

Abstract

Efficient delivery of therapeutics across the cell membrane to the interior of the cell remains a challenge both in vitro and in vivo. Here, we demonstrate that vesicles derived from cellular membranes can be efficiently loaded with cargo that can then be delivered to the interior of the cell. These vesicles demonstrated cell-targeting specificity as well as the ability to deliver a wide range of different cargos. We utilized this approach to deliver both lipophilic and hydrophilic cargos including therapeutics and DNA in vitro. We further demonstrated in vivo targeting and delivery using fluorescently labeled vesicles to target tumor xenografts in an animal. Cell-derived vesicles can be generated in high yields and are easily loaded with a variety of cargos. The ability of these vesicles to specifically target the same cell type from which they originated provides an efficient means of delivering cargo, such as therapeutics, both in vitro and in vivo.
Introduction
The effective delivery of cargos such as fluorescent markers, genetic material, therapeutics, and proteins to the interior of the cell is essential for the development of new therapeutics and for understanding biological function.1−4 Despite advances in areas such as gene delivery,5 targeted therapeutics, vesicle-based delivery systems,6,7 and the use of cell-penetrating peptides,8 the efficient transport of cargo across the cell membrane remains one of the primary challenges to the development of therapeutics. The most common strategies for accessing the interior of the cell utilize endocytic pathways. While this provides a relatively efficient means of crossing the cell membrane, it results in the trapping of cargo in endosomal vesicles. The cargo must then escape from these vesicles, however, lowering both the efficiency and the potential efficacy of therapeutics.9 This increases the complexity of cargo delivery for both cell culture-based and in vivo applications. Ideal delivery vehicles would allow for the direct transport of cargo to the interior of the cell, bypassing endocytosis altogether.
Vesicles composed of phospholipid bilayers have shown promise as therapeutic delivery vectors capable of encapsulating the cargo and delivering it to the interior of target cells.10,11 Synthetic vesicles such as liposomes composed of phospholipid membranes are relatively easy to load and have shown promise as in vitro and in vivo intracellular delivery devices.7,12−14 However, applications are limited by a lack of biocompatibility, as liposomes are not capable of avoiding the immune system for in vivo delivery. Naturally occurring vesicles produced by cells are an attractive alternative. For example, exosomes have received significant attention as therapeutic delivery vehicles to transport cargo across cell membranes because they are both nonimmunogenic and specifically target select cell types.11,15−18 While cell specificity addresses a major problem with targeted therapeutic delivery, the application of exosomes as cellular delivery devices is limited by low production efficiency and difficulty in loading with cargo. Despite these limitations, exosomes have been utilized for in vitro delivery of therapeutics and for gene delivery. Recently, vesicles generated from the membranes of organelles within the cells were used as exosome-mimics and retained several of the targeting properties seen with exosomes.19−22
Several key factors need to be considered for the development of intracellular delivery vectors. Loading of the cargo must be efficient and easy and the vehicle must also be compatible with a wide range of cargo. Here, we develop cell-derived vesicles that can be easily loaded with cargo and exhibit cell-targeting capabilities. We harness these vesicles as general cell-delivery vehicles that can deliver a wide range of cargos including genetic material, therapeutics, protein, and fluorescent markers to the interior of cells both in vitro and in vivo.
Results and Discussion
Characterization of Cell-Derived Vesicles
We generated vesicles through nitrogen cavitation23 where cells in solution are subjected to high-pressure N2. The pressure is rapidly released resulting in the formation of gas bubbles that fragment the cellular membranes. These small fragments then reform to generate enclosed vesicles. We separate vesicles from the remaining cell debris through a series of centrifugation steps. A schematic of the vesicle generation and isolation process is shown in Scheme 1. One advantage of this approach is that the solution containing the cells during cavitation is encapsulated in the vesicles. Thus, therapeutics or other cargos are entrapped in the vesicles with high efficiency at the time of vesicle formation. An image of vesicles generated using nitrogen cavitation from human embryonic kidney 293 cells (HEK) formed in the presence of fluorescein, a fluorescent dye, is shown in Figure 1A. The fluorescence image shows punctate regions indicating that the fluorophore is trapped inside the vesicles verifying encapsulation. The cargo, encapsulated by a phospholipid bilayer, is safeguarded from free solution. To illustrate the versatility of this approach we performed a series of studies using vesicles from HEK, human colorectal cancer (HCT 116), human lung cancer (A549), and macrophage-like cell lines (RAW 264.7).
Scheme 1. Schematic of Vesicle Generation, Loading, and Isolation.

Cultured cells undergo nitrogen cavitation in the presence of cargo in free solution followed by serial centrifugation to generate purified vesicles. Vesicles serve as nanocarriers for hydrophilic cargo encapsulated during cavitation on the interior or for lipophilic cargo that can be embedded in the vesicle membrane.
Figure 1.
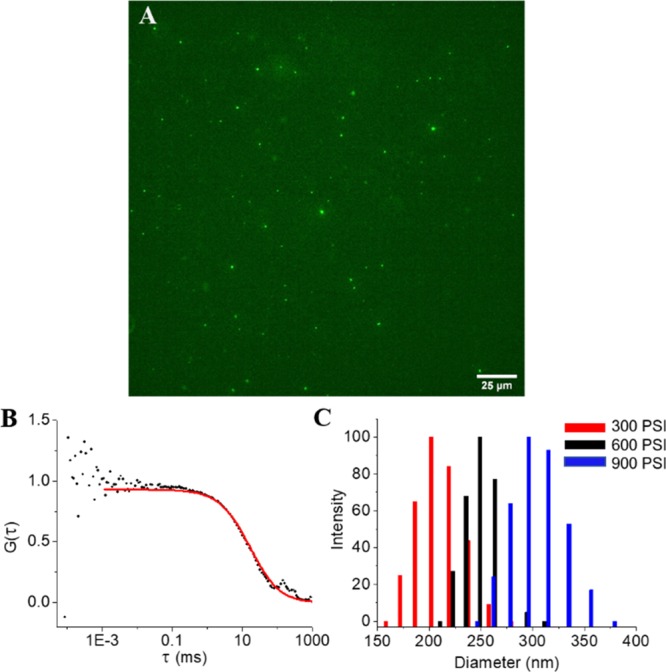
Cell-derived vesicle characterization. (A) Wide-field fluorescence image of vesicles loaded with the fluorescent dye fluorescein. (B) Fluorescence correlation spectroscopy correlogram of vesicles used to determine vesicle concentration and relative yield. (C) Plot of vesicle size distribution at different cavitation pressures as determined by dynamic light scattering.
The collection of vesicles endogenously expressed by cells, such as exosomes, suffers from relatively poor yields.24,25 To determine the yield of our preparation, we performed fluorescence correlation spectroscopy (FCS). Vesicles were generated from approximately 40 million A549 cells in culture using nitrogen cavitation. After the vesicles were isolated, they were labeled through the incorporation of a lipophilic dialkylcarbocyanine fluorophore (DiI), which is nonfluorescent in an aqueous solution but emits brightly when embedded in a lipid bilayer. FCS tracks fluctuations in fluorescence as vesicles diffuse through the focal volume.26,27 Both the diffusion time and the average number of molecules can be extracted from the autocorrelation curve (Figure 1B). The FCS focal volume was calibrated using commercial tetraspeck beads leading to a determination that our preparations yielded ∼4 × 1011 vesicles per mL, which is approximately 1.3 × 1011 vesicles per preparation (40 million cells). Thus, we were able to generate a relatively large number of vesicles from a modest number of cells.
To further characterize cell-derived vesicles, we performed dynamic light scattering (DLS) to determine the diameter of vesicles generated via nitrogen cavitation. Figure 1C shows the distribution of vesicle diameters of a typical preparation at different pressures. The 200 nm observed at 300 psi is slightly larger than standard exosomes (100 to 150 nm) but is within a similar range that is unlikely to affect cell delivery. To determine stability, we also measured the surface charge (zeta potential) of cell-derived vesicles suspended in a PBS buffer. Vesicles preparations exhibited a surface charge of −2.5 mV. DLS measurements of vesicles in solution after 6 h showed no changes in size distributions over time. To determine how pressure influenced the diameter of vesicles, we generated vesicles with nitrogen cavitation pressures of 300 (red), 600 (black), and 900 psi (blue) (Figure 1C). Interestingly, 300 psi yields the smallest vesicles whereas 900 psi yielded the largest. Vesicle size was clearly dependent on the cavitation pressure. In addition to yielding tunable vesicle diameters, one advantage of cavitation over other techniques to fracture the membrane is that it does not generate heat that can damage samples or alter the chemical composition of the cell medium. This results in the formation of relatively uniform vesicles likely due to all the cells in solution being exposed to the same pressure conditions.28
Determining Vesicle Targeting Specificity across Different Cell Types
Previous studies have shown that exosomes and vesicles generated from cancer cells preferentially target the same cell type from which they were derived.19,29 To determine the degree of targeting specificity of vesicles generated via nitrogen across cell types, we performed a series of studies comparing the delivery of labeled vesicles to the cell type from which the vesicles originated versus alternate cell types. We first generated vesicles from HEK cells and labeled them with DiI. We then determined the efficiency of delivery to both HEK and A549 cells by measuring the fluorescence signal at time points over 4 h. We added 5 × 109 vesicles to each cell culture condition and allowed them to incubate with the cells. Vesicles were then rinsed from the cells, and the cells were subsequently imaged using wide-field microscopy. Most cell types showed a clear targeting specificity for the cell type where they originated. At the 2 h time point of HEK vesicle delivery, HEK cells exhibited ∼10 times as much fluorescence as A549 cells after incubation with the same number of vesicles for the same time period (Figure 2A). Similarly, RAW vesicles were much more efficiently delivered (8×) to RAW cells as they were to A549 cells (Figure 2B). We also compared the delivery of different vesicles to the same cell type. While HCT vesicles were approximately 3 times more efficient at delivering cargo to HCT cells as compared to RAW vesicles, the RAW vesicles still exhibited targeting properties for the HCT cells (Figure 2C). Combined results show that vesicles tend to have an affinity for the delivery of cargo to the same cell type from which they originated. The wide-field image comparing RAW vesicle delivery to RAW cells versus delivery to A549 cells after 2.5 h (Figure 2D,E) shows a clear preference for delivery to RAW cells. However, the ability of RAW vesicles to effectively target cancer cells with only a 3-fold deficit as compared to cancer vesicles could allow macrophage vesicles to be used as a general delivery vehicle. The use of cancer vesicles for clinical application is limited due to the likelihood for them to increase the metastatic potential in vivo. Additionally, the tumor microenvironment consists of a heterogenous mixture of cells including large numbers of macrophages. Thus, macrophages offer better long-term potential for clinical applications. These studies illustrate the ability of nanoscale cell-derived vesicles to deliver lipophilic cargo to cells.
Figure 2.

Cell-targeting specificity. (A) Comparison of HEK vesicles delivered to HEK cells (black) versus HEK vesicles delivered to A549 cells (red). (B) Comparison of RAW vesicles delivered to RAW cells (black) versus RAW vesicles delivered to A549 cells (red). (C) Comparison of HCT vesicles delivered to HCT cells (black) versus RAW vesicles delivered to HCT cells (red). (D) Wide-field fluorescence image of DiI-labeled RAW vesicles delivered to RAW cells after 2.5 h showing clear loading. (E) Wide-field fluorescence image of DiI-labeled RAW vesicles delivered to A549 cells after 2.5 h showing limited cellular uptake. Norm ID is the integrated density of the image normalized to the time 0 value. Each data point is the average of five experiments. A Student’s t-test was used to determine significance between end points. Each end point was significant with a p value of <.001.
Determining in Vitro Efficacy for Therapeutic Delivery
The efficient delivery of therapeutics to the interior of the cell is one of the primary challenges of cell delivery vehicles. We performed a set of experiments to measure the efficiency of drug loading into cell-derived vesicles and then the efficiency of delivery to cancer cells. We generated vesicles from A549 cells in the presence of 8.33 mM cisplatin. To verify that the vesicles successfully encapsulated cisplatin, we measured the drug concentration in the vesicles directly after formation and separation from free cisplatin. The cisplatin in vesicles as measured by ICP-OES for 1.3 × 1011 vesicles was ∼3 μg of total cisplatin. This effectively shows that the vesicles generated from nitrogen cavitation can encapsulate cisplatin at the time of formation, protecting it from the environment outside of the vesicle. To further verify that stability of encapsulation over time, we performed the same ICP-OES studies at intervals over 3 days after drug encapsulation on the supernatant to measure any cisplatin leakage from the vesicles. Vesicles maintain roughly the same concentration at each time point showing the stability of encapsulation with a total decrease of 23 ng of cisplatin from day 1 through day 3. Cell-derived vesicles are able to efficiently encapsulate a large concentration of chemotherapeutics and remain stable over 2 days with no apparent leakage. We then performed cell delivery experiments to determine if we could utilize drug-loaded vesicles for therapeutic delivery by comparing the efficacy of free cisplatin to that of cisplatin encapsulated in vesicles.
We first performed a control study to determine whether the vesicles themselves had any effect on cell growth. We generated unloaded A549-derived vesicles and delivered them to A549 cells. We determined cell viability using a fluorescence assay (alamar blue), where the fluorescence intensity scales with the population of live cells. Comparing cell growth and viability to that of untreated cells, empty vesicles appeared to have no effect on cell growth (Figure 3). With a clear indication that vesicles targeted the same cells from which they originated (Figure 2A–C) and that vesicles did not alter cell viability, we compared the effect of free cisplatin and cisplatin-loaded vesicles on A549 cell proliferation. We chose cisplatin because of its hydrophilic nature and because it is a first-line therapeutic for lung cancer. Vesicles were loaded through nitrogen cavitation in the presence of cisplatin (8.33 mM). Vesicles were then added to A549 cells in culture, and we compared the cell proliferation to cells alone and those in the presence of free cisplatin in solution at the same levels (3 μg) as we measured in the vesicle solution. This low concentration of free cisplatin resulted in no apparent cell death at 24 h while cisplatin-loaded vesicles resulted in 70% cell death. By the 48 h time point, loaded cell-derived vesicles resulted in 90% cell death, while free cisplatin resulted in 15% cell death. At 72 h,, free cisplatin led to 50% cell death while vesicles maintained approximately 90% cell death. Vesicles (1 × 1010) containing .3 μg of cisplatin (∼4 μM) delivered to A549 cells were much more effective at limiting cell viability than the same concentration of free cisplatin. This verified the capability of the vesicles to more efficiently deliver the drug across the cell membrane.
Figure 3.
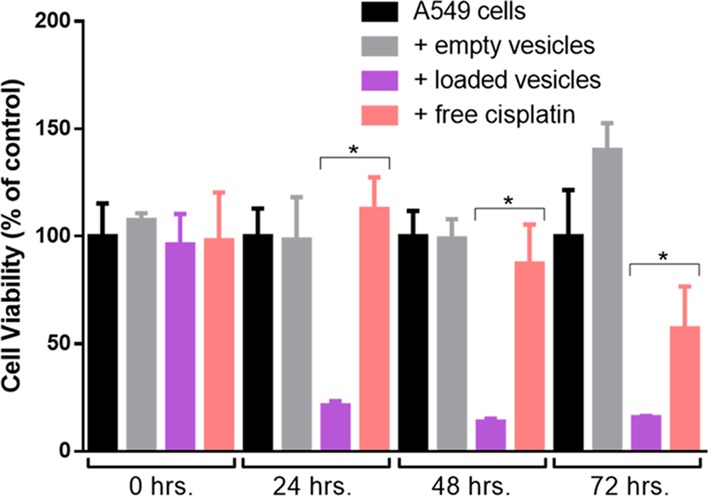
Efficacy of cisplatin-loaded vesicles. Comparison of cell growth at time 0, 24, 48, and 72 h for A549 cells with no treatment (black), treated with empty vesicles (gray), with cisplatin-loaded vesicles (purple), and free cisplatin in solution (pink). Empty vesicles have no effect on cell growth while both free cisplatin and loaded vesicles show similar efficacy in killing A549 cells. Each data point is the average of five experiments. A Student’s t-test was used to determine significance. The asterisk “*” indicates a p value of <.001.
Cell Derived Vesicles for Gene Delivery
To demonstrate the versatility of vesicle-based delivery, we also encapsulated plasmid DNA for gene delivery. Current strategies for gene delivery primarily use transfection reagents such as cationic lipids for efficient delivery. We generated vesicles in the presence of a plasmid that encoded for the fluorescent protein Dendra2. Vesicles were loaded via the same nitrogen cavitation approach and then incubated with HEK cells for 48 h. The cells were then visualized using wide-field fluorescence microscopy to identify cells expressing the fluorescent protein. Virtually none of the control cells showed any fluorescence. The two cells exhibiting any fluorescence (Figure 4A) in the control sample correspond to autofluorescence from dead cells in the field of view and the rest of the live cells show no fluorescence as indicated in the bright-field image shown in the Supporting Information (Figure S1A). The majority of HEK cells in the vesicle-treated sample had taken up the plasmid and produced a characteristic green fluorescence. The bright-field image is shown in Supporting Information (Figure S1B). Here we used ∼1 × 1010 vesicles for delivery. As can be seen from the similar level of fluorescence across most cells, the delivery in culture and loading of vesicles appeared to be homogenous across the sample. This verifies that vesicles are capable of delivering DNA across the cell membrane to the interior of the cell.
Figure 4.
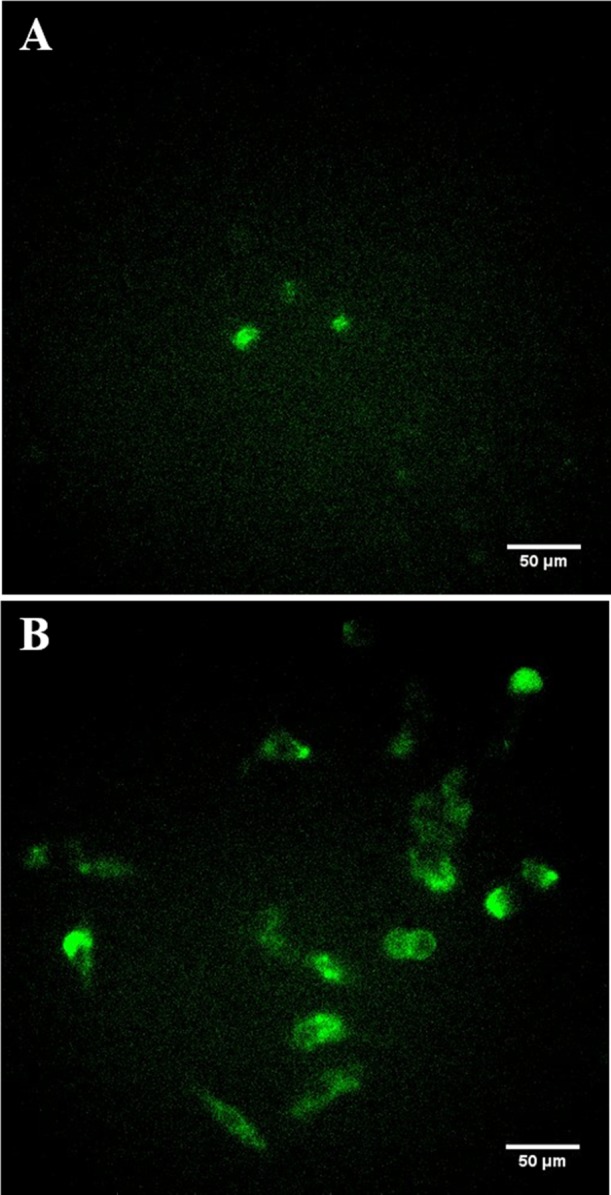
Vesicle-based gene delivery. (A) Wide-field image of HEK cells in the absence of plasmid. (B) Wide-field image of HEK cells after exposure to Dendra2 plasmid-loaded vesicles showing clear cellular delivery based on the expression of the fluorescent protein.
Delivery of Hydrophilic Cargo
To visualize the delivery of cargo to the cell interior, we generated vesicles while simultaneously encapsulating the green emitting fluorophore, fluorescein. After generating the vesicles, we then labeled the vesicle membrane with DiD, a lipophilic dye. This allowed us to observe the delivery of cargo within the vesicles to the interior of the cell as well as observe integration of the vesicle membrane into the membrane of the target cells. After 45 min of exposure, vesicles were rinsed from the cells and we performed confocal imaging. Clear fluorescein fluorescence permeated the interior of the cell indicating its presence in the cytosol (Figure 5). At the same time, isolated vesicles were observed on the cell surface. The presence of fluorescein in the cytosol after only 45 min verifies the encapsulation and delivery of hydrophilic cargo. Individual vesicles labeled with DiD were observed on the cell surface.
Figure 5.
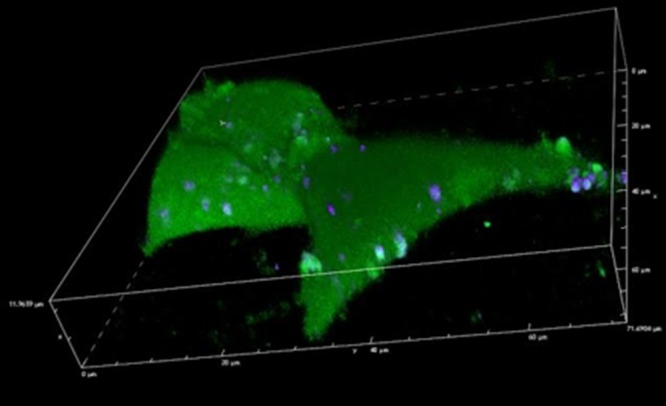
Confocal imaging of vesicle delivery. Confocal image of HEK 293 cells after the delivery of vesicles loaded with fluorescein (interior) and DiD (cell membrane). The interior of the cell is filled with fluorescein and vesicles can be seen on the cell surface.
In Vivo Targeting and Delivery of Vesicles
We also performed a set of studies to determine if cell-derived vesicles could be used to target a specific tissue in a live animal. We generated vesicles from HEK cells, A549 cells, and RAW 264.7 cells and labeled each separately with DiR to enable tracking to specific sites within the animal. RAW cells were used because the tumor environment contains large numbers of macrophages, RAW vesicles showed some targeting affinity for cancer cells, and targeting delivery with cancer cell-derived vesicles is not feasible for clinical applications. There is concern for the potential of cancer vesicles to lead to an increase in the spread of cancer throughout the body. We used a male outbred athymic nude (nu/nu) mouse from Jackson Laboratories implanted with a subcutaneous tumor xenograft composed of A549 or HCT116 cells. After the tumor reached at least 100 mm3, we injected labeled vesicles (∼2 × 1011), systemically, through the tail vein. In vivo imaging was performed 24 h postinjection using an IVIS whole animal imager. Injection of DiR alone lead to nonspecific accumulation, while HEK vesicles accumulated in the area of the bladder (Figure 6A,B). Clear fluorescence was observed in the tumor site for the delivery of A549 and RAW vesicles (Figure 6C,D). These experiments verify the in vivo targeting capability of both A549 vesicles and RAW vesicles for the tumor xenograft.
Figure 6.

Mice bearing A549 xenografts on the right shoulder (dashed blue ovals) were injected with (A) dye (DiR) alone, (B) dye-labeled vesicles derived from HEK cells, (C) dye-labeled vesicles derived from A549 cells, and (D) dye-labeled vesicles derived from RAW264.7 cells demonstrating RAW vesicles specifically targeted the A549 xenograft. Free domain photograph courtesy of JRM.
Conclusions
These studies demonstrate that cell-derived vesicles can be efficiently loaded with a wide variety of cargos. They also exhibit similar properties as exosomes in that they specifically target the cell from which they originated. However, cell-derived vesicles can be easily loaded and generated at higher yields than exosomes. These properties can be harnessed to efficiently deliver cargos ranging from therapeutics to DNA across the cell membrane. These vesicles demonstrated targeting efficiency in animals as shown through their delivery to tumor sites. Overall, cell-derived vesicles can be used as a versatile cell delivery vehicle both in vitro and in vivo.
Experimental Section
Cisplatin Delivery
A549 cells (60 million) were used to generate vesicles for delivery. Cells were scraped from culture in 20 mL of sucrose buffer containing protease inhibitor. The cell solution was collected in a 50 mL conical tube and pelleted at 2000 rpm at 25 °C for 2 min. The solution was aspirated off, and the pellet was resuspended in 8 mL of 8.33 mM cisplatin in sucrose buffer solution or 20 mg cisplatin/8 mL sucrose buffer solution plus protease inhibitor. The cell solution was fragmented using N2 cavitation at 300 psi at 4 °C for 5 min. The resulting cell slurry was centrifuged at 4000g for 10 min at 4 °C. The supernatant from centrifugation was transferred to a 25 mL ultracentrifuge tube for 10,000g centrifugation for 20 min at 4 °C. The supernatant from the UCF tube was then transferred to a new 25 mL ultracentrifuge tube and centrifuged at 100,000g for 60 min at 4 °C. The pellet in the UCF tube was washed with 500 μL of sucrose solution, and the residual solution was pipetted out and discarded. Seven hundred fifty microliters of sucrose buffer solution was added to the UCF tube, and the pellet was resuspended via pipetting. Empty vesicles were generated in the exact same way except in the absence of cisplatin.
A549 cells (30,000) were plated in each well of a 96-well plate and allowed to seed for 24 h. During that time, the growth media was exchanged for 250 μL of fresh HEK media containing either cisplatin in solution (4.17 mM), cisplatin-loaded vesicles, empty vesicles, or untreated media cisplatin solution in HEK media. The media was aspirated off. Two hundred microliters of Optimem was added followed by 20 μL of alamar blue. The plate was left to incubate at 37 °C with gentle tapping every 10 min for 40–45 min to ensure uniform turnover to a brilliant purple. The plate was read using a FlexStation plate reader.
Gene Delivery
HEK cells (32 million) were scraped with 10 mL of sucrose buffer solution with protease inhibitor. All of the cell solution was collected into a 15 mL conical tube and pelleted at 2000 rpm at 25 °C for 2 min. The solution was aspirated off and resuspended in 3 mL sucrose buffer solution plus protease inhibitor. Plasmid (75 μg of Dendra2) was added to the N2 cavitation chamber. The solution was fragmented using N2 cavitation at 600 psi at 4 °C for 5 min. The cell slurry was centrifuged at 4000g for 10 min at 4 °C. The supernatant from centrifugation was transferred to a 25 mL ultracentrifuge tube and centrifuged at 10,000g for 20 min at 4 °C. The supernatant from the UCF tube was then transferred to a new 25 mL ultracentrifuge tube and centrifuged at 100,000g for 60 min at 4 °C. The pellet in the UCF tube was washed with 500 μL sucrose solution, and the residual solution was pipetted out and discarded. One milliliter of sucrose buffer solution was added to the UCF tube, and the pellet was resuspended via pipetting. Two hundred fifty microliters of this solution was added to 30,000 HEK cells plated on glass bottom dishes. After 24 h, the cells were rinsed and then imaged at 48 h using a wide-field microscope with a 488 nm band-pass filter for excitation.
Vesicle Delivery
Vesicles were prepared as described above. The vesicle solution was mixed in an Eppendorf tube (1 mL) with 2 μL of 2 mM DiI and left to incubate for 30 min at 37 °C. Labeled vesicles were separated from free fluorescein/DiI using a PD MidiTrap equilibrated with sucrose buffer solution. One hundred eighty microliters of the vesicle solution was added to cells plated on glass bottom dishes. Cells were imaged using an excitation wavelength of 561 nm after 2 h of incubation at 37 °C. Control studies to determine the leaching of encapsulated and membrane-bound fluorophores were performed. Vesicles with either DiI or fluorescein were incubated in solution for 4 h to mimic the conditions of cell labeling. After 4 h, the vesicles were pelleted using ultracentrifugation. The supernatant was then added to the cell culture to determine the presence of any free dye. No visible fluorescence was observed for control studies with fluorescein or with DiI.
In Vivo Xenograft
Athymic nude mice were injected with A549 cells (NSCLC, immortalized) in the right shoulder and monitored for 3–4 weeks until measurable xenograft tumors were observed. RAW cells were cultured in vitro by standard methods. Cell-derived vesicles were manufactured by the Richards lab as described above and loaded with DiR near-infrared fluorescent dye. Prepared vesicles were administered, and mice were then imaged approximately 24, 48, and 72 hours later. Mice were anesthetized for imaging using isoflurane. Epifluorescence was measured using an IVIS Spectrum In Vivo Imaging System (PerkinElmer). Vesicles were labeled with DiR near-infrared fluorescent dye, which was excited at 710 nm, with emission measured at 780 nm. Fluorescent signal intensity (i.e., total radiant efficiency) within regions of interest (ROI) were quantified using Living Image software (PerkinElmer), correcting for background fluorescence using distal site ROI within the same mouse.
RAW264.7 cells (100 million) were scraped in 40 mL of sucrose buffer solution with protease inhibitor. Vesicles were prepared as described above. The cells were labeled using DiR at 2 μM for 30 min at 37 °C. Samples were injected intravenously to the mouse, through the tail vein. Approximately 1 × 1011 vesicles were delivered per injection. All animal experiments were repeated three times.
Determining Cisplatin Concentration
Vesicles were generated as described above in the presence of 8.33 mM cisplatin in sucrose buffer solution. To release cisplatin from the vesicles, they were treated with 5 μL (1%) Triton X-100 followed by 500 μL of 70% nitric acid and incubated on a heat block at 60 °C for 2 h. The solution was diluted to 5 mL of 1% nitric acid and analyzed using ICP-OES (Varian Vista Pro).
To determine the amount of leakage into the solution from the vesicles, the supernatant of the vesicle solution was collected after the vesicles were pelleted using ultracentrifugation. The supernatant was then diluted in 5 mL of 1% nitric acid and analyzed using ICP-OES. Separately, vesicles were stored for 1, 2, and 3 days. At each time point, the vesicles were pelleted, and the supernatant was collected and analyzed for cisplatin using ICP-OES. A standard curve was generated using standard concentrations of platinum in 1% nitric acid ranging from 1 ppm to 10 ppb. Ytterbium was used as in the internal standard to compensate for the internal drift of the instrument.
Dynamic Light Scattering
Vesicles were prepared as described above. The vesicle solution was then diluted (1:20) and analyzed using DLS.
Cell Specificity
HEK vesicles onto HEK and A549 cells: 64 million HEK were used to generate vesicles for delivery. Cells were scraped from culture in 20 mL of sucrose buffer containing protease inhibitor. The cell solution was collected in a 50 mL conical tube and pelleted at 2000 rpm at 25 °C for 2 min. The solution was aspirated off such that the final volume was 10 mL. The cell solution was fragmented using N2 cavitation at 300 psi at 4 °C for 5 min. The resulting cell slurry was centrifuged at 4000g for 10 min at 4 °C. The supernatant from centrifugation was transferred to a 25 mL ultracentrifuge tube for 10,000g centrifugation for 20 min at 4 °C. The supernatant from the UCF tube was then transferred to a new 25 mL ultracentrifuge tube and centrifuged at 100,000g for 60 min at 4 °C. The pellet in the UCF tube was washed with 500 μL of sucrose solution, and the residual solution was pipetted out and discarded. One thousand microliters of sucrose buffer solution was added to the UCF tube, and the pellet was resuspended via pipetting. Two microliters of 1 mM DiI was added to the resuspension and left to incubate for 30 min at 37 °C. After this time, the vesicles were purified from the free dye using a PD MidiTrap. Fifty microliters of the purified vesicles was added into each glass bottom dish containing 90,000 HEK or A549 cells plated 24 h prior.
This procedure was repeated for RAW vesicles on RAW and A549 cells using 54 million cells to generate vesicles.
The above protocol was repeated for HCT vesicles on HCT cells and RAW vesicles on HCT cells using 70.4 and 56.8 million cells, respectively, to generate vesicles. Since HCT cells grow at a faster rate than most other cell types we used, we plated 50,000 cells onto glass bottom dishes instead of the traditional 90,000 cells.
Fluorescence Correlation Spectroscopy
Forty million A549 cells were scraped from the culture in 20 mL of sucrose buffer containing protease inhibitor. The cell solution was collected in a 50 mL conical tube and pelleted at 2000 rpm at 25 °C for 2 min. The solution was aspirated off such that the final volume was 10 mL. The cell solution was fragmented using N2 cavitation at 250 psi at 4 °C for 5 min. The resulting cell slurry was centrifuged at 4000g for 10 min at 4 °C. The supernatant from centrifugation was transferred to a 25 mL ultracentrifuge tube for 10,000g centrifugation for 20 min at 4 °C. The supernatant from the UCF tube was then transferred to a new 25 mL ultracentrifuge tube and centrifuged at 100,000g for 60 min at 4 °C. The pellet in the UCF tube was washed with 1 mL of PBS, and the residual solution was pipetted out and discarded. Three hundred microliters of PBS was added to the UCF tube, and the pellet was resuspended via pipetting. DiI (2 μM) was added to the resuspension and left to incubate for 40 min at 37 °C. After this time, the vesicles were purified from the free dye using a 450 nm Corning sterile syringe filter only after the filter was pre-saturated with 150 μL of PBS. The solution was then diluted 1:2, and then 20 μL was placed onto a coverslip mounted on a 60× water objective. A 532 nm laser (45 mW) was used as an excitation source. A picoquant PicoHarp 300 photon counting system was used to time tag photon arrival events.
Acknowledgments
We thank Meagan Combs at the Environmental Research Training Laboratories at UK for assistance with ICP-OES. We also thank the Light Microscopy Core at UK for confocal microscopy experiments. This work was supported by funding from the NIH (DA038817).
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsomega.9b01353.
Bright-field image of cells used for gene delivery (PDF)
Author Contributions
A.A.S. and C.I.R. wrote the manuscript. A.A.S., K.R.N., J.R.M., X.F., F.H.M., and E.B.C. performed the experiments. C.I.R. and J.K. designed the experiments. J.R.M. took all photographs.
The authors declare no competing financial interest.
Supplementary Material
References
- Mout R.; Ray M.; Tay T.; Sasaki K.; Yesilbag Tonga G.; Rotello V. M. General Strategy for Direct Cytosolic Protein DeliveryviaProtein–Nanoparticle Co-engineering. ACS Nano 2017, 11, 6416–6421. 10.1021/acsnano.7b02884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitragotri S.; Burke P. A.; Langer R. Overcoming the challenges in administering biopharmaceuticals: formulation and delivery strategies. Nat. Rev. Drug Discovery 2014, 13, 655–72. 10.1038/nrd4363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu Z.; Biswas A.; Zhao M.; Tang Y. Tailoring nanocarriers for intracellular protein delivery. Chem. Soc. Rev. 2011, 40, 3638–55. 10.1039/c0cs00227e. [DOI] [PubMed] [Google Scholar]
- Moonschi F. H.; Hughes C. B.; Mussman G. M.; Fowlkes J. L.; Richards C. I.; Popescu I. Advances in micro- and nanotechnologies for the GLP-1-based therapy and imaging of pancreatic beta-cells. Acta diabetologica 2018, 55, 405–418. 10.1007/s00592-017-1086-7. [DOI] [PubMed] [Google Scholar]
- Xiong Q.; Lee G. Y.; Ding J.; Li W.; Shi J. Biomedical applications of mRNA nanomedicine. Nano Res. 2018, 11, 5281–5309. 10.1007/s12274-018-2146-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z.; Ling L.; Du Y.; Yao C.; Li X. Reduction responsive liposomes based on paclitaxel-ss-lysophospholipid with high drug loading for intracellular delivery. Int. J. Pharm. 2019, 564, 244. 10.1016/j.ijpharm.2019.04.060. [DOI] [PubMed] [Google Scholar]
- Sercombe L.; Veerati T.; Moheimani F.; Wu S. Y.; Sood A. K.; Hua S. Advances and Challenges of Liposome Assisted Drug Delivery. Front. Pharmacol. 2015, 6, 286. 10.3389/fphar.2015.00286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel S. G.; Sayers E. J.; He L.; Narayan R.; Williams T. L.; Mills E. M.; Allemann R. K.; Luk L. Y. P.; Jones A. T.; Tsai Y. H. Cell-penetrating peptide sequence and modification dependent uptake and subcellular distribution of green florescent protein in different cell lines. Sci. Rep. 2019, 9, 6298. 10.1038/s41598-019-42456-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stewart M. P.; Sharei A.; Ding X.; Sahay G.; Langer R.; Jensen K. F. In vitro and ex vivo strategies for intracellular delivery. Nature 2016, 538, 183–192. 10.1038/nature19764. [DOI] [PubMed] [Google Scholar]
- Allen T. M. Long-circulating (sterically stabilized) liposomes for targeted drug delivery. Trends Pharmacol. Sci. 1994, 15, 215–20. 10.1016/0165-6147(94)90314-X. [DOI] [PubMed] [Google Scholar]
- Ha D.; Yang N.; Nadithe V. Exosomes as therapeutic drug carriers and delivery vehicles across biological membranes: current perspectives and future challenges. Acta Pharm Sin B 2016, 6, 287–96. 10.1016/j.apsb.2016.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akbarzadeh A.; Rezaei-Sadabady R.; Davaran S.; Joo S. W.; Zarghami N.; Hanifehpour Y.; Samiei M.; Kouhi M.; Nejati-Koshki K. Liposome: classification, preparation, and applications. Nanoscale Res. Lett. 2013, 8, 102–102. 10.1186/1556-276X-8-102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Green A. E.; Rose P. G. Pegylated liposomal doxorubicin in ovarian cancer. Int. J. Nanomed. 2006, 1, 229–239. [PMC free article] [PubMed] [Google Scholar]
- Sharma A.; S. Sharma U. Liposomes in drug delivery: Progress and limitations. Int. J. Pharm. 1997, 154, 123–140. 10.3389/fphar.2015.00286. [DOI] [Google Scholar]
- Zhu L.; Oh J. M.; Gangadaran P.; Kalimuthu S.; Baek S. H.; Jeong S. Y.; Lee S. W.; Lee J.; Ahn B. C. Targeting and therapy of glioblastoma in a mouse model using exosomes derived from Natural Killer cells. Front. Pharmacol. 2018, 9, 824. 10.3389/fimmu.2018.00824. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Li S. P.; Lin Z. X.; Jiang X. Y.; Yu X. Y. Exosomal cargo-loading and synthetic exosome-mimics as potential therapeutic tools. Acta Pharmacol. Sin. 2018, 39, 542–551. 10.1038/aps.2017.178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choo Y. W.; Kang M.; Kim H. Y.; Han J.; Kang S.; Lee J. R.; Jeong G. J.; Kwon S. P.; Song S. Y.; Go S.; Jung M.; Hong J.; Kim B. S. M1 Macrophage-Derived Nanovesicles Potentiate the Anticancer Efficacy of Immune Checkpoint Inhibitors. ACS Nano 2018, 12, 8977–8993. 10.1021/acsnano.8b02446. [DOI] [PubMed] [Google Scholar]
- Hon K. W.; Abu N.; Ab Mutalib N.-S.; Jamal R. Exosomes As Potential Biomarkers and Targeted Therapy in Colorectal Cancer: A Mini-Review. Front. Pharmacol. 2017, 8, 583. 10.3389/fphar.2017.00583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhai Y.; Su J.; Ran W.; Zhang P.; Yin Q.; Zhang Z.; Yu H.; Li Y. Preparation and Application of Cell Membrane-Camouflaged Nanoparticles for Cancer Therapy. Theranostics 2017, 7, 2575–2592. 10.7150/thno.20118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao J.; Wang S.; Wang Z. High yield, scalable and remotely drug-loaded neutrophil-derived extracellular vesicles (EVs) for anti-inflammation therapy. Biomaterials 2017, 135, 62–73. 10.1016/j.biomaterials.2017.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao J.; Chu D.; Wang Z. Cell membrane-formed nanovesicles for disease-targeted delivery. J. Controlled Release 2016, 224, 208–216. 10.1016/j.jconrel.2016.01.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeong D.; Jo W.; Yoon J.; Kim J.; Gianchandani S.; Gho Y. S.; Park J. Nanovesicles engineered from ES cells for enhanced cell proliferation. Biomaterials 2014, 35, 9302–10. 10.1016/j.biomaterials.2014.07.047. [DOI] [PubMed] [Google Scholar]
- Simpson R. J. Disruption of cultured cells by nitrogen cavitation. Cold Spring Harbor protocols 2010, 2010, pdb-prot5513. 10.1101/pdb.prot5513. [DOI] [PubMed] [Google Scholar]
- Taylor D. D.; Shah S. Methods of isolating extracellular vesicles impact down-stream analyses of their cargoes. Methods 2015, 87, 3–10. 10.1016/j.ymeth.2015.02.019. [DOI] [PubMed] [Google Scholar]
- Fang X.; Duan Y.; Adkins G. B.; Pan S.; Wang H.; Liu Y.; Zhong W. Highly Efficient Exosome Isolation and Protein Analysis by an Integrated Nanomaterial-Based Platform. Anal. Chem. 2018, 90, 2787–2795. 10.1021/acs.analchem.7b04861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moonschi F. H.; Effinger A. K.; Zhang X.; Martin W. E.; Fox A. M.; Heidary D. K.; DeRouchey J. E.; Richards C. I. Cell-Derived Vesicles for Single-Molecule Imaging of Membrane Proteins. Angew Chem Int Ed Engl 2015, 31, 201408707. 10.1002/ange.201408707. [DOI] [PubMed] [Google Scholar]
- Štimac A.; Tokić M.; Ljubetič A.; Vuletić T.; Šekutor M.; Požar J.; Leko K.; Hanževački M.; Frkanec L.; Frkanec R. Functional self-assembled nanovesicles based on β-cyclodextrin, liposomes and adamantyl guanidines as potential nonviral gene delivery vectors. Org. Biomol. Chem. 2019, 17, 4640. 10.1039/C9OB00488B. [DOI] [PubMed] [Google Scholar]
- Gottlieb R. A.; Adachi S. Nitrogen Cavitation for Cell Disruption to Obtain Mitochondria from Cultured Cells. Apoptosis 2000, 213–221. 10.1016/S0076-6879(00)22022-3. [DOI] [PubMed] [Google Scholar]
- Zhu J.-Y.; Zheng D.-W.; Zhang M.-K.; Yu W.-Y.; Qiu W.-X.; Hu J.-J.; Feng J.; Zhang X.-Z. Preferential Cancer Cell Self-Recognition and Tumor Self-Targeting by Coating Nanoparticles with Homotypic Cancer Cell Membranes. Nano Lett. 2016, 16, 5895–5901. 10.1021/acs.nanolett.6b02786. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.