

Abstract
A well-behaved model chemistry previously validated for the study of the chemical reactivity of peptides was considered for the calculation of the molecular properties and structures of a group of five new antifungal tripeptides, namely (2R)-2-[(2S)-2-[(2S)-2-amino-3-phenylpropanamido]propanamido]-5-[(diaminomethylidene)amino]pentanoic acid, (2S)-2-[(2S)-2-[(2S)-2-amino-3-phenyl propanamido]propanamido]-3-(4-hydroxyphenyl)propanoic acid, (2S)-2-[(2S)-2-[(2S)-2-amino-3-phenylpropanamido]-3-methylbutanamido]-3-(4-hydroxyphenyl)propanoic acid, (2R)-2-[(2S)-2-[(2S)-2-amino-3-phenylpropanamido]-3-(1H-indol-3-yl)propanamido]-3-sulfanylpropanoic acid, and (2S)-2-[(2S)-2-[(2S)-2-amino-3-phenylpropanamido]-3-(1H-indol-3-yl)propanamido]-3-(4-hydroxyphenyl)propanoic acid, according to their amino acid sequences. A methodology based on conceptual density functional theory was chosen for the determination of the reactivity descriptors. The molecular active sites were associated with the active regions of the molecules that were associated with the nucleophilic, electrophilic, and radical Fukui functions. Additionally, the pKa values for the different peptides are predicted with great accuracy, which constitutes a useful knowledge for the process of drug design. Finally, the bioactivity scores for the new antifungal peptides are predicted through a homology methodology relating them with the calculated reactivity descriptors.
Introduction
It is well known nowadays that, within medicinal chemistry research, the design and development of new pharmaceutical drugs have a preponderant place. These drugs act by producing changes in some physiological process or function, exerting their effect by interacting specifically with some macromolecular structure of an organism which is called the receptor. In this way, the interaction of each drug with its respective receptor or site of action initiates the biochemical and physiological changes that are characteristic of that drug.1−3
Starting from the knowledge of the receptor associated to each disease, a protocol composed of a series of steps, generally known as computer-aided drug design, has been devised by researchers in this field over the past decades allowing us to understand the way by which the potential drugs interact with the corresponding sites of action, leading to the name of druggable receptors. Computers play a significant role in this protocol and, together with their associated software, have become an indispensable tool for performing these tasks with a guarantee of success.1−3
There are actually two variants of this protocol depending on whether the molecular structure of the druggable receptor is or is not known. When the structure of the receptor is already known, the following process is called structure-based drug design and has a long-standing record of already developed pharmaceutical drugs. However, when this knowledge is not available, the process is called ligand-based drug design, and it is associated with some computer-related techniques like pharmacophore modeling, quantitative structure activity relationship (QSAR), and three-dimensional (3D)-QSAR.
QSAR is one of the most important methodologies that can be used in the process of drug design and development.4−6 Its utility resides in the idea that the biological activity of a given molecule candidate, to be considered as a potential drug, can be expressed in the form of a mathematical formula relating this activity with a series of parameters called descriptors. Although there are hundreds of descriptors available from the specialized literature, for the objectives of the present work, we will be more interested in those properly called chemical reactivity descriptors which arise naturally from the conceptual density functional theory (CDFT), which allow to understand the interaction between molecules as well to have a comprehensive picture on the way that chemical reactions proceed.7−9
The reasons for this election are based on the assumption that relates the potential bioactivity of drugs with their chemical reactivity from the molecular point of view10,11 and also that when a group of descriptors cannot be cast in a QSAR mathematical formulation because of the limited availability of information, the knowledge of the chemical reactivity of a small group of molecules may still provide useful results that can serve as a guide for the purpose of the research.
One of the most important groups of molecules which have arisen during the last years as potential candidates to be considered as pharmaceutical drugs are that of peptides of marine origin. Although there have been some controversies in the literature about the possibilities of considering small peptides as drugs because of their limited bioavailability, it is also true that there has been an explosion of research in this field since the beginning of the 21st century because of the potential importance that peptides may have in the practice of medicinal chemistry.12−14
Assuming that an understanding of the chemical interactions is essential for the development of new pharmaceutical drugs, several recently synthesized small tripeptides have been chosen as the object of this study by considering that they could be the basis for the development of new therapeutic antifungal peptides.15 Thus, in this work, we will be studying the reactivity properties of five new antifungal tripeptides, namely (2R)-2-[(2S)-2-[(2S)-2-amino-3-phenylpropanamido]propanamido]-5-[(diaminomethylidene)amino]pentanoic acid, (2S)-2-[(2S)-2-[(2S)-2-amino-3-phenyl propanamido]propanamido]-3-(4-hydroxyphenyl)propanoic acid, (2S)-2-[(2S)-2-[(2S)-2-amino-3-phenylpropanamido]-3-methylbutanamido]-3-(4-hydroxyphenyl)propanoic acid, (2R)-2-[(2S)-2-[(2S)-2-amino-3-phenylpropanamido]-3-(1H-indol-3-yl)propanamido]-3-sulfanylpropanoic acid, and (2S)-2-[(2S)-2-[(2S)-2-amino-3-phenylpropanamido]-3-(1H-indol-3-yl)propanamido]-3-(4-hydroxyphenyl)pro-panoic acid, by resorting to the CDFT methodology, which will allow the determination of the global properties as well as the local properties for the prediction of the active reaction sites, both electrophilic and nucleophilic. The corresponding short names for these tripeptides are FAR, FAY, FVY, FWC, and FWY. In a similar way, the pKa values for each of the tripeptides will be calculated by resorting to a methodology previously considered by us,16 and the descriptors of bioactivity (bioactivity scores) will be established through a procedure described in the literature,17,18 trying to relate them with the global and local CDFT reactivity descriptors that result from a calculation protocol based on DFT already validated by our group in previous research.19−26
Computational Methodology
Following the methodology considered in our previous works,19−26 similar computations have been performed in this work by resorting to the Gaussian 09 software.27 The full methodological procedure is explained in detail at the beginning of the Results and Discussion section. In a similar way as it was done in the referenced works, the MN12SX density functional28 was considered because of the fact that it is a well-behaved density functional for our purposes according to our proposed KID (for Koopmans in DFT) criteria,19−26 related to the approximate validity of the Koopmans theorem within DFT.29−33 For the calculation of the electronic properties, a model chemistry has been considered on the basis of the MN12SX density functional associated to the Def2TZVP basis set, whereas a smaller Def2SVP was considered for the prediction of the most stable structures.34,35 All calculations were performed using water, which is the universal biological solvent, simulated with the Solvent Model Density (SMD) model.36
Results and Discussion
The molecular structures of the antifungal tripeptides FAR, FAY, FVY, FWC, and FWY drawn by scratch, as depicted in Figure 1, were optimized in gas phase by resorting to the Density Functional Tight Binding Model A (DFTBA) model, through the consideration of the five most stable conformers chosen from a preoptimization accomplished by means of molecular mechanics techniques37−41 using the conformer search engine available in the Marvin View 17.15 program, which can be regarded as an advanced chemical viewer (https://www.chemaxon.com). All the resulting conformers were processed, as is customary within computational chemistry, by means of a new reoptimization with the MN12SX density functional mentioned before, together with the Def2SVP basis set and the SMD solvent model, using water as the solvent. Once it has been verified that every structure belonged to the minimum energy conformation by means of a frequency calculation analysis, the corresponding electronic properties were calculated with the Def2TZVP basis set instead of that used for the geometry optimization.
Figure 1.

Graphical sketches of the molecular structures of the five antifungal tripeptides showing the numbering of the atoms: (a) FAR; (b) FAY; (c) FVY; (d) FWC; and (e) FWY, where green is used for C, red for O, blue for N, and gray for H.
As it has been mentioned recently by Becke42 and also by Baerends et al.,43 it can be said that the lowest excitation energy can be associated with the highest occupied molecular orbital (HOMO)–lowest unoccupied molecular orbital (LUMO) gap of the ground state.44 Therefore, in this work, the determination of the maximum wavelength absorption of the five antifungal tripeptides was done by conducting ground-state calculations with the aforementioned density functional at the same level of model chemistry and theory and then determining the HOMO–LUMO gaps from which the maximum absorption wavelengths λmax were obtained (Table 1).
Table 1. HOMO and LUMO Orbital Energies (in eV), the HOMO–LUMO Gap (Also in eV), and the Maximum Absorption Wavelengths λmax (in nm) of the Five Antifungal Tripeptides, FAR, FAY, FVY, FWC, and FWY, Predicted by the MN12SX/Def2TZVP/H2O Model Chemistry.
| HOMO | LUMO | HOMO–LUMO gap | λmax | |
|---|---|---|---|---|
| FAR | –7.051 | –2.838 | 4.213 | 294 |
| FAY | –6.223 | –3.285 | 2.938 | 422 |
| FVY | –6.254 | –1.753 | 4.501 | 275 |
| FWC | –5.634 | –1.733 | 3.901 | 318 |
| FWY | –5.634 | –3.184 | 2.450 | 506 |
Calculation of Global Reactivity Descriptors
According to the results obtained when studying melanoidins19−25 as well as peptides from marine sources,26 it can be said that the calculations performed with the MN12SX density functional render HOMO and LUMO energies that satisfy the approximate Koopmans theorem. Thus, the application of the KID procedure will be justified. The global reactivity descriptors, electronegativity χ,7,9 global hardness η,7,9 electrophilicity ω,45 electrodonating power ω–,46 electroaccepting power ω+,46 and net electrophilicity Δω±47 were calculated by resorting to the HOMO and LUMO energies determined with the MN12SX density functional, with the results being presented in Table 2. The interested reader in the mathematical formulations of these reactivity descriptors is referred to the original works and to our previous research on the field.19−26
Table 2. Global Reactivity Descriptors of the Five New Antifungal Tripeptides FAR, FAY, FVY, FWC, and FWY (in eV), Calculated with the MN12SX/Def2TZVP/H20 Model Chemistry.
| molecule | electronegativity | global hardness | electrophilicity |
|---|---|---|---|
| FAR | 4.945 | 4.212 | 2.902 |
| FAY | 4.754 | 2.938 | 3.846 |
| FVY | 4.003 | 4.501 | 1.780 |
| FWC | 3.683 | 3.901 | 1.739 |
| FWY | 4.409 | 2.450 | 3.968 |
| molecule | electrodonating power | electroaccepting power | net electrophilicity |
|---|---|---|---|
| FAR | 8.540 | 3.595 | 12.135 |
| FAY | 10.252 | 5.498 | 15.750 |
| FVY | 5.844 | 1.840 | 7.684 |
| FWC | 5.563 | 1.880 | 7.442 |
| FWY | 10.294 | 5.885 | 16.178 |
As expected from the molecular structure of these species, their electrodonating ability is more important than their electroaccepting character as can be seen from the values of the electrodonating and electroaccepting powers and their comparison through the net electrophilicity. However, an interesting comparison can be performed by taking into account the values for the global hardness which is a measure of the deformability of the molecular electron density and, hence, of the chemical reactivity. In this case, it can be observed that FVY and FWC are much more reactive than the other tripeptides. This is corroborated by the lower values of the global electrophilicity, that is, the balance between the chemical electronegativity and the global hardness, for those peptides.
Local Reactivity Descriptor Calculation
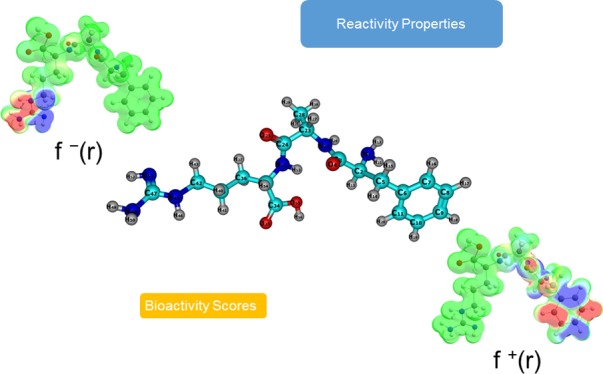
We now turn our attention to the local descriptors of chemical reactivity, namely the electrophilic Fukui function f–(r),7−9 the nucleophilic Fukui function f+(r),7−9 and the dual descriptor (DD) Δf(r).48−52 As for the case of the global reactivity descriptors, the interested reader in the mathematical formulations of these reactivity descriptors is referred to the original works and to our previous research on the field.19−26
The electrophilic Fukui functions f–(r) and nucleophilic Fukui functions f+(r) for the five new antifungal tripeptides FAR, FAY, FVY, FWC, and FWY are shown in Figure 2.
Figure 2.

Graphical representation of the electrophilic Fukui functions f–(r) (left column) and nucleophilic Fukui functions f+(r) (right column) of the five antifungal tripeptides.
Martínez-Araya has explained in a recent research52 that the condensed expression for DD as Δfk will be more useful for the prediction of the preferred sites of reaction than the condensed Fukui functions alone. For this reason, we have decided to present the results for the condensed DD Δfk in comparison with the nucleophilic and electrophilic Parr functions, Pk+ and Pk–, proposed by Domingo et al.53,54 through the consideration of atomic spin densities that result from the Mulliken population analysis (MPA).
The definitions for the Parr functions are as follows:
Nucleophilic Parr function53,54
Electrophilic Parr function53,54
where ρsrc(r) and ρs(r) are related to the atomic spin density of the radical cation or anion of the considered system, respectively.55
The results for the calculation of these local reactivity descriptors for the five antifungal tripeptides FAR, FAY, FVY, FWC, and FWY are presented in Table 3 where the condensed DD Δfk has been determined by localizing the corresponding Fukui functions over the atomic sites, employing a charge scheme based on MPA as it was done for the Parr functions. It must be noticed that we are presenting the results only for those atomic sites where Δfk are maxima in absolute value. The values of Δfk are multiplied by 100 for easier comparison.
Table 3. Local Reactivity Descriptors for the Antifungal Tripeptides FAR, FAY, FVY, FWC, and FWY, Calculated with the MN12SX/Def2TZVP/H2O Model Chemistry: Condensed DD Δfk, Nucleophilic Parr Function Pk+, and Electrophilic Parr Function Pk–
| atom | Δfk | Pk+ | Pk– |
|---|---|---|---|
| FAR | |||
| 11C | 26.62 | 0.383 | 0.000 |
| 51N | –51.22 | 0.000 | 0.713 |
| FAY | |||
| 29C | –22.96 | 0.013 | 0.459 |
| 41C | 12.68 | 0.316 | 0.126 |
| FVY | |||
| 8C | 8.23 | 0.145 | 0.002 |
| 45C | –23.25 | 0.038 | 0.384 |
| FWC | |||
| 29C | –22.55 | 0.001 | 0.434 |
| 41C | 15.14 | 0.325 | 0.084 |
| FWY | |||
| 29C | –22.96 | 0.013 | 0.459 |
| 41C | 12.68 | 0.316 | 0.126 |
As can be seen from Table 3, there is a good agreement between the results that come from the condensed DD Δfk and those obtained through the nucleophilic and electrophilic Parr functions Pk+ and Pk–. Thus, it can be expected that the methodology used in this work could be the basis for the study of the chemical reactivity of therapeutic peptides of larger size. Moreover, by comparing the results from Table 3 and the graphics in Figure 2, it can be concluded that there is a perfect match for both kinds of analysis.
Calculation of the pKa’s of the Five Antifungal Tripeptides
In a recent work, the relationship between the pKa’s of small peptides and the chemical hardness was developed in our group,16 leading to the conclusion that it represented a starting point for the prediction of the pKa of bigger peptides which could be of interest for the development of new therapeutic drugs.
According to the methodology employed in our previous work, we have applied the mentioned relationship of the form pKa = 16.3088 – 0.8268η to the calculation of the pKa of the new tripeptides, with the η values presented in Table 4.
Table 4. pKa’s of the Antifungal Tripeptides FAR, FAY, FVY, FWC, and FWY.
| molecule | pKa |
|---|---|
| FAR | 12.83 |
| FAY | 13.88 |
| FVY | 12.59 |
| FWC | 13.08 |
| FWY | 11.44 |
It is good to see from these results that this approximate relationship allows us to discriminate between the pKa results for the tripeptides. This could be of interest for the development of pharmaceutical drugs starting from these molecules, enabling at the same time to obtain an explanation about the mechanisms of action and drug delivery procedures. Moreover, they can be shown as an additional application of the results of the calculation of the global reactivity descriptors to the new field of computational peptidology56 and as a possible basis for explaining the solubilities of the peptides.
Bioactivity Scores
The properties of the molecules that are identified with the idea of their ability to behave as a pharmaceutical drug are those which are related to the proposal by Lipinski et al.57,58 for the prediction of druggability and have been determined by associating the SMILES notations corresponding to each tripeptide with the MolInspiration software which is readily available online (Slovensky Grob, Slovak Republic https://www.molinspiration.com).
Indeed, this criterion which is called the Lipinski Rule of Five, or Ro5 for short, does not apply to peptides. Then, a homology modeling approach was considered by searching for structures with known pharmacological properties that could be compared with those that are the object of our study. Thus, a series of descriptors called bioactivity scores which are an indication of the capacity of the potential drugs to interact with different receptors, such as G protein-coupled receptor (GPCR) ligands or kinase inhibitors, to act as ion channel modulators, or to interact with enzymes and nuclear receptors. It must be recalled that GPCRs are the largest family of signaling receptors in the human genome and also are the largest class of targets of approved drugs. The values of the bioactivity scores for the five antimicrobial peptides are presented in Table 5.
Table 5. Bioactivity Scores of the Antimicrobial Peptides FAR, FAY, FVY, FWC, and FWY Calculated on the Basis of the GPCR Ligand, Ion Channel Modulator, Nuclear Receptor Ligand, Kinase Inhibitor, Protease Inhibitor, and Enzyme Inhibitor Interactions.
| molecule | GPCR ligand | ion channel modulator | kinase inhibitor | nuclear receptor ligand | protease inhibitor | enzyme inhibitor |
|---|---|---|---|---|---|---|
| FAR | 0.42 | 0.24 | 0.08 | 0.08 | 0.64 | 0.47 |
| FAY | 0.35 | 0.18 | 0.10 | 0.17 | 0.67 | 0.38 |
| FVY | 0.32 | 0.15 | 0.07 | 0.15 | 0.67 | 0.36 |
| FWC | 0.43 | 0.29 | 0.09 | 0.07 | 1.09 | 0.51 |
| FWY | 0.39 | 0.21 | 0.07 | 0.11 | 0.78 | 0.35 |
The interpretation of the bioactivity scores is based on a scheme that tends to classify them as active, moderately active, or inactive, depending on the obtained values. If the bioactivity score is >0, it will correspond to the first case; if the bioactivity score lies between −5.0 and 0.0, it will belong to the second case; and if the bioactivity score is <−5.0, it will be assigned to the third case. It can be seen that although the antifungal tripeptides considered in this work may act as GPCR ligands and as ion channel modulators, their main bioactivity is related to their ability to act as protease inhibitors. It is also evident that for the FWC peptide, this ability is larger than for the other tripeptides. Although this behavior cannot be cast into a QSAR equation because the number of results is low, it can be however related to the values of the global descriptors for this particular tripeptide. From Table 2, it can be seen that for FWC, the values of the electronegativity χ, the electrophilicity ω, the electrodonating and electroaccepting powers, ω– and ω+, and the net electrophilicity Δω± attain minimum values. Thus, it can be said that, as an approximation, the protease inhibition ability for these antifungal tripeptides has a behavior related to the inverse of the mentioned global reactivity descriptors.
Conclusions
In this work, the chemical reactivity of a group of five recently synthesized antifungal tripeptides with therapeutic potential, FAR, FAY, FVY, FWC, and FWY, was studied by resorting to CDFT as a tool to explain the molecular interactions.
The information about the global and local reactivity descriptors of the tripeptides acquired in this work could be helpful to assist in the design of new pharmaceutical drugs based on these compounds.
Among the many descriptors that could be useful for the development of new medicines, pKa is of paramount importance because it is related to the water solubility of the drugs. Thus, when the experimental values of pKa are unknown, the approximate QSAR relationship employed in this work could be a useful predictive tool for the determination of the pKa’s of small and large peptides.
Finally, the molecular properties related to the bioavailability and the descriptors used for the quantification of the bioactivity allowed the characterization of the studied antifungal tripeptides, establishing some relationships between the bioactivity properties and the calculated global reactivity descriptors.
Acknowledgments
N.F.-H. and D.G.-M. are researchers of CIMAV and CONACYT from which the partial support through grant 219566/2014 is gratefully acknowledged. D.G.-M. conducted this work as a Visiting Lecturer at the University of the Balearic Islands. This work was also cofunded by the Ministerio de Economía y Competitividad (MINECO) and the European Fund for Regional Development (FEDER) through grant CTQ2014-55835-R.
Author Contributions
D.G.-M. conceived and designed the research and headed, wrote, and revised the manuscript, and J.F. and N.F.-H. contributed to the writing and the revision of the article.
The authors declare no competing financial interest.
References
- Young D. C.Computational Drug Design—A Guide for Computational and Medicinal Chemists; John Wiley & Sons: Hoboken, NJ, 2009. [Google Scholar]
- Drug Design and Discovery—Methods and Protocols; Satyanarayanajois S. D., Ed.; Humana Press: New York, 2011. [Google Scholar]
- Computational Medicinal Chemistry for Drug Discovery; Bultinck P., De Winter H., Langenaeker W., Tollenaere J. P., Eds.; Marcel Dekker, Inc: New York, 2004. [Google Scholar]
- Roy K.; Kar S.; Das N.. A Primer on QSAR/QSPR Modeling—Fundamental Concepts; Springer: Cham, Switzerland, 2015. [Google Scholar]
- Advances in QSAR Modeling—Applications in Pharmaceutical, Chemical, Food, Agricultural and Environmental Sciences; Roy K., Ed.; Springer International Publishing AG: Cham, Switzerland, 2017. [Google Scholar]
- Roy K.; Kar S.; Das R. N.. Understanding the Basics of QSAR for Applications in Pharmaceutical Sciences and Risk Assessment; Academic Press: London, U.K., 2015. [Google Scholar]
- Parr R.; Yang W.. Density-Functional Theory of Atoms and Molecules; Oxford University Press: New York, 1989. [Google Scholar]
- Chermette H. Chemical Reactivity Indexes in Density Functional Theory. J. Comput. Chem. 1999, 20, 129–154. . [DOI] [Google Scholar]
- Geerlings P.; De Proft F.; Langenaeker W. Conceptual Density Functional Theory. Chem. Rev. 2003, 103, 1793–1874. 10.1021/cr990029p. [DOI] [PubMed] [Google Scholar]
- Rekka E.; Kourounakis P.. Chemistry and Molecular Aspects of Drug Design and Action; CRC Press: Boca Raton, 2008. [Google Scholar]
- Náray-Szabó G.; Warshel A.. Computational Approaches to Biochemical Reactivity; Kluwer Academic Publishers: New York, 2002. [Google Scholar]
- Peptides as Drugs; Groner B., Ed.; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2009. [Google Scholar]
- Peptide Drug Discovery and Development—Translational Research in Academy and Industry; Castanho M., Santos N. C., Eds.; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2011. [Google Scholar]
- Peptide Chemistry and Drug Design; Dun B. M., Ed.; John Wiley & Sons, Inc: Hoboken, NJ, 2015. [Google Scholar]
- Gill K.; Kumar S.; Xess I.; Dey S. Novel Synthetic Anti-fungal Tripeptide Effective against Candida krusei. Indian J. Med. Microbiol. 2015, 33, 110–116. 10.4103/0255-0857.148404. [DOI] [PubMed] [Google Scholar]
- Frau J.; Hernández-Haro N.; Glossman-Mitnik D. Computational Prediction of the pKas of Small Peptides through Conceptual DFT Descriptors. Chem. Phys. Lett. 2017, 671, 138–141. 10.1016/j.cplett.2017.01.038. [DOI] [Google Scholar]
- Gupta G. K.; Kumar V.. Chemical Drug Design; Walter de Gruyter GmbH: Berlin, 2016. [Google Scholar]
- Gore M.; Jagtap U. B.. Computational Drug Discovery and Design; Springer Science+Business Media, LLC: New York, 2018. [Google Scholar]
- Frau J.; Glossman-Mitnik D. Molecular Reactivity and Absorption Properties of Melanoidin Blue-G1 through Conceptual DFT. Molecules 2018, 23, 559–615. 10.3390/molecules23030559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frau J.; Glossman-Mitnik D. Conceptual DFT Study of the Local Chemical Reactivity of the Dilysyldipyrrolones A and B Intermediate Melanoidins. Theor. Chem. Acc. 2018, 137, 67. 10.1007/s00214-018-2244-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frau J.; Glossman-Mitnik D. Conceptual DFT Study of the Local Chemical Reactivity of the Colored BISARG Melanoidin and Its Protonated Derivative. Front. Chem. 2018, 6, 136. 10.3389/fchem.2018.00136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frau J.; Glossman-Mitnik D. Molecular Reactivity of some Maillard Reaction Products Studied through Conceptual DFT. Contemp. Chem. 2018, 1, 1–14. [Google Scholar]
- Frau J.; Glossman-Mitnik D. Computational Study of the Chemical Reactivity of the Blue-M1 Intermediate Melanoidin. Comput. Theor. Chem. 2018, 1134, 22–29. 10.1016/j.comptc.2018.04.018. [DOI] [Google Scholar]
- Frau J.; Glossman-Mitnik D. Chemical Reactivity Theory Applied to the Calculation of the Local Reactivity Descriptors of a Colored Maillard Reaction Product. Chem. Sci. Int. J. 2018, 22, 1–14. 10.9734/csji/2018/41452. [DOI] [Google Scholar]
- Frau J.; Glossman-Mitnik D. Blue M2: An Intermediate Melanoidin Studied via Conceptual DFT. J. Mol. Model. 2018, 24, 138. 10.1007/s00894-018-3673-0. [DOI] [PubMed] [Google Scholar]
- Frau J.; Flores-Holguín N.; Glossman-Mitnik D. Chemical Reactivity Properties, pKa Values, AGEs Inhibitor Abilities and Bioactivity Scores of the Mirabamides A–H Peptides of Marine Origin Studied by Means of Conceptual DFT. Mar. Drugs 2018, 16, 302–319. 10.3390/md16090302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frisch M. J.; et al. Gaussian 09, Revision E.01; Gaussian Inc.: Wallingford CT, 2016.
- Peverati R.; Truhlar D. G. Screened-Exchange Density Functionals with Broad Accuracy for Chemistry and Solid-State Physics. Phys. Chem. Chem. Phys. 2012, 14, 16187–16191. 10.1039/c2cp42576a. [DOI] [PubMed] [Google Scholar]
- Borghi G.; Ferretti A.; Nguyen N. L.; Dabo I.; Marzari N. Koopmans-compliant Functionals and Their Performance Against Reference Molecular Data. Phys. Rev. B: Condens. Matter Mater. Phys. 2014, 90, 075135. 10.1103/physrevb.90.075135. [DOI] [Google Scholar]
- Dabo I.; Ferretti A.; Poilvert N.; Li Y.; Marzari N.; Cococcioni M. Koopmans’ Condition for Density-Functional Theory. Phys. Rev. B: Condens. Matter Mater. Phys. 2010, 82, 115121. 10.1103/physrevb.82.115121. [DOI] [Google Scholar]
- Kar R.; Song J.-W.; Hirao K. Long-Range Corrected Functionals Satisfy Koopmans’ Theorem: Calculation of Correlation and Relaxation Energies. J. Comput. Chem. 2013, 34, 958–964. 10.1002/jcc.23222. [DOI] [PubMed] [Google Scholar]
- Salzner U.; Baer R. Koopmans’ Springs to Life. J. Chem. Phys. 2009, 131, 231101. 10.1063/1.3269030. [DOI] [PubMed] [Google Scholar]
- Vanfleteren D.; Van Neck D.; Ayers P. W.; Morrison R. C.; Bultinck P. Exact Ionization Potentials From Wavefunction Asymptotics: the Extended Koopmans’ Theorem, Revisited. J. Chem. Phys. 2009, 130, 194104. 10.1063/1.3130044. [DOI] [PubMed] [Google Scholar]
- Weigend F.; Ahlrichs R. Balanced Basis Sets of Split Valence, Triple Zeta Valence and Quadruple Zeta Valence Quality for H to Rn: Design and Assessment of Accuracy. Phys. Chem. Chem. Phys. 2005, 7, 3297–3305. 10.1039/b508541a. [DOI] [PubMed] [Google Scholar]
- Weigend F. Accurate Coulomb-fitting basis sets for H to Rn. Phys. Chem. Chem. Phys. 2006, 8, 1057–1065. 10.1039/b515623h. [DOI] [PubMed] [Google Scholar]
- Marenich A. V.; Cramer C. J.; Truhlar D. G. Universal Solvation Model Based on Solute Electron Density and a Continuum Model of the Solvent Defined by the Bulk Dielectric Constant and Atomic Surface Tensions. J. Phys. Chem. B 2009, 113, 6378–6396. 10.1021/jp810292n. [DOI] [PubMed] [Google Scholar]
- Halgren T. A. Merck Molecular Force Field. I. Basis, Form, Scope, Parameterization, and Performance of MMFF94. J. Comput. Chem. 1996, 17, 490–519. . [DOI] [Google Scholar]
- Halgren T. A. Merck Molecular Force Field. II. MMFF94 van der Waals and Electrostatic Parameters for Intermolecular Interactions. J. Comput. Chem. 1996, 17, 520–552. . [DOI] [Google Scholar]
- Halgren T. A. MMFF VI. MMFF94s Option for Energy Minimization Studies. J. Comput. Chem. 1999, 20, 720–729. . [DOI] [PubMed] [Google Scholar]
- Halgren T. A.; Nachbar R. B. Merck Molecular Force Field. IV. Conformational Energies and Geometries for MMFF94. J. Comput. Chem. 1996, 17, 587–615. . [DOI] [Google Scholar]
- Halgren T. A. Merck Molecular Force field. V. Extension of MMFF94 Using Experimental Data, Additional Computational Data, and Empirical Rules. J. Comput. Chem. 1996, 17, 616–641. . [DOI] [Google Scholar]
- Becke A. D. Vertical Excitation Energies From the Adiabatic Connection. J. Chem. Phys. 2016, 145, 194107. 10.1063/1.4967813. [DOI] [PubMed] [Google Scholar]
- Baerends E. J.; Gritsenko O. V.; van Meer R. The Kohn-Sham Gap, the Fundamental Gap and the Optical Gap: The Physical Meaning of Occupied and Virtual Kohn-Sham Orbital Energies. Phys. Chem. Chem. Phys. 2013, 15, 16408–16425. 10.1039/c3cp52547c. [DOI] [PubMed] [Google Scholar]
- van Meer R.; Gritsenko O. V.; Baerends E. J. Physical Meaning of Virtual Kohn-Sham Orbitals and Orbital Energies: An Ideal Basis for the Description of Molecular Excitations. J. Chem. Theory Comput. 2014, 10, 4432–4441. 10.1021/ct500727c. [DOI] [PubMed] [Google Scholar]
- Parr R. G.; Szentpály L. v.; Liu S. Electrophilicity Index. J. Am. Chem. Soc. 1999, 121, 1922–1924. 10.1021/ja983494x. [DOI] [Google Scholar]
- Gázquez J. L.; Cedillo A.; Vela A. Electrodonating and Electroaccepting Powers. J. Phys. Chem. A 2007, 111, 1966–1970. 10.1021/jp065459f. [DOI] [PubMed] [Google Scholar]
- Chattaraj P. K.; Chakraborty A.; Giri S. Net Electrophilicity. J. Phys. Chem. A 2009, 113, 10068–10074. 10.1021/jp904674x. [DOI] [PubMed] [Google Scholar]
- Morell C.; Grand A.; Toro-Labbé A. New Dual Descriptor for Chemical Reactivity. J. Phys. Chem. A 2005, 109, 205–212. 10.1021/jp046577a. [DOI] [PubMed] [Google Scholar]
- Morell C.; Grand A.; Toro-Labbé A. Theoretical Support for Using the Δf(r) Descriptor. Chem. Phys. Lett. 2006, 425, 342–346. 10.1016/j.cplett.2006.05.003. [DOI] [Google Scholar]
- Martínez-Araya J. I. Revisiting Caffeate’s Capabilities as a Complexation Agent to Silver Cation in Mining Processes by means of the Dual Descriptor – A Conceptual DFT Approach. J. Mol. Model. 2012, 18, 4299–4307. 10.1007/s00894-012-1405-4. [DOI] [PubMed] [Google Scholar]
- Martínez-Araya J. I. Explaining Reaction Mechanisms Using the Dual Descriptor: A Complementary Tool to the Molecular Electrostatic Potential. J. Mol. Model. 2013, 19, 2715–2722. 10.1007/s00894-012-1520-2. [DOI] [PubMed] [Google Scholar]
- Martínez-Araya J. I. Why is the Dual Descriptor a More Accurate Local Reactivity Descriptor than Fukui Functions?. J. Math. Chem. 2015, 53, 451–465. 10.1007/s10910-014-0437-7. [DOI] [Google Scholar]
- Domingo L. R.; Pérez P.; Sáez J. A. Understanding the Local Reactivity in Polar Organic Reactions through Electrophilic and Nucleophilic Parr Functions. RSC Adv. 2013, 3, 1486–1494. 10.1039/c2ra22886f. [DOI] [Google Scholar]
- Chamorro E.; Pérez P.; Domingo L. R. On the Nature of Parr Functions to Predict the Most Reactive Sites along Organic Polar Reactions. Chem. Phys. Lett. 2013, 582, 141–143. 10.1016/j.cplett.2013.07.020. [DOI] [Google Scholar]
- Domingo L.; Ríos-Gutiérrez M.; Pérez P. Applications of the Conceptual Density Functional Theory Indices to Organic Chemistry Reactivity. Molecules 2016, 21, 748. 10.3390/molecules21060748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Computational Peptidology; Zhou P., Huang J., Eds.; Humana Press: New York, 2015. [Google Scholar]
- Lipinski C. A.; Lombardo F.; Dominy B. W.; Feeney P. J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings 1PII of original article: S0169-409X(96)00423-1. The article was originally published in Advanced Drug Delivery Reviews 23 (1997) 3-25. 1. Adv. Drug Delivery Rev. 2001, 46, 3–26. 10.1016/s0169-409x(00)00129-0. [DOI] [PubMed] [Google Scholar]
- Leeson P. Chemical beauty contest. Nature 2012, 481, 455–456. 10.1038/481455a. [DOI] [PubMed] [Google Scholar]