

Abstract
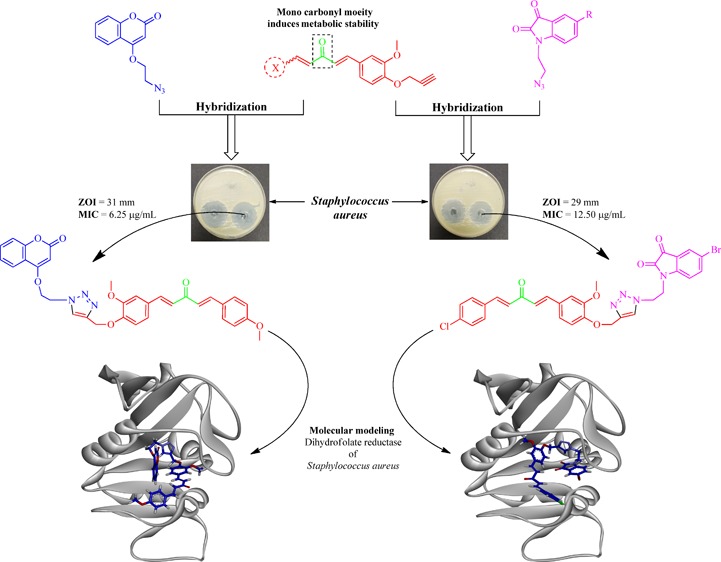
Keeping in view various pharmacological attributes of curcumin, coumarin, and isatin derivatives, triazole-tethered monocarbonyl curcumin–coumarin and curcumin–isatin molecular hybrids have been synthesized and evaluated for their antibacterial potential against Gram-positive (Enterococcus faecalis and Staphylococcus aureus) and Gram-negative (Pseudomonas aeruginosa and Escherichia coli) human pathogenic bacterial strains. Among all hybrid molecules, A-4 and B-38 showed the most potent antibacterial activity with inhibition zones of 29 and 31 mm along with MIC values of 12.50 and 6.25 μg/mL, respectively. Structure–activity relationship that emerged from biological data revealed that the two-carbon alkyl chain between triazole and coumarin/isatin moiety is well tolerable for the activity. Bromo substitution at the fifth position of isatin, para-cholo substitution in the case of curcumin–isatin, and para-methoxy in the case of curcumin–coumarin hybrids on ring A of curcumin are most suitable groups for the antibacterial activity. Various types of binding interactions of A-4 and B-38 within the active site of dihydrofolate reductase (DHFR) of S. aureus are also streamlined by molecular modeling studies, suggesting their capability in completely blocking DHFR.
Introduction
Microbial resistance has prominently emerged as a serious threat to human health within the last few decades and becomes the most difficult and serious challenge to healthcare experts and researchers across the globe. Surveillance data of the World Health Organization (WHO) revealed that approximately 50,000 people across the globe including male, female, and children are dying every day due to microbial infections in both high- and low-income countries.1 A report released by WHO states that 16 million people died in 1990 due to microbial infections; the figure reduced to 15 million in 2010 and is expected to be 13 million in 2050, suggesting that the mortality rate due to microbial infections is declining but very slowly, which may be due to the sudden increase in population as well as microbial resistance.2 Recent efforts in the development of antimicrobial agents were incapable of meeting expectations due to microbial resistance and inadequate antimicrobial activity accompanied with toxicity, which suggests that more rigorous efforts in the search of novel antimicrobial agents are needed.3 In the last few years, various research strategies have been successfully utilized to tackle the multidrug-resistant microbial infection. Numerous studies suggested that drug resistance is more prevalent in solo targeting agents, which is the major reason for the failure of expected drug candidate.4 Drugs targeting more than a single target or multiple sites of a single target exhibited higher potency, minimum resistance, and lesser toxicity as compared to solo targeting agents.5−8 Molecular hybridization technique is dedicated to the development of such multitargeting agents.
In molecular hybridization approach, two different active pharmacophores are combined together with or without the help of a linker to get the desired hybrid.9 In 2006, FDA approved vorinostat, a potent histone deacetylase inhibitor (HDAC inhibitor) for cutaneous T-cell lymphoma treatment. CUDC-907, a hybrid molecule of vorinostat and GDC-0941 (a phosphoinositide 3-kinase (PI3K) inhibitor), is more efficient against cancer in both in vitro and in vivo models without showing any systemic toxicity and resistance. This dual HDAC/PI3K inhibitor has recently passed the phase I trial for advanced solid tumor, lymphoma, or multiple myeloma treatment and entered phase II trials.10 Thus, hybrid molecules can lower the risk of multiple drug resistance along with drug–drug interactions, which could be very beneficial for humanity in confronting microbial resistance.
Curcumin, a polyphenolic compound and major constituent of Curcuma longa L., is well known for its countless biological activities including antioxidant, neuroprotective, anticancer, anti-inflammatory, antiviral, and antimicrobial activities.11,12 Besides its potent biological activities, curcumin suffers from pharmacokinetic issues such as low solubility, poor absorption, and lower bioavailability along with instant metabolism in the human body that limits its clinical use. The α,β-unsaturated β-diketone moiety of curcumin undergoes keto–enol tautomerism, forming a hydrogen bond containing a six-membered ring. The six-membered ring so formed is a potential substrate for the aldoketo reductase enzyme in the human body, resulting in its rapid metabolism. The monocarbonyl architecture of curcumin has been developed by eliminating its sensitive β-diketone moiety (responsible for its pharmacokinetic limitations) via several modifications in its parent structure to elevate its stability as well as bioavailability. Some recently designed monocarbonyl analogs of curcumin (1–3) displayed potent antimicrobial activity with improved solubility and stability13 (Figure 1).
Figure 1.

(a) Rational architecture of monocarbonyl curcumin. (b) Monocarbonyl curcumin analogs emerged as potent antibacterial agents with improved stability and potent antimicrobial activity (1–3).
Isatin is an indole derivative and pharmacologically active nucleus with a wide range of biological applications. It is widely distributed in a number of natural products including fungal metabolites as well as plant and marine natural products especially alkaloids. Numerous reports have been available, describing the antimicrobial potential of isatin-based hybrids including propylene-tethered ciprofloxacin–isatin hybrids (4) and indole–isatin hybrids (5).14,15
Coumarin is well known for its wide range of biological activities.16−20 The promising antimicrobial potential of coumarin-based hybrid molecules such as coumarin–theophylline hybrids (6), coumarin–thiazolyl hybrids (7), coumarin–sulfonamide hybrids (8), and coumarin–imidazole hybrids (9) has been widely reported by numerous researchers18,21−27 (Figure 2).
Figure 2.

Recently reported various hybrid molecules as antimicrobial agents containing isatin (4 and 5) and coumarin (6–9) nuclei.
Triazole is a vital heterocyclic moiety that plays a significant role in medicinal chemistry and has been employed in architecting various biologically active molecules including antimicrobial, antiviral, antioxidant, anticancer, anti-inflammatory, and anticonvulsant.14,15,28−36 This elite moiety has been successfully incorporated in various molecules that exhibit admirable antimicrobial profile.35−40 Due to the excellent profile of this moiety in the antimicrobial area, it has been selected to incorporate into targeted hybrid molecules as a linker to join curcumin–coumarin and curcumin–isatin.
Considering the global health issue of antimicrobial resistance as a major limitation of currently available antimicrobial drugs and emergency need of novel potent antimicrobial agents, the present study targets the synthesis and characterization of triazole-tethered monocarbonyl curcumin–isatin and curcumin–coumarin hybrids by using the click chemistry approach (Figure 3) along with their evaluations against various bacterial strains. The zones of inhibition of all synthesized compounds were determined, and the minimum inhibitory concentration values of potent compounds were calculated. Furthermore, various binding interactions of potent compounds were explored by using molecular modeling studies.
Figure 3.

Design strategy.
Results and Discussion
Synthesis of targeted hybrid molecules was carried out via a series of chemical reactions (Scheme 1), initiating to form vanillin. Vanillin was reacted with acetone in the presence of 40% KOH at 25 °C to form vinyldenacetone (VA), characterized by two doublets at 6.5 and 7.5 ppm with a J value higher than 16 Hz in 1H NMR, designating two doublets for each proton. The obtained VA was then reacted with propargyl bromide in K2CO3 at 25 °C in DMF as a solvent to get propargylated vinyldenacetone (PVA), which was characterized by identifying singlets, one at 2.5 for one proton and another at 4.8 for two protons in 1H NMR. Various substituted aldehydes were reacted with PVA in the presence of 5% NaOH at 25 °C in methanol as a solvent to get various propargylated analogs of monocarbonyl curcumin.
Scheme 1. Synthesis of Triazole-Tethered Monocarbonyl Curcumin–Coumarin and Curcumin–Isatin Hybrids.

Reagents and conditions: (a) Acetone, 40% KOH, 2 h, stir, rt; (b) propargyl bromide, K2CO3, DMF, 2 h, stir, rt; (c) various substituted aromatic aldehydes, 5% NaOH, MeOH, 2 h, stir, rt; (d) dibromoalkane, K2CO3, DMF, 2 h, stir, rt; (e) NaN3, DMF, 1 h, stir, rt; (f) dibromoethane, K2CO3, DMF, 2 h, stir, rt; (g) sodium ascorbate, CuSO4, DMF, 15 to 30 min, rt.
On the other hand, various 1,2-dibromoalkanes were treated with 4-hydroxy coumarin in the presence of K2CO3 at 25 °C in DMF as a solvent to get the desired products (4-(2-bromoalkoxy)-2H-chromen-2-ones), which were further treated with NaN3 at 25 °C in DMF to get 4-(2-azidoalkoxy)-2H-chromen-2-ones.
Simultaneously, differently substituted isatins were subjected to treatment with 1,2-dibromoethane in the presence of K2CO3 at 25 °C in DMF as a solvent. The obtained crude products were purified using column chromatography to get N-bromoethyl isatin, which was further treated with NaN3 at 25 °C in DMF to get N-azidoethyl isatins.
The obtained 4-(2-azidoethoxy)-2H-chromen-2-ones and N-azidoethyl isatins were treated with various propragylated analogs of monocarbonyl curcumin in the presence of the catalytic amount of copper sulfate and reducing agent sodium ascorbate at 25 °C in DMF to yield various triazole-linked monocarbonyl curcumin–coumarin and curcumin–isatin hybrids.
Antibacterial Activity
All the synthesized hybrids were evaluated to check their antibacterial potential against two of both Gram-positive (Enterococcus faecalis and Staphylococcus aureus) and Gram-negative (Pseudomonas aeruginosa and Escherichia coli) human pathogenic bacterial strains. Almost all of the synthesized hybrids were active against the tested bacterial strains (Tables 1 and 2). Among the screened bacterial strains, S. aureus was the most sensitive one, and E. coli was the most resistant strain to the hybrid molecules. Compound A-4 from curcumin–coumarin hybrids and B-38 from curcumin–isatin hybrids emerged as the most potent hybrids with inhibition zones of 29 and 31 mm against S. aureus, respectively (Figure 4). From Tables 1 and 2, an interesting structure–activity relationship has been established for both curcumin–coumarin and curcumin–isatin hybrid molecules. (Figure 5).
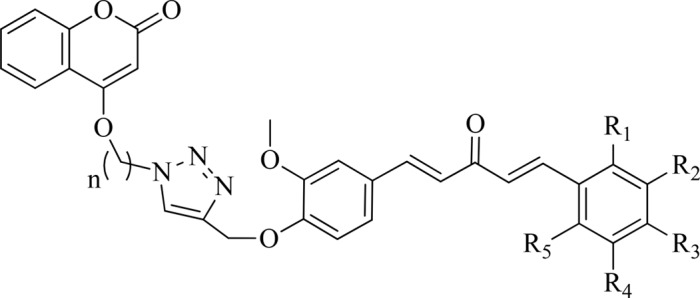
Table 1. Various Substituted Monocarbonyl Curcumin–Coumarin Hybrids with Their Inhibition Zones against Four Bacterial Strains.

Values are means of results obtained from three individual experiments; hyphen denotes no activity.
Table 2. Various Substituted Monocarbonyl Curcumin–Isatin Hybrids with Their Inhibition Zones against Four Bacterial Strains.


Values are means of results obtained from three individual experiments; hyphen denotes no activity.
Figure 4.
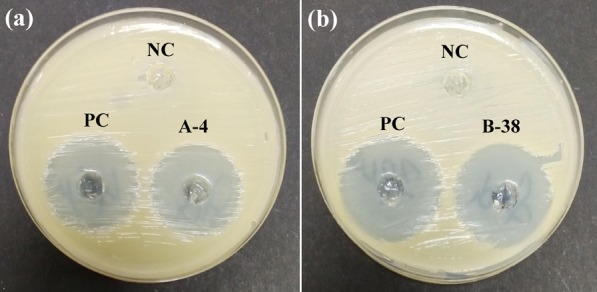
(a) Zone of inhibition (29 mm) shown by hybrid A-4 along with positive control (PC) levofloxacin and negative control (NC) DMSO. (b) Zone of inhibition (31 mm) shown by hybrid B-38 with similar standard and negative control, both against S. aureus.
Figure 5.

Structure–activity relationship.
In the case of curcumin–coumarin hybrids, a methoxy-substituted phenyl ring (ring X) was found to favor antibacterial potential. Inhibition zones observed for A-2, A-3, A-4, A-5, and A-6 against S. aureus were 13, 17, 29, 24, and 22 mm, respectively. This suggests that mono-methoxy substitution at the para position on the phenyl ring as ring X is most suitable followed by meta- and ortho-methoxy substitution. As the number of methoxy substitutions increased around ring X, the activity decreased. An unsubstituted phenyl ring as ring X (A-1) showed moderate activity with an inhibition zone of 12 mm. Naphthyl as ring X (A-7) further diminishes the activity (11 mm), suggesting that polycyclic rings are not suitable for antimicrobial potential. Ring X as heteroaryl ring-like furan (A-8) and thiophene (A-9) was not a suitable companion for antimicrobial potential. Thus, the overall preference order for ring X is as follows: 4-methoxy phenyl > 3-methoxy phenyl > 2-methoxy phenyl > 2,3-dimethoxy phenyl > 2,3,4-trimethoxy phenyl > phenyl > naphthyl > furan = thiophene. Data also revealed that, with an increment of carbon number in the chain joining triazole and coumarin, the antibacterial activity decreased, suggesting a preference order of chain length as n = 2 > 3 > 4 > 5. Thus, the two-carbon chain length joining triazole and coumarin is a most suitable linker.
Taking lead from the structure–activity relationship of curcumin–coumarin hybrids, the two-carbon chain length between triazole and isatin has been employed in curcumin–isatin hybrids. In curcumin–isatin hybrid molecules, the electronegative environment around isatin and ring X of monocarbonyl curcumin significantly affect the antimicrobial activity. Inhibition zones observed for B-38, B-39, B-40, B-41, and B-42 against S. aureus were 31, 28, 21, 12, and 10 mm, respectively, suggesting that para-chloro substitution on the phenyl ring as ring X was most suitable followed by para-bromo and meta-bromo substitutions. With more electronegative substitutions, as in the case of B-41 (para-nitro) and B-42 (meta-nitro), the activity decreased significantly. Inhibition zones observed for methoxy-substituted compounds, that is, B-30, B-31, B-32, B-33, and B-34, against S. aureus were 8, 9, 12, 10, and 9 mm, respectively, suggesting that para-methoxy substitution is somewhat found favorable followed by meta-methoxy, 2,3-dimethoxy, and bulky 2,3,4-trimethoxy substitutions but were much lower as compared to B-38. Therefore, methoxy substitutions at the phenyl ring as ring X are not the substitutions of choice. The unsubstituted phenyl ring as ring X (B-29) showed a much lower inhibition zone of 11 mm that was further lowered when exchanged with a naphthyl ring (B-35). Ring X as heteroaryl ring-like furan (B-36) and thiophene (B-37) as in curcumin-isatin hybrids showed a moderate activity with 18 mm inhibition zones. Thus, the overall preference order for ring X is as follows: 4-chloro phenyl > 4-bromo phenyl > 3-bromo phenyl > furan = thiophene > 4-methoxy phenyl = 4-nitro phenyl > phenyl > 3-nitro phenyl = 3-methoxy phenyl > 2-methoxy phenyl = 2,3-dimethoxy phenyl > 2,3,4-trimethoxy phenyl = naphthyl. The preference order for substituent R on isatin is as follows: Br > Cl > H.
MIC values of the most potent hybrid molecules (A-4 and B-38) against S. aureus have been determined by the broth microdilution method. Both A-4 and B-38 possess decent MIC values (12.50 and 6.25 μg/mL, respectively), which were much close to the levofloxacin (3.12 μg/mL) and better than ciprofloxacin (49 μg/mL) and cefuroxime (98 μg/mL).41
Molecular Modeling Studies
DHFR in S. aureus catalyzes the reduction process of dihydrofolate to tetrahydrofolate through NADPH, which is therefore important in pathways for production of intracellular purines (adenine and guanine). Inhibition of DHFR completely blocks DNA replication in S. aureus, leading to bacterial death. Therefore, DHFR is a validated target for antibacterial drug development especially against S. aureus. Molecular modeling studies were performed to elucidate interactions of the most potent compounds (A-4 and B-38) with the dihydrofolate reductase enzyme of S. aureus.42 For that purpose, the X-ray crystallographic structure of the dihydrofolate reductase (DHFR) enzyme of S. aureus, complexed with 7-aryl-2,4-diaminoquinazoline (PDB entry: 3SRQ; resolution, 1.69 Å), was employed. The accuracy of the docking program was validated by docking cocrystallized ligand 7-aryl-2,4-diaminoquinazoline in its binding site. The program was able to reproduce best fit confirmation of 7-aryl-2,4-diaminoquinazoline with a root mean square deviation (RMSD) of 0.523, indicating the reliability of the docking protocol. After that, A-4 and B-38 were individually docked into the 7-aryl-2,4-diaminoquinazoline binding site, and their best poses with −4.3014 and −6.1337 scores having ΔG values of −39 and −30 kJ/mol, respectively, were selected for discussion (Figure 6).
Figure 6.

(a) Dihydrofolate reductase (DHFR) enzyme of S. aureus complexed with 7-aryl-2,4-diaminoquinazoline (PDB entry: 3SRQ; resolution, 1.69 Å); (b) A-4 docked on the binding site of DHFR; (c) B-38 docked on the binding site of DHFR; (d) 3D view of interactions of A-4 with residues of binding site of DHFR; (e) 3D view of interactions of B-38 with residues of binding site of DHFR; (f) 2D view of interactions of A-4 with residues of binding site of DHFR; (g) 2D view of interactions of B-38 with residues of binding site of DHFR.
The overall binding mode of A-4 and B-38 with residues of the binding site suggests that both compounds fit well in the cavity and are well stabilized by various electrostatic interactions. In the case of A-4, major interactions with DHFR include π–σ, π–alkyl, carbon–hydrogen bond, π–donor hydrogen bond, and conventional hydrogen bond interaction. The coumarin moiety is well positioned in a cavity formed by Ile51, Phe93, Leu55, and Leu29 (hydrophobic residues) and perfectly sandwiched between the aliphatic side chains of Leu21, Leu29, Val32, and Ile51, stabilized by π–σ and π–alkyl interactions. The backbone −NH of Ala8 shows a H-bond interaction with the nitrogen group present in the triazole ring (H-bond acceptor; d = 2.843 Å). The triazole ring is also sandwiched between aliphatic side chains of Val7 and Leu21, stabilized by π–alkyl interactions. The curcumin moiety is surrounded by the cavity formed by Thr47, Lys53, Ser50, Gln20 (polar residues), and Leu21 (hydrophobic residue). Ring B showed π–σ interaction with Leu21 and π–donor hydrogen bond interaction with Ser50. The methoxy group on ring A forms a carbon–hydrogen bond with Lys53. Apart from these interactions’ alkyl carbon, hydrogens around the triazole ring also form carbon–hydrogen bonds with Asp28 (polar residue) and Phe93 (hydrophobic residue).
B-38 also interacts with DHFR by π–σ, π–alkyl, carbon–hydrogen bond, π–donor hydrogen bond, and conventional hydrogen bond interactions just like A-4. The curcumin moiety is well positioned in the cavity formed by Ala8, Val32, Leu29, Leu55, Phe93, Gly94, Val7, Ile51, Phe99 (hydrophobic residues), Asp28, and Ser50 (polar residues). Ring B has shown π–alkyl interactions with Val32 and Ala8 as well as π–donor hydrogen bond interaction with Asp28. The oxygen of the methoxy group at ring B shows unfavorable acceptor–acceptor interaction, while hydrogen shows carbon–hydrogen bond interaction with Asp28. The oxygen of the ether linkage between rings B and C shows H-bond interaction with the −NH backbone of Ala8 (H-bond acceptor; d = 2.875 Å). The triazole ring is well positioned between Leu21, Ile15, Val7, and Ala8, stabilized through π–alkyl interactions. The isatin moiety is sandwiched between Thr47, Ser50, and Leu21, stabilized by π–donor hydrogen bond and π–alkyl interactions. Rings D and E show π–donor hydrogen bond interaction with Thr47 and Ser50, respectively, while both rings interact with Leu21 through π–σ bonds. Hydrogens of alkyl carbons around the triazole ring interact with Ile15, Phe93, and Leu6 through carbon–hydrogen bonds. The overall study seems to confirm that both A-4 and B-38 are sufficiently decorated with small, rigid, and planar groups representing optimum scaffolds that are able to fulfill the pharmacophoric requirements for S. aureus DHFR inhibition.
Conclusions
In the present study, triazole-tethered monocarbonyl curcumin–coumarin and curcumin–isatin molecular hybrids were designed, synthesized, and characterized using 1H and 13C NMR. All synthesized hybrids were assessed for antibacterial potential against two Gram-positive (E. faecalis and S. aureus) and two Gram-negative (P. aeruginosa and E. coli) human pathogenic bacterial strains. Among all hybrids, A-4 and B-38 emerged as the most potent hybrids with potent antibacterial activity against all screened strains, with the highest activity against S. aureus. Both A-4 and B-38 showed inhibition zones of 29 and 31 mm with MIC values of 12.50 and 6.25 μg/mL, respectively. Structure–activity relationship revealed that the two-carbon alkyl chain between triazole and coumarin/isatin moiety is well tolerable. Bromo substitution on the fifth position of isatin, para-chloro substitution in the case of curcumin–isatin, and para-methoxy in the case of curcumin–coumarin hybrids on ring A of curcumin are most suitable for the antibacterial activity. Various binding interactions of A-4 and B-38 with the active site of S. aureus DHFR are also streamlined to justify their potential in blocking DHFR. Thus, the overall study concluded that these hybrid molecules could act as the hit lead for further development of antibacterial agents.
Experimental Section
Materials and Measurements
Chemicals used in the synthesis were procured from CDH, Sigma Aldrich, and Loba, India. Characterization of synthesized compounds was done by spectroscopic techniques such as 1H NMR, 13C NMR, and elemental analysis. Recording of NMR spectra was done by using an Avance III HD 500 MHz Bruker Biospin and JEOL AL 300 MHz. The spectra were recorded using CDCl3 and DMSO-d6 relative to tetramethylsilane (TMS) (0.00 ppm). Chemical shifts in 1H NMR were reported in δ values using an internal standard (TMS) with a number of protons, multiplicities (s, singlet; d, doublet; t, triplet; m, multiplet), and coupling constants (J) in hertz (Hz). Open capillaries were employed for determining the melting points and were uncorrected.
Synthesis and Characterization
Targeted triazole-tethered monocarbonyl curcumin–isatin and curcumin–coumarin hybrid molecules were synthesized by employing previously described standardized procedures43,44 summarized below, and their characterization data are provided in the Supporting Information.
Synthesis of 4-(2-Bromoalkoxy)-2H-chromen-2-one
To a solution of 4-hydroxy coumarin (1 equiv) in DMF, K2CO3 (1.5 equiv) was added and allowed to stir for 15 min. After 15 min, dibromoethane (1 equiv) was added, and the obtained reaction mixture was stirred at room temperature until it was completed (examined by TLC). After completion, the reaction mixture was poured in crushed ice, set aside for a while (until the ice converted into water completely), filtered, and dried. The obtained crude product was purified via column chromatography using hexane/ethylacetate (9.5:0.5) to get 4-(2-bromoethoxy)-2H-chromen-2-ones.43 The same procedure mentioned above was used to synthesize the remaining 4-(2-bromoalkoxy)-2H-chromen-2-ones by taking different types of dibromoalkanes.
Synthesis of 4-(2-Azidoalkoxy)-2H-chromen-2-one (ACE)
To a solution of 4-(2-bromoethoxy)-2H-chromen-2-one (1 equiv) in DMF, sodium azide (1 equiv) was added, and the obtained reaction mixture was stirred at room temperature until it was completed (examined by TLC). After completion, the reaction mixture was poured in crushed ice, set aside for a while (until the ice converted into water completely), filtered, and dried to get pure 4-(2-azidoethoxy)-2H-chromen-2-one (ACE).43
Synthesis of (E)-4-(4-Hydroxy-3-methoxyphenyl)but-3-en-2-one (VA)
To a solution of 4-hydroxy-3-methoxybenzaldehyde (20 g) in acetone (50 mL), 40% KOH was added and stirred at 0 °C for 10 min. After 10 min, the reaction mixture was further stirred at room temperature until it was completed (examined by TLC). After completion, pH of the reaction mixture was changed to 7 using dilute hydrochloric acid. The reaction mixture was then poured on crushed ice, filtered, and dried to get (E)-4-(4-hydroxy-3-methoxyphenyl)but-3-en-2-one.43
Synthesis of (E)-4-(3-Methoxy-4-(prop-2-ynyloxy)phenyl)but-3-en-2-one (PVA)
To a solution of (E)-4-(4-hydroxy-3-methoxyphenyl)but-3-en-2-one (1 equiv) in DMF, K2CO3 (1.5 equiv) was added and allowed to stir for 15 min. After 15 min, propargyl bromide (1equiv) was added, and the reaction mixture was stirred at room temperature until it was completed (examined by TLC). After completion, reaction mixture was poured in crushed ice, set aside for a while (until the ice converted into water completely), filtered, and dried to get (E)-4-(3-methoxy-4-(prop-2-ynyloxy)phenyl)but-3-en-2-one.43
Synthesis of (1E,4E)-1-(3-Methoxy-4-(prop-2-ynyloxy)phenyl)-5-(3,4-dimethoxyphenyl)penta-1,4-dien-3-one (PMCC)
To a solution of (E)-4-(3-methoxy-4-(prop-2-ynyloxy)phenyl)but-3-en-2-one (1 equiv) in methanol, 5% NaOH (0.5 mL) was added and stirred for 15 min. After 15 min, 3,4-dimethoxy benzaldehyde (1 equiv) was added, and the reaction mixture was further allowed to stir at room temperature until it was completed (examined by TLC). After completion, the reaction mixture was poured in crushed ice, set aside for a while (until the ice converted into water completely), filtered, and dried to get (1E,4E)-1-(3-methoxy-4-(prop-2-ynyloxy)phenyl)-5-(3,4-dimethoxy phenyl)penta-1,4-dien-3-one.43 The same procedure mentioned above was used to synthesize the remaining propargylated monocarbonyl curcumin analogs by taking different types of aromatic aldehydes.
Synthesis of Triazole-Tethered Monocarbonyl Curcumin–Coumarin Hybrids
Catalytic quantities of copper sulfate and sodium ascorbate were added to a solution of propargylated monocarbonyl curcumin (1 equiv) and 4-(2-azidoalkoxy)-2H-chromen-2-one (1 equiv). The reaction mixture was stirred at room temperature until it was completed (examined by TLC). After completion, the reaction mixture was poured in crushed ice to eliminate extra copper sulfate and sodium ascorbate, kept aside for some time (until the ice converted into water completely), filtered, and dried to get triazole-tethered monocarbonyl curcumin–coumarin hybrids.43
Synthesis of N-Bromoethyl Isatins
To a solution of isatin (1 equiv) in DMF, K2CO3 (1.5 equiv) was added and allowed to stir for 15 min. After 15 min, dibromoethane (1 equiv) was added, and the reaction mixture was stirred at room temperature until it was completed (examined by TLC). After completion, the reaction mixture was poured in crushed ice, set aside for some time (until the ice converted into the water completely), filtered, and dried. The obtained crude product was purified via column chromatography using hexane/ethylacetate (9.5:0.5) to get N-bromoethyl isatin.44 The same procedure mentioned above was used to synthesize the remaining N-bromoethyl isatins by taking different types of substituted isatins.
Synthesis of N-Azidoethyl Isatins
To a solution of N-bromoethyl isatin (1 equiv) in DMF, sodium azide (1 equiv) was added, and the obtained reaction mixture was allowed to stir at room temperature until it was completed (examined by TLC). After completion, the reaction mixture was poured in crushed ice, kept aside for some time (until the ice converted into water completely), filtered, and dried. The obtained crude product was purified via column chromatography using hexane/ethylacetate (9.5:0.5) to get pure N-azidoethyl isatin.44 The same procedure mentioned above was used to synthesize the remaining N-azidoethyl isatins by taking different types of substituted N-bromoethyl isatins.
Synthesis of Triazole-Tethered Monocarbonyl Curcumin–Isatin Hybrids
Catalytic quantities of copper sulfate and sodium ascorbate were added to a solution of propargylated monocarbonyl curcumin (1 equiv) and N-azidoethyl isatin (1 equiv). The reaction mixture was stirred at room temperature until it was completed (examined by TLC). After completion, the reaction mixture was poured in crushed ice to eliminate extra copper sulfate and sodium ascorbate, set aside for a while (until the ice converted into water completely), filtered, and dried to get triazole-tethered monocarbonyl curcumin–isatin hybrids.44
Antibacterial Activity
In vitro antibacterial activity of synthesized hybrids were evaluated against two Gram-positive (E. faecalis and S. aureus) and two Gram-negative (P. aeruginosa and E. coli) human pathogenic bacterial strains by using agar well diffusion and broth microdilution methods.
Agar Well Diffusion Method
Mueller-Hinton and MacConkey agar plates were employed in the antibacterial screening of 78 synthesized hybrids. Ten milligrams of test hybrids and standard drug (levofloxacin) was dissolved in 10 mL DMSO to get a final solution of 1 mg/mL. One hundred microliters of each hybrid, standard, and negative control (DMSO) was introduced into the wells of agar plates, which were seeded with 100 μL of activated bacterial cultures. Plates were incubated for 24–48 h at 37 °C under aerobic conditions. Clear zones of inhibition around the wells were measured and compared with standard and negative control.45
Broth Microdilution Method
Bacteria grown in Mueller-Hinton and MacConkey agar plates were diluted with the same corresponding broth media. Test hybrids and the standard drug were dissolved in DMSO to get the stock solution of 1 mg/mL, and 2-fold serial dilutions (100, 50, 25, 12.5, 6.25, 3.12, and 1.56 μg/mL) of the stock solution were added to the wells. Then, the wells were inoculated with bacterial suspensions such that the final concentration of the inoculum in wells was 100 CFU/mL. Plates were incubated for 24 h at 37 °C under aerobic conditions. The lowest concentrations of hybrids with no visible turbidity were recorded as MIC values.46
Molecular Modeling Studies
The crystal structure of S. aureus DHFR (PDB entry: 3SRQ; resolution, 1.69 Å) was downloaded from Protein Data Bank. Preparation of the structure was done by using the drug design platform LeadIT. Cocrystalized ligand 7-aryl-2,4-diaminoquinoline was used for defining the binding site with the radius of 6.50 Å.47 Structures of A-4 and B-38 were drawn on ChemDraw Ultra (2013), and their energies were minimized by employing MM2 force field in Chem3D Ultra software.48 Prepared compounds were used as protonated in aqueous solution and docked into a prepared binding site using the FlexX docking module in LeadIT. All FlexX solutions obtained were scored using a consensus scoring function (CScore) and ranked accordingly. The top best pose with the highest score was selected for investigation of interactions, HYDE assessment, and calculation of free energy of binding (ΔG).49−51 3D enzyme–hybrid interactions were visualized using Discovery Studio Visualizer.52
Acknowledgments
The authors are grateful to the University Grants Commission for providing funds under Rajiv Gandhi National Fellowship (RGNF), Council of Scientific and Industrial Research (CSIR), and Women Scientists Scheme-A (WOS-A: DST). The authors are also thankful to Guru Nanak Dev University, Amritsar for providing various facilities to carry out the research work.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsomega.9b01109.
Additional figures illustrating characterization of synthesized compounds by 1H and 13C NMR spectra (PDF)
Author Contributions
# A.S. and J.V.S contributed equally to this work.
The authors declare no competing financial interest.
Supplementary Material
References
- https://www.who.int/antimicrobial-resistance/en/ (retrieved on 4 April 2019)
- Dye C. After 2015: infectious diseases in a new era of health and development. Phil. Trans. R. Soc. B. 2014, 369, 20130426. 10.1098/rstb.2013.0426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marepu N.; Yeturu S.; Pal M. 1,2,3-Triazole fused with pyridine/pyrimidine as new template for anti-microbial agents: Regioselective synthesis and identification of potent N-heteroarenes. Bioorg. Med. Chem. Lett. 2018, 28, 3302–3306. 10.1016/j.bmcl.2018.09.021. [DOI] [PubMed] [Google Scholar]
- El-Gohary N.-S.; Shaaban M.-I. Design, synthesis, anti-microbial, antiquorum-sensing and antitumor evaluation of new series of pyrazolopyridine derivatives. Eur. J. Med. Chem. 2018, 157, 729–742. 10.1016/j.ejmech.2018.08.008. [DOI] [PubMed] [Google Scholar]
- Gao F.; Yang H.; Lu T.; Chen Z.; Ma L.; Xu Z.; Schaffer P.; Lu G. Design, synthesis and anti-mycobacterial activity evaluation of benzofuran-isatin hybrids. Eur. J. Med. Chem. 2018, 159, 277–281. 10.1016/j.ejmech.2018.09.049. [DOI] [PubMed] [Google Scholar]
- Wang J.-Q.; Wang X.; Wang Y.; Tang W.-J.; Shi J.-B.; Liu X.-H. Novel curcumin analogue hybrids: Synthesis and anticancer activity. Eur. J. Med. Chem. 2018, 156, 493–509. 10.1016/j.ejmech.2018.07.013. [DOI] [PubMed] [Google Scholar]
- Maestro A.; Martín-Encinas E.; Alonso C.; de Marigorta E. M.; Rubiales G.; Vicario J.; Palacios F. Synthesis of novel antiproliferative hybrid bis-(3-indolyl)methane phosphonate derivatives. Eur. J. Med. Chem. 2018, 158, 874–883. 10.1016/j.ejmech.2018.09.011. [DOI] [PubMed] [Google Scholar]
- Chopra R.; Chibale K.; Singh K. Pyrimidine-chloroquinoline hybrids: Synthesis and antiplasmodial activity. Eur. J. Med. Chem. 2018, 148, 39–53. 10.1016/j.ejmech.2018.02.021. [DOI] [PubMed] [Google Scholar]
- Parkes A. L.; Yule I. A. Hybrid antibiotics – clinical progress and novel designs. Expert Opin. Drug Discovery 2016, 11, 665–680. 10.1080/17460441.2016.1187597. [DOI] [PubMed] [Google Scholar]
- a Klahn P.; Brönstrup M. Bifunctional antimicrobial conjugates and hybrid antimicrobials. Nat. Prod. Rep. 2017, 34, 832–885. 10.1039/C7NP00006E. [DOI] [PubMed] [Google Scholar]; b Qian C.; Lai C.-J.; Bao R.; Wang D.-G.; Wang J.; Xu G.-X.; Atoyan R.; Qu H.; Yin L.; Samson M.; Zifcak B.; Ma A. W. S.; Rocca S. D.; Borek M.; Zhai H.-X.; Cai X.; Voi M. Cancer network disruption by a single molecule inhibitor targeting both histone deacetylase activity and phosphatidylinositol 3-kinase signaling. Clin. Cancer Res. 2012, 18, 4104–4113. 10.1158/1078-0432.CCR-12-0055. [DOI] [PubMed] [Google Scholar]; c Wu C.-P.; Hsieh Y.-J.; Hsiao S.-H.; Su C.-Y.; Li Y.-Q.; Huang Y.-H.; Huang C.-W.; Hsieh C.-H.; Yu J.-S.; Wu Y.-S. Human ATP-binding cassette transporter ABCG2 confers resistance to CUDC-907, a dual inhibitor of histone deacetylase and phosphatidylinositol 3-kinase. Mol. Pharmaceutics 2016, 13, 784–794. 10.1021/acs.molpharmaceut.5b00687. [DOI] [PubMed] [Google Scholar]; d Lai C.J.; Bao R.; Tao X.; Wang J.; Atoyan R.; Qu H.; Wang D. G.; Yin L.; Samson M.; Forrester J.; Zifcak B.; Xu G. X.; DellaRocca S.; Zhai H. X.; Cai X.; Munger W. E.; Keegan M.; Pepicelli C. V.; Qian C. CUDC-101, a multitargeted inhibitor of histone deacetylase, epidermal growth factor receptor, and human epidermal growth factor receptor 2, exerts potent anticancer activity. Cancer Res. 2010, 70, 3647–3656. 10.1158/0008-5472.CAN-09-3360. [DOI] [PubMed] [Google Scholar]; e Shimizu T.; LoRusso P. M.; Papadopoulos K. P.; Patnaik A.; Beeram M.; Smith L. S.; Rasco D. W.; Mays T. A.; Chambers G.; Ma A.; Wang J.; Laliberte R.; Voi M.; Tolcher A. W. Phase I first-in-human study of CUDC-101, a multitargeted inhibitor of HDACs, EGFR, and HER2 in patients with advanced solid tumors. Clin. Cancer Res. 2014, 20, 5032–5040. 10.1158/1078-0432.CCR-14-0570. [DOI] [PubMed] [Google Scholar]; f Wang J.; Pursell N. W.; Samson M. E. S.; Atoyan R.; Ma A. W.; Selmi A.; Xu W.; Cai X.; Voi M.; Savagner P.; Lai C.-J. Potential advantages of CUDC-101, a multitargeted HDAC, EGFR, and HER2 inhibitor, in treating drug resistance and preventing cancer cell migration and invasion. Mol. Cancer Ther. 2013, 12, 925–936. 10.1158/1535-7163.MCT-12-1045. [DOI] [PubMed] [Google Scholar]; g Seo S.-Y. Multi-targeted hybrids based on HDAC inhibitors for anti-cancer drug discovery. Arch. Pharmacal Res. 2012, 35, 197–200. 10.1007/s12272-012-0221-9. [DOI] [PubMed] [Google Scholar]; h Cai X.; Zhai H. X.; Wang J.; Forrester J.; Qu H.; Yin L.; Lai C. J.; Bao R.; Qian C. Discovery of 7-(4-(3-ethynylphenylamino)-7-methoxyquinazolin-6-yloxy)-N-hydroxyheptanamide (CUDC-101) as a potent multi-acting HDAC, EGFR, and HER2 inhibitor for the treatment of cancer. J. Med. Chem. 2010, 53, 2000–2009. 10.1021/jm901453q. [DOI] [PubMed] [Google Scholar]
- Amalraj A.; Pius A.; Gopi S.; Gopi S. Biological activities of curcuminoids, other biomolecules from turmeric and their derivatives – A review. J. Tradit. Complement. Med. 2017, 7, 205–233. 10.1016/j.jtcme.2016.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu S.; Maiti P.; Ma Q.; Zuo X.; Jones M. R.; Cole G. M.; Frautschy S. A. Clinical development of curcumin in neurodegenerative disease. Expert. Rev. Neurother. 2015, 15, 629–637. 10.1586/14737175.2015.1044981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Kumar P.; Kandi S. K.; Manohar S.; Mukhopadhyay K.; Rawat D. S. Monocarbonyl curcuminoids with improved stability as Antibacterial agents against Staphylococcus aureus and their mechanistic studies. ACS Omega 2019, 4, 675–687. 10.1021/acsomega.8b02625. [DOI] [Google Scholar]; b Liang G.; Yang S.; Jiang L.; Zhao Y.; Shao L.; Xiao J.; Ye F.; Li Y.; Li X. Synthesis and anti-bacterial properties of mono-carbonyl analogues of curcumin. Chem. Pharm. Bull. 2008, 56, 162–167. 10.1248/cpb.56.162. [DOI] [PubMed] [Google Scholar]; c da Silva C. C.; Pacheco B. S.; das Neves R. N.; Dié Alves M. S.; Sena-Lopes A.; Moura S.; Borsuk S.; de Pereira C. M. P. Antiparasitic activity of synthetic curcumin monocarbonyl analogues against Trichomonas vaginalis. Biomed. Pharmacother. 2019, 111, 367–377. 10.1016/j.biopha.2018.12.058. [DOI] [PubMed] [Google Scholar]; d Baldwin P. R.; Reeves A. Z.; Powell K. R.; Napier R. J.; Swimm A. I.; Sun A.; Giesler K.; Bommarius B.; Shinnick T. M.; Snyder J. P.; Liotta D. C.; Kalman D. Monocarbonyl analogs of curcumin inhibit growth of antibiotic sensitive and resistant strains of Mycobacterium tuberculosis. Eur. J. Med. Chem. 2015, 92, 693–699. 10.1016/j.ejmech.2015.01.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang R.; Yin X.; Zhang Y.; Yan W. Design, synthesis and antimicrobial evaluation of propylene-tethered ciprofloxacin-isatin hybrids. Eur. J. Med. Chem. 2018, 156, 580–586. 10.1016/j.ejmech.2018.07.025. [DOI] [PubMed] [Google Scholar]
- Almutairi M. S.; Zakaria A. S.; Ignasius P. P.; Al-Wabli R. I.; Joe I. H.; Attia M. I. Synthesis, spectroscopic investigations, DFT studies, molecular docking and antimicrobial potential of certain new indole-isatin molecular hybrids: Experimental and theoretical approaches. J. Mol. Struct. 2018, 1153, 333–345. 10.1016/j.molstruc.2017.10.025. [DOI] [Google Scholar]
- Bouckaert C.; Serra S.; Rondelet G.; Dolušić E.; Wouters J.; Dogné J.-M.; Frédérick R.; Pochet L. Synthesis, evaluation and structure-activity relationship of new 3-carboxamide coumarins as FXIIa inhibitors. Eur. J. Med. Chem. 2016, 110, 181–194. 10.1016/j.ejmech.2016.01.023. [DOI] [PubMed] [Google Scholar]
- Shen F.-Q.; Wang Z.-C.; Wu S.-Y.; Ren S.-Z.; Man R.-J.; Wang B.-Z.; Zhu H.-L. Synthesis of novel hybrids of pyrazole and coumarin as dual inhibitors of COX-2 and 5-LOX. Bioorg. Med. Chem. Lett. 2017, 27, 3653–3660. 10.1016/j.bmcl.2017.07.020. [DOI] [PubMed] [Google Scholar]
- Chavan R. R.; Hosamani K. M.; Kulkarni B. D.; Joshi S. D. Molecular docking studies and facile synthesis of most potent biologically active N-tert-butyl-4-(4-substituted phenyl)-2-((substituted-2-oxo-2H-chromen-4-yl)methylthio)-6-oxo-1,6-dihydropyrimidine-5-carboxami de hybrids: An approach for microwave-assisted syntheses and biological evaluation. Bioorg. Chem. 2018, 78, 185–194. 10.1016/j.bioorg.2018.03.007. [DOI] [PubMed] [Google Scholar]
- Pérez-Cruz K.; Moncada-Basualto M.; Morales-Valenzuela J.; Barriga-González G.; Navarrete-Encina P.; Núñez-Vergara L.; Squella J. A.; Olea-Azar C. Synthesis and antioxidant study of new polyphenolic hybrid-coumarins. Arabian J. Chem. 2018, 11, 525–537. 10.1016/j.arabjc.2017.05.007. [DOI] [Google Scholar]
- Ashraf R.; Hamidullah; Hasanain M.; Pandey P.; Maheshwari M.; Singh L. R.; Saddiqui M. Q.; Konwar R.; Sashidhara K. V.; Sarkar J. Coumarin-chalcone hybrid instigates DNA damage by minor groove binding and stabilizes p53 through post translational modifications. Sci. Rep. 2017, 7, 45287. 10.1038/srep45287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mangasuli S. N.; Hosamani K. M.; Devarajegowda H. C.; Kurjogi M. M.; Joshi S. D. Synthesis of coumarin-theophylline hybrids as a new class of anti-tubercular and anti-microbial agents. Eur. J. Med. Chem. 2018, 146, 747–756. 10.1016/j.ejmech.2018.01.025. [DOI] [PubMed] [Google Scholar]
- Mangasuli S. N.; Hosamani K. M.; Satapute P.; Joshi S. D. Synthesis, molecular docking studies and biological evaluation of potent coumarin–carbonodithioate hybrids via microwave irradiation. Chem. Data Collect. 2018, 15-16, 115–125. 10.1016/j.cdc.2018.04.001. [DOI] [Google Scholar]
- Singh L. R.; Avula S. R.; Raj S.; Srivastava A.; Palnati G. R.; Tripathi C. K. M.; Pasupuleti M.; Sashidhara K. V. Coumarin-benzimidazole hybrids as a potent antimicrobial agent: synthesis and biological elevation. J. Antibiot. 2017, 70, 954–961. 10.1038/ja.2017.70. [DOI] [PubMed] [Google Scholar]
- Hu Y.; Shen Y.; Wu X.; Tu X.; Wang G.-X. Synthesis and biological evaluation of coumarin derivatives containing imidazole skeleton as potential antibacterial agents. Eur. J. Med. Chem. 2018, 143, 958–969. 10.1016/j.ejmech.2017.11.100. [DOI] [PubMed] [Google Scholar]
- Osman H.; Yusufzai S. K.; Khan M. S.; Razik B. M. A.; Sulaiman O.; Mohamad S.; Gansau J. A.; Ezzat M. O.; Parumasivam T.; Hassan M. Z. New thiazolyl-coumarin hybrids: Design, synthesis, characterization, X-ray crystal structure, antibacterial and antiviral evaluation. J. Mol. Struct. 2018, 1166, 147–154. 10.1016/j.molstruc.2018.04.031. [DOI] [Google Scholar]
- Basanagouda M.; Shivashankar K.; Kulkarni M. V.; Rasal V. R.; Patel H.; Mutha S. S.; Mohite A. A. Synthesis and antimicrobial studies on novel sulfonamides containing 4-azidomethyl coumarin. Eur. J. Med. Chem. 2010, 45, 1151–1157. 10.1016/j.ejmech.2009.12.022. [DOI] [PubMed] [Google Scholar]
- Vukovic N.; Sukdolak S.; Solujic S.; Milosevic T. Synthesis and Antimicrobial Evaluation of Some Novel 2-Aminothiazole Derivatives of 4-Hydroxy-chromene-2-one. Arch. Pharm. 2008, 341, 491–496. 10.1002/ardp.200700215. [DOI] [PubMed] [Google Scholar]
- Kraljević T. G.; Harej A.; Sedić M.; Pavelić S.K.; Stepanić V.; Drenjanečević D.; Talapko J.; Raić-Malić S. Synthesis, in vitro anticancer and antibacterial activities and in silico studies of new 4-substituted 1,2,3-triazole-coumarin hybrids. Eur. J. Med. Chem. 2016, 124, 794–808. 10.1016/j.ejmech.2016.08.062. [DOI] [PubMed] [Google Scholar]
- Riveiro M. E.; Moglioni A.; Vazquez R.; Gomez N.; Facorro G.; Piehl L.; de Celis E. R.; Shayo C.; Davio C. Structural insights into hydroxycoumarin-induced apoptosis in U-937 cells. Bioorg. Med. Chem. 2008, 16, 2665–2675. 10.1016/j.bmc.2007.11.038. [DOI] [PubMed] [Google Scholar]
- Ostrov D. A.; Prada J. A. H.; Corsino P. E.; Finton K. A.; Le N.; Rowe T. C. Discovery of novel DNA gyrase inhibitors by high-throughput virtual screening. Antimicrob. Agents Chemother. 2007, 51, 3688–3698. 10.1128/AAC.00392-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gali R.; Banothu J.; Gondru R.; Bavantula R.; Velivela Y.; Crooks P.A. One-pot multicomponent synthesis of indole incorporated thiazolylcoumarins and their antibacterial, anticancer and DNA cleavage studies. Bioorg. Med. Chem. Lett. 2015, 25, 106–112. 10.1016/j.bmcl.2014.10.100. [DOI] [PubMed] [Google Scholar]
- Arshad A.; Osman H.; Bagley M. C.; Lam C. K.; Mohamad S.; Zahariluddin A. S. M. Synthesis and antimicrobial properties of some new thiazolyl coumarin derivatives. Eur. J. Med. Chem. 2011, 46, 3788–3794. 10.1016/j.ejmech.2011.05.044. [DOI] [PubMed] [Google Scholar]
- Maria J. M.; Saleta V.-R.; Lourdes S.; Eugenio U.; Cristina F.-E.; Ysabel S.; Angeles M.-C. Looking for new targets: simple coumarins as antibacterial agents. Med. Chem. 2012, 8, 1140–1145. 10.2174/157340612804075205. [DOI] [PubMed] [Google Scholar]
- Shi Y.; Zhou C.-H. Synthesis and evaluation of a class of new coumarin triazole derivatives as potential antimicrobial agents. Bioorg. Med. Chem. Lett. 2011, 21, 956–960. 10.1016/j.bmcl.2010.12.059. [DOI] [PubMed] [Google Scholar]
- Yadav P.; Lal K.; Kumar L.; Kumar A.; Kumar A.; Paul A. K.; Kumar R. Synthesis, crystal structure and antimicrobial potential of some fluorinated chalcone-1,2,3-triazole conjugates. Eur. J. Med. Chem. 2018, 155, 263–274. 10.1016/j.ejmech.2018.05.055. [DOI] [PubMed] [Google Scholar]
- Ashok D.; Gundu S.; Aamate V. K.; Devulapally M. G.; Bathini R.; Manga V. Dimers of coumarin-1,2,3-triazole hybrids bearing alkyl spacer: Design, microwave-assisted synthesis, molecular docking and evaluation as antimycobacterial and antimicrobial agents. J. Mol. Struct. 2018, 1157, 312–321. 10.1016/j.molstruc.2017.12.080. [DOI] [Google Scholar]
- Lal K.; Yadav P.; Kumar A.; Kumar A.; Paul A. K. Design, synthesis, characterization, antimicrobial evaluation and molecular modeling studies of some dehydroacetic acid-chalcone-1,2,3-triazole hybrids. Bioorg. Chem. 2018, 77, 236–244. 10.1016/j.bioorg.2018.01.016. [DOI] [PubMed] [Google Scholar]
- Kant R.; Kumar D.; Agarwal D.; Gupta R. D.; Tilak R.; Awasthi S. K.; Agarwal A. Synthesis of newer 1,2,3-triazole linked chalcone and flavone hybrid compounds and evaluation of their antimicrobial and cytotoxic activities. Eur. J. Med. Chem. 2016, 113, 34–49. 10.1016/j.ejmech.2016.02.041. [DOI] [PubMed] [Google Scholar]
- Ruddrraju R. R.; Murugulla A. C.; Kotla R.; Tirumalasetty M. C. B.; Wudayagiri R.; Donthabakthuni S.; Maroju R.; Baburao K.; Parasa L. S. Design, synthesis, anticancer, antimicrobial activities and molecular docking studies of theophylline containing acetylenes and theophylline containing 1,2,3-triazoles with variant nucleoside derivatives. Eur. J. Med. Chem. 2016, 123, 379–396. 10.1016/j.ejmech.2016.07.024. [DOI] [PubMed] [Google Scholar]
- a Kant R.; Singh V. P.; Nath G.; Awasthi S. K.; Agarwal A. Design, synthesis and biological evaluation of ciprofloxacin tethered bis-1,2,3-triazole conjugates as potent antibacterial agents. Eur. J. Med. Chem. 2016, 124, 218–228. 10.1016/j.ejmech.2016.08.031. [DOI] [PubMed] [Google Scholar]; b Singh H.; Singh J. V.; Gupta M. K.; Saxena A. K.; Sharma S.; Nepali K.; Bedi P. M. S. Triazole tethered isatin-coumarin based molecular hybrids as novel antitubulin agents: Design, synthesis, biological investigation and docking studies. Bioorg. Med. Chem. Lett. 2017, 27, 3974–3979. 10.1016/j.bmcl.2017.07.069. [DOI] [PubMed] [Google Scholar]; c Singh J.; Sharma S.; Saxena A. K.; Nepali K.; Bedi P. M. S. Synthesis of 1,2,3-triazole tethered bifunctional hybrids by click chemistry and their cytotoxic studies. Med. Chem. Res. 2013, 22, 3160–3169. 10.1007/s00044-012-0312-7. [DOI] [Google Scholar]; d Sharma M.; Sharma S.; Buddhiraja A.; Saxena A. K.; Nepali K.; Bedi P. M. S. Synthesis and cytotoxicity studies of 3,5-diaryl N-acetyl pyrazoline-isatin hybrids. Med. Chem. Res. 2014, 23, 4337–4344. 10.1007/s00044-014-1001-5. [DOI] [Google Scholar]
- Trotsko N.; Kosikowska U.; Paneth A.; Plech T.; Malm A.; Wujec M. Synthesis and antibacterial activity of new thiazolidine-2,4-dione-based chlorophenylthiosemicarbazone hybrids. Molecules 2018, 23, 1023. 10.3390/molecules23051023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayashi M.; Kinjo T.; Koseki Y.; Bourne C. R.; Barrow W. W.; Aoki S. Identification of novel potential antibiotics against Staphylococcus using structure-based drug screening targeting dihydrofolate reductase. J. Chem. Inf. Model. 2014, 54, 1242–1253. 10.1021/ci400686d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh H.; Kumar M.; Nepali K.; Gupta M. K.; Saxena A. K.; Sharma S.; Bedi P. M. S. Triazole tethered C5-curcuminoid-coumarin based molecular hybrids as novel antitubulin agents: Design, synthesis, biological investigation and docking studies. Eur. J. Med. Chem. 2016, 116, 102–115. 10.1016/j.ejmech.2016.03.050. [DOI] [PubMed] [Google Scholar]
- Sharma S.; Gupta M. K.; Saxena A. K.; Bedi P. M. S. Triazole linked mono carbonyl curcumin-isatin bifunctional hybrids as novel anti tubulin agents: Design, synthesis, biological evaluation and molecular modeling studies. Bioorg. Med. Chem. 2015, 23, 7165–7180. 10.1016/j.bmc.2015.10.013. [DOI] [PubMed] [Google Scholar]
- Suhas R.; Chandrashekar S.; Gowda D. C. Synthesis of elastin based peptides conjugated to benzisoxazole as a new class of potent antimicrobials-A novel approach to enhance biocompatibility. Eur. J. Med. Chem. 2011, 46, 704–711. 10.1016/j.ejmech.2010.12.005. [DOI] [PubMed] [Google Scholar]
- Jose G.; Kumara T. H. S.; Sowmya H. B. V.; Sriram D.; Row T. N. G.; Hosamani A. A.; More S. S.; Janardhan B.; Harish B. G.; Telkar S.; Ravikumar Y. S. Synthesis, molecular docking, antimycobacterial and antimicrobial evaluation of new pyrrolo[3,2-c]pyridine Mannich bases. Eur. J. Med. Chem. 2017, 131, 275–288. 10.1016/j.ejmech.2017.03.015. [DOI] [PubMed] [Google Scholar]
- Li X.; Hilgers M.; Cunningham M.; Chen Z.; Trzoss M.; Zhang J.; Kohnen L.; Lam T.; Creighton C.; Kedar G. C.; Nelson K.; Kwan B.; Stidham M.; Brown-Driver V.; Shaw K. J.; Finn J. Structure-based design of new DHFR-based antibacterial agents: 7-aryl-2,4-diaminoquinazolines. Bioorg. Med. Chem. Lett. 2011, 21, 5171–5176. 10.1016/j.bmcl.2011.07.059. [DOI] [PubMed] [Google Scholar]
- ChemDraw Ultra 6.0 and Chem3D Ultra; Cambridge Soft Corporation: Cambridge, USA.
- Tonelli M.; Naesens L.; Gazzarrini S.; Santucci M.; Cichero E.; Tasso B.; Moroni A.; Costi M. P.; Loddo R. Host dihydrofolate reductase (DHFR)-directed cycloguanil analogues endowed with activity against influenza virus and respiratory syncytial virus. Eur. J. Med. Chem. 2017, 135, 467–478. 10.1016/j.ejmech.2017.04.070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emami S.; Shojapour S.; Faramarzi M. A.; Samadi N.; Irannejad H. Synthesis, in vitro antifungal activity and in silico study of 3-(1,2,4-triazol-1-yl)flavanones. Eur. J. Med. Chem. 2013, 66, 480–488. 10.1016/j.ejmech.2013.06.008. [DOI] [PubMed] [Google Scholar]
- LeadIT version 2.3.2; BioSolveIT GmbH: Sankt Augustin, Germany, 2017.
- Dassault Systemes BIOVIA , Discovery Studio Modeling Environment; Release 2017, San Diego, Dassault Systemes: 2016.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.