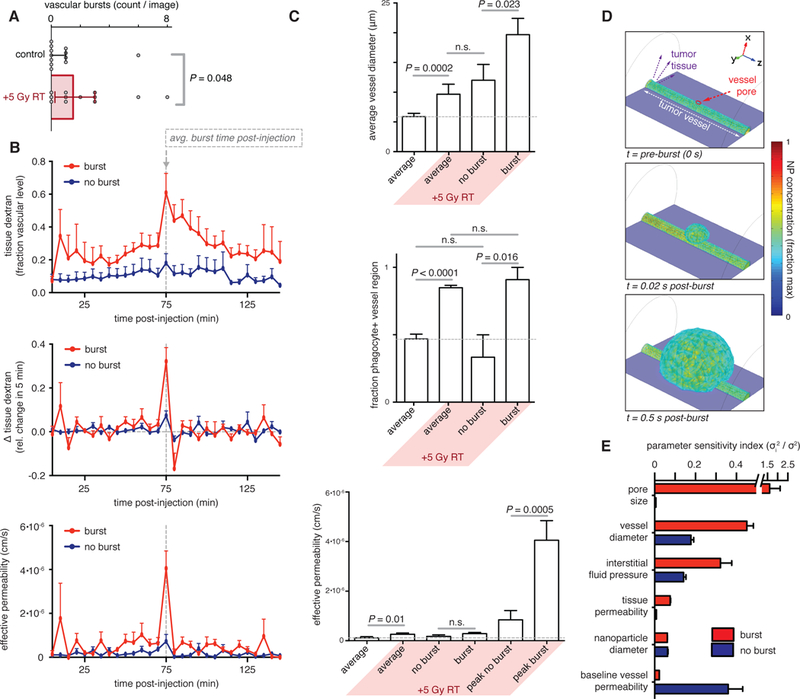
a) Plot of vascular bursts per image assessed 3 days post-RT (n ≥ 4; two-tailed Mann-Whitney test; data are median ± IQR). b) Tissue dextran (top), change in dextran (middle), and effective permeability (bottom) within RT-treated tumors after injection. Burst regions were compared to regions with consistently low (no burst) permeability (n ≥ 10; data are means ± standard error). c) Vessel diameter (top), phagocyte proximity (middle), and permeability (bottom) were averaged across all imaged tumors or across specific regions where bursts did or did not occur (n ≥ 10; data are means ± standard error). For permeability, measurements were calculated as averaged over the entire imaging time course (2 hrs) or at the time of peak extravasation. Two-tailed student’s t-test used, except for phagocyte proximity, which used Mann-Whitney. d) 3D simulation of vascular burst showing the initial opening of a pore on the vessel wall as a function of time. e) Parameter sensitivity analysis of 3D pore-opening simulations showing the degree to which physicochemical parameters influence the change in effective permeability of the vessel (mean ± standard error, n > 5).
Computational modeling provides a mechanistic basis for burst features.
To mechanistically comprehend how dynamic bursts may be occurring, we developed a three-dimensional, non-linear partial differential equation (PDE) model of macromolecular and TNP transport and extravasation. The PDE model was parameterized using published data (table S1) combined with the imaging data acquired here, and then in silico experiments were performed to test the effect of various features on burst behavior (Fig. 4d). We first tested the effect of pore size and found that bursts were most prominent with a dynamic vessel opening of >1 μm in diameter (fig. S7e). This result matches imaging data showing initial dextran protrusion from the vasculature with a diameter ranging from 5–10 μm (Fig. 3). Such length scales suggest large intercellular gaps in the vascular wall (for example as seen by TEM, fig. S3b), rather than intracellular transcytosis mechanisms, which occur on a smaller length scale(20). PDE modeling demonstrated that larger vessels seen with RT treatment (Fig. 1c) lead to greater bursting due to the higher fluid volume source (fig. S7e), although higher luminal fluid pressures in these vessels may also contribute. The PDE modeling likewise matched the imaging data in showing that bursts occur on a very rapid time-scale (Fig. 4d). Finally, decreases in interstitial fluid pressure that are consistent with past RT reports (21) and imaging evidence of reduced tumor cellular density relative to TAM (Fig. 2) also lead to enhanced bursting, as predicted by PDE modeling (fig. S7e). We used PDE modeling to predict the impact of NP diameter on vascular bursting and observed little effect (fig. S7e). To compare the relative importance of all parameters, we performed a sensitivity analysis by quantifying how vessel bursting changes in response to alterations in underlying rate constants and physicochemical properties. This result showed that extravasation at a vessel burst site (red bars) depends most on the size of the vessel pore; outside of burst sites (blue bars), the burst’s pore size did not impact extravasation behavior (Fig. 4e). Vessel diameter and interstitial fluid pressure both highly influenced extravasation, more so at burst sites compared to elsewhere in the vessel; in contrast, permeability of the extravascular tumor tissue (hydraulic conductivity) and NP diameter had less substantial influences (Fig. 4e; S7e). Overall, these results provide a mechanistic explanation of how microscopy observations, such as increased vessel diameter (Fig. 4c), contribute to enhanced vascular bursting.
TAM depletion reverts global RT-mediated shifts in the tumor microenvironment.
We next tested whether TAM are required for the effects of RT on tumor vasculature, dynamic bursting, and enhanced TNP uptake. Mice bearing irradiated tumors were co-treated with clodronate liposomes (clod-lip) to systemically deplete phagocytes, and tumors were imaged and quantitatively analyzed just as in Fig. 1. To ascertain general patterns in tumor microenvironmental features, we hierarchically bi-clustered data in an unsupervised manner and found that RT-treated tumors only clustered distinctly in the absence of clod-lip (fig. S8a). As expected, clod-lip treatment reduced the amount of perivascular phagocytes. Additionally, other RT-increased features including vessel diameter, permeability, and number of bursts also substantially decreased with clod-lip co-treatment (fig. S8a). A statistical classifier combining all 16 tumor features to differentiate irradiated from un-irradiated tumors inferred that co-treatment with clod-lip completely reverted the irradiation phenotype (Fig. 5a). We next examined the effect of clod-lip on RT-mediated TNP uptake using three model TNPs: fluorescent liposomes, Pt-NP, and dextran-coated NPs. As previously shown, we observed 2.6-fold enhanced TNP uptake within the bulk mass of irradiated tumors (Fig. 5b). In contrast, no difference in uptake was observed between irradiated and un-irradiated tumors in clod-lip treated mice (Fig. 5b). Thus, TAM and perivascular phagocytes are required for RT-mediated changes to the tumor microenvironment and corresponding improvements in TNP delivery.
Figure 5. Radiation effects on vascular structure and NP uptake require TAM.
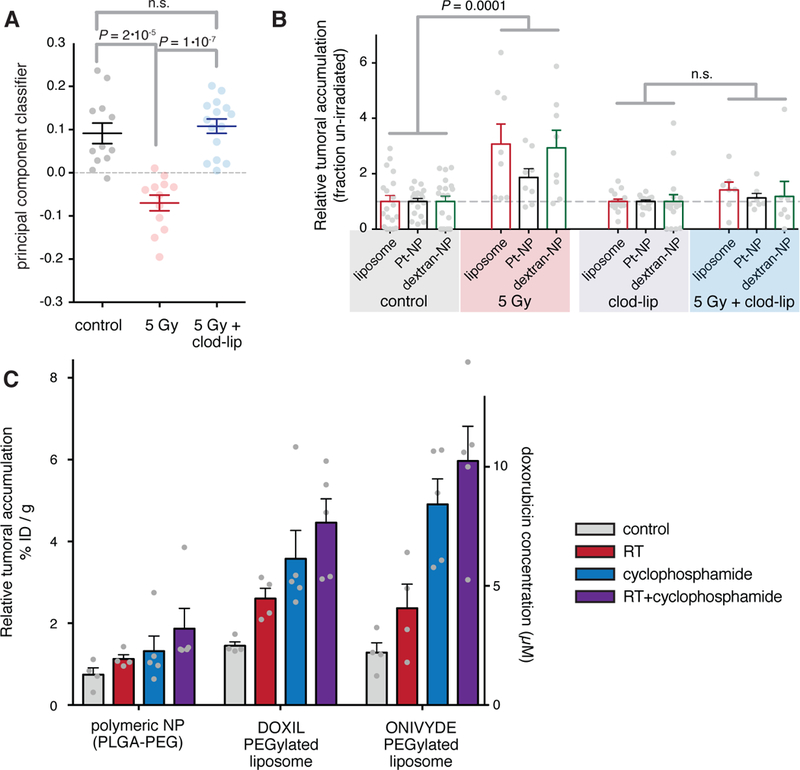
a) Tumor-bearing animals were irradiated 3-days prior, with or without co-treatment using clod-lip to deplete TAM, and analyzed by IVM as in Fig. 1. Machine-learning classification of tumors based on principal components analysis are shown (student’s two-tailed t-test; data are means ± standard error, n ≥ 12). b-c) Relative tumoral accumulation of fluorescent liposomes, polymeric micelle Pt-NP, dextran-NP (b) along with PEGylated liposomal doxorubicin and PEGylated liposomal irinotecan (c) in tumors injected 3 days post-RT. Tumors were excised 24 hrs after injection and were analyzed by whole-tumor fluorescence and HPLC (for doxorubicin, right axis) to quantify drug delivery. Data are means ± standard error; sample sizes n are shown by grey dots.
Radiation combined with cyclophosphamide enhances dynamic vessel bursting and TNP delivery.
To determine the potential of RT combination therapy with other tumor-priming treatments, we co-treated tumors with RT and the DNA alkylating agent cyclophosphamide. Previous reports show that cyclophosphamide enhances TNP delivery to tumors several days after treatment, primarily through decreasing tumor cellularity, decreasing interstitial fluid pressure, and therefore facilitating drug extravasation and penetration(22). We hypothesized that such influences on the tumor microenvironment could be beneficially combined with RT effects, and indeed found that cyclophosphamide together with RT yielded the greatest increase in TNP uptake compared to either treatment alone (Fig. 5c; P<0.002, two-tailed t-test, n≥12). For liposomal irinotecan, the measured liposome vehicle accumulating in the tumor increased from 1% to 6% ID/g. Delivery of the polymeric NP vehicle (PLGA-PEG labeled with PLGA-BODIPY630) was lower compared the liposome formulations, possibly owing to a shorter plasma half-life (fig. S2), but nevertheless increased with RT and cyclophosphamide treatment (Fig. 5c). For liposomal DOXIL, the doxorubicin payload itself was measured by HPLC to increase from 1% to 4%, which corresponds with an increase in tumor concentration to 8 μM (Fig. 5c). A similar 600% increase was seen in the 4T1 orthotopic, syngeneic model of breast cancer, where combined RT and cyclophosphamide increased tumor concentration to 7 μM (fig. S8b). Average tumor mass at time of TNP treatment ranged from 150 mg (Fig. 5c, 6a) to 250 mg (fig. S8b), and therefore 1–1.5% ID accumulated in the combined cyclophosphamide and RT treated tumors. For all three nanoformulations in the HT1080 model (Fig. 5c), pooled two-way ANOVA showed significant independent effects of both RT (P = 0.03) and cyclophosphamide (P < 0.001; n ≥ 12) on increasing TNP tumor deposition.
Figure 6. RT-enhanced drug efficacy depends on nano-encapsulation and TAM.
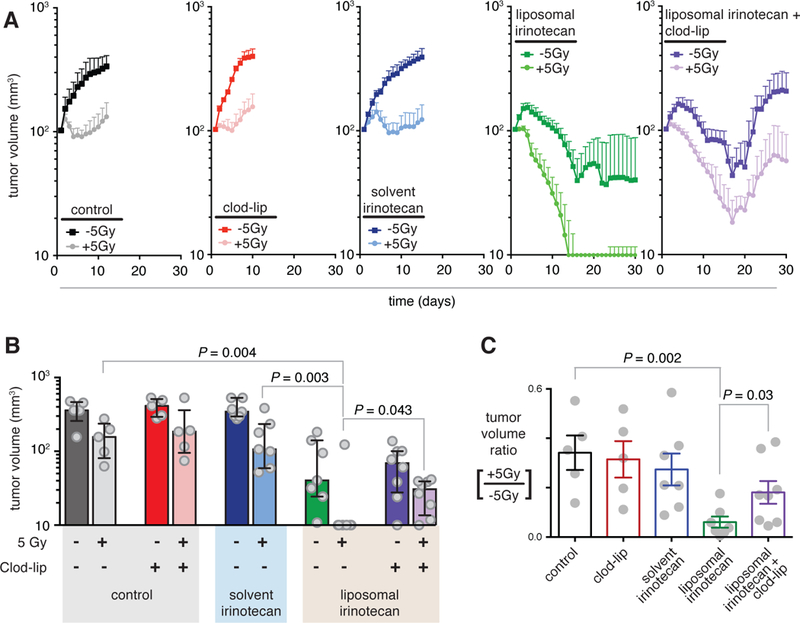
a-c) Tumor volume as measured by caliper over time. 2.5 weeks post-implantation, half of bilateral tumors were irradiated on day 0 with or without clod-lip co-treatment. 3 days post-RT, either solvent or liposomal irinotecan (20 mg/kg) was i.v. injected(a; n ≥ 5; data are means ± standard error). b) Individual tumor volumes (grey points) were measured on day 18 (median ± IQR; two-tailed Mann-Whitney test). c) Minimum tumor volume ratios for irradiated vs. non-irradiated bilateral tumors treated with or without clod-lip, solvent, or liposomal irinotecan (mean ± standard error; student’s two-tailed t-test).
Increased TNP delivery with RT and cyclophosphamide corresponded with tumor microenvironment changes. For instance, in agreement with previous observations using cyclophosphamide we found that combined treatment with RT and cyclophosphamide substantially decreased tumor cellularity by 38%, while not substantially decreasing phagocyte density (fig. S8c–d). Of note, the correspondingly enhanced bursts of vessel permeability were observed to be of larger area than as seen with RT alone (fig. S8c–f). Indeed, computational PDE modeling agrees with these data (fig. S7e) and predicts the area of burst extravasation to increase with decreased tumor cellularity and interstitial fluid pressure. Supporting this model, interstitial fluid pressure has been directly measured to decrease by ~30% with cyclophosphamide treatment (22).
RT synergistically improves longitudinal drug efficacy in a nano-encapsulation and TAM-dependent manner.
We next addressed the questions of how RT impacts longitudinal response to TNP treatment, and whether these effects depend on either nano-encapsulation or TAM. We irradiated one of two bilateral HT1080 tumors and then monitored tumor growth over time. By itself, RT stalled tumor growth (Fig. 6a–b; P = 0.011, two-tailed t-test, n=5). Consistent with previous reports using this tumor model (11), clod-lip treatment alone impacted neither untreated nor RT-treated tumors. Next, we examined a standard solvent-based irinotecan formulation and the recently FDA-approved liposomal irinotecan (Onivyde) combined with RT in blocking tumor growth. Consistent with previous reports (23), we found solvent irinotecan to be relatively ineffective; furthermore, combined solvent-irinotecan and RT provided no discernible benefit over RT alone. In contrast, liposomal irinotecan blocked tumor growth in un-irradiated tumors, and completely eliminated the majority of RT-treated tumors. Although TAM depletion had little impact on tumor growth in the absence of TNP, TAM depletion using clod-lip significantly reduced the efficacy of the nano-encapsulated irinotecan both in un-irradiated (P = 0.047, two-tailed t-test, n > 6) and irradiated (P = 0.02, two-tailed t-test, n > 6) tumors. As a metric of synergistic tumor killing, irradiated tumors shrunk most compared to un-irradiated tumors when liposomal irinotecan was used, especially in the absence of TAM depletion (Fig. 6c). We used two-way ANOVA tests to quantify synergy and found significant synergistic interaction between liposomal irinotecan and RT (P = 0.03, two-way ANOVA interaction term, n > 5). With TAM depletion, no significant synergy was found between RT and TNP treatment, nor was synergy found between RT and the solvent-based irinotecan formulation (P > 0.05, two-way ANOVA interaction term, n > 5). Overall, these results are consistent with above experiments showing nano-encapsulated drugs accumulate to a greater degree in irradiated tumors through TAM-dependent changes in the tumor microenvironment.
Discussion
In this work, we use a combination of computational tools and in vivo microscopy to find new principles of how RT can be used as a neoadjuvant therapy to prime the tumor microenvironment and enhance TNP delivery. Despite efforts to optimize TNP physicochemical properties such as size, shape, and binding affinity, extensive heterogeneity in drug delivery across patients and tumors (3) has remained a barrier to efficacy and clinical translation. Rather than focus on engineering TNPs to specifically accumulate in certain types of tumors, here we hypothesized that therapeutic intervention could re-wire the tumor microenvironment to generally promote delivery of multiple nanomedicines already in the clinic. Previous work has examined the influence of RT on vascular architecture, angiogenic pathways, and vessel permeability. Yet, in general there has been a gap between disparate anatomical observations, cellular and immunological behaviors, TNP cellular uptake and redistribution, and longitudinal disease progression. By using semi-automated multivariate methods at several levels of analysis --- from image segmentation to downstream network-level statistical interpretation --- we identified key features of how the tumor microenvironment responds to RT and which variables facilitate TNP delivery. In particular, we found that RT-induced vessel thickening, tortuous vascular branching, and perivascular TAM localization all combine in enhancing vessel permeability via dynamic bursts of extravasation. TAM depletion reverts RT-induced vascular changes, blocks dynamic bursting, and reduces the tumor-killing benefits of combining TNP and RT.
Our results are consistent with a series of past observations describing RT-induced morphologic changes in vasculature (24–26). Across the 16 features of the tumor microenvironment measured, we found average vessel volume to increase most. Past reports have used electron microscopy, histology, and the genetically-engineered MMTV breast cancer mouse model to observe enlarged vessel lumens after RT, most prominently noted several days after treatment (24). Interestingly, this past report found that RT led to a sinusoidal-like endothelium and the development of sub-micron holes in the microvascular network formed by transluminal pillars, which was posited to be the result of intussusceptive angiogenesis (24) and may contribute to the vascular bursting described herein. As a complementary process, small-diameter capillaries have been reported as more radiosensitive, so that vasculature shifts to larger diameter vessel distributions following RT when the small capillaries are destroyed (25). Large fenestrated and tortuous vasculature has long been associated with VEGF and nitric oxide (NO) signaling (27), and both have found upregulated in irradiated endothelium (26). Angiogenic factors, along with reactive oxygen and nitrogen species (ROS/RNS) including nitric oxide, likely contribute to the enhanced permeability observed here. Past studies have shown that anti-VEGF treatment blocks vascular bursting (8), and clinical trials have examined the combination of anti-VEGF therapies with RT (28). In this work, we found that RT-induced vessel enlargement, enhanced tortuosity, and increased vascular bursting could be co-opted for selective TNP delivery and improved tumor-killing.
We found that TAM are required for RT-enhanced vascular bursting and drug delivery, which is supported by recent reports describing a role for bone-marrow derived myeloid cells in the post-RT tumor microenvironment (29). In particular, studies demonstrate that Tie-2+ monocytes and TAM interact with irradiated tumor endothelium via paracrine signaling to facilitate endothelial cell survival, vascular remodeling, and angiogenesis (8, 30). Leukocyte interaction with endothelium is further modulated via the upregulation of cell adhesion molecules such as ICAM-1 (31) and galectins (32) following irradiation. Through a variety of computational approaches and validation with TAM-depletion experiments, we showed that perivascular TAM, particularly on larger branched vessels, are required for enhanced vascular bursting and permeability. We and others observed that RT stimulates a significant increase in TAM relative to tumor cells (Fig. 2; ref. (10, 15)), and the clinical relevance of this finding is further supported here using a new analysis of 56 matched pre- and post-RT patient biopsies (Fig. 2g).
The applicability of combined RT and TNP treatment is broad: more than half of all cancer patients undergo RT at some point, ongoing clinical trials for TNP and immunotherapy frequently use RT, and advanced RT techniques including proton therapy, image-guided RT, and intensity modulated RT are continually improving. Immune-modulating and DNA-damaging treatments besides RT also can improve TNP delivery, for instance as has been noted for the alkylating agent cyclophosphamide (22). The effective combination of RT, cyclophosphamide, and TNP tested here is highly translatable, considering ongoing clinical trials are evaluating the triple therapy. Importantly, our results have implications for how such studies are designed with respect to sequencing RT and TNP administration. RT may also combine with other approaches for improving TNP delivery, including local hyperthermia, ultrasonication, and blood pressure modulation. Given the prominent role TAM and other myeloid-derived phagocytes play in nanotherapeutic delivery, it is likely the technique will apply to other NP platforms, including those based on carbon nanotubes, quantum dots, dendrimers, albumin, and transition metals such as gold. Clinically it is often difficult to treat tumors with high TAM amounts (33), and combined RT and TNP may be a successful approach in such cases.
Despite their advantages, combined RT and TNP have some limitations. RT is less common for tumors that are difficult to irradiate without affecting neighboring radiosensitive tissue. Especially at higher doses (≥15 Gy), RT carries risks including fibrosis, infertility, and bowel damage. Encouragingly, we found that the lower 5 Gy dose was still effective in enhancing TNP delivery. Cancer cells may become radioresistant, which could be problematic considering we found that tumor cell response was a component of microenvironment priming. Although RT dose fractionation was studied here in one example (Fig. 2g), most analysis used a model one-time dosing regimen. Thus, future studies may explore how repeated rounds of treatment, fractionated RT, and the potential emergence of resistance, may influence the tumor microenvironment. In this work we found that TAM were beneficial for drug delivery, yet this observation may depend on the tumor type and particular nano-formulation. For instance, in some cases systemic phagocyte ablation may actually improve tumor delivery by decreasing hepatic clearance and extending systemic PK (34). Ideally, future studies may develop strategies to combine RT and TAM enrichment with inhibition of phagocytic uptake in the liver.
Overall, the integration of imaging and computational methodologies presented here, together with the principles of tumor-priming via TAM-dependent vascular bursting, have general application to multiple clinical therapeutic settings.
Materials and Methods
Study Design:
The hypothesis was that RT pre-treatment would enhance the accumulation of TNP in tumors. Image studies and drug accumulation measurements were designed to measure the effects of RT on vascular structure, phagocyte content, and drug uptake. Tumor caliper measurements assessed the long-term impact of combined RT and TNP treatment on tumor progression. All experiments were performed with at least three independent replicates, as specified in figure legends; data collection methodology and randomized treatment group assignment procedure was predetermined; and no outliers were excluded. Cohort sizes were informed by group sizes of previously performed experiments and their corresponding power analyses (3, 14). Researchers were blinded to groups during caliper measurements, and additional automated computational measurements were performed using equally applied algorithm parameters. Further materials and methods details can be found in the Supplementary Materials and Methods.
Animal and cell models:
Experiments were performed with female mice that were 5–10 weeks old at the start of the experiment. For human HT1080 tumors, 2 × 106 cells were bilaterally implanted subcutaneously into nu/nu mice (Cox7, Massachusetts General Hospital); once tumors reached 6 mm in diameter (2.5 weeks later) treatment was initiated for imaging and/or flow cytometry analysis. Every other day, mice were weighed and caliper measurements were averaged from two independent researchers, and tumor volumes were calculated according to a spherical model (V = 4/3 π r3). In all experiments, animals were euthanized once tumors reached size over 10 mm in diameter or health deteriorated (body condition score 2 or less). For the breast cancer model, 5 × 105 4T1 cells were bilaterally implanted into the thoracic mammary fat pads of BALB/c mice (the Jackson Laboratory); about 7 days later, RT and/or TNP treatment was initiated once tumors were palpable. Purchased in 2012 from ATCC, the mouse cell line 4T1, along with the human cell line HT1080, were authenticated by the vendor and routinely tested for mycoplasma and MAP contamination. Cell culture followed manufacturer guidelines. Transgenic HT1080 cells (53BP1-mApple) were produced as described previously (35). All animal research was performed in accordance with the guidelines from the Institutional Subcommittee on Research Animal Care.
Radiation Therapy:
For all RT, a dual source 137Cs Gammacell 40 Exactor (Best Theratronics) was used with a custom-built lead shield comprising two 4 cm thick lead slabs, a lucite immobilization platform, and a metal frame for positioning within the Gammacell center RT field (fig. S2). 3 cm corner posts separated the two slabs from each other to accommodate the lucite platform, and sides did not require shielding given the photon source directionality. A 2 cm aperture was cut through each lead slab to allow local irradiation. Prior to use, dosimetric studies using standard film-based and thermoluminescent dosimeter techniques showed the average dose in the center of the aperture to be 68.5% ± 3% (mean ± S.D.) of that delivered with a fully open field. Dose at 2 cm inferior to the aperture was 95.6% lower than in the aperture center. The Gammacell holds a published dose rate accuracy of ± 15%. Immediately prior to RT, mice were anesthetized via 87.5 mg/kg ketamine and 12.5 mg/kg xylazine intraperitoneally (i.p.), immobilized on the lucite platform with surgical tape, and irradiated individually according to the calibrated Gammacell dose rate of 0.6 Gy min−1.
Intravital microscopic imaging:
An Olympus FV1000 confocal-multiphoton imaging system was used with a XLUMPLFLN 20x water immersion objective (NA 1.0; Olympus America); 2x digital zoom; sequential scanning using 405-nm, 473-nm, 559-nm, and 635-nm diode lasers and a DM405/473/559/635-nm dichroic beam splitter; and collection of emitted light using beam splitters (SDM473, SDM560, and/or SDM 640) and emission filters BA430–455, BA490–540, BA575–620, and BA655–755 (all Olympus America). Dorsal window chamber imaging was performed following previously described procedures (35); briefly, 2 × 106 HT1080–53BP1-mApple cells were suspended in 50 μ;l PBS, subcutaneously injected in nu/nu mice (Cox7, MGH) 30 min after surgical chamber implantation, and imaged and irradiated 2 weeks later. Ferumoxytol labeled with VT680XL was used to visualize TAM as previously described (3), and was injected i.v. at least 3 hr prior to tumor imaging. 70 kDa dextran labeled with 50 μ;l (1 mg/ml) Oregon Green dye (Thermo-Fisher) was injected i.v. approximately 10 min after initiating time-course acquisition of 50 −150 μm z-stacks at 3–4 distinct tumor regions, with stacks imaged on a 5 min cycle. In some cases, vasculature was labeled with 50 μ;l fluorescein-labeled tomato lectin (2 mg/ml; Vector Laboratories) 30 min prior to imaging. Plasma half-life measurements were calculated from time-lapse images in the window-chamber and ear of tumor-bearing mice as described previously (3). 30 μ;l AngioSPARK-680 (PerkinElmer) was used to validate dextran vasculature imaging, and was co-injected within 10 min of image acquisition. All NPs were injected via tail-vein catheter immediately after mixing to a final 1x PBS solution. During imaging, animals were anesthetized by isoflurane, immobilized on an aluminum window-chamber mount, and heated on a robotic stage.
Statistical analysis:
Statistical analyses were performed using MATLAB (Mathworks), Prism (GraphPad), and Excel (Microsoft). Measurement statistics and error bars are described in figure legends. Student’s t-test was used unless stated otherwise. All tests were two-tailed with thresholds for false-positive of α = 0.05. Individual-level data are shown in table S2.
Supplementary Material
Acknowledgments:
We thank Jim Mohan, Dianne Mathis, and Christophe Benoist for the MerTK reporter mouse model, and David Pirovich for technical assistance. We thank Diane Capen and Dennis Brown (MGH) for TEM assistance.
Funding: This work was supported in parts by the NIH UO1CA206997, K99CA207744, R01EB010011 and P50GM107618. C.P. was supported in part by the MGH ECOR Tosteson and Fund for Medical Discovery Fellowship. Electron microscopy was performed in the Microscopy Core of the Center for Systems Biology/Program in Membrane Biology (MGH), which is partially supported by an Inflammatory Bowel Disease Grant DK043351 and a Boston Area Diabetes and Endocrinology Research Center (BADERC) Award DK057521.
Footnotes
Competing interests: The authors declare no competing interests. R.W. is a co-founder of T2Biosystems and Lumicell. He serves as a scientific advisor for ModeRNA Therapeutics, Tarveda Therapeutics and Alivio Therapeutics. M.J.P. has served as a consultant for Baxalta, Deciphera Pharmaceuticals, Forma Therapeutics, Incyte Pharmaceuticals, Jounce Therapeutics, KSQ Therapeutics, Secarna, Siamab and Syndax. None of these activities are related to the manuscript.
Data and materials availability: Materials and data are available upon request by contacting the corresponding author. All cell lines were obtained through material transfer agreements. Parental cell lines (4T1, HT1080) are available from ATCC (Manassas, VA) under a material transfer agreement with ATCC. Requests for collaboration involving materials used in this research will be fulfilled provided that a written agreement is executed in advance between Massachusetts General Hospital and the requesting parties.
References:
- 1.Min Y, Caster JM, Eblan MJ, Wang AZ, Clinical Translation of Nanomedicine. Chem Rev 115, 11147 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Prabhakar U, Maeda H, Jain RK, Sevick-Muraca EM, Zamboni W, Farokhzad OC, Barry ST, Gabizon A, Grodzinski P, Blakey DC, Challenges and key considerations of the enhanced permeability and retention effect for nanomedicine drug delivery in oncology. Cancer Res 73, 2412 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Miller MA, Gadde S, Pfirschke C, Engblom C, Sprachman MM, Kohler RH, Yang KS, Laughney AM, Wojtkiewicz G, Kamaly N, Bhonagiri S, Pittet MJ, Farokhzad OC, Weissleder R, Predicting therapeutic nanomedicine efficacy using a companion magnetic resonance imaging nanoparticle. Sci Transl Med 7, 314ra183 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wilhelm S, Tavares AJ, Dai Q, Ohta S, Audet J, Dvorak HF, Chan WCW, Analysis of nanoparticle delivery to tumours. Nat Rev Mat 16014, 1 (2016). [Google Scholar]
- 5.LoRusso P, Krop I, Miller K, Ma C, Shields AF, Molnar I, Wickham T, Reynolds J, Campbell K, Hendriks B, McClure T, Moyo V, Munster P, Abstract CT234: A phase I study of MM-302, a HER2-targeted PEGylated liposomal doxorubicin, in patients with HER2+ metastatic breast cancer. Cancer Research 75, CT234 (2015). [Google Scholar]
- 6.Von Hoff DD, Mita MM, Ramanathan RK, Weiss GJ, Mita AC, LoRusso PM, Burris HA, Hart LL, Low SC, Parsons DM, Zale SE, Summa JM, Youssoufian H, Sachdev JC, Phase I Study of PSMA-Targeted Docetaxel-Containing Nanoparticle BIND-014 in Patients with Advanced Solid Tumors. Clin Cancer Res 22, 3157 (2016). [DOI] [PubMed] [Google Scholar]
- 7.Matsumoto Y, Nichols JW, Toh K, Nomoto T, Cabral H, Miura Y, Christie RJ, Yamada N, Ogura T, Kano MR, Matsumura Y, Nishiyama N, Yamasoba T, Bae YH, Kataoka K, Vascular bursts enhance permeability of tumour blood vessels and improve nanoparticle delivery. Nat Nanotechnol 11, 533 (2016). [DOI] [PubMed] [Google Scholar]
- 8.Harney AS, Arwert EN, Entenberg D, Wang Y, Guo P, Qian BZ, Oktay MH, Pollard JW, Jones JG, Condeelis JS, Real-Time Imaging Reveals Local, Transient Vascular Permeability, and Tumor Cell Intravasation Stimulated by TIE2hi Macrophage-Derived VEGFA. Cancer Discov 5, 932 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Stapleton S, Jaffray D, Milosevic M, Radiation effects on the tumor microenvironment: Implications for nanomedicine delivery. Adv Drug Deliv Rev (2016). [DOI] [PubMed] [Google Scholar]
- 10.Shiao SL, Ruffell B, DeNardo DG, Faddegon BA, Park CC, Coussens LM, TH2-Polarized CD4(+) T Cells and Macrophages Limit Efficacy of Radiotherapy. Cancer Immunol Res 3, 518 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Miller MA, Zheng YR, Gadde S, Pfirschke C, Zope H, Engblom C, Kohler RH, Iwamoto Y, Yang KS, Askevold B, Kolishetti N, Pittet M, Lippard SJ, Farokhzad OC, Weissleder R, Tumour-associated macrophages act as a slow-release reservoir of nano-therapeutic Pt(IV) pro-drug. Nat Commun 6, 8692 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Peng H, Bria A, Zhou Z, Iannello G, Long F, Extensible visualization and analysis for multidimensional images using Vaa3D. Nat Protoc 9, 193 (2014). [DOI] [PubMed] [Google Scholar]
- 13.Miller MA, Weissleder R, Imaging the pharmacology of nanomaterials by intravital microscopy: Toward understanding their biological behavior. Adv Drug Deliv Rev (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Meng Y, Beckett MA, Liang H, Mauceri HJ, van Rooijen N, Cohen KS, Weichselbaum RR, Blockade of tumor necrosis factor alpha signaling in tumor-associated macrophages as a radiosensitizing strategy. Cancer Res 70, 1534 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Klug F, Prakash H, Huber PE, Seibel T, Bender N, Halama N, Pfirschke C, Voss RH, Timke C, Umansky L, Klapproth K, Schäkel K, Garbi N, Jäger D, Weitz J, Schmitz-Winnenthal H, Hämmerling GJ, Beckhove P, Low-dose irradiation programs macrophage differentiation to an iNOS⁺/M1 phenotype that orchestrates effective T cell immunotherapy. Cancer Cell 24, 589 (2013). [DOI] [PubMed] [Google Scholar]
- 16.Yuan F, Leunig M, Huang SK, Berk DA, Papahadjopoulos D, Jain RK, Microvascular permeability and interstitial penetration of sterically stabilized (stealth) liposomes in a human tumor xenograft. Cancer Res 54, 3352 (1994). [PubMed] [Google Scholar]
- 17.Iwakawa M, Ohno T, Imadome K, Nakawatari M, Ishikawa K, Sakai M, Katoh S, Ishikawa H, Tsujii H, Imai T, The radiation-induced cell-death signaling pathway is activated by concurrent use of cisplatin in sequential biopsy specimens from patients with cervical cancer. Cancer Biol Ther 6, 905 (2007). [DOI] [PubMed] [Google Scholar]
- 18.Horton JK, Siamakpour-Reihani S, Lee CT, Zhou Y, Chen W, Geradts J, Fels DR, Hoang P, Ashcraft KA, Groth J, Kung HN, Dewhirst MW, Chi JT, FAS Death Receptor: A Breast Cancer Subtype-Specific Radiation Response Biomarker and Potential Therapeutic Target. Radiat Res 184, 456 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Newman AM, Liu CL, Green MR, Gentles AJ, Feng W, Xu Y, Hoang CD, Diehn M, Alizadeh AA, Robust enumeration of cell subsets from tissue expression profiles. Nat Methods 12, 453 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dvorak AM, Kohn S, Morgan ES, Fox P, Nagy JA, Dvorak HF, The vesiculo-vacuolar organelle (VVO): a distinct endothelial cell structure that provides a transcellular pathway for macromolecular extravasation. J Leukoc Biol 59, 100 (1996). [PubMed] [Google Scholar]
- 21.Stapleton S, Milosevic M, Tannock IF, Allen C, Jaffray DA, The intra-tumoral relationship between microcirculation, interstitial fluid pressure and liposome accumulation. J Control Release 211, 163 (2015). [DOI] [PubMed] [Google Scholar]
- 22.Geretti E, Leonard SC, Dumont N, Lee H, Zheng J, De Souza R, Gaddy DF, Espelin CW, Jaffray DA, Moyo V, Nielsen UB, Wickham TJ, Hendriks BS, Cyclophosphamide-Mediated Tumor Priming for Enhanced Delivery and Antitumor Activity of HER2-Targeted Liposomal Doxorubicin (MM-302). Mol Cancer Ther 14, 2060 (2015). [DOI] [PubMed] [Google Scholar]
- 23.Kalra AV, Kim J, Klinz SG, Paz N, Cain J, Drummond DC, Nielsen UB, Fitzgerald JB, Preclinical activity of nanoliposomal irinotecan is governed by tumor deposition and intratumor prodrug conversion. Cancer Res 74, 7003 (2014). [DOI] [PubMed] [Google Scholar]
- 24.Hlushchuk R, Riesterer O, Baum O, Wood J, Gruber G, Pruschy M, Djonov V, Tumor recovery by angiogenic switch from sprouting to intussusceptive angiogenesis after treatment with PTK787/ZK222584 or ionizing radiation. Am J Pathol 173, 1173 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Dimitrievich GS, Fischer-Dzoga K, Griem ML, Radiosensitivity of vascular tissue. I. Differential radiosensitivity of capillaries: a quantitative in vivo study. Radiat Res 99, 511 (1984). [PubMed] [Google Scholar]
- 26.Sonveaux P, Brouet A, Havaux X, Grégoire V, Dessy C, Balligand JL, Feron O, Irradiation-induced angiogenesis through the up-regulation of the nitric oxide pathway: implications for tumor radiotherapy. Cancer Res 63, 1012 (2003). [PubMed] [Google Scholar]
- 27.Drake CJ, Little CD, Exogenous vascular endothelial growth factor induces malformed and hyperfused vessels during embryonic neovascularization. Proc Natl Acad Sci U S A 92, 7657 (1995). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yoon SS, Duda DG, Karl DL, Kim TM, Kambadakone AR, Chen YL, Rothrock C, Rosenberg AE, Nielsen GP, Kirsch DG, Choy E, Harmon DC, Hornicek FJ, Dreyfuss J, Ancukiewicz M, Sahani DV, Park PJ, Jain RK, Delaney TF, Phase II study of neoadjuvant bevacizumab and radiotherapy for resectable soft tissue sarcomas. Int J Radiat Oncol Biol Phys 81, 1081 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Engblom C, Pfirschke C, Pittet MJ, The role of myeloid cells in cancer therapies. Nat Rev Cancer 16, 447 (2016). [DOI] [PubMed] [Google Scholar]
- 30.Kozin SV, Kamoun WS, Huang Y, Dawson MR, Jain RK, Duda DG, Recruitment of myeloid but not endothelial precursor cells facilitates tumor regrowth after local irradiation. Cancer Res 70, 5679 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hallahan D, Kuchibhotla J, Wyble C, Cell adhesion molecules mediate radiation-induced leukocyte adhesion to the vascular endothelium. Cancer Res 56, 5150 (1996). [PubMed] [Google Scholar]
- 32.Upreti M, Jamshidi-Parsian A, Apana S, Berridge M, Fologea DA, Koonce NA, Henry RL, Griffin RJ, Radiation-induced galectin-1 by endothelial cells: a promising molecular target for preferential drug delivery to the tumor vasculature. J Mol Med (Berl) 91, 497 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zhang QW, Liu L, Gong CY, Shi HS, Zeng YH, Wang XZ, Zhao YW, Wei YQ, Prognostic significance of tumor-associated macrophages in solid tumor: a meta-analysis of the literature. PLoS One 7, e50946 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Diagaradjane P, Deorukhkar A, Gelovani JG, Maru DM, Krishnan S, Gadolinium chloride augments tumor-specific imaging of targeted quantum dots in vivo. ACS nano, 4, 4131 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Miller MA, Askevold B, Yang KS, Kohler RH, Weissleder R, Platinum compounds for high-resolution in vivo cancer imaging. ChemMedChem 9, 1131 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Cuccarese MF, Dubach JM, Pfirschke C, Engblom C, Garris C, Miller MA, Pittet MJ, Weissleder R, Heterogeneity of macrophage infiltration and therapeutic response in lung carcinoma revealed by 3D whole organ imaging. Nat Commun 8, 14293 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Jain RK, Determinants of tumor blood flow: a review. Cancer Res 48, 2641 (1988). [PubMed] [Google Scholar]
- 38.Albright CF, Graciani N, Han W, Yue E, Stein R, Lai Z, Diamond M, Dowling R, Grimminger L, Zhang SY, Behrens D, Musselman A, Bruckner R, Zhang M, Jiang X, Hu D, Higley A, Dimeo S, Rafalski M, Mandlekar S, Car B, Yeleswaram S, Stern A, Copeland RA, Combs A, Seitz SP, Trainor GL, Taub R, Huang P, Oliff A, Matrix metalloproteinase-activated doxorubicin prodrugs inhibit HT1080 xenograft growth better than doxorubicin with less toxicity. Mol Cancer Ther 4, 751 (2005). [DOI] [PubMed] [Google Scholar]
- 39.Bao A, Goins B, Klipper R, Negrete G, Phillips WT, Direct 99mTc labeling of pegylated liposomal doxorubicin (Doxil) for pharmacokinetic and non-invasive imaging studies. J Pharmacol Exp Ther 308, 419 (2004). [DOI] [PubMed] [Google Scholar]
- 40.Soundararajan A, Bao A, Phillips WT, Perez R, Goins BA, [(186)Re]Liposomal doxorubicin (Doxil): in vitro stability, pharmacokinetics, imaging and biodistribution in a head and neck squamous cell carcinoma xenograft model. Nucl Med Biol 36, 515 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.