

Abstract
Symphyocladia latiuscula (Harvey) Yamada is a red alga with a myriad of bromophenols accompanied by a diverse array of biological activities. The main purpose of the present study was to characterize the anti-Alzheimer’s disease activity of bromophenols from S. latiuscula via inhibition of cholinesterases (AChE and BChE), β-site amyloid precursor protein cleaving enzyme 1 (BACE1), and glycogen synthase kinase-3β (GSK-3β). The results of enzyme inhibition assays demonstrated 2,3,6-tribromo-4,5-dihydroxybenzyl alcohol (1), 2,3,6-tribromo-4,5-dihydroxybenzyl methyl ether (2), and bis-(2,3,6-tribromo-4,5-dihydroxybenzyl) ether (3) as potent inhibitors of aforementioned enzymes. Among the tested bromophenols, 3 showed multifold higher inhibition of all of the tested enzymes. Enzyme kinetics revealed different modes of inhibition, and in silico molecular docking simulation demonstrated the importance of the 7–OH group and bromine number for H-bond and halogen-bond interactions, respectively. Similarly, 1–3 at 20 μM concentration showed more than 50% inhibition of self-induced Aβ25–35 aggregation. These results suggest that bromophenols from S. latiuscula, especially highly brominated (3), may represent a novel class of anti-Alzheimer’s disease drugs.
Introduction
Neurodegenerative diseases, such as Alzheimer’s disease (AD) and Parkinson’s disease (PD), are major causes of death worldwide and are characterized by a progressive loss of specific neuronal cell populations due to the accumulation of aggregated proteins within neurons. AD represents a degenerative brain disease that is characterized by a decline in cognitive function, memory, and understanding. Various structural and functional damages in specific regions of the brain occur in AD, leading to a decline in neural connectivity within those regions.1 Of the changes in the brain that are associated with AD, the two main changes are β-amyloid plaques, which are protein fragments that accumulate outside neurons and contribute to cell death by interfering with neuron-to-neuron communication at synapses, and tau tangles, clusters of abnormal tau proteins inside neurons that block the transport of nutrients and other essential molecules inside neurons.2 Researchers suggest that the brain change that occurs in AD may begin approximately 20 years before the appearance of symptoms.3−5 No pharmacological treatments at present stop or slow the progression of nerve damage that leads to AD, and the U.S. FDA-approved drugs (donepezil, galantamine, memantine, rivastigmine, and memantine combined with donepezil and tacrine) only temporarily improve symptoms by increasing the amount of neurotransmitters in the brain. Similarly, as of 2012, out of 244 drugs tested, only memantine passed the clinical trial and received FDA approval. Therefore, the discovery of anti-AD candidates is still challenging.
The marine biosphere is the richest source of structurally diverse and unique compounds and is known as a treasure house of natural bioactive secondary metabolites. In recent years, red and brown algae have been shown to contain diverse groups of secondary metabolites (especially bromophenols and phlorotannins) with high therapeutic potentials. Natural brominated compounds have been reported to exhibit a variety of biological activities including antibacterial, antidiabetic, antifungal, antiviral, antioxidant, antitumor, anti-inflammatory, and enzymatic activity through protein kinase and acetylcholinesterase inhibition.6−10 There have been limited reports of natural bromophenols showing anti-AD disease activity. So far, it has been proven difficult to identify a selective, safe, and effective new drug from marine-derived bromophenols. Due to the limited amount of bromophenols in marine algae, immediate in vivo investigations have been hindered. However, reports on semisynthetic and synthetic brominated compounds have been increasing.11,12 The bioactivity of bromophenols from marine sources, their structure modification via synthetic and semisynthetic routes, and optimization of pharmacokinetic and pharmacodynamics parameters to discover leads for drug development have become crucial subjects of investigation. With the aim to discover anti-AD disease treatment candidates from marine sources, we conducted the present study on a red alga, Symphyocladia latiuscula (Harvey) Yamada.
S. latiuscula (Harvey) Yamada is a marine red algal species with a high content of bromophenols,13 and is widely distributed in Korea, Japan, and northern China. As constituents with a highly conserved biosynthetic pathway, bromophenols from S. latiuscula typically contain at least one 2,3,6-tribromo-4,5-dihydroxybenzyl moiety linked with different groups (aconitic acids, diketopiperazines, glutamines, pyrrolidin-2-ones, sulfoxides, sulfones, sulfates, and ureas) and have been reported to exhibit aldose reductase inhibitory,14 antibacterial,15 anticancer,16 antifungal,17 antiviral,18 antidiabetic,19 antityrosinase,20 and free radical scavenging activities.21 The presence of a number of electron-donating hydroxyl groups in the 2,3,6-tribromo-4,5-dihydroxybenzyl moiety supports the pronounced antioxidant effect of bromophenols from S. latiuscula.22,23 We previously isolated three bromophenol derivatives from S. latiuscula and reported their antidiabetic19 and antityrosinase20 activities. Recently, S. latiuscula-derived polyphenols showed a neuroprotective effect by ameliorating streptozotocin-induced diabetic peripheral neuropathy in rats.24 To the best of our knowledge, there are no reports of anti-AD activity of bromophenols from S. latiuscula. Therefore, we performed the first investigation of the anti-AD activity of three 2,3,6-tribromo-4,5-dihydroxybenzyl derivatives from S. latiuscula via inhibition of cholinesterases (ChEs), β-site amyloid precursor protein (APP) cleaving enzyme 1 (BACE1), and glycogen synthase kinase-3β (GSK-3β) and explored the inhibition mechanism by enzyme kinetics and molecular docking simulation. In addition, their effect on self-induced Aβ25–35 aggregation is also evaluated here.
Results
Inhibition of Multiple Enzyme Targets by a MeOH Extract of S. latiuscula and Isolated Bromophenols
The anti-Alzheimer’s disease activities of a methanol extract of S. latiuscula and three isolated 2,3,6-tribromo-4,5-dihydroxybenzyl derivatives (Figure 1) were evaluated via measuring AChE, BChE, BACE1, and GSK-3β inhibition. Figure 2 shows the concentration-dependent inhibition of enzymes by the MeOH extract and reference compounds. Table 1 shows the 50% inhibitory concentrations of the extract, test compounds, and reference compounds. As shown in Figure 2, the MeOH extract inhibited BChE and BACE1 activity by comparable magnitudes, which were greater than its inhibition of AChE and GSK-3β.
Figure 1.

Structures of bromophenols isolated from the EtOAc fraction of S. latiuscula.
Figure 2.

Concentration-dependent enzyme inhibition by the MeOH extract of S. latiuscula and reference compounds.
Table 1. Anti-Alzheimer’s Disease Activity of Bromophenols from S. latiusculaa.
| 50% enzyme inhibition concentration (IC50, μM) (mean ± SD)b |
||||
|---|---|---|---|---|
| compounds | AChE (n = 3) | BChE (n = 3) | BACE1 (n = 3) | GSK-3β (n = 3) |
| MeOH extractd | 195.71 ± 5.07 | 103.16 ± 3.51 | 109.59 ± 3.69 | 219.05 ± 6.21 |
| 2,3,6-tribromo-4,5-dihydroxybenzyl alcohol | 7.31 ± 0.25f | 8.95 ± 2.18g | 5.16 ± 0.60f | 229.42 ± 12.05e |
| 2,3,6-tribromo-4,5-dihydroxybenzyl methyl ether | 9.61 ± 0.35e | 14.41 ± 0.27f | 4.79 ± 0.82g | 140.01 ± 15.08f |
| bis-(2,3,6-tribromo-4,5-dihydroxybenzyl) ether | 2.66 ± 0.24g | 4.03 ± 0.15h | 2.32 ± 0.10h | 56.46 ± 2.48g |
| berberinec | 1.17 ± 0.09h | 26.15 ± 0.27e | ||
| quercetinc | 25.21 ± 3.12e | |||
| luteolinc | 5.42 ± 0.19h | |||
| SB-415286c | 0.11 ± 0.01i | |||
Note: Means with different superscripts (e−i) within a column are significantly different with Duncan’s test at p < 0.05.
The 50% inhibitory concentration (IC50) values were calculated from a log dose inhibition curve and expressed as mean ± SD of triplicate experiments.
Used as reference controls (SB-415286; 3-[(3-chloro-4-hydroxyphenyl)-amino]-4-(2-nitrophenyl)-1H-pyrrol-2,5-dione).
IC50 values are expressed in μg/mL.
All of the test compounds, 2,3,6-tribromo-4,5-dihydroxybenzyl alcohol (1), 2,3,6-tribromo-4,5-dihydroxybenzyl methyl ether (2), and bis-(2,3,6-tribromo-4,5-dihydroxybenzyl) ether (3) exhibited significant (p < 0.05) inhibition on ChE and BACE1 activity (Table 1). Among them, 3, with an IC50 value of 2.66 ± 0.24 μM, was the most potent AChE inhibitor, followed by 1 (IC50; 7.31 ± 0.25 μM) and 2 (IC50; 9.61 ± 0.35 μM). To validate the assay result, berberine was used as a reference compound, which had an IC50 value of 1.17 ± 0.09 μM. In BChE enzyme inhibition, all test compounds showed superior activity to berberine (IC50; 26.15 ± 0.27 μM). Again, with the lowest IC50 value of 4.03 ± 0.15 μM, 3 dominated 1 (IC50; 8.95 ± 2.18μM) and 2 (IC50; 14.41 ± 0.27 μM) in BChE inhibition. Interestingly, the BACE1 inhibition potential of tested bromophenols was 5–10-fold higher than the reference drug, quercetin (IC50; 25.21 ± 3.12 μM). Unlike AChE and BChE inhibition, the BACE1 inhibitory potential of 2 was greater than that of 1, but 3 dominated the others by 2-fold. Furthermore, bromophenols showed mild to moderate inhibition of GSK-3β. Luteolin and SB-415286 were used as natural and synthetic reference inhibitors of GSK-3β to validate our result, which showed inhibition of enzyme activity by 50% at 5.42 ± 0.19 and 0.11 ± 0.01 μM, respectively. By inhibiting 50% of enzyme activity at 56.46 ± 2.48 μM, 3 showed moderate activity against GSK-3β. However, 1 and 2 exhibited mild inhibition of GSK-3β, with IC50 values of 229.42 ± 12.05 and 140.01 ± 15.08 μM, respectively. The dimer form of (3) exhibited multifold higher activity than its monomer (2) in all of the tested enzymes.
Enzyme Kinetics of Bromophenols
To explore enzyme inhibition mechanisms, we analyzed the kinetics of the studied enzymes at different substrate concentrations. Since all of the tested bromophenol derivatives exhibited potent inhibition of AChE, BChE, and BACE1 activity, their inhibition modes and the enzyme kinetic parameters were investigated via Lineweaver–Burk plots and Dixon plots. Figures 3–5 and Table 2 show the results. Bromophenols 1–3 were mixed-type inhibitors of AChE and competitive inhibitors of BChE enzymes, with Ki values of 0.58, 0.71, and 0.64 μM for AChE inhibition and 1.15, 0.51, and 0.37 μM for BChE inhibition, respectively. In the mixed type of enzyme inhibition (Figure 3D–F), the plots of 1/V versus 1/[S] produced a family of straight lines with a common intercept in the second quadrant, indicating an increased Km value and a decreased Vmax value. As shown in Figure 4D–F, the Lineweaver–Burk plots (1/V vs 1/[S]) revealed that all of the lines intersected at the same point on the y-axis, suggesting that Km increased with increasing concentrations of 1–3, while 1/Vmax did not change. Similarly, the mode of BACE1 inhibition by 1 and 2 was noncompetitive (the Vmax value decreased in a concentration-dependent manner without changing the Km value), with Ki values of 3.10 and 0.70 μM (Figure 5D,E). However, for 3, the inhibition mode was competitive, with a Ki value of 1.24 μM (Figure 5F).
Figure 3.

Dixon plots (A–C) and Lineweaver–Burk plots (D–F) for AChE inhibition by bromophenols 1–3.
Figure 5.

Dixon plots (A–C) and Lineweaver–Burk plots (D–F) for BACE1 inhibition by bromophenols 1–3.
Table 2. Kinetic Studies of Bromophenols from S. latiuscula.
| AChE |
BChE |
BACE1 |
||||
|---|---|---|---|---|---|---|
| compounds | inhibition typea | Ki (μM)b | inhibition typea | Ki (μM)b | inhibition typea | Ki (μM)b |
| 1 | mixed | 0.58 | competitive | 1.15 | noncompetitive | 3.10 |
| 2 | mixed | 0.71 | competitive | 0.51 | noncompetitive | 0.70 |
| 3 | mixed | 0.64 | competitive | 0.37 | competitive | 1.24 |
Inhibition type was determined by the Lineweaver–Burk plot.
Inhibition constant was determined by the Dixon plot.
Figure 4.

Dixon plots (A–C) and Lineweaver–Burk plots (D–F) for BChE inhibition by bromophenols 1–3.
Molecular Docking Simulation of AChE Inhibition
Molecular docking simulation was performed to elucidate the binding mode of bromophenols 1–3 in the active gorge of the Tetronarce californica AChE enzyme (1acj), using AutoDock 4.2 (the Scripps Research Institute, La Jolla, CA). Figure 6 shows the binding pattern of the respective bromophenols in the active site cavity of an enzyme, and Table 3 shows the binding energies along with the interacting residues. In Figure 6, the chemical structures of compounds 1, 2, and 3 are shown in yellow, orange, and green colored sticks, respectively. Similarly, H-bond and halogen contacts between 1–3 and enzyme residues are shown in blue and red lines, respectively. As shown in Table 3, the reference catalytic inhibitor (tacrine) bound to the catalytic active site of the AChE enzyme via the H-bond interaction with His440 and π–π interactions involving Phe330 and Trp84, with a binding energy of −9.80 kcal/mol. Similarly, donepezil (the reference allosteric inhibitor) bound to the peripheral active site of the AChE enzyme through H-bond interactions with Phe288, Arg289, and Asp72 (salt bridge) along with π–π interactions involving Phe330, Trp279, and Trp84. Three test bromophenols were involved in the interaction with His440 at the catalytic active site. In particular, 1 and 3 showed the H-bond interaction with His440 (Figure 6A,C), whereas 2 interacted with His440 via the Br–O bond (Figure 6B). In addition, 1–3 bound the peripheral anionic site through interactions involving Tyr334, Asp72, Trp84, and Tyr121. Compared to the monomers (1 and 2), the dimer (3) displayed more interactions with respect to the bromine number. Therefore, 1–3 showed mixed-type inhibition mode binding to both the catalytic active site (CAS) and peripheral active site (PAS) of the AChE enzyme, with binding energies −7.47, −7.37, and −10.9 kcal/mol, respectively.
Figure 6.

Molecular docking results of bromophenols (1–3) from S. latiuscula in the active site of the Tetronarce californica acetylcholinesterase enzyme (1acj). Chemical structures of bromophenols 1 (A), 2 (B), and 3 (C) are shown with yellow, orange, and green colored sticks, respectively. The H-bond and halogen contacts between 1–3 and enzyme residues are shown in dotted blue and solid red lines.
Table 3. Binding Energy and Interaction Residues of Bromophenols from S. latiuscula against Acetylcholinesterase (1acj).
| compounds | binding energy (kcal/mol)a | H-bond interactions | other interactions |
|---|---|---|---|
| 1 | –7.47 | His440, Tyr334, Asp72 | Trp432, Ile439, Phe330, Tyr442, Trp84 |
| 2 | –7.37 | Asp72 (2 bonds) | His440 (Br–O bond), Trp84, Phe330, Trp432 |
| 3 | –10.9 | His440, Gln69 | Tyr121, Tyr130, Leu127, Trp84 (π–π stacked), His440, Tyr442, Trp432, Tyr334, Phe330 (π–π stacked), Gly80 (Br–O bond) |
| tacrineb | –9.80 | His440 | Trp84 (π–π stacked, π–cation), Phe330 (π–π stacked, π–cation) |
| donepezilb | –10.10 | Phe288, Arg289, Asp72 (salt bridge) | Phe330 (π–π stacked), Trp279 (π–π T-shaped), Trp84 (π–π stacked), Ile287 (π–alkyl) |
Estimated binding energy of the ligand–receptor complex.
Reference ligands.
Molecular Docking Simulation of BChE Inhibition
To study the inhibition mechanism of ligands inside the catalytic domain of BChE, the chemical interactions of bromophenols 1–3 with the active site amino acid residues were investigated. Figure 7 shows the positions of the ligands and the polar (blue dotted lines) and halogen-bond (solid red lines) interactions between the ligands and the amino acids. Similarly, Table 4 shows the binding energies and interacting amino acid residues. Tacrine was used as a reference catalytic inhibitor to validate the docking results, which showed H-bond interaction with a π-system of the ring (His438) of BChE. Bromophenols 1–3 showed catalytic inhibition of BChE with low binding energies of −6.17, −6.44, and −9.80 kcal/mol, respectively. Bromophenol 1 was involved in an H-bond interaction with Pro285 and halogen-bond interactions with His438 (π–π T-shaped), Trp231, Leu286, Phe329, and Gly116. At the same time, 2 displayed four H-bond interactions [His438, Ser198, and Pro285(×2)], along with nonpolar interactions with Trp231, Ala199, Val288, Leu286, and Phe329. Interestingly, 3, which had the lowest binding energy among the tested bromophenols and the reference compound, displayed multiple bond interactions with His438 (two H-bonds and one nonpolar interaction) and Trp82 (three H-bonds and one nonpolar interaction). Furthermore, 3 also had nonpolar interactions with Ala328, Trp430, Phe329, and Gly116. Altogether, through interactions with catalytic active site residues, 1–3 displayed competitive mode BChE enzyme inhibition.
Figure 7.

Molecular docking results of bromophenols (1–3) from S. latiuscula in the active site of the human butyrylcholinesterase enzyme (4bds). Chemical structures of bromophenols 1 (A), 2 (B), and 3 (C) are shown with yellow, orange, and green colored sticks, respectively. H-bond and halogen contacts between 1–3 and enzyme residues are shown in dotted blue and solid red lines.
Table 4. Binding Energy and Interaction Residues of Bromophenols from S. latiuscula against Butyrylcholinesterase (4bds).
| compounds | binding energy (kcal/mol)a | H-bonds interactions | other interactions |
|---|---|---|---|
| 1 | –6.17 | Pro285 | His438 (π–π T-shaped), Trp231, Leu286, Phe329, Gly116 |
| 2 | –6.44 | His438, Ser198, Pro285 (2 bonds) | Trp231, Ala199, Val288, Leu286, Phe329 (π–π T-shaped) |
| 3 | –9.80 | His438 (2 bonds), Trp82 (3 bonds) | Ala328, Trp430, Phe329, Gly116, His438, Trp82 |
| tacrineb | –8.60 | His438 | Glu197 (attractive charge), Trp82 (π–cation, π–π stacked, π–alkyl), Ala328 (π–alkyl) |
Estimated binding energy of the ligand–receptor complex.
Reference ligand.
Molecular Docking Simulation of BACE1 Inhibition
To gain a better understanding of the BACE1 enzyme inhibitory mechanism in detail, the interactions of bromophenols 1–3 with the BACE1 structure were evaluated via molecular docking simulation. Binding energies and interacting residues, which include H-bond interactions and other interactions with bromine atoms, are listed in Table 5 and depicted in Figure 8. To validate the docking results, we employed 3,5,7,3′,4′-pentamethoxyflavone as allosteric and 2-amino-3-{(1R)-1-cyclohexyl-2-[(cyclohexylcarbonyl)amino]ethyl}-6-phenoxyquinazolin-3-ium (QUD) as catalytic reference inhibitors. As shown in the results, with bridging of H-bond interacting residues (Ser10, Gly11, Thr232, and Gln304) and other interactions involving bromine atoms (Arg307, Pro308, Ala335, Glu339, Val336, and Ser10) denoted by red solid lines, 1–2 bound to the active allosteric site of BACE1 with low energies (−6.59 and −5.98 kcal/mol, respectively). However, these interacting residues are not involved in the binding of 3 to the enzyme. Instead, the interaction showed the involvement of the catalytic Asp dyad (Asp32–Asp228). With the lowest binding energy among the test compounds (−8.79 kcal/mol), 3 displayed five H-bond interactions (Ser36, Ile126 × 2, Gly230, and Asp228), as shown by blue dotted lines in Figure 8C. In addition, bromine atoms interacted with Ser36, Val69, Asp228, Trp76, and Tyr71. The determining factors for competitive inhibition by 3 were the interactions with the catalytic Asp dyad (Asp32–Asp228), Ser36, Ile126, and Gly230, which were not observed for 1 and 2.
Table 5. Binding Energy and Interaction Residues of Bromophenols from S. latiuscula against BACE1 (2wjo).
| compounds | binding energy (kcal/mol)a | H-bond interactions | other interactions |
|---|---|---|---|
| 1 | –6.59 | Ser10, Gly11, Thr232, Gln304 | Arg307, Pro308, Ala335, Glu339 (π–anion) |
| 2 | –5.98 | Gly11, Gln304 (2 bonds) | Arg307, Val336, Ala335, Pro308, Glu339 (π–anion), Ser10 |
| 3 | –8.79 | Ser36, Ile126 (2 bonds), Gly230, Asp228 | Asp32 (π–anion), Val69, Tyr71, Trp76, Arg128 (π–alkyl) |
| QUDb | –11.20 | Asp32, Asp228 (2 bonds), Gly230 | Tyr71 (π–π T-shaped), Phe108 (π–π T-shaped), Val332 |
| PMFb | –6.50 | Ser10 | Gly156 (amide−π stacked), Pro308, Ala168 (π–σ), Thr232, Glu339 (π–anion), Arg307, Ala335, Val170, Gln304 |
Estimated binding energy of the ligand–receptor complex.
Reference ligands (QUD, 2-amino-3-{(1R)-1-cyclohexyl-2-[(cyclohexylcarbonyl)amino]ethyl}-6-phenoxyquinazolin-3-ium and PMF, 3,5,7,3′,4′-pentamethoxyflavone).
Figure 8.

Molecular docking results of bromophenols (1–3) from S. latiuscula in the active site of the human BACE1 enzyme (2wjo). Chemical structures of bromophenols 1 (A), 2 (B), and 3 (C) are shown with yellow, orange, and green colored sticks, respectively. The H-bond and halogen contacts between 1–3 and enzyme residues are shown in dotted blue and solid red lines.
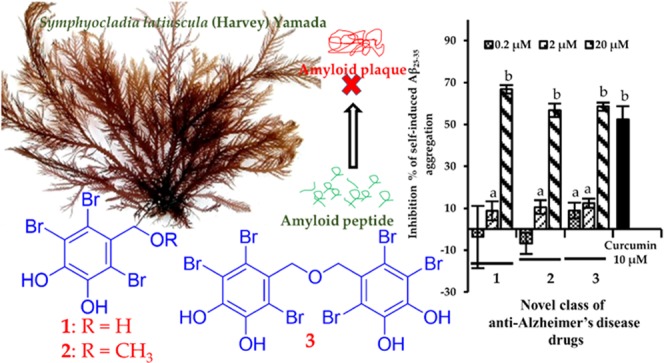
Self-Induced Aβ25–35 Aggregation
Extracellular aggregation of Aβ peptide is the major clinical hallmark of AD and Aβ25–35 is produced in aged brains from proteolytic cleavage of soluble racemized Aβ1–40 peptides. Therefore, we evaluated the effect of bromophenols 1–3 on the self-induced aggregation of Aβ25–35. As shown in Figure 9, 1–3 displayed concentration-dependent inhibition on amyloid aggregation. At 20 μM concentration, 1–3 displayed more than 50% inhibition on amyloid aggregation. Particularly, 1 showed higher activity (66.79% inhibition) followed by 3 (58.49%) and 2 (56.69%) at 20 μM concentration. Curcumin was used as a reference drug that inhibited aggregation by 52.40% at 10 μM.
Figure 9.

Inhibition of Aβ25–35 self-aggregation bromophenols (1–3) from S. latiuscula and a reference compound, curcumin. The data represent the mean ± SD of three independent experiments. Different letters in graphs are statistically different with Duncan’s test at p < 0.05.
Drug-likeness and ADME Prediction
In addition to good efficacy, an acceptable ADME profile is of the utmost importance in drug discovery. Therefore, we predicted the drug-like behavior and ADME properties of bromophenols, and Table 6 lists the predictions. In the experiments, 1 showed nondrug-like behavior, whereas 2 and 3 showed mid-structure behavior according to the MDDR-like rule,25 which characterizes a molecule as drug-like, mid-structure, or nondrug-like on the basis of the numbers of rings, rigid bonds, and rotatable bonds. Similarly, according to Lipinski’s rule [any orally active drug can have no more than one violation of H-bond donors (≤5), H-bond acceptors (≤10), molecular weight (≤500 Da), and log P (≤5)],261 and 2 were suitable, but 3 was not.
Table 6. Drug-likeness and ADME Characteristics as Determined by PreADMET.
| drug-likeness |
ADME
characteristics |
|||||||
|---|---|---|---|---|---|---|---|---|
| compounds | MDDR-like rule | Lipinski’s rule | log Po/wa | PPBb | HIAc | in vitro Caco2 permeability (nm/s)d | in vitro MDCK cell permeability (nm/s)e | in vivo BBB penetration ([brain]/[blood])f |
| 1 | nondrug-like | suitable | 2.96 | 97.13 | 92.70 | 18.84 | 0.48 | 3.55 |
| 2 | mid-structure | suitable | 3.45 | 94.49 | 94.62 | 24.87 | 0.46 | 4.52 |
| 3 | mid-structure | violated | 6.74 | 100 | 94.78 | 22.36 | 0.52 | 6.44 |
The log of the coefficient of solvent partitioning between 1-octanol and water.
Plasma protein binding (PPB) (weak binding: <90%; strong binding: >90%).
Human intestinal absorption (HIA) (poor: 0–20%; moderate: 20–70%: good: 70–100%).
Permeability across Caco2 cells (low: 0–10 nm/s; medium: 10–100 nm/s; high: >100 nm/s).
Permeability across MDCK cells.
Absorption by the central nervous system (low: <0.1; moderate: 0.1–2.0; high: >2.0).
Interestingly, the ADME predictions of these bromophenols revealed an excellent percentage of plasma protein binding: 100% for 3, 97.13% for 1, and 94.49% for 2, representing strong binding. Similarly, human intestinal absorption was higher than 90% for all bromophenol derivatives, indicating good absorption. The blood–brain barrier (BBB) penetration values ([brain]/[blood]) ranged from 3.55 to 6.44%, indicating high absorption by the central nervous system. In addition, the prediction of intestinal permeability in human epithelial colorectal adenocarcinoma cells (Caco-2) and Madin-Darby Canine Kidney (MDCK) cells showed moderate permeability for the tested bromophenols. The overall results of the prediction provide insight for optimizing drug-like properties.
Discussion
Alzheimer’s disease drug development has proven to be unusually difficult, as evidenced by the 99.6% failure rate from 2002 to 2012;27 despite continuous research, the success rate has not increased. In a recent research framework, Khachaturian et al.28 have conclusively summarized the reasons for the poor performance in AD drug development, which include inappropriate/wrong drug molecule, dose, target, study design, analytical methods, outcome measures, stages of disease (too late or too early), intolerability, disease model, and poor study conduct. Therefore, AD drug discovery and development are crucial and challenging.
The exact pathogenesis of AD is still unknown. On the basis of causative factors, research institutes and pharmaceutical companies collectively have proposed five hypotheses, the cholinergic hypothesis, the amyloid hypothesis, the tau hypothesis, the calcium hypothesis, and isoprenoid change.29 Of these, the cholinergic hypothesis is the most accepted because AD is accompanied by degeneration in cholinergic neurotransmission in the CNS, and the cholinergic deficit is a consistent finding in AD.30 In addition, research on the amyloid and tau hypotheses is ongoing and the focus of much interest. Therefore, our attempt to characterize bromophenol derivatives as inhibitors of ChE, BACE1, and GSK-3β extends these rationales for neuronal drug discovery.
The process of drug discovery and development begins with pharmacophore identification from natural sources or development through synthetic and semisynthetic routes. As a treasure house of diverse functional secondary metabolites, the marine biosphere has attracted much interest, and investigation has reached a peak level as evidenced by frequent publications. In particular, brown and red algae, which are rich in bromophenols and phlorotannins, have attracted the attention of research scientists due to their profound biological activities. To continue our efforts to discover a novel class of anti-AD drugs from natural sources, we designed this study. We found that all tested bromophenols are potent inhibitors of ChEs and BACE1 and moderate inhibitors of GSK-3β. Depending on the structure and the test enzyme, the 50% inhibitory concentration for bromophenols on each enzyme was significantly different from each other with Duncan’s test at p < 0.05. All of the tested bromophenols are fully substituted by different groups and highly brominated, and have a 2,3,6-tribromo-4,5-dihydroxy unit in common. These bromophenols showed a potent inhibitory effect toward ChEs and BACE1. The potency pattern of bromophenols toward ChE inhibition (IC50 range: 2.6–14.4 μM) was 3 > 1 > 2, whereas for BACE1 (IC50 range: 2.3–5.1 μM) and GSK-3β (IC50 range: 56.4–229.4 μM) inhibition, it was 3 > 2 > 1. In particular, toward BChE and BACE1 inhibition, these bromophenols were more potent than reference controls berberine and quercetin. It seems that bromines at C2, C3, and C6, and hydroxyl groups at C4 and C5 are necessary for inhibition and substituents at C1 determines the potency. However, it cannot be concluded unless the study is carried out on a larger number of derivatives with various numbers and positions of bromine and hydroxyl groups in the phenol ring. The pattern of AChE, BChE, BACE1, and GSK-3β inhibition by 1–3 in the present study was similar to the pattern of PTP1B and α-glucosidase inhibition in our recent work,19 which varied with a substituent at C7. When the C7–OH group of bromophenol 1 was substituted with −OCH3 (as in 2), its activity against AChE slightly decreased; however, BChE inhibition decreased by almost half. Interestingly, replacement of the C7–OCH3 group of 2 with another 2,3,6-tribromo-4,5-dihydroxyl methyl ether moiety, linked via an O-linkage to give the bis form (3), enhanced its activity multifold. In BACE1 and GSK-3β enzyme inhibition, bromophenol 2, with its C7–OCH3 group, showed better activity than bromophenol 1, with its C7–OH group. Again, 3 showed multifold more potent activity than 1 and 2. Although the number of test compounds is not sufficient to draw conclusions about the structure–activity relationship, an increase in numbers of phenol rings and bromines greatly enhanced the activity. Zhang et al.31 reported a similar result with PTP1B inhibition.
The binding mode of bromophenols (1–3) with AChE, BChE, and BACE1 was examined through in silico studies to explore the key residues involved in the bromophenol–enzyme interaction. The active pocket of the AChE enzyme (AChE gorge) from Torpedo californica contains a pair of regions for enzyme catalysis: the catalytic active site (CAS) at the bottom of the gorge, 20 Å deep, comprising a catalytic triad (Ser200-His440-Glu327), and a peripheral anionic site (PAS) at the upper part of the gorge, comprising 14 highly conserved amino acid residues.32 For allosteric regulation of AChE, ACh should bind to the PAS residues, which would lead to conformational changes in the active center. Bromophenols 1–3 were stably bound to the active site of the AChE enzyme, encompassing both the CAS and PAS. In the CAS, bromophenols demonstrated H-bonding or halogen-bonding with His440. In particular, 3 interacted with His440 via both H-bond and halogen-bond interactions and also displayed multiple halogen bonds corresponding to the bromine number. In the PAS, the primary interactions observed were via Phe330, Asp72, Tyr121, and Trp84 for the tested bromophenols. The higher binding affinity compared to the lowest binding energy of 3 might be attributed to multiple bond interactions parallel to the bromine number. The mixed-type inhibition mode of 1–3 can be explained by the bidirectional binding modes (CAS and PAS). Similarly, the binding site of the hBChE protein is characterized by an acyl pocket at the bottom of a deep catalytic gorge comprising the oxyanion hole (Gly118, Gly119, and Ala201); the peripheral site at the edge of the gorge; and the choline-binding site (Trp84) within the gorge.33 Bromophenols 1–3 inhibited catalysis by forming H-bond interactions with Pro285, His438, Trp82, and Asp70 at a bond distance of 1.8–2.8 Å. His438 is a member of a catalytic triad, and 1 and 2 showed single-bond interactions with this residue. However, 3 interacted via multiple bonds, as shown in Figure 7. In addition, other interactions with Trp82, Ala328, Val288, and Leu286 were observed in the active catalytic site. This explains the lowest binding energy and highest binding affinity of 3 at the active catalytic site of an enzyme. Similarly, electrostatic interactions between 3 and the side chains of the catalytic aspartates (Asp32 and Asp228) of BACE1 were observed via hydrogen and halogen bonds. However, 1 and 2 showed interactions with the allosteric residues Ser10, Arg307, Ala335, Pro308, Glu339, and Gln304 of the BACE1 enzyme without involving catalytic dyads. Thus, from the docking result, 1 and 2 were characterized as noncompetitive inhibitors and 3 as a competitive inhibitor of BACE1.
Numerous studies have demonstrated that aberrant regulation of GSK-3 is involved in the etiology of neurodegenerative diseases such as AD, PD, amyotrophic lateral sclerosis, and multiple sclerosis.34−37 In addition, GSK-3 likely activates a variety of immune response targets, such as cyclic-AMP response element binding protein, toll-like receptors, transcription factor NF-κB, and proteins involved in cytokine production.38,39 Thus, inhibition of hyperactivity of GSK-3 to a normal level emerges as a promising therapy for the treatment of neurodegenerative and behavior disorders. A review by Avrahami et al.40 stated that “Mild inhibition of GSK-3 is favored because this decreases the exacerbated GSK-3 function in the tissue affected with minimum deleterious effects on healthy tissues.” The three bromophenol derivatives tested in the present study showed mild to moderate inhibition of GSK-3β. Therefore, these derivatives could be of additional benefit in treating neurodegenerative disorders. However, how these compounds affect the aforementioned signaling pathways is not known. Similarly, in the amyloid aggregation assay, all of the tested bromophenols at 20 μM concentration displayed prominent inhibition on self-induced Aβ25–35 peptide aggregation. Aβ25–35 represents the biologically active region of Aβ peptide because it is the shortest fragment with large β-sheet aggregated structures and retains the toxicity of the full-length peptide.41 Therefore, the inhibition of Aβ25–35 self-assembly contributes to the management of AD and highlights rational drug design.
Halogen bonding has attracted much interest recently due to its important role in molecular recognition in biological systems. Voth et al.42 showed halogen bonding to be a stabilizing factor and a conformational determinant in protein–ligand and DNA structures. Halogens facilitate the crossing of the BBB and prolong the half-life of a drug molecule by filling hydrophobic cavities in the binding sites.43,44 Therefore, these brominated phenols might be of particular interest in the design and discovery of neuronal drugs. In addition, we predicted several pharmacokinetic parameters of 1–3. Although in silico ADME predictions cannot fully replace well-established in vitro cell-based approaches or in vivo assays, they can provide significant insights. The abilities of the drug to reach the appropriate receptors in the target tissue, and remain metabolically stable to exert the desired effect are important criteria for drug development. For this, the drug must have excellent plasma binding and BBB penetration. Our predictions of the ADME characteristics of bromophenols 1–3 showed excellent plasma binding (94.4–100%), human intestinal absorption (92.7–94.7%), and BBB penetration (3.5–6.4%). These results provide evidence of improved delivery to the CNS. Jitareanu et al.45 found a similar result: halogenation led to improvements in membrane binding, permeation, and diffusion. Similarly, Gentry et al.46 found that addition of bromine and chlorine to peptide drugs significantly enhanced in vitro BBB permeation, showing improved CNS delivery. Furthermore, a recent review by Gribble,6 demonstrated superior antimicrobial, anticancer, antimalarial, anti-inflammatory, and antioxidant activity for brominated compounds, highlighting their advantage in drug discovery.
In the present study, three 2,3,6-tribromo-4,5-dihydroxybenzyl derivatives (1–3) from S. latiuscula were characterized as potent inhibitors of ChEs, BACE1, and GSK-3β, and emphasized the importance of the 7–OH group and bromine number in ligand–enzyme interaction. In addition, these bromophenols exhibited good inhibition of self-induced Aβ25–35 aggregation. Overall, the results of the present study show that these bromophenol derivatives could be promising drug candidates against AD, and these compounds should be tested in vivo for neuronal drug discovery, especially targeting AD.
Materials and Methods
Chemicals and Reagents
All chemicals and reagents used in this study were obtained from commercial sources at the highest available grade, and list of chemicals with vendors are mentioned alongside. Acetylcholine iodide (ACh), butyrylthiocholine chloride (BCh), electric eel AChE (EC 3.1.1.7), horse serum BChE (EC 3.1.1.8), 5,5′-dithiobis[2-nitrobenzoic acid] (DTNB), adenosine 5-triphosphate (ATP) disodium salt hydrate, ammonium hydroxide, 4-(2-hydroxyethyl)piperazine-1-ethanesulfonic acid (HEPES), ethylene glycol-bis(aminoethylether)-N,N,N,N-tetra acetic acid tetrasodium salt, ethylenediaminetetraacetic acid, magnesium acetate tetrahydrate, amyloid-β protein fragments (Aβ25–35), 3-[(3-chloro-4-hydroxyphenyl)-amino]-4-(2-nitrophenyl)-1H-pyrrol-2,5-dione (SB-415286), 1,1,1,3,3,3-hexafluoro-2-propanol (HFIP) and thioflavin T (Sigma-Aldrich: St. Louis, MO); human recombinant GSK-3β (ProSpec: ProSpec-Tany TechnoGene Ltd., Ness-Ziona, Israel), glycogen muscle synthase (Merck Millipore: Millipore Corporation, CA); Kinase-Glo kits (Promega: Promega Corporation, Madison, WI); and BACE1 FRET assay kits (Invitrogen: Life Technologies, Carlsbad).
Plant Material Extraction and Isolation
The plant material (leafy thalli of S. latiuscula (Harvey) Yamada) was collected in January 2016 from the Cheongsapo region of Busan, S. Korea, and authenticated by Dr K. W. Nam (an algologist) from the Department of Marine Biology, Pukyong National University. A voucher specimen (No. 20160140) has been deposited in the laboratory of Prof. J. S. Choi, Pukyong National University.
Extraction of clean and dried leafy thali of S. latiuscula, along with chromatographic steps and detailed procedures to isolate 2,3,6-tribromo-4,5-dihydroxybenzyl alcohol (1), 2,3,6-tribromo-4,5-dihydroxybenzyl methyl ether (2), and bis-(2,3,6-tribromo-4,5-dihydroxybenzyl) ether (3), is reported in our recent publication.19
2,3,6-Tribromo-4,5-dihydroxybenzyl alcohol (1): obtained as a pale yellow solid. 1H NMR (CD3OD, 600 MHz) δ 4.80 (2H, s, H-7); 13C NMR (CD3OD, 150 MHz) δ 147.42 (C-5), 145.05 (C-4), 128.65 (C-1), 119.34 (C-6), 114.68 (C-3), 113.96 (C-2), and 74.41 (C-7). HRFAB MS m/z 373.7743 calculated for C7H5Br3O3.
2,3,6-Tribromo-4,5-dihydroxybenzyl methyl ether (2): obtained as an amorphous light brown powder; 1H NMR (CD3OD, 600 MHz) δ 4.89 (2H, s, H-7), 3.39 (3H, s, H-8); 13C NMR (CD3OD, 150 MHz) δ 146.87 (C-5), 144.89 (C-4), 129.26 (C-1), 119.12 (C-6), 114.59 (C-3), 114.12 (C-2), 76.52 (C-7), and 58.39 (CH3, H-8). HRFAB MS m/z 387.7904 calculated for C8H7Br3O3.
Bis-(2,3,6-tribromo-4,5-dihydroxybenzyl) ether (3): obtained as an amorphous light brown powder; 1H NMR (CD3OD, 600 MHz) δ 4.95 (4H, s, H-7 and H-7′); 13C NMR (CD3OD, 150 MHz) δ 146.79 (C-5 and C-5′), 144.69 (C-4 and C-4′), 129.22 (C-1 and C-1′), 119.59 (C-6 and C-6′), 115.03 (C-3 and C-3′), 114.09 (C-2 and C-2′), and 74.44 (C-7 and C-7′). HRESIMS [M + Na]+m/z 752.5364 calculated for C14H8Br6O5.
Cholinesterase Enzyme Inhibition Assay
The potential of bromophenols to inhibit ChE enzymes was determined following our previously reported procedure.47 AChE and BChE enzyme inhibition was evaluated using ACh and BCh substrates, respectively. Bromophenols and positive control (berberine) were dissolved in 10% analytical grade DMSO, and each enzymatic reaction was conducted in triplicate in a 96-well microplate. Briefly, 140 μL of sodium phosphate buffer (pH 8.0), 20 μL of different concentrations of bromophenols or berberine, and 20 μL of either AChE or BChE solution (0.36 U/mL) were mixed and incubated at room temperature for 15 min. Then the reaction was initiated by adding 10 μL of DTNB and 10 μL of substrate (either AChI or BCh), and further incubated at RT for 10 min. Finally, the hydrolysis of AChI or BCh (formation of the yellow 5-thio-2-nitrobenzoate anion) was monitored at 412 nm using a VersaMax microplate reader (Molecular Devices, Sunnyvale, CA). Percentage inhibition of ChEs was calculated as (1 – T/C) × 100, where T and C are the enzyme activities in the presence or absence of inhibitors, respectively and are expressed as IC50 values calculated from the log dose–inhibition curve.
BACE1 Enzyme Inhibition Assay
The ability of bromophenols to inhibit the BACE1 enzyme was evaluated in vitro using human recombinant β-secretase and a BACE1 fluorescence resonance energy transfer assay kit (Life Technologies, Carlsbad, CA). All of the experimental conditions and procedures were similar to those in our previous report.48 In brief, 10 μL of assay buffer (pH 4.5), 10 μL of BACE1 (1.0 U/mL), 10 μL of the BACE1 substrate (750 nM), and 10 μL of different bromophenols/quercetin concentrations were mixed in 384-well back plates and incubated for 60 min at RT in the dark. After incubation, mixtures in wells were irradiated at 545 nm and emission intensity at 585 nm was recorded using the SpectraMax Gemini XPS microplate spectrofluorometer (Molecular Devices, Sunnyvale, CA). The percentage (%) inhibition of BACE1 activity was calculated from the following equation: % inhibition = [1 – (S60 – S0)/(C60 – C0)] × 100%, where S0 and C0 are the initial fluorescence of the test samples and control groups, and S60 and C60 are the final (after 60 min) fluorescence of the test samples and control groups, respectively, and are expressed as IC50 values calculated from the log dose–inhibition curve.
GSK-3β Enzyme Inhibition Assay
The potential of 1–3 to inhibit GSK-3β enzyme activity was evaluated in vitro using hGSK-3β and a Kinase-Glo reagent kit, following our previous report with some modifications.49 Test samples were prepared by serial dilution in assay buffer (pH 7.5) (concentration of DMSO in the final reaction mixture did not exceed 5%). Briefly, the reaction was initiated by mixing 5 μL of a sample, 5 μL of ATP (1 μM final concentration), 5 μL of 50 μM GSM, and 5 μL of 20 ng GSK-3β per well in a 384-well black plate, and incubated at 37 °C for 30 min. After incubation, 20 μL of Kinase-Glo reagent was added into each well to terminate the reaction and glow-type luminescence was recorded in the Synergy HTX multi-mode microplate reader (BioTek Instruments, Winooski, VT). The method was validated using luteolin and 3-[(3-chloro-4-hydroxyphenyl)-amino]-4-(2-nitrophenyl)-1H-pyrrol-2,5-dione (SB-415286) as reference compounds having maximum inhibition, and the maximum enzyme activity was achieved in the absence of inhibitors (5 μL of sample replaced by 5 μL of buffer).
Enzyme Kinetics
The kinetic study of AChE inhibition by bromophenol 1 (1.25, 2.5, and 5 μM), 2 (2.5, 5, and 10 μM), and 3 (0.5, 1 and 5 μM) was performed with the assay method in Section 2.3 in the presence of varying concentrations of the ACh substrate (0.1, 0.3, and 0.6 nM). For BChE enzyme kinetics, bromophenols 1 and 2 were tested at the same concentrations as for the AChE kinetics study, and 3 was tested at 1.25, 2.5, and 5 μM with BCh substrate concentrations of 0.1, 0.3, and 0.6 nM. Similarly, bromophenols 1 (1.25, 2.5, and 5 μM), 2 (1.25, 2.5, and 5 μM) and 3 (0.625 and 1.25) were tested with different substrate concentrations (250, 375, and 750 nM) following an assay method for BACE1 enzyme inhibition. Lineweaver–Burk plots were derived from the double reciprocal plots of enzyme kinetic data. Ki values were calculated from the Dixon plots.
Docking Studies
Molecular docking analysis was carried out using AutoDock 4.2.50 X-ray crystallography of AChE–tacrine (PDB ID: 1ACJ),32 BChE–tacrine (PDB ID: 4BDS),51 and BACE1–QUD (PDB ID: 2WJO)52 complexes were obtained from the Research Collaboratory for Structural Bioinformatics Protein Data Bank website (http://www.rcsb.org/) at resolutions of 2.80, 2.10, and 2.50 Å, respectively. Rotatable bonds in the inhibitors and positive controls were assigned by AutoDockTools. The 3D structures of compounds 2,3,6-tribromo-4,5-dihydroxybenzyl alcohol (1), 2,3,6-tribromo-4,5-dihydroxybenzyl methyl ether (2), and bis-(2,3,6-tribromo-4,5-dihydroxybenzyl) ether (3) were constructed using Chem3D Pro (v12.0, CambridgeSoft Inc., Cambridge, MA) and adjusted to pH 7 using MarvinSketch (ChemAxon, Budapest, Hungary). Three-dimensional structures of 3,5,7,3′,4′-pentamethoxyflavone and donepezil were obtained from the National Center for Biotechnology Information PubChem, with the respective compound identification numbers of 97332 and 3152. A molecular mechanics 2 force field was used to minimize the energy of each ligand. AutoDock 4.2 was used for docking simulations and AutoGrid was used to generate grid maps (catalytic sites and allosteric sites). The docking protocol for rigid and flexible ligand docking comprised 15 independent genetic algorithms. Docking results were analyzed and visualized using UCSF Chimera (http://www.cgl.ucsf.edu/chimera/).
Self-Induced Aβ25–35 Aggregation Inhibition Assay
Bromophenols 1–3 were accessed for their ability to inhibit self-induced Aβ25–35 aggregations via the thioflavin T (ThT) fluorescence method,53 with slight modifications.54 In brief, Aβ25–35, lyophilized from 1mg/mL hexafluoro-2-propanol (HFIP) solution, was dissolved in 65% acetonitrile to obtain 0.4 mM stock solution. Then the stock solution of Aβ25–35 was diluted by 2-fold in 69 mM phosphate buffer (pH 7.0). To 72.5 μL of the amyloid solution, 2.5 μL of different concentrations of 1–3 (0.2, 2, and 20 μM) and/or curcumin (10 μM) was added. After 24 h of incubation at 4 °C, 675 μL of a 25 μM thioflavin T in 50 mM glycine–NaOH buffer (pH 8.5) was added and after 5 min the fluorescence emission at 490 nm (ex = 446 nm) was measured by the SpectraMax Gemini XPS microplate spectrofluorometer (Molecular Devices, Sunnyvale, CA). The fluorescence intensities were compared and the percent inhibition due to the presence of the inhibitor was calculated by the following formula: 100 – (Fi/Fc × 100), where Fi and Fc were the fluorescence intensities obtained for Aβ25–35 in the presence and in the absence of inhibitors, respectively.
Drug-likeness and ADME Prediction
Drug-likeness and ADME predictions were carried out using PreADMET (v2.0, YONSEI University, Seoul, Korea).
Statistical Analysis
All of the results are expressed as the mean ± standard deviation of three independent experiments and one-way analysis of variance and Duncan’s test (v23.0, Systat Inc., Evanston, IL) were used to calculate the statistical significance. A p-value <0.05 was considered to be significant.
Acknowledgments
This research was supported by the Basic Science Research Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Science (2012R1A6A1028677).
Glossary
Abbreviations
- Aβ
amyloid β
- AD
Alzheimer’s disease
- ADT
AutoDockTools
- BACE1
β-site amyloid precursor protein (APP) cleaving enzyme 1
- BBB
blood–brain barrier
- CAS
catalytic active site
- ChEs
cholinesterases
- DTNB
5,5′-dithiobis[2-nitrobenzoic acid]
- GSK-3β
glycogen synthase kinase-3β
- PAS
peripheral anionic site
- QUD
2-amino-3-{(1R)-1-cyclohexyl-2-[(cyclohexylcarbonyl)amino]ethyl}-6-phenoxyquinazolin-3-ium
- ThT
thioflavin T
The authors declare no competing financial interest.
References
- Dai Z.; Lin Q.; Li T.; Wang X.; Yuan H.; Yu X.; He Y.; Wang H. Disrupted structural and functional brain networks in Alzheimer’s disease. Neurobiol. Aging 2019, 75, 71–82. 10.1016/j.neurobiolaging.2018.11.005. [DOI] [PubMed] [Google Scholar]
- 2018 Alzheimer’s disease facts and figures. Alzheimer’s Dementia 2018, 14, 367–429. 10.1016/j.jalz.2018.02.001. [DOI] [Google Scholar]
- Villemagne V. L.; Burnham S.; Bourgeat P.; Brown B.; Ellis K. A.; Salvado O.; Szoeke C.; Macaulay S. L.; Martins R.; Maruff P.; Ames D. Amyloid β deposition, neurodegeneration, and cognitive decline in sporadic Alzheimer’s disease: a prospective cohort study. Lancet Neurol. 2013, 12, 357–367. 10.1016/S1474-4422(13)70044-9. [DOI] [PubMed] [Google Scholar]
- Reiman E. M.; Quiroz Y. T.; Fleisher A. S.; Chen K.; Velez-Pardo C.; Jimenez-Del-Rio M.; Fagan A. M.; Shah A. R.; Alvarez S.; Arbelaez A.; Giraldo M. Brain imaging and fluid biomarker analysis in young adults at genetic risk for autosomal dominant Alzheimer’s disease in the presenilin 1 E280A kindred: a case-control study. Lancet Neurol. 2012, 11, 1048–1056. 10.1016/S1474-4422(12)70228-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bateman R. J.; Xiong C.; Benzinger T. L.; Fagan A. M.; Goate A.; Fox N. C.; Marcus D. S.; Cairns N. J.; Xie X.; Blazey T. M.; Holtzman D. M. Clinical and biomarker changes in dominantly inherited Alzheimer’s disease. N. Engl. J. Med. 2012, 367, 795–804. 10.1056/NEJMoa1202753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gribble G. W. Biological activity of recently discovered halogenated marine natural products. Mar. Drugs 2015, 13, 4044–4136. 10.3390/md13074044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayer A.; Rodríguez A.; Taglialatela-Scafati O.; Fusetani N. Marine pharmacology in 2009–2011: Marine compounds with antibacterial, antidiabetic, antifungal, anti-inflammatory, antiprotozoal, antituberculosis, and antiviral activities; affecting the immune and nervous systems, and other miscellaneous mechanisms of action. Mar. Drugs 2013, 11, 2510. 10.3390/md11072510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eguchi K.; Kato H.; Fujiwara Y.; Losung F.; Mangindaan R. E.; de Voogd N. J.; Takeya M.; Tsukamoto S. Bastadins, brominated-tyrosine derivatives, suppress accumulation of cholesterol ester in macrophages. Bioorg. Med. Chem. Lett. 2015, 25, 5389–5392. 10.1016/j.bmcl.2015.09.024. [DOI] [PubMed] [Google Scholar]
- Asolkar R. N.; Singh A.; Jensen P. R.; Aalbersberg W.; Carte B. K.; Feussner K. D.; Subramani R.; DiPasquale A.; Rheingold A. L.; Fenical W. Marinocyanins, cytotoxic bromo-phenazinone meroterpenoids from a marine bacterium from the streptomycete clade MAR4. Tetrahedron 2017, 73, 2234–2241. 10.1016/j.tet.2017.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin X.; Liu M. Bromophenols from marine algae with potential anti-diabetic activities. J. Ocean Univ. China 2012, 11, 533–538. 10.1007/s11802-012-2109-1. [DOI] [Google Scholar]
- Behçet A.; Çağlılar T.; Celepci D. B.; Aktaş A.; Taslimi P.; Gök Y.; Aygün M.; Kaya R.; Gülçin İ. Synthesis, characterization and crystal structure of 2-(4-hydroxyphenyl) ethyl and 2-(4-nitrophenyl) ethyl substituted benzimidazole bromide salts: Their inhibitory properties against carbonic anhydrase and acetylcholinesterase. J. Mol. Struct. 2018, 1170, 160–169. 10.1016/j.molstruc.2018.05.077. [DOI] [Google Scholar]
- Fais A.; Kumar A.; Medda R.; Pintus F.; Delogu F.; Matos M. J.; Era B.; Delogu G. L. Synthesis, molecular docking and cholinesterase inhibitory activity of hydroxylated 2-phenylbenzofuran derivatives. Bioorg. Chem. 2019, 84, 302–308. 10.1016/j.bioorg.2018.11.043. [DOI] [PubMed] [Google Scholar]
- Xu X.; Yang H.; Khalil Z. G.; Yin L.; Xiao X.; Neupane P.; Bernhardt P. V.; Salim A. A.; Song F.; Capon R. J. Chemical diversity from a Chinese marine red alga Symphyocladia latiuscula. Mar. Drugs 2017, 15, 374. 10.3390/md15120374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang W.; Okada Y.; Shi H.; Wang Y.; Okuyama T. Structures and aldose reductase inhibitory effects of bromophenols from the red alga Symphyocladia latiuscula. J. Nat. Prod. 2005, 68, 620–622. 10.1021/np040199j. [DOI] [PubMed] [Google Scholar]
- Xu X.; Song F.; Fan X.; Fang N.; Shi J. A novel bromophenol from marine red alga Symphyocladia latiuscula. Chem. Nat. Compd. 2009, 45, 811–813. 10.1007/s10600-010-9501-0. [DOI] [Google Scholar]
- Lee J. H.; Park S. E.; Hossain M. A.; Kim M. Y.; Kim M. N.; Chung H. Y.; Choi J. S.; Yoo Y. H.; Kim N. D. 2, 3, 6-Tribromo-4, 5-dihydroxybenzyl methyl ether induces growth inhibition and apoptosis in MCF-7 human breast cancer cells. Arch. Pharm. Res. 2007, 30, 1132–1137. 10.1007/BF02980248. [DOI] [PubMed] [Google Scholar]
- Xu X.; Yin L.; Gao J.; Gao L.; Song F. Antifungal bromophenols from marine red alga Symphyocladia latiuscula. Chem. Biodiversity 2014, 11, 807–811. 10.1002/cbdv.201300239. [DOI] [PubMed] [Google Scholar]
- Park H. J.; Kurokawa M.; Shiraki K.; Nakamura N.; Choi J. S.; Hattori M. Antiviral activity of the marine alga Symphyocladia latiuscula against herpes simplex virus (HSV-1) in vitro and its therapeutic efficacy against HSV-1 infection in mice. Biol. Pharm. Bull. 2005, 28, 2258–2262. 10.1248/bpb.28.2258. [DOI] [PubMed] [Google Scholar]
- Paudel P.; Seong S. H.; Park H. J.; Jung H. A.; Choi J. S. Anti-diabetic activity of 2, 3, 6-tribromo-4, 5-dihydroxybenzyl derivatives from Symphyocladia latiuscula through PTP1B downregulation and α-glucosidase inhibition. Mar. Drugs 2019, 17, 166. 10.3390/md17030166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paudel P.; Wagle A.; Seong S. H.; Park H. J.; Jung H. A.; Choi J. S. A new tyrosinase inhibitor from the red alga Symphyocladia latiuscula (Harvey) Yamada (Rhodomelaceae). Mar. Drugs 2019, 17, 295. 10.3390/md17050295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi J. S.; Park H. J.; Jung H. A.; Chung H. Y.; Jung J. H.; Choi W. C. A cyclohexanonyl bromophenol from the red alga Symphyocladia latiuscula. J. Nat. Prod. 2000, 63, 1705–1706. 10.1021/np0002278. [DOI] [PubMed] [Google Scholar]
- Chung H. Y.; Choi H. R.; Park H. J.; Choi J. S.; Choi W. C. Peroxynitrite scavenging and cytoprotective activity of 2,3,6-tribromo-4,5-dihydroxybenzyl methyl ether from the marine alga Symphyocladia latiuscula. J. Agric. Food Chem. 2001, 49, 3614–3621. 10.1021/jf0101206. [DOI] [PubMed] [Google Scholar]
- Duan X. J.; Li X. M.; Wang B. G. Highly brominated mono- and bis-phenols from the marine red alga Symphyocladia latiuscula with radical-scavenging activity. J. Nat. Prod. 2007, 70, 1210–1213. 10.1021/np070061b. [DOI] [PubMed] [Google Scholar]
- Samaddar S.; Koneri R. Polyphenols of marine red macroalga Symphyocladia latiuscula ameliorate diabetic peripheral neuropathy in experimental animals. Heliyon 2019, 5, e01781 10.1016/j.heliyon.2019.e01781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oprea T. I. Property distribution of drug-related chemical databases. J. Comput.-Aided Mol. Des. 2000, 14, 251–264. 10.1023/A:1008130001697. [DOI] [PubMed] [Google Scholar]
- Lipinski C. A.; Lombardo F.; Dominy B. W.; Feeney P. J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Delivery Rev. 1997, 23, 3–25. 10.1016/S0169-409X(96)00423-1. [DOI] [PubMed] [Google Scholar]
- Cummings J. L.; Morstorf T.; Zhong K. Alzheimer’s disease drug-development pipeline: few candidates, frequent failures. Alzheimers Res. Ther. 2014, 6, 37. 10.1186/alzrt269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khachaturian A. S.; Hayden K. M.; Mielke M. M.; Tang Y.; Lutz M. W.; Gustafson D. R.; Kukull W. A.; Mohs R.; Khachaturian Z. S. Future prospects and challenges for Alzheimer’s disease drug development in the era of the NIA-AA Research Framework. Alzheimers Dement. 2018, 14, 532–534. 10.1016/j.jalz.2018.03.003. [DOI] [PubMed] [Google Scholar]
- Singh M.; Kaur M.; Kukreja H.; Chugh R.; Silakari O.; Singh D. Acetylcholinesterase inhibitors as Alzheimer therapy: from nerve toxins to neuroprotection. Eur. J. Med. Chem. 2013, 70, 165–188. 10.1016/j.ejmech.2013.09.050. [DOI] [PubMed] [Google Scholar]
- Mehta M.; Adem A.; Sabbagh M.. New acetylcholinesterase inhibitors for Alzheimer’s disease Int. J. Alzheimers Dis. 2012, 2012. 10.1155/2012/728983. [DOI] [PMC free article] [PubMed]
- Zhang R.; Yu R.; Xu Q.; Li X.; Luo J.; Jiang B.; Wang L.; Guo S.; Wu N.; Shi D. Discovery and evaluation of the hybrid of bromophenol and saccharide as potent and selective protein tyrosine phosphatase 1B inhibitors. Eur. J. Med. Chem. 2017, 134, 24–33. 10.1016/j.ejmech.2017.04.004. [DOI] [PubMed] [Google Scholar]
- Harel M.; Schalk I.; Ehret-Sabatier L.; Bouet F.; Goeldner M.; Hirth C.; Axelsen P.; Silman I.; Sussman J. Quaternary ligand binding to aromatic residues in the active-site gorge of acetylcholinesterase. Proc. Natl. Acad. Sci. U.S.A. 1993, 90, 9031–9035. 10.1073/pnas.90.19.9031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sussman J. L.; Harel M.; Frolow F.; Oefner C.; Goldman A.; Toker L.; Silman I. Atomic structure of acetylcholinesterase from Torpedo californica: a prototypic acetylcholine-binding protein. Science 1991, 253, 872–879. 10.1126/science.1678899. [DOI] [PubMed] [Google Scholar]
- Gómez-Sintes R.; Hernández F.; Lucas J. J.; Avila J. GSK-3 mouse models to study neuronal apoptosis and neurodegeneration. Front. Mol. Neurosci. 2011, 4, 45. 10.3389/fnmol.2011.00045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hernández F.; Avila J. The role of glycogen synthase kinase 3 in the early stages of Alzheimers’ disease. FEBS Lett. 2008, 582, 3848–3854. 10.1016/j.febslet.2008.10.026. [DOI] [PubMed] [Google Scholar]
- Terwel D.; Muyllaert D.; Dewachter I.; Borghgraef P.; Croes S.; Devijver H.; Van Leuven F. Amyloid activates GSK-3β to aggravate neuronal tauopathy in bigenic mice. Am. J. Pathol. 2008, 172, 786–798. 10.2353/ajpath.2008.070904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palomo V.; I Perez D.; Gil C.; Martínez A. The potential role of glycogen synthase kinase 3 inhibitors as amyotrophic lateral sclerosis pharmacological therapy. Curr. Med. Chem. 2011, 18, 3028–3034. 10.2174/092986711796391697. [DOI] [PubMed] [Google Scholar]
- Beurel E.; Michalek S. M.; Jope R. S. Innate and adaptive immune responses regulated by glycogen synthase kinase-3 (GSK3). Trends Immunol. 2010, 31, 24–31. 10.1016/j.it.2009.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H.; Brown J.; Martin M. Glycogen synthase kinase 3: a point of convergence for the host inflammatory response. Cytokine 2011, 53, 130–140. 10.1016/j.cyto.2010.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Avrahami L.; Licht-Murava A.; Eisenstein M.; Eldar-Finkelman H. GSK-3 inhibition: Achieving moderate efficacy with high selectivity. Biochim. Biophys. Acta, Proteins Proteomics 2013, 1834, 1410–1414. 10.1016/j.bbapap.2013.01.016. [DOI] [PubMed] [Google Scholar]
- Sato K.; Wakamiya A.; Maeda T.; Noguchi K.; Takashima A.; Imahori K. Correlation among secondary structure, amyloid precursor protein accumulation, and neurotoxicity of amyloid β (25–35) peptide as analyzed by single alanine substitution. J. Biochem. 1995, 118, 1108–1111. 10.1093/oxfordjournals.jbchem.a124994. [DOI] [PubMed] [Google Scholar]
- Voth A. R.; Hays F. A.; Ho P. S. Directing macromolecular conformation through halogen bonds. Proc. Natl. Acad. Sci. U.S.A. 2007, 104, 6188–6193. 10.1073/pnas.0610531104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cavallo G.; Metrangolo P.; Milani R.; Pilati T.; Priimagi A.; Resnati G.; Terraneo G. The halogen bond. Chem. Rev. 2016, 116, 2478–2601. 10.1021/acs.chemrev.5b00484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolář M. H.; Hobza P. Computer modeling of halogen bonds and other σ-hole interactions. Chem. Rev. 2016, 116, 5155–5187. 10.1021/acs.chemrev.5b00560. [DOI] [PubMed] [Google Scholar]
- Jitareanu A.; Tataringa G.; Zbancioc A.-M.; Trifan A. Bromination-A versatile tool for drugs optimization. Med. Surg. J. 2018, 122, 614–626. [Google Scholar]
- Gentry C.; Egleton R.; Gillespie T.; Abbruscato T.; Bechowski H.; Hruby V.; Davis T. The effect of halogenation on blood-brain barrier permeability of a novel peptide drug. Peptides 1999, 20, 1229–1238. 10.1016/S0196-9781(99)00127-8. [DOI] [PubMed] [Google Scholar]
- Shrestha S.; Seong S. H.; Paudel P.; Jung H. A.; Choi J. S. Structure related inhibition of enzyme systems in cholinesterases and BACE1 in vitro by naturally occurring naphthopyrone and its glycosides isolated from Cassia obtusifolia. Molecules 2018, 23, 69. 10.3390/molecules23010069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu T.; Paudel P.; Seong S. H.; Kim J. A.; Jung H. A.; Choi J. S. Computational insights into β-site amyloid precursor protein enzyme 1 (BACE1) inhibition by tanshinones and salvianolic acids from Salvia miltiorrhiza via molecular docking simulations. Comput. Biol. Chem. 2018, 74, 273–285. 10.1016/j.compbiolchem.2018.04.008. [DOI] [PubMed] [Google Scholar]
- Paudel P.; Seong S. H.; Zhou Y.; Park C. H.; Yokozawa T.; Jung H. A.; Choi J. S. Rosmarinic acid derivatives’ inhibition of glycogen synthase kinase-3β is the pharmacological basis of Kangen-Karyu in Alzheimer’s disease. Molecules 2018, 23, 2919. 10.3390/molecules23112919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodsell D. S.; Morris G. M.; Olson A. J. Automated docking of flexible ligands: applications of AutoDock. J. Mol. Recognit. 1996, 9, 1–5. . [DOI] [PubMed] [Google Scholar]
- Nachon F.; Carletti E.; Ronco C.; Trovaslet M.; Nicolet Y.; Jean L.; Renard P. Y. Crystal structures of human cholinesterases in complex with huprine W and tacrine: elements of specificity for anti-Alzheimer’s drugs targeting acetyl-and butyryl-cholinesterase. Biochem. J. 2013, 453, 393–399. 10.1042/BJ20130013. [DOI] [PubMed] [Google Scholar]
- Nicholls A.; McGaughey G. B.; Sheridan R. P.; Good A. C.; Warren G.; Mathieu M.; Muchmore S. W.; Brown S. P.; Grant J. A.; Haigh J. A.; Nevins N. Molecular shape and medicinal chemistry: a perspective. J. Med. Chem. 2010, 53, 3862–3886. 10.1021/jm900818s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naldi M.; Fiori J.; Pistolozzi M.; Drake A. F.; Bertucci C.; Wu R.; Mlynarczyk K.; Filipek S.; De Simone A.; Andrisano V. Amyloid β-peptide 25-35 self-assembly and its inhibition: a model undecapeptide system to gain atomistic and secondary structure details of the Alzheimer’s disease process and treatment. ACS Chem. Neurosci. 2012, 3, 952–962. 10.1021/cn3000982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paudel P.; Seong S. H.; Zhou Y.; Ha M. T.; Min B. S.; Jung H. A.; Choi J. S. Arylbenzofurans from the root bark of Morus alba as triple inhibitors of cholinesterase, β-site amyloid precursor protein cleaving enzyme 1, and glycogen synthase kinase-3β: Relevance to Alzheimer’s disease. ACS Omega 2019, 4, 6283–6294. 10.1021/acsomega.9b00198. [DOI] [PMC free article] [PubMed] [Google Scholar]