

Abstract
Deoxynucleotide misincorporation efficiencies can span a wide 104-fold range, from ~10−2 to ~10−6, depending principally on polymerase (pol) identity and DNA sequence context. We have addressed DNA pol fidelity mechanisms from a transition-state (TS) perspective using our “tool-kit” of dATP-and dGTP-β,γ substrate analogues in which the pyrophosphate leaving group (pKa4 = 8.9) has been replaced by a series of bisphosphonates covering a broad acidity range spanning pKa4 values from 7.8 (CF2) to 12.3 [C(CH3)2]. Here, we have used a linear free energy relationship (LFER) analysis, in the form of a Brønsted plot of log(kpol) versus pKa4, for Y-family error-prone pol η and X-family pols λ and β to determine the extent to which different electrostatic active site environments alter kpol values. The apparent chemical rate constant (kpol) is the rate-determining step for the three pols. The pols each exhibit a distinct catalytic signature that differs for formation of right (A·T) and wrong (G·T) incorporations observed as changes in slopes and displacements of the Brønsted lines, in relation to a reference LFER. Common to this signature among all three pols is a split linear pattern in which the analogues containing two halogens show kpol values that are systematically lower than would be predicted from their pKa4 values measured in aqueous solution. We discuss how metal ions and active site amino acids are responsible for causing “effective” pKa4 values that differ for dihalo and non-dihalo substrates as well as for individual R and S stereoisomers for CHF and CHCl.
Graphical Abstract
DNA polymerase η (pol η), a member of the Y family of pols, is encoded by the xeroderma pigmentosum variant (XPV) gene.1 Pol η plays an essential role in the avoidance of skin cancer by copying past ultraviolet-induced cyclobutene dimers that block normal DNA replication.1,2 It also plays an important role in generating antibody diversity in B cells, by making mutations at A·T sites during somatic hypermutation (SHM) of immunoglobulin genes.3 Whether catalyzing translesion synthesis on distorted template DNA or copying undamaged DNA with remarkably low fidelity,4,5 the active site of pol η appears to favor “relaxed” specificity in the deoxynucleotide incorporation transition state (TS). How dNTP substrate selection occurs in the TS is fundamental to understanding the molecular mechanisms of pol fidelity.
Here, we use single-turnover kinetics to investigate base selection occurring in the TS from a leaving group perspective by measuring the rate of incorporation of the natural dNTPs along with β,γ-bridging O-substituted dNTP analogues6–10 (Figure 1).
Figure 1.
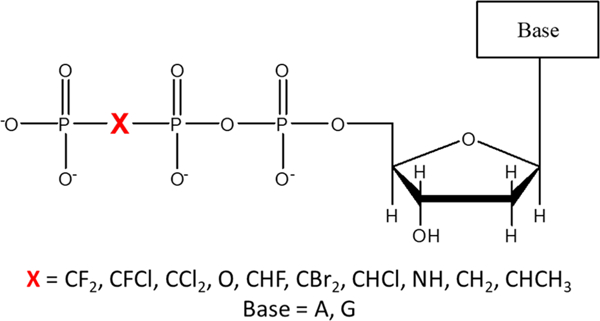
Generic structure of dNTP analogues used in this study. The pKa4 values, experimentally determined in solution, of the corresponding biosphosphonate leaving groups are 7.8 for CF2, 8.4 for CFCl, 8.8 for CCl2, 8.9 for O (pyrophosphate), 9.0 for CHF, 9.3 for CBr2, 9.5 for CHCl, 9.7 for NH, 10.5 for CH2, and 11.6 for CHCH3.9 The individual stereoisomers of CHF and CHCl have the same pKa4 because the leaving group is the same bisphosphonate compound regardless of the R or S identity of the starting dNTP.
These analogues undergo the standard α,β-P−O−P cleavage reaction whereby each atom involved in both bond making and breaking at the nucleotide Pα remains the same as in the natural dNTP. The chemical reaction mechanism is in principal conserved, but the chemical step, as embodied in the deoxynucleotide incorporation rate (kpol) for either right (e.g., A·T) or wrong (G·T) reactions, is sensitive to the extent to which each β,γ-bridging oxygen substituent stabilizes the triphosphate group negative charge in the TS. If the rate of the polymerization reaction of any pol is determined mainly by this (i.e., chemical) TS, the dependence of deoxynucleotide right, e.g., (A·T) and wrong (G·T) incorporation rates on PPi and PPi bisphosphonate leaving group electronic and steric properties provides a sensitive probe of the DNA synthesis reaction.6 In a series of studies with pol β,6,7,9,10 we found that nucleotide incorporation rates were decreased with increasing pKa4 values, with each dNTP analogue becoming less reactive to nucleophilic attack at Pα as the leaving group becomes increasingly harder to displace. A key observation was that a linear free energy relationship (LFER) in the form of a Brønsted plot, which describes the linear dependence of log(kpol) on leaving group pKa4, was typically observed. However, different pols are very likely to impose distinctive active site constraints that can strongly modify incorporation specificities in the TS and thereby influence fidelity.11,12 One striking example is pol η, the X-ray structure of which reveals an active site that stabilizes a wobble G·T base mispair at a 3′-AT template motif.13
We have investigated right (A·T) versus wrong (G·T) base pair selection occurring in the TS using LFERs for pol η and pol λ, an X-family pol that is principally involved in base excision repair and nonhomologous end-joining processes.14–16 The differences in Brønsted slopes and Brønsted line displacements along the pKa4 axis depict pol-specific differential stabilization of product relative to reactant states occurring in the TS. A comparison of LFER profiles for pols η and λ reveals large slope and displacement differences, along with subtle, yet significant, differences for individual R and S dNTP β,γ-substituted stereoisomers. Differences in LFER profiles for correct incorporation of A opposite T are strongly accentuated for the misincorporation of G opposite T.
An LFER for the analogous non-enzymatic base-catalyzed hydrolysis reaction in aqueous solution has a slope of approximately −0.9.17,18 An analysis of the differences between the slopes of pol-generated Brønsted lines and their locations along the pKa4 axis in relation to the aqueous reference line provides a way to investigate the influence of pol active site architecture on the chemical reaction mechanism and nucleotide incorporation fidelity occurring in the TS. In this paper, we compare the Brønsted profiles for pols η and λ with LFER data reported previously for X-family pol β.7,9,10
METHODS
DNA Synthesis, Purification, Radiolabeling, and Annealing.
Two sequence contexts were used in this study. The B sequence consists of a primer (5′-TAT TAC CGC GCT GAT GCG C), a template (5′-GCG TTG TTC CGA M TG CGC ATC AGC GCG GTA ATA, where M = C for all pol λ and pol β reactions as well as A opposite T pol η reactions, or G for G opposite T for pol η reactions only), and 5′-phosphorylated downstream (5′-GTC GGA ACA ACG C, used for only pol λ and pol β reactions); the W sequence consists of a primer (5′-GTG CCT AGC GTA T) and a template (5′-GAG TCA TGT ATA CGC TAG GCA C). The nearest neighbor had to be changed for the B sequence with pol η to prevent primer slippage when misincorporating G opposite T. Oligomers were synthesized by solid phase synthesis, purified by 16% polyacrylamide gel electrophoresis, and desalted prior to use. Radiolabeling reaction mixtures consisted of 1 molar equivalent of primer 5′-end-labeled with 0.4 unit/μL T4 polynucleotide kinase and 0.7 molar equivalent of [γ−32P]ATP with the supplied buffer. Reactions were performed at 37 °C for 30 min, followed by heat inactivation at 95 °C for 10 min, and then mixtures annealed by being mixed with 1.2 molar equivalents of template and 1.5 molar equivalents of downstream oligo if it was used. The mixture was heated to 95 °C and cooled slowly to room temperature.
Buffer and Protein Preparation.
Pol η reaction buffer consisted of 40 mM Tris (pH 8.0), 50 mM NaCl, 2 mM MgCl2, 10 mM dithiothreitol (DTT), and 4% glycerol. Pol λ and pol β reaction buffers contained 50 mM Tris (pH 8.0), 20 mM KCl, 20 mM NaCl, 10 mM MgCl2, 1 mM DTT, and 6% glycerol. Truncated human pol λ and wild-type human pol β were provided by S. H. Wilson’s lab. The expression plasmid and strain for N-terminally His-tagged pol η were provided by R. Woodgate’s lab (The Eunice Kennedy Shriver National Institute of Child Health and Human Development) and purified as previously described.19
Synthesis of β,γ-NH-dNTPs.
In our previous studies with DNA polymerase β, we utilized a tool-kit of dNTP-mimicking probes constructed from a series of CXY-substituted bisphosphonates (BPs) (pCXYp; where X and Y = Br, Cl, F, H, CH3, and N3) with varying pKa4 values.9 Here, we have expanded the tool-kit to include novel β,γ-pNHp-dNTP (dA, 3; dG, 4) analogues prepared by reacting the tris-(triethylamine) salt of imidodiphosphoric acid with 5′-dNMP-N-methylimidazolide in anhydrous acetonitrile (freshly prepared from 5′-dNMP) (Scheme 1).
Scheme 1.
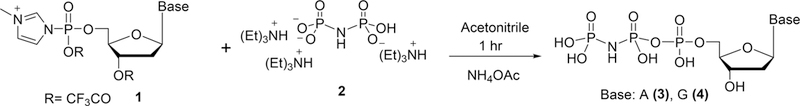
General Synthetic Route to β,γ-pNHp-dNTP Analogues
To estimate relative leaving group aptitude in the β,γ-CXY dNTP probes, we determined pKa4 values of the relevant pCXYp bisphosphonic acid derivatives by potentiometric titration using a Schott Instruments Titrator Basic and fitting the data to pKa1–4 values using Hyperquad2008 software. The same methods were used here to determine pKa1–4 values for imidodiphosphoric acid in a 0.1 M KCl solution at 25 °C, titrated with ~0.1 M KOH in CO2-free H2O: 2.01, 2.81, 7.11, and 9.70.
Pre-Steady-State Reactions and Data Analysis.
Radiolabeled DNA (100 nM) was incubated with 400 nM pol η, 400 nM pol λ, or 600 nM pol β, in reaction buffer (2× mixture) for 3 min at 37 °C. Equal volumes of the DNA/pol mixture and a 2× solution of the dNTP analogue in reaction buffer at different concentrations were rapidly combined using a KinTek model RQF-3 quench flow apparatus. After the appropriate reaction time, the reaction was quenched with 0.5 M EDTA (pH 8.0). For times of >20 s, reactions were initiated and quenched by manual mixing. Reaction products were separated by 20% denaturing polyacrylamide gel electrophoresis (39 cm × 33 cm × 0.4 mm). Dehydrated gels were exposed to a phosphor screen and detected by phosphorescence emission. All reactions were carried out in triplicate.
For each time point, the percentage of extension is plotted opposite the reaction time for each reaction and the data for each analogue concentration are fit to the exponential
| (1) |
where a is the amplitude and k is the observed rate constant for that analogue concentration (kobs). The observed rate constant is then plotted opposite the corresponding analogue concentration, and the data are fit to the rectangular hyperbola
| (2) |
where kpol and Kd are the pre-steady-state parameters for the rate of incorporation and dNTP binding, respectively. The fidelity ratio, F, numerically describes the difference in fidelity between the two sequences used in this study and is given in eq 3:
| (3) |
Potentiometric Titrations.
The stability constants of the Mg(II) complexes (Table S5) were determined at 25 °C by pH-metric titration of 25 mL samples. A semimicro combined glass electrode (Metrohm 6.0234.100) and precision pH-meter (Orion 720A+) were calibrated with at least two buffer standard solutions and subsequent strong acid-strong base titration. CO2-free deionized water (18 MΩ cm−1) was used throughout for all stock solutions. Titrations were performed with KOH solutions of known concentrations (~0.2 M) under a purified argon atmosphere to avoid interference from the oxygen and carbon dioxide in air. The Mg2+ stock was prepared from MgCl2-6H2O and standardized by EDTA titration. Methylenediphosphonic acid, sodium pyrophosphate, imidodiphosphate sodium salt, and anhydrous tetrasodium difluorobisphosphonate were used as received. A precise amount of HCl (~4 equiv) was added to the titration vial when the tetrasodium salt of bisphosphonic acid was used as a starting material.
A representative titration was performed as follows. The acidic bisphosphonate solution (0.002–0.008 M) containing various amounts of Mg2+ (0–0.008 M) was titrated with standardized CO2-free KOH (~0.2 M) in a 25 °C thermostated titration vial under an Ar atmosphere. Metal:-ligand ratios in solutions were 0:1, 1:1, 1:2, and 1:4. The ionic strength was kept constant at 0.4 M with KCl. Solutions were titrated in the pH range of 2.3–11.2 or until precipitation. The experimental data points were analyzed using the PSEQUAD computer program,20 and details are provided in the Supporting Information.
The reversibility of the complexation reactions was checked by back-titration, i.e., by titrating samples from basic pH (>10) with HCl solutions of known concentrations. Equilibrium was reached in all of these solutions within 5 min. Experiments were performed in duplicate, and in all cases, the reproducibility of the titration curves was within 0.005 pH unit.
RESULTS
Dependence of Pol η Fidelity on Sequence Context for Parent dNTP and CHF dNTP Analogue Substrates.
A study by Yang and colleagues13 showed that pol η exhibits a strong bias toward the misincorporation of dGMP opposite a template T when the 3′ primer terminus is either A or T, stabilizing a T·dGTP wobble base pair in the catalytically competent conformation. We have designated the abbreviation “W” for the 3′ primer terminus T sequence context, in comparison with a sequence used extensively for LFER studies of pol β fidelity, which contains C at the 3′ primer end (see Methods for sequences), abbreviated here as the “B” sequence context.
Pol η incorporates A opposite T with essentially no sequence context bias (Figure 2A). The apparent kpol and apparent Kd values are 4.7 ± 1.0 s−1 and 7.1 ± 2.1 μM for W and 4.1 ± 1.2 s−1 and 9.4 ± 3.1 μM for B, respectively (Table S1). There is also minimal bias for the apparent dissociation constants for misincorporation of G opposite T (Figure 2B), with an apparent Kd of 20 ± 4 μM for W compared to a Kd of 30 ± 3 μM for B. There is, however, an ~4-fold increase in the chemical misincorporation rate constant for W (kpol = 0.86 ± 0.02 s−1) versus B (kpol = 0.21 ± 0.02 s−1). The fidelity ratio (eq 3) F = 0.25. This 4-fold reduction in the fidelity of pol η in the W sequence context agrees with data reported in ref 13.
Figure 2.
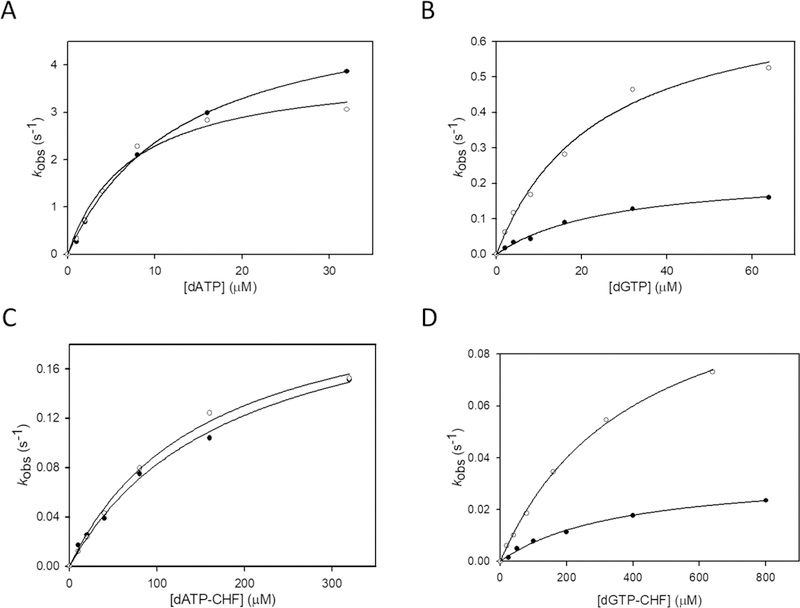
Hyperbolic fits for incorporation of dNTPs by pol η for different sequences. Filled circles correspond to B sequence, and empty circles to W sequence. (A) Correct incorporation of dATP opposite T. (B) Incorrect incorporation of dGTP opposite T. (C) Correct incorporation of dATP-CHF opposite T. (D) Incorrect incorporation of dGTP-CHF opposite T. For both the parent and the CHF analogue, the incorporation is sequence-independent for the correct incorporation (panels A and C) but very different for the misincorporation (panels B and D). In both cases, the W sequence results in a kpol for G opposite T significantly higher than that obtained with the B sequence while the Kd remains essentially unchanged.
A modification in leaving group acidity (pKa4) will modify the incorporation rate constant (kpol) for pols whose rate-determining step (RDS) is the chemical step for either right or wrong β,γ-dNTP analogues in accord with LFER Brønsted plots, as illustrated in a series of studies with pol β.6,7,9,10 To determine if the presence of the bisphosphonate leaving group influences polymerase fidelity, we have repeated the sequence-dependent fidelity measurement with pol η using dATP and dGTP analogues with CHF replacing the leaving group β−γ-bridging O (Figure 1, X = CHF). Among the nine β,γ-dNTP analogues (Figure 1), the CHF bisphosphonate leaving group has a pKa4 in solution that is closest to the parent pyrophosphate leaving group pKa4 (9.0 for CHF vs 8.9 for O).6
A Michaelis-Menten plot for the sequence context dependence to form either an A·T base pair (Figure 2C) or a G·T mispair (Figure 2D) using the CHF analogue appears qualitatively similar to those of the parent dATP (Figure 2A) and dGTP (Figure 2B) substrates, where kpol values for the CHF analogues are virtually identical for the formation of A·T in either W or B (Figure 2C) but dramatically different for the formation of G·T. G misincorporation is strongly favored in the W context compared to B (Figure 2D), as was observed for the parent dGTP (Figure 2B). However, unexpectedly, there are sizable quantitative differences in the chemical step for the parent versus the analogue, where there is a reduction in the range of 5–30-fold for dNTP analogue kpol values compared to those of the parent dNTPs. These large reductions in the observed rate constants were surprising on the basis of our previous data with pol β, where CHF kpol values are virtually indistinguishable from those of the parent dNTP substrates (Figure 3C for A·T and Figure 4C for G·T).10
Figure 3.
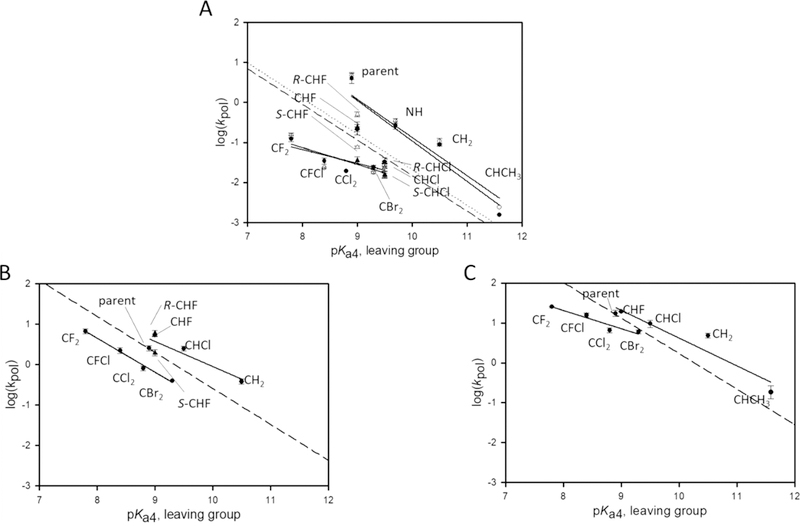
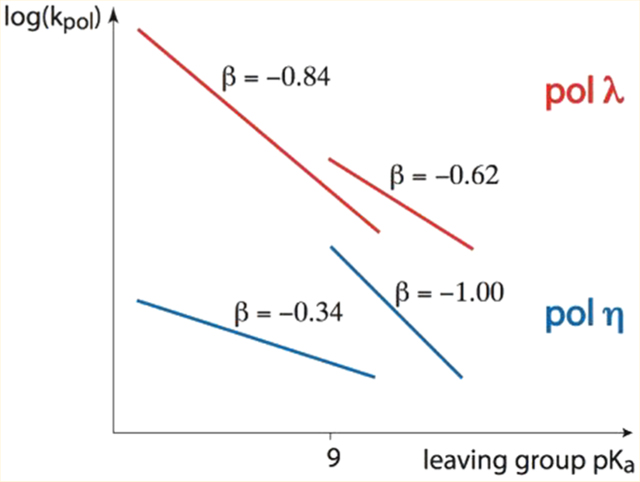
LFER plots for A opposite T. Dashed/dotted lines correspond to the theoretical Brønsted lines for each enzyme as described in the text. A full set of data was collected for pol β,8–10 and a subset was collected for pols η and λ. (A) Overlay of pol η plots for both sequences. The slopes of the lines are as follows: B sequence (dashed theoretical line, filled circles), −1.0 (upper) and −0.34 (lower); W sequence (dotted theoretical line and empty circles), −0.95 (upper) and −0.42 (lower). (B) Pol λ with the B sequence. The slopes are −0.62 (upper) and −0.84 (lower). (C) Pol β with the B sequence. The slopes of the lines are −0.69 (upper) and −0.44 (lower).
Figure 4.
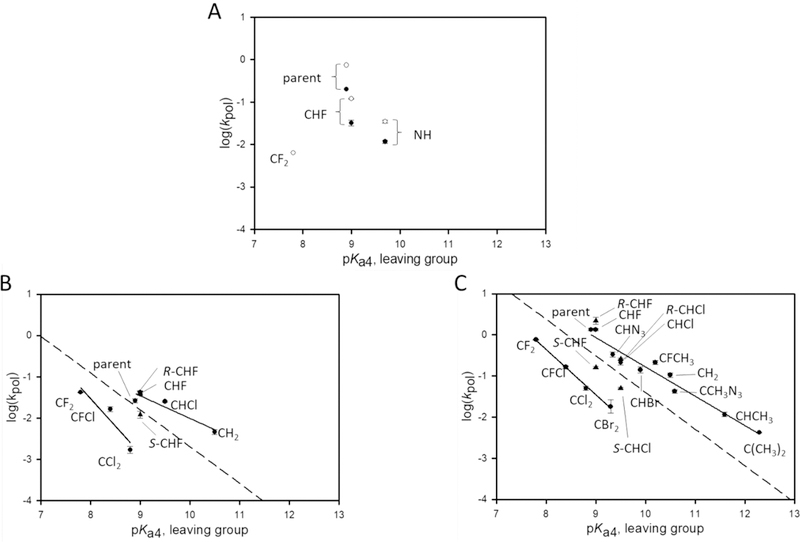
LFER plots for G opposite T. Dashed lines correspond to the theoretical Brønsted line as described in the text. (A) Overlay of pol η plots for both sequences. Because the analogues are not well used by pol η, only three compounds were incorporated for the B sequence, parent, CHF, and NH. In addition to these three, CF2 was incorporated for the W sequence context. In each case, the W sequence resulted in a ~3-fold higher kpol, a marked difference from the correct incorporations (Figure 3A). (B) Pol λ with the B sequence. The slopes are −0.55 and −1.3 for the upper and lower LFER lines, respectively. (C) Pol β with the B sequence.7,8,10 The slopes of the lines are −0.71 (upper) and −1.1 (dihalo compounds).
The sequence context fidelity ratio as calculated with eq 3 for the CHF analogue of 0.37 is slightly higher than for the parent substrate (0.25). Thus, despite the large reductions in chemical rate constants for dATP and dGTP CHF substrates compared to those of the parent dATP and dGTP, there is a roughly comparable reduction in fidelity in W versus B sequence contexts, 2.7-fold for CHF versus 4-fold for the parent dNTP substrates. The apparent Kds for substrate binding are much greater for the CHF-dNTP analogue than for the parent dNTP substrates, in a range of 12–20-fold (Table S1). However, binding of CHF does not depend on sequence context: Kd (A·T) = 140 ± 20 and 190 ± 80 μM in W and B sequences, respectively, and Kd (G·T) = 390 ± 20 and 370 ± 60 μM in W and B sequences, respectively (Table S1). Therefore, as found for the parent dNTP substrates, the principal contribution to decreased pol η fidelity in W compared to B for the CHF dNTP substrates comes from a 3.8-fold increase in the G·T misincorporation rate constant (kpol of 0.12 ± 0.00 s−1 for W vs 0.032 ± 0.005 s−1 for B) (Table S1).
Using LFER To Probe the TS of Pols η, λ, and β during A·T and G·T Base Pair Synthesis.
The key to deciphering the mechanisms underlying DNA synthesis fidelity involves determining deoxynucleotide selection occurring in the TS. A measurement of deoxynucleotide incorporation as a function of leaving group PPi and bisphosphonate PPi analogues basicity provides a sensitive way to measure right versus wrong base pair selection taking place in the TS, which depends on the active site architecture of individual pols. We have investigated pol-specific TS selection by generating Brønsted plots, log(kpol) versus leaving group pKa4, for formation of A·T base pairs (Figure 3) and G·T base mispairs (Figure 4), with pols η, λ, and β, for the parent dNTPs and bisphosphonate nucleotide analogue substrates shown in Figure 1.
The dashed lines shown in Figures 3 and 4 serve as a reference for comparing Brønsted plots for each of the pols to a theoretically derived LFER calculated for a non-enzymatic base-catalyzed methyl triphosphate hydrolysis reaction in aqueous solution.18 This reference non-enzymatic LFER has a single pol-independent slope of −0.89.18 The magnitude of this slope, which is similar to the non-enzymatic slope of −0.94 ± 0.05 observed for hydrolysis of methyl phenyl phosphate diesters,17 will be used as a reference for discussing the observed enzymatic LFER properties. The individual intercepts of the non-enzymatic reference lines are given by average kpol values for each enzyme acting on a specific p/t DNA, which are not germane to the TS analysis per se. However, the locations of the intercepts determine the separation along the x-axis of the pol and non-enzymatic Brønsted lines, which is essential when estimating “effective” pKa values, pKaeff, for the β,γ-dNTP analogues in each pol active site.
A·T Base Pair Brønsted Plots.
Brønsted plots for pol η in B and W sequence contexts for A·T base pairs are virtually identical (Figure 3A). Therefore, the absence of a sequence context dependence in the chemical nucleotide incorporation step (kpol) for correct base pair formation generalizes our initial observation for dATP (Figure 2A) and dATP-CHF (Figure 2C) to all of the β,γ-dNTP analogues (Figure 1) spanning a 4-log range of leaving group basicity, from CF2 (pKa4 = 7.8) to CHCH3 (pKa4 =11.6).
We compared the A·T base pair LFER profiles for the three pols in the B sequence context, pol η (Figure 3A, filled circles), pol λ (Figure 3B), and pol β (Figure 3C). The LFER profiles of the two closely related family X pols λ and β are more similar to each other than to that of pol η. These similarities include the typical members of the lower (dihalo bisphosphonates) and upper (the remaining leaving groups) LFER lines,6 nearly identical slopes of the upper lines, a small separation (<1 pKa unit) of the upper and lower LFER lines, and the parent compound falling on the reference non-enzymatic LFER line. The largest dissimilarity involves the substantially less negative slope of the lower LFER line in pol β (−0.44) than in pol λ (−0.84).
The LFER profile of pol η showed a large decrease in slope upon going from the upper LFER line (−0.98, averaged over W and B sequences) to the lower line (slope = −0.38), and the separation of the two LFER lines in pol η increased dramatically to ~2 pKa units. In another anomaly, kpol for the monohalo compounds was significantly smaller than would correspond to their pKa value when compared to that of the non-halo compounds, resulting in β,γ-CHCl-dATP appearing to fall on the lower “dihalo” line and β,γ-CHF-dATP becoming the largest outlier from the upper LFER line. A feature common to all pols is a favored incorporation of R over S separated stereoisomers, CHF and CHCl for pol η (Figure 3A) and CHF for pol λ (Figure 3B). Previous data with pol β also show favored incorporation of R versus S, for a variety of stereoisomers.8
G·T Base Mispair Brønsted Plots.
The crystal structures from the study of Zhao et al.13 show there is little structural difference for different DNA sequences but significant differences for the W sequence context versus non-W sequence contexts. Our data are consistent with these observations, with no difference in the LFERs for the two sequences for the correct pairing of A opposite T (Figure 3A) but large-scale differences for the mispairing of G opposite T (Figure 4A). There are no LFER profiles that emerge from measurements of kpol values for the formation of G·T mispairs by pol η (Figure 4A), primarily due to slow misincorporation rates, although we cannot rule out poor binding also playing a part.
What does emerge, however, is the retention of a sequence context effect, strongly favoring kpol for the misincorporation of G opposite T in the W context, which accommodates the presence of a stable G-T wobble structure in the active site of pol η.13 There is a 3–4-fold increase in kpol observed for W (empty circles) compared to that of B (filled circles) for dGTP, CHF-dG, and NH-dG (Figure 4A). Misincorporation of the CF2 analogue could barely be detected in the W sequence context (Figure 4A) but could be not be detected in the B context. There is a sizable ~ 115-fold reduction in kpol for CF2-β,γ dGTP compared to dGTP for GT mispair formation in the W context, which is accompanied by a much smaller ~6-fold reduction in the apparent binding constant (Table S1). We speculate that the inability to detect incorporation of the CF2-dG analogue opposite T in the B context is probably caused by an inability of pol η to catalyze G·T bond formation and not to an inability to bind analogues. The incorporation of the remaining analogues, which were observed for the formation of A·T base pairs (Figure 3A), could not be observed for G·T mispairs.
The G·T mispair data for pol λ separate into non-dihalo and dihalo lines (Figure 4B), as observed for formation of A·T base pairs (Figure 3B). Consistent with only small differences in the A·T LFER profiles comparing pol λ and pol β (panels B and C of Figure 3, respectively), the Brønsted lines for the G·T mispair are closely similar for the X-family pols (Figure 4B,C). The non-dihalo slopes are −0.55 (pol λ) and −0.71 (pol β); the dihalo slopes are −1.3 (pol λ) and −1.1 (pol β), and the slopes of both lines are roughly similar to the slope of −0.89 for the non-enzymatic reference line (Figure 4B,C, dashed line).
Effective pKa4 in the Polymerase Active Site.
To quantify equilibrium electrostatic effects that are presumably dominating the observed LFER splitting, we have defined an “effective” pKa4 parameter, pKaeff. This represents the magnitude of pKa4 that each analogue would have to have for its measured kpol to fall on the reference line (Table 1 for correct base pairs and Table S4 for base mispairs).
Table 1.
Effective pKa Calculations for Correct Pairings
| pol η |
pol λ |
po1 β |
|||||||
|---|---|---|---|---|---|---|---|---|---|
| B sequence |
W sequence |
B sequence |
B sequence |
||||||
| compound | pKa4 (soln) | pKeff | ΔPKa4,eff | pKeff | ΔPKa4,eff | pKeff | ΔPKa4,eff | pKeff | ΔPKa4,eff |
| CF2 | 7.8 | 9.0 | 1.2 | 9.0 | 1.2 | 8.4 | 0.6 | 8.7 | 0.9 |
| CFCl | 8.4 | 9.6 | 1.2 | 9.8 | 1.4 | 8.9 | 0.5 | 8.9 | 0.5 |
| CCl2 | 8.8 | 9.9 | 1.1 | 9.4 | 0.6 | 9.3 | 0.5 | ||
| O | 8.9 | 7.3 | −1.6 | 7.2 | −1.7 | 8.9 | 0.0 | 8.9 | 0.0 |
| CHF | 9.0 | 8.7 | −0.3 | 8.7 | −0.3 | 8.5 | −0.5 | 8.8 | −0.2 |
| R-CHF | 9.0 | 8.6 | −0.4 | 8.3 | −0.7 | 8.5 | −0.5 | ||
| S-CHF | 9.0 | 9.6 | 0.6 | 9.2 | 0.2 | 9.0 | 0.0 | ||
| CBr2 | 9.3 | 9.8 | 0.5 | 9.9 | 0.6 | 9.8 | 0.5 | 9.4 | 0.1 |
| CHCl | 9.5 | 9.6 | 0.1 | 9.7 | 0.2 | 8.9 | −0.6 | 9.2 | −0.3 |
| R-CHCl | 9.5 | 9.6 | 0.1 | 9.6 | 0.1 | ||||
| S-CHCl | 9.5 | 10.0 | 0.5 | 9.9 | 0.4 | ||||
| NH | 9.7 | 8.6 | −1.1 | 8.5 | −1.2 | ||||
| CH2 | 10.5 | 9.1 | −1.4 | 9.0 | −1.5 | 9.8 | −0.7 | 9.5 | −1.0 |
| CHCH3 | 11.59 | 11.1 | −0.5 | 10.9 | −0.7 | 11.1 | −0.5 | ||
The magnitude of the pKa shifts approximately doubles for the correct insertion in pol η (the dihalo pKaeff difference is 1.04, and the non-dihalo pKaeff difference −0.94) compared to pol λ and β (the dihalo pKaeff differences are 0.58 for pol λ and 0.55 pol β and −0.60 and −0.43, respectively, for non-dihalo). It also increases for G·T misincorporation in both pol λ and β (the dihalo pKaeff difference is 0.94 for both, whereas the non-dihalo pKaeff differences are −0.77 and −0.70, respectively). As a result of having substantially reduced overall deoxynucleotide incorporation “efficiencies” (kpol/Kd) for the β,γ-modified dNTP substrates, Brønsted lines could not be identified for G·T misincorporations (Figure 4A).
pKa Shifts Caused by Formation of [Mg(II)·bi-sphosphonate]2− Complexes in Aqueous Solution.
The leaving groups of dNTP and β,γ-modified dNTP substrates are bound in the polymerase active site during the phosphodiester bond formation reaction to the “structural” Mg2+ ion.21–24 On the basis of potentiometric titrations in aqueous solution (at 25 °C and I = 0.4 M KCl), the dNTP-Mg2+ complexation exerts a strong effect on the acidity of PPi and three representative bisphosphonates, where X = CH2, CF2, and NH (Table 2). The positive charge of the Mg2+ ion stabilizes the deprotonated form of the bisphosphonate, which leads to a decrease in its pKa value of ~2 pH units. The magnitude of this decrease is the largest when X = O and CH2 and smallest when X = NH (Table 2).
Table 2.
pKa4 Values Measured for Bisphosphonate Analogues with and without Magnesium Ions in Aqueous Solution
DISCUSSION
The DNA polymerase transition state is, for all intents and purposes, a “black box”. At present, there are no TS analogues for any pol. We have instead turned to TS probes in an attempt to make the TS somewhat less opaque. These probes are composed of a broad family containing more than 50 dNTP substrate analogues where the pyrophosphate leaving group (pKa4 = 8.9) has been replaced by a series of bisphosphonates covering a broad acidity range spanning pKa4 values from 7.8 (CF2) to 12.3 [C(CH3)2].6,7,9 To maintain optimal internal consistency, we have measured solution pKa4 values for each β,γ-bisphosphonate analogue.6,7,9,10,25,26 These pKa4 values can and indeed likely will be different in the pol active site, modified by electrostatic perturbations imposed by surrounding amino acid residues and metal ions. Elegant studies by Boxer and colleagues have shown that externally imposed electric fields can profoundly influence enzyme catalytic rates.27,28 Additionally, the measurements described in Table 2 show the difference magnesium ions can make in the solution pKa values.
An LFER analysis in the form of a Brønsted plot of log(kpol) versus pKa4 shows that pols η, λ, and β exhibit distinctive catalytic (kpol) signatures that differ for formation of A·T base pairs and G·T mispairs. Pol-specific influences on kpol can be characterized by the changes in slopes and displacements of the Brønsted lines in relation to a reference line for the non-enzymatic base-catalyzed hydrolysis reactions in aqueous solution18 (Figures 3 and 4, dashed lines). The enzymatic LFER profiles are determined by pol-specific TS properties and by changes in the equilibria of leaving groups upon going from aqueous solution to the enzyme active site. This latter effect, which is reflected in a Brønsted line shift parameter pKa,eff is likely to be strongly influenced by the electrostatic environment in the active site.
The LFER profiles are “well-behaved” for the three pols, in the sense that deoxynucleotide incorporation rate constants (kpol) are grouped along two straight lines (Figures 3 and 4). The lower line typically contains dihalo methylene β,γ bridges, although this exclusivity is compromised in pol η, where the lower LFER line for the formation of the A·T base pair also includes CHCl, while the monohalo CHF is the largest outlier from the upper LFER line. Our studies with pol β have shown that the split between LFER lines is not due to steric hindrance within the active site of the enzyme by two methods. First, crystallographic studies10,24 have shown little to no perturbation in the active site of the enzyme when comparing the sterically large methyl-substituted analogues with the small methylene (CH2) analogues, and second, the methyl-substituted analogues continue to fall on the upper line instead of the lower line with the traditionally bulky dihalogen compounds. The persistence of Brønsted LFER behavior in DNA polymerases implies that the net effect of the series of complex electrostatic perturbations involving active site amino acids and Mg ions tends to either stabilize or destabilize reaction products and TS structures to approximately the same extent on the free energy surface.29
Each pol exhibits a split linear pattern for A·T base pairs, in which the analogues containing two halogens show kpol values systematically lower than would be predicted from their pKa4 values measured in aqueous solution (Figure 3). For G·T mispairs, a split pattern is also observed for pols λ and β (Figure 4B,C) but not for pol η, presumably because it cannot misincorporate many of the analogues (Figure 4A). The split LFER pattern may be caused by an increase in TS free energy that can be directly attributed to the dihalo substituents and to an increase in pKa,eff for each dihalo in the pol active site. Changes in pKa,eff determine the displacement of each Brønsted line relative to the solution LFER reference line, while changes in TS free energies can alter Brønsted slopes and displacements.
Electrostatic perturbations involving metal ions and active site amino acids are likely to determine product-state equilibria and may be principally responsible for the displaced dihalo and non-dihalo LFERs. Because Mg ions in solution cause significant alterations in pKa for the bisphosphonates (Table 2), then by interacting differently with dihalo and non-dihalo substituents in the pol active site they could contribute to LFER splitting. Although in the polymerase active site the Mg2+ ions do not appear to form direct interactions with atoms belonging to the β,γ-bridging group,30 they could still shift pKa indirectly, for example, by changing the Pβ–X–Pγ angle or by modifying interactions of structurally equivalent arginine 183, 420, or 61 in pol β, λ, or η, respectively, with the β, γ-bridging group. Another interaction could involve the scissile bond O atom of bisphosphonates with a third Mg2+ ion that was observed in crystal structures of the pol fi product state31 and pol β product and pre-TS states.32 Electrostatically induced splitting also occurs for individual CHF and CHCl R and S stereoisomers (Figures 3 and 4). In contrast to the family of β, γ-substituted dNTP analogues, with each member having a distinct pKa4, the leaving groups of the R and S stereoisomers are the same compound; yet, pKa,eff values for the incorporation of the S stereoisomers exhibit large increases in magnitude of ~0.5–1.0 pK unit (and reductions in kpol). On the contrary, pKa,eff (and kpol) values for the R stereoisomers are closely coincident with the diastereomer mix.
Pol-catalyzed incorporation involves concerted phospho-diester bond formation as the PPi/bisphosphonate leaving group bonds are broken via a TS plateau that is formed during a nucleophilic attack of the deprotonated O3′ terminus of the primer on the Pα atom of the dNTP substrate complexed with two Mg2+ ions.29,33,34 The Brønsted slope provides a “snapshot” of the extent of Pα–O3′ bond formation or, more precisely, the TS charge distribution.35–38 A slope of near zero indicating little if any dependence on the leaving group pKa would be consistent with a nonchemical rate-limiting step, for example, a protein conformational change. Shallow slopes (between −0.2 and −0.6) suggest an “early” TS in which leaving group charges are close to their values in dNTP or dNTP analogue reactants, while steep slopes (greater than −1.0) are indicative of a “late” TS with leaving group charges comparable to those of PPi or bisphosphonate products. A slope of approximately −0.9 (non-enzymatic reference slope) suggests that the TS charge is balanced evenly between initial substrate and product forms.17,39
With pol η, the Brønsted slopes for formation of A·T base pairs range between −0.34 (dihalo) and −1.0 (non-dihalo), with those for pols λ and β lying between those values (Figure 3). For G·T mispairs (Figure 4B,C), the slopes for pol λ lie between −1.3 (dihalo) and −0.55 (non-dihalo), and those for pol β, −1.1 (dihalo) and −0.71 ( non-dihalo), are closely comparable to the reference slope. The chemical alteration of the ability of the leaving group to stabilize its negative charge, which is reflected by its pKa4, affects kpol differently for each pol. For example, because the Brønsted slope to form A·T base pairs with the non-dihalo substrates is steep for pol η, approximately −1.0, its TS is likely to be “product-like”, whereas pols λ and β, ~0.6−0.7, should appear more like the substrates. In contrast, pol η kinetics are characterized by a small slope, ~0.4 for the incorporation of dihalo-β,γ-dATP and β,γ-CHCl-ATP compounds (Figure 3A), which suggests a mechanistic change from a late-to-early TS caused by the presence of a halogen atom in the bridging methylene group.
Pol η stands apart from many non-Y-family pols by copying DNA with low fidelity.4 Pol η was shown by Yang and colleagues13 to form G·T/A·T with 4-fold lower fidelity by stabilizing a T·dGTP wobble structure in its active site when the 3’ primer terminus is either A or T. A 4-fold lower fidelity was observed with pol η, when comparing the formation of A·T versus G·T in W (3′ primer A) and B (3′ primer C) sequence contexts with the bisphosphonate dATP-CHF and dGTP-CHF substrates, as with the parent dNTP (Figure 2). Using LFER to observe the formation of A·T versus G·T in the two sequence contexts, the split Brønsted lines show virtually identical pKa,eff shifts for the dihalo and non-dihalo incorporation of A opposite T (Figure 3A). In contrast, a clear sequence context effect is observed for incorporation of G opposite T, where the dGTP-CHF and dGTP-NH analogues and dGTP show a 4-fold higher misincorporation opposite T in the W sequence context.
Looking toward the future, we note that the TS and product-state differences between pol η and family X polymerases, deduced from the LFER analysis, are interesting in light of the similarity of their active site structures near the triphosphate moiety of bound dNTP substrates for correct base pairs.40,41 We suggest that the LFER approach prescribes a path toward using polymerase active site-directed mutagenesis to investigate systematically the ways in which individual amino acid electrostatic perturbations act to modulate pol chemistry and fidelity in the TS. By providing fresh insight into how nuances in the structures of pol active sites can selectively regulate chemistry, we suggest that these data make the pol TS a bit less opaque and can serve as “grist for the mill” of computational chemistry calculations and further structural analyses.
Supplementary Material
Acknowledgments
Funding
This research was supported by National Institutes of Health (NIH) Grant 1U19CA177547 and by the Division of Intramural Research of the NIH, National Institute of Environmental Health Sciences, Projects Z01 ES050158 and ES050159.
Footnotes
ASSOCIATED CONTENT
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.bio-chem.9b00087.
Description of the synthesis of β,γ-dNTP analogues, tables of kinetic parameters, effective pKa data, and a description of potentiometric calculations (PDF)
The authors declare no competing financial interest.
REFERENCES
- (1).Johnson RE, Prakash S, and Prakash L (1999) Efficient bypass of a thymine-thymine dimer by yeast DNA polymerase, Pol η. Science 283, 1001–1004. [DOI] [PubMed] [Google Scholar]
- (2).Washington MT, Johnson RE, Prakash L, and Prakash S (2001) Accuracy of lesion bypass by yeast and human DNA polymerase η. Proc. Natl. Acad. Sci. U. S. A. 98, 8355–8360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Zeng X, Winter DB, Kasmer C, Kraemer KH, Lehmann AR, and Gearhart PJ (2001) DNA polymerase η is an A-T mutator in somatic hypermutation of immunoglobulin variable genes. Nat. Immunol. 2, 537–541. [DOI] [PubMed] [Google Scholar]
- (4).Matsuda T, Bebenek K, Masutani C, Hanaoka F, and Kunkel TA (2000) Low fidelity DNA synthesis by human DNA polymerase η. Nature 404, 1011–1003. [DOI] [PubMed] [Google Scholar]
- (5).Washington MT, Helquist SA, Kool ET, Prakash L, and Prakash S (2003) Requirement of Watson-Crick Hydrogen Bonding for DNA Synthesis by Yeast DNA Polymerase η. Mol Cell. Biol. 23, 5107–5112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Sucato CA, Upton TG, Kashemirov BA, Batra VK, Martinek V, Xiang Y, Beard WA, Pedersen LC, Wilson SH, McKenna CE, Florián J, Warshel A, and Goodman MF (2007) Modifying the β,γ leaving-group bridging oxygen alters nucleotide incorporation efficiency, fidelity, and the catalytic mechanism of DNA polymerase β. Biochemistry 46, 461–471. [DOI] [PubMed] [Google Scholar]
- (7).Sucato CA, Upton TG, Kashemirov BA, Osuna J, Oertell K, Beard WA, Wilson SH, Florián J, Warshel A, McKenna CE, and Goodman MF (2008) DNA polymerase β fidelity: Halomethylene-modified leaving groups in pre-steady-state kinetic analysis reveal differences at the chemical transition state. Biochemistry 47, 870–879. [DOI] [PubMed] [Google Scholar]
- (8).Oertell K, Wu Y, Zakharova VM, Kashemirov BA, Shock DD, Beard WA, Wilson SH, McKenna CE, and Goodman MF (2012) Effect of β,γ-CHF- and β,γ-CHCl-dGTP halogen atom stereochemistry on the transition state of DNA polymerase β. Biochemistry 51, 8491–8501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Oertell K, Chamberlain BT, Wu Y, Ferri E, Kashemirov BA, Beard WA, Wilson SH, McKenna CE, and Goodman MF (2014) Transition state in DNA polymerase β catalysis: rate-limiting chemistry altered by base-pair configuration. Biochemistry 53, 1842–1848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Oertell K, Kashemirov BA, Negahbani A, Minard C, Haratipour P, Alnajjar KS, Sweasy JB, Batra VK, Beard WA, Wilson SH, McKenna CE, and Goodman MF (2018) Probing DNA Base-Dependent Leaving Group Kinetic Effects on the DNA Polymerase Transition State. Biochemistry 57, 3925–3933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Petruska J, and Goodman MF (2017) Relating DNA base-pairing in aqueous media to DNA polymerase fidelity. Nat. Rev. Chem. 1, 0074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Wu W-J, Yang W, and Tsai M-D (2017) How DNA polymerases catalyse replication and repair with contrasting fidelity. Nat. Rev. Chem. 1, 0068. [Google Scholar]
- (13).Zhao Y, Gregory MT, Biertumpfel C, Hua Y-J, Hanaoka F, and Yang W (2013) Mechanism of somatic hypermutation at the WA motif by human DNA polymerase η. Proc. Natl. Acad. Sci. U. S. A. 110, 8146–8151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Braithwaite EK, Prasad R, Shock DD, Hou EW, Beard WA, and Wilson SH (2005) DNA polymerase λ mediates a back-up base excision repair activity in extracts of mouse embryonic fibroblasts. J. Biol. Chem. 280, 18469–18475. [DOI] [PubMed] [Google Scholar]
- (15).Braithwaite EK, Kedar PS, Lan L, Polosina YY, Asagoshi K, Poltoratsky VP, Horton JK, Miller H, Teebor GW, Yasui A, and Wilson SH (2005) DNA polymerase λ protects mouse fibroblasts against oxidative DNA damage and is recruited to sites of DNA damage/repair. J. Biol. Chem. 280, 31641–31647. [DOI] [PubMed] [Google Scholar]
- (16).Bebenek K, Pedersen LC, and Kunkel TA (2014) Structure-function studies of DNA polymerase λ. Biochemistry 53, 2781–2792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Zalatan JG, and Herschlag D (2006) Alkaline phosphate mono- and diesterase reactions: Comparative transition state analysis. J.Am. Chem. Soc. 128, 1293–1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Zhang Z, Eloge J, and Florián J (2014) Quantum Mechanical Analysis of Nonenzymatic Nucleotidyl Transfer Reactions: Kinetic and Thermodynamic Effects of β−γ Bridging Groups of dNTP Substrates. Biochemistry 53, 4180–4191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Frank EG, McDonald JP, Karata K, Huston D, and Woodgate R (2012) A strategy for the expression of recombinant proteins traditionally hard to purify. Anal. Biochem. 429, 132–139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Zekany L, and Nagypal I (1985) Computational Methods for the Determination of Stability Constants In Computational Methods for the Determination of Stability Constants (Leggett D, Ed.) pp 291–355, Plenum Press, New York. [Google Scholar]
- (21).Sawaya MR, Prasad R, Wilson SH, Kraut J, and Pelletier H (1997) Crystal structures of human DNA polymerase β complexed with gapped and nicked DNA: evidence for an induced fit mechanism. Biochemistry 36, 11205–11215. [DOI] [PubMed] [Google Scholar]
- (22).Pelletier H, Sawaya MR, Kumar A, Wilson SH, and Kraut J (1994) Structure of ternary complexes of rat DNA polymerase β, a DNA template-primer, and ddCTP. Science 264, 1891–1903. [PubMed] [Google Scholar]
- (23).Doublie S, Sawaya MR, and Ellenberger T (1999) An open and closed case for all polymerases. Structure 7, R31–R35. [DOI] [PubMed] [Google Scholar]
- (24).Batra VK, Oertell K, Beard WA, Kashemirov BA, McKenna CE, Goodman MF, and Wilson SH (2018) Mapping functional substrate-enzyme interactions in the pol β active site through chemical biology: Structural responses to acidity modification of incoming dNTPs. Biochemistry 57, 3934–3944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Kadina AP (2017) Deoxyribonucleoside triphosphate analogues for inhibition of therapeutically important enzymes. Ph.D. Thesis, University of Southern California, Los Angeles. [Google Scholar]
- (26).Negahbani A (2017) Nucleophilic fluorination of bi-sphosphonates and its application in PET imaging. Ph.D. Thesis, University of Southern California, Los Angeles. [Google Scholar]
- (27).Fried SD, Bagchi S, and Boxer SG (2014) Extreme electric fields power catalysis in the active site of ketosteroid isomerase. Science 346, 1510–1514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Fried SD, and Boxer SG (2017) Electric Fields and Enzyme Catalysis. Annu. Rev. Biochem. 86, 387–415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Klvaňa M, Bren U, and Florián J (2016) Uniform Free-Energy Profiles of the P-O Bond Formation and Cleavage Reactions Catalyzed by DNA Polymerases β and λ. J. Phys. Chem. B 120, 13017–13030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).McKenna CE, Kashemirov BA, Upton TG, Batra VK, Goodman MF, Pedersen LC, Beard WA, and Wilson SH (2007) (R)-β,γ-Fluoromethylene-dGTP-DNA Ternary Complex with DNA Polymerase β. J. Am. Chem. Soc. 129, 15412–15413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Freudenthal BD, Beard WA, Shock DD, and Wilson SH (2013) Observing a DNA polymerase choose right from wrong. Cell 154, 157–168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Gao Y, and Yang W (2016) Capture of a third Mg(2)(+) is essential for catalyzing DNA synthesis. Science 352, 1334–1337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Florián J, Goodman MF, and Warshel A (2003) Computer simulation of the chemical catalysis of DNA polymerases: discriminating between alternative nucleotide insertion mechanisms for T7 DNA polymerase. J. Am. Chem. Soc. 125, 8163–8177. [DOI] [PubMed] [Google Scholar]
- (34).Yoon H, and Warshel A (2017) Simulating the fidelity and the three Mg mechanism of pol η and clarifying the validity of transition state theory in enzyme catalysis. Proteins: Struct., Funct., Genet. 85, 1446–1453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Bourne N, and Williams A (1984) Effective Charge on Oxygen in Phosphoryl Group Transfer from an Oxygen Donor. J. Org. Chem. 49, 1200–1204. [Google Scholar]
- (36).Davis AM, Hall AD, and Williams A (1988) Charge description of base-catalyzed alcoholysis of aryl phosphodiesters: A ribonuclease model. J. Am. Chem. Soc. 110, 5105–5108. [Google Scholar]
- (37).Williams A (2003) Free Energy Relationships in Organic and Bio-Organic Chemistry, Royal Society of Chemistry, Cambridge, U.K. [Google Scholar]
- (38).Lassila JK, Zalatan JG, and Herschlag D (2011) Biological Phosphoryl-Transfer Reactions: Understanding Mechanism and Catalysis. Annu. Rev. Biochem. 80, 669–702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Zhang Z, Eloge J, and Florián J (2014) Quantum mechanical analysis of nonenzymatic nucleotidyl transfer reactions: kinetic and thermodynamic effects of β−γ bridging groups of dNTP substrates. Biochemistry 53, 4180–4191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Patra A, Nagy LD, Zhang Q, Su Y, Muller L, Guengerich FP, and Egli M (2014) Kinetics, structure, and mechanism of 8-oxo-7,8-dihydro-2’-deoxyguanosine bypass by human DNA polymerase η. J. Biol. Chem. 289, 16867–16882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Biertumpfel C, Zhao Y, Kondo Y, Ramon-Maiques S, Gregory M, Lee JY, Masutani C, Lehmann AR, Hanaoka F, and Yang W (2010) Structure and mechanism of human DNA polymerase η. Nature 465, 1044–1048. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.