

Abstract

Repetitive DNA sequences are abundant in the genome of most biological species. These sequences are naturally “preamplified”, which makes them a preferential target for a variety of biological assays. Current methods to detect specific DNA sequences are based on the quantitative polymerase chain reaction (PCR), which relies on target amplification by Taq polymerase and uses a fluorescent resonance energy transfer (FRET)-based probe. Here, to rapidly detect a repetitive DNA sequence, we combine a highly sensitive magnetic modulation biosensing (MMB) system and a modified double-quenched FRET-based probe. The high numbers of copies of the female-specific XhoI sequence of the domestic chicken (Gallus gallus), combined with the low background fluorescence levels of the modified double-quenched probe and the high sensitivity of the MMB system, allow us to determine the chick sex in ovo within 13 min, with 100% sensitivity and specificity. Compared to quantitative PCR, the presented assay is 4–9 times faster. More broadly, by specifically tailoring the primers and probe, the proposed assay can detect any target DNA sequence, either repetitive or nonrepetitive.
Introduction
Shaped through billions of years of evolution, the genomes of almost all living creatures—from bacteria to plants and animals—have accumulated substantial quantities of repetitive nucleic acid sequences.1−3 These sequences can appear throughout the genome in exceptionally high numbers of copies, ranging from hundreds to tens of thousands of repetitions. They are divided into groups, primarily based on their size, location, number of copies, and mobility.3 Some of them (e.g., ribosomal genes) are fully functional genes, highly conserved, and serve a specific purpose.4 Others have no specific function and may be either mobile or dispersed through the entire genome (e.g., transposable elements) or distributed in blocks in one or more defined locations on the chromosomes (e.g., tandem repeats). Some structural elements of the chromosomes, such as centromeres and telomeres, are also composed of short repetitive elements.3
To date, significant parts of the genomes that contain long stretches of repetitive motifs remain poorly mapped.5 These areas are frequently regarded as “junk” DNA and are either marked as “undefined” or completely omitted from the genome map. Nevertheless, apparent lack of function does not necessarily mean that such junk DNA is entirely useless. For instance, modern forensic science extensively relies on the detection of short tandem repeats polymorphism for DNA “fingerprinting”6 and paternity tests.7 Moreover, specific repetitive DNA sequences can be used to discriminate closely related species8 and to perform sex-typing of birds.9
Specific DNA sequences are commonly detected using conventional polymerase chain reaction (PCR), which is highly sensitive, but presents several challenges. First, conventional PCR is very slow (e.g., target amplification and analysis of the results by gel electrophoresis takes up to 2.5 h).10 Second, possible polymorphisms of the target sequence or amplification of unrelated sequences similar in length to the target can result in false identification.11 Third, when repetitive sequences are used as targets for PCR amplification, amplicons of multiple lengths are expected, thereby making it difficult to conclusively interpret the results.12,13
A TaqMan quantitative PCR (qPCR) assay overcomes some of these challenges.14 Unlike conventional PCR, a TaqMan assay requires three independent binding events (two primers and a probe), which significantly improves the specificity of the assay and reduces the probability of false identification. In a TaqMan assay, a fluorescent resonance energy transfer (FRET)-based probe (i.e., a short oligonucleotide labeled with a fluorescent molecule at the 5′ end and a quencher molecule at the 3′ end) is degraded by advancing Taq polymerase, which physically separates the fluorescent molecule from the quencher. Subsequently, the fluorescent signal increases with each PCR cycle. Direct optical detection of the fluorescent signal eliminates the need for additional analytical steps, such as gel electrophoresis, and thereby shortens the duration of the test.
In general, in a standard FRET-based probe, the fluorescent and the quencher molecules are located 20–23 bases apart. Hence, the quenching efficiency, which is inversely proportional to the sixth power of the distance between the fluorescent and quencher molecules, is low.15 Even in its quenched state, a standard FRET-based probe has a relatively high fluorescent background. Shortening the probes (e.g., to 15–18 bases) may improve the quenching efficiency, but will compromise the probe specificity. Recently, Wilson et al. have employed a novel FRET-based probe, termed a double-quenched ZEN probe,16,17 which uses two quenchers instead of a single quencher, thus reducing the distance between the fluorophore and the quencher to only nine bases. Compared to the standard single-quenched FRET probes, the double-quenched ZEN probe has ∼90% lower background fluorescence, which facilitates the detection of target DNA sequences and requires one to two fewer amplification cycles.17
Here, we propose a modified double-quenched ZEN probe that further reduces the number of PCR cycles required to detect repetitive nucleic acid sequences. The modified double-quenched probe has a biotin on the same 5′ nucleotide as the fluorescent molecule. Hence, following separation of the fluorescent molecule from the quenchers by Taq polymerase activity, the fluorescent molecule remains attached to the biotin molecule. At this point, streptavidin-coupled magnetic beads are added to the sample and capture the biotinylated fluorescent molecules. To increase the sensitivity of fluorescence detection, an oscillating external magnetic field gradient is applied to the sample, attracting the beads from the entire solution volume and concentrating them into a small detection area. To separate the fluorescent signal from the background noise of unbound fluorescent molecules, the oscillating magnetic field gradient moves the bead aggregate from one side to other, in and out of a laser beam. The resulting flashing signal is clearly distinguishable from the constant background noise, and the amplitude of the oscillating signal is proportional to the concentration of target molecules. Aggregating the magnetic beads attached to target molecules and modulating them to increase sensitivity are the basic principles of a novel optical detection technology, termed magnetic modulation biosensing (MMB). While the principles and characteristics of the MMB system have been demonstrated before,18,19 it has never been used to detect repetitive DNA sequences or to work with a modified double-quenched probe.
To demonstrate the advantages of the modified double-quenched probe in rapid and sensitive detection of repetitive DNA sequences, in this work, we target the female-specific repetitive sequences of the chick embryo and address the challenges of chick sexing in ovo. In most bird species, visually determining the sex of newly hatched chicks is challenging.20 This problem is particularly acute in the laying hens industry, where male chicks are sorted out and disposed of on the day of hatching.21 The cruelty of the methods utilized for the disposal of male chicks has created a widely discussed ethical problem.22 This issue can be resolved by determining the chick’s sex in ovo in the early stages of embryonic development and then disposing of unhatched eggs containing male embryos.
In birds, males are the homogametic sex, having two Z chromosomes, whereas females are the heterogametic sex, having one Z chromosome and one W chromosome.23 The majority of the W chromosome (up to 90%) is occupied by an extremely high number of repetitive sequences.24,25 Because repetitive sequences of the W chromosome (e.g., XhoI, EcoRI) are found in more than 5000 copies per female genome, they can be used as potential targets26 for a chick sexing assay. Compared to the standard ZEN probe, the modified double-quenched probe combined with the MMB detection technology allows using four to five fewer PCR cycles and can reliably detect the chick sex in less than 15 min.
Results and Discussion
The results of adult bird sexing by detecting a fragment of the XhoI repetitive sequence using an MMB system and a qPCR system are presented in Figure 1. Using the MMB system, the fluorescent signal after five amplification cycles was similar for both male and female samples. However, after 10 amplification cycles, the fluorescent signal was 100% higher for females than for males. The extrapolated threshold that allows discrimination between male and female birds is ca. 6–8 PCR cycles (Figure 1a). Using the qPCR system, a significant difference in signal between males and females is detected after at least ca. 12–13 cycles (Figure 1b). No significant signal amplification is observed for any of the male or negative control samples.
Figure 1.
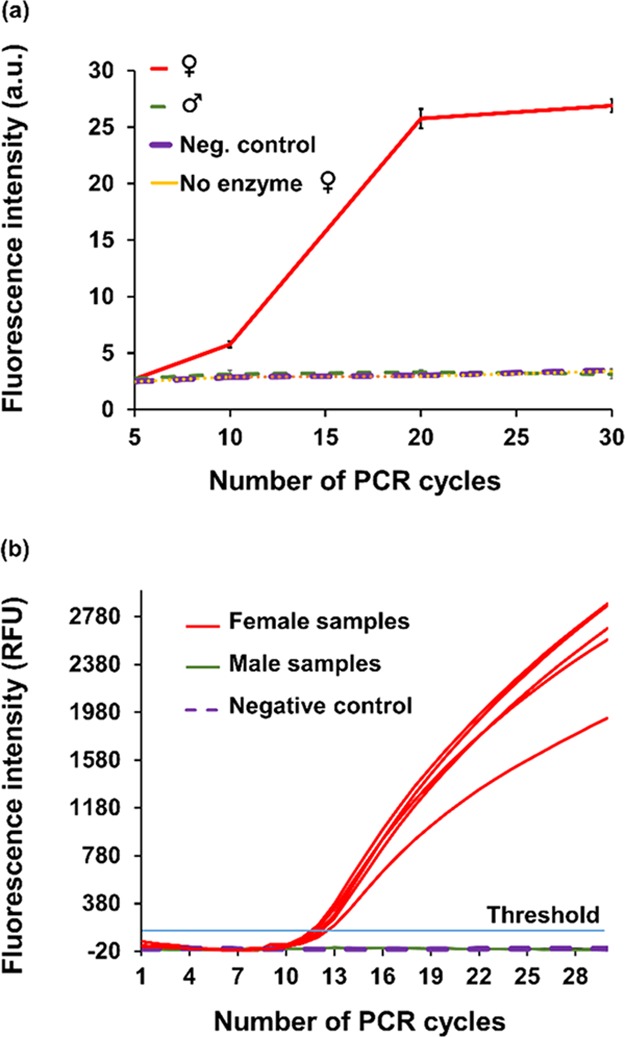
Adult bird sexing by detecting a fragment of the XhoI repetitive sequence using modified double-quenched probes. Fluorescent signal as a function of the number of PCR cycles, measured using (a) the MMB system and (b) qPCR system. The total number of samples tested was 12. Shown are five adult females (solid red), five adult males (dashed green), a negative control without target DNA (dashed violet), and a negative control without Taq polymerase enzyme (dotted orange). The error bars for the male and female samples represent the standard deviation of five independent samples (n = 5). The error bars for the negative controls represent the standard deviation of three measurements (n = 3).
Sex determination of 16 chick embryos samples by conventional PCR, which is the gold standard of molecular-based chick sexing, is presented in Figure 2. All of the amplified sequences were at the expected size of the female-specific nonrepetitive HUR0417 sequence (642 bp).27 Samples with a band of the expected size were considered as female. No nonspecific amplification was observed in any of the male or negative control samples. Overall, out of 25 samples tested, 13 were identified as females and 12 were identified as males.
Figure 2.
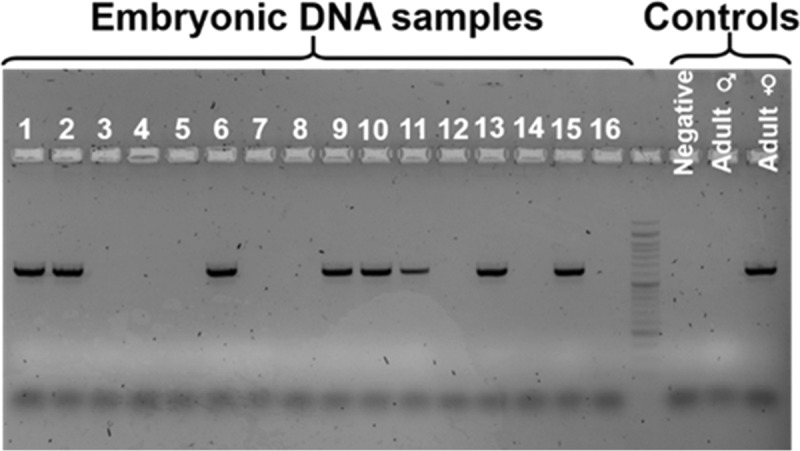
Sex determination of chick embryos using conventional PCR. A PCR test, targeting a female-specific nonrepetitive HUR0417 sequence, is used as a gold standard to determine the chick embryos’ sex. All of the amplified sequences were at the expected size (642 bp). Samples with a band at the expected size were considered as female. No nonspecific amplification was observed in any of the control samples. A total of 16 samples are presented (out of 25 samples that were tested).
Figure 3 presents comparative results for sex determination of chick embryos using qPCR and the MMB system. Using qPCR, to positively identify all female samples, at least 13 cycles are required (Figure 3a). Using the MMB system, sex determination with 100% specificity (all of the female samples are identified as females) and 100% selectivity (no male samples are identified as females) is achieved after only eight cycles (Figure 3b). These results are identical to the results obtained using the adult birds’ DNA (Figure 1b), in which a minimum of 13 cycles were required for qPCR to achieve 100% specific sex determination.
Figure 3.
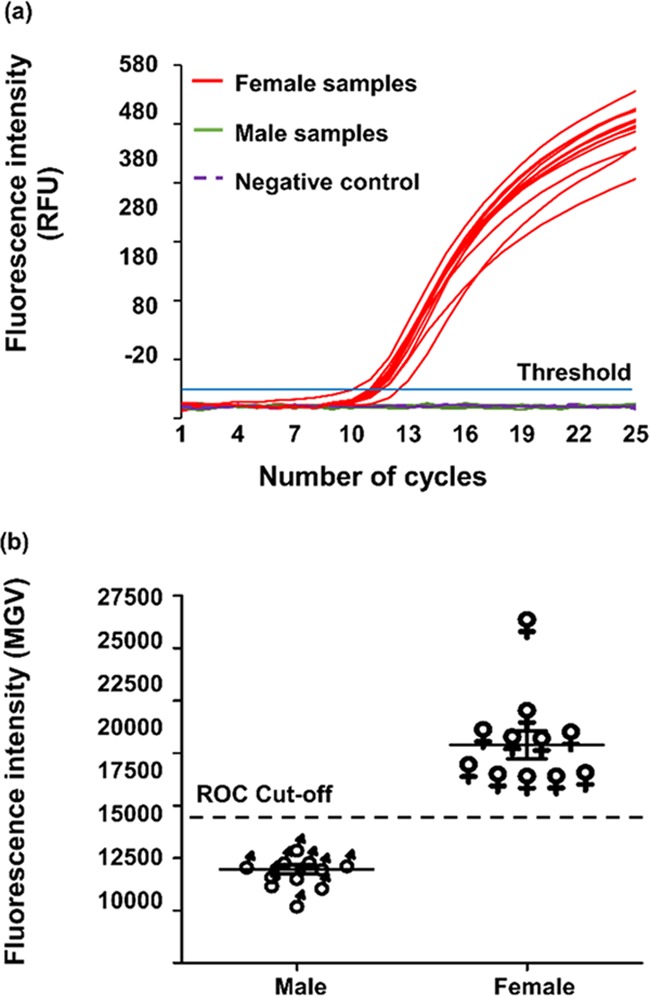
Sex determination of chick embryos using qPCR and the MMB system. A total of 12 male and 13 female samples were tested using (a) qPCR and (b) the MMB system. Using qPCR, to positively identify all female samples, at least 13 cycles are required. Using the MMB system, after eight cycles, the receiver operating characteristic cutoff value allows sex determination with 100% sensitivity (all of the female samples are identified as females) and 100% specificity (all of the male samples are identified as males).
Detection of repetitive DNA sequences is required in various fields of biological research, such as clinical diagnosis and agriculture.28,29 The natural “preamplification” of the target DNA sequence facilitates detection of the target sequence at low concentrations, either without or with a minimal number of PCR amplification cycles, and it subsequently shortens the total duration of the assay. Quantitative PCR combined with a TaqMan-based assay is relatively simple and does not require additional analytical steps (e.g., gel electrophoresis). To further improve the sensitivity of qPCR, double-quenched ZEN probes were developed. These probes reduce the background noise caused by the suboptimal quenching of the single quencher in a standard TaqMan assay.
Here, to further reduce the detection time of repetitive DNA sequences, we introduced a modified double-quenched FRET-based assay that incorporates two major modifications to the established TaqMan methodology. First, similar to the double-quenched ZEN probe, the modified double-quenched probe has two quenching molecules that reduce the background noise. Second, the modified double-quenched probe has a biotin molecule on the 5′ end, which allows it to be conjugated to streptavidin-coupled magnetic beads and be detected by the MMB system. The MMB system significantly improves the detection sensitivity and further reduces the number of PCR amplification cycles required for reliable detection of the repetitive DNA sequences. The lower number of amplification cycles also reduces the possibility of nonspecific amplification and subsequently the probability of false-positive results.
The MMB system, combined with the modified double-quenched probe, identified the chick sex in ovo with 100% sensitivity and specificity after eight PCR amplification cycles. In comparison, the gold standard qPCR discriminated female from male chicks after 13 PCR amplification cycles. In practice, for qPCR, due to the requirement to complete the entire testing program (∼30 cycles) prior to performing the final analysis of the results, the testing time is 1–2 h. For example, in a recent study that used qPCR for chick sexing and targeted the XhoI repetitive sequence, the total assay time was ∼2 h.30
Amplification of a long-target DNA sequence, such as the 717 bp XhoI sequence, is time-consuming. Amplification of a short portion of the target is faster, but may compromise the specificity of the assay. The intrinsic high specificity of FRET-based assays, which include two primers and one probe (e.g., TaqMan or ZEN probes), enable amplification and detection of shorter-target DNA sequences (in the 120–140 bp range) without compromising specificity. High processivity of the modern Taq polymerases (∼20 bases/s) facilitates the amplification of such short stretches of DNA in a matter of seconds. Therefore, in the modified double-quenched FRET-based assay, the length of a single PCR cycle was reduced to 25 s and the total PCR process was completed in less than 6 min. Consequently, the total time of the assay was ∼13 min, which is 4–9 times shorter than other conventional PCR- or qPCR-based chick sexing assays reported to date.
Similar to other PCR-based methods, the proposed assay is sensitive to the presence of PCR inhibitors in blood or tissue samples. Therefore, the DNA samples were extracted and purified using the designated kits. However, commonly used commercial DNA kits employ extraction protocols that are lengthy, laborious, and require approximately 45 min. To make the proposed assay applicable for commercial use, the DNA extraction procedure must be simplified. Rapid DNA extraction methods31,32 can be adapted for use with avian blood samples and will reduce the extraction time to 2 min, bringing the overall testing time from blood collection to final sex determination to approximately 15 min. The automation of the process can reduce the overall assay duration even further.
The proposed assay can also be used to detect nonrepetitive sequences. However, without the natural preamplification of the repetitive sequences, the number of cycles needed for detection of nonrepetitive sequences may be higher. Nevertheless, the advantages of the MMB-based assay relative to qPCR are expected to be maintained.
Conclusions
The modified double-quenched FRET-based assay detects repetitive target DNA sequences in less than 15 min, which is 4–9 times faster than qPCR-based assays. The widespread presence of repetitive DNA motifs in the genomes of almost all branches of life, sometimes in exceptionally high numbers of copies, makes this method a valuable tool for a variety of scientific and diagnostic tasks that require rapid detection and high sensitivity. By specifically tailoring the primers and probe, the proposed assay can be used to detect any target DNA sequence, either repetitive or nonrepetitive.
Experimental Section
Ethical Statement
All experiments involving collection of biological material (blood samples) from adult birds and in ovo embryos of domestic chicken (Gallus gallus) were performed according to the guidelines and protocols approved by the Bar-Ilan University Institutional Animal Care and Use Committee. Internal research number: 53-08-2015
Blood Sample Collection from Adult Birds and in Ovo Embryos
Whole blood samples (1.5 mL) from 20 adult Gallus gallus chickens (10 males and 10 females) were obtained at a local breeding farm (Yoram Weissman, Sitria, Israel) using heparin-flushed disposable syringes (Heparin Lock Flush Solution, Kamada, Beit Kama, Israel). Immediately after collection, the whole blood samples were aliquoted (0.5 mL/Eppendorf tube) and transferred on dry ice to Bar-Ilan University. Upon arrival at the laboratory, one aliquot of each sample was used for DNA extraction and the rest of the aliquoted samples were frozen at −80 °C until further use.
A total of 25 fertilized eggs, also obtained from the Weissman farm, were incubated at 37 °C and 60% relative humidity for 16 days. On day 17, the pointed pole of the egg’s shell was carefully removed and 50 μL of blood was extracted from the embryo’s peripheral blood vessels using a heparin-flushed insulin disposable syringe (Insumed 31G, Artsana S.p.a, Grandate, Italy).
DNA Extraction
For the initial testing of the assay, genomic DNA was extracted from 0.5 mL of the adult chickens’ whole blood, using a QIAGEN QIAamp DNA Blood Maxi Kit (QIAGEN, GmbH, Hilden, Germany) according to the manufacturer’s protocol. For the in ovo assay validation, genomic DNA was extracted from 5 μL of whole blood that was collected from each fertilized egg. The DNA extraction was performed using a QIAGEN QIAamp DNA Blood Mini Kit (QIAGEN, GmbH, Hilden, Germany) according to the manufacturer’s protocol. Extracted DNA samples were aliquoted and stored at −80 °C until further use.
Bioinformatics Analysis
Bioinformatics analysis was performed using the NCBI BLAST+ web service and the most recent available version of the domestic chicken genome (Gallus_gallus-5.0; GCA_000002315.3).33 To determine their relative abundance and chromosomal affiliation, known female-specific repetitive sequences26 were verified against the genomic data. Highly repetitive sequences located within the female-specific W chromosome (XhoI and RhoI) were chosen as potential targets. Specifically, the repetitive sequence XhoI (717 bp) was further analyzed and the inner portion (120 bp) of the sequence was selected as a final target for the development of the modified double-quenched FRET-based assay.
Oligonucleotides
All of the oligonucleotides (i.e., FRET-based probes and primers) in the current research were designed using the PrimerQuest Tool web service from Integrated DNA Technologies, Inc. (IDT, Skokie, IL) and purchased from the same vendor. Lyophilized oligonucleotides of the standard and modified ZEN assay were reconstituted in DNase-free ultrapure water (Biological Industries, Beit HaEmek, Israel) to a final concentration of 100 μM. The resulting solutions were aliquoted and frozen at −20 °C until further use. The modified ZEN probe (23b long), targeting the selected portion of the XhoI repetitive sequence, included a biotin and a fluorophore (Alexa532, Thermo Fisher Scientific, Waltham, MA) on the first nucleotide at the 5′ end, a dark quencher (ZEN quencher) at the ninth nucleotide, and another dark quencher (Iowa Black FQ) at the 3′ end. The synthetic oligonucleotides used in this research are described in Table 1.
Table 1. Probe and Primers Used for the Development of the Assay.
| oligonucleotide | sequence (5′–3′) |
|---|---|
| XhoI forward primer | 5′-CTGCACTTCCTTCCCGAAA-3′ |
| XhoI reverse primer | 5′-CGGCTGAAAGGTGGTACTT-3′ |
| ZEN probea | 5′-/Alex532N//iBiodT/CGCTTCACT/ZEN/CACAAAGCACGC/3IABkFQ/-3′ |
| HUR0417 forward primer | 5′-ACACTATGTTTTCTGCCCGC-3′ |
| HUR0417 reverse primer | 5′-CTGAGGGTTGACCTTTCCAA-3′ |
ZEN probe includes two black quenchers: ZEN quencher (proprietary to IDT) at the ninth nucleotide and an Iowa Black Quencher (3IABkFQ) at 3′ end of the probe.
Adult Bird Sexing Using a Magnetically Modulated FRET-Based Assay
To estimate the number of PCR cycles required to positively differentiate between male and female chicks using the MMB system, we took genomic DNA samples from five adult male and five adult female birds. For each DNA sample, four PCRs with a final volume of 25 μL/reaction were prepared. Each reaction contained 1 μL of genomic DNA (50 ng/μL), 12.5 μL of JumpStart Taq ReadyMix (Sigma-Aldrich, MO), 0.5 μL (500 pmol) of forward and 0.5 μL (500 pmol) of reverse primers, 10 μL (10 pmol) of a modified double-quenched probe, and 0.5 μL of DNase/RNase-free ultrapure water.
Using the Mastercycler Nexus Thermal Cycler (Eppendorf, Hamburg, Germany), the four PCR mixtures were subjected to 2 min at 94 °C, followed by 5, 10, 20, or 30 PCR cycles of 30 s at 94 °C, 30 s at 54 °C, and 30 s at 68 °C. The total duration of each cycle was 90 s. Subsequently, the reaction tubes were removed from the thermocycler and their content was supplemented with 75 μL of ultrapure water for a final volume of 100 μL and then analyzed using the MMB system.
We prepared two groups of negative controls, in which either the genomic DNA (1 μL) or the JumpStart Taq ReadyMix (12.5 μL) was replaced with ultrapure water (upH2O). Each group of negative controls consisted of four PCRs that were subjected to the same number of incubation cycles as the experimental reactions. Following incubation, the negative controls were analyzed using the MMB system.
Adult Bird Sexing Using qPCR
To estimate the number of PCR cycles required to positively differentiate between male and female chicks using qPCR, we used the same genomic DNA samples from five adult male and five adult female birds. For each DNA sample, a PCR with a final volume of 25 μL/reaction was prepared. Each reaction contained 1 μL of genomic DNA (50 ng/μL), 12.5 μL of JumpStart Taq ReadyMix (Sigma-Aldrich, MO), 0.5 μL (500 pmol) of forward and 0.5 μL (500 pmol) of reverse primers, 10 μL (10 pmol) of a modified double-quenched probe, and 0.5 μL of DNase/RNase-free ultrapure water. Using the CFX96Touch Real-Time PCR detection system (Bio-Rad, Hercules, CA), the PCR mixtures were subjected to 2 min at 94 °C, followed by 30 PCR cycles of 30 s at 94 °C, 30 s at 54 °C, and 30 s at 68 °C. The total duration of each cycle was 95 s, including the 5 s imaging step. As a negative control, we prepared a PCR mixture in which the genomic DNA (1 μL) was replaced with ultrapure water (upH2O).
Chick Sexing of Embryos Using Conventional PCR
To determine the chick sex in ovo, we used conventional PCR, targeting the 642 bp female-specific nonrepetitive sequence (HUR0417).27 Genomic DNA samples extracted from 25 fertilized eggs were used. For each DNA sample, we prepared a reaction mixture containing 1 μL of DNA (50 ng/μL), 12.5 μL of JumpStart Taq ReadyMix (Sigma-Aldrich, MO), 0.5 μL (500 pmol) of forward and 0.5 μL (500 pmol) of reverse primers (see Table 1), and 10.5 μL of DNase/RNase-free ultrapure water.
The 25 PCR mixtures were subjected to 60 s at 94 °C, followed by 30 cycles of 30 s at 94 °C, 30 s at 52 °C, and 30 s at 68 °C. The total duration of each cycle was 90 s, and the total duration of the PCR phase (including heating and cooling) was ∼50 min. Subsequently, the reaction tubes were removed from the thermocycler and their content was analyzed using agarose gel electrophoresis. Agarose gel was prepared by dissolving 1% (w/v) agarose (Benchmark Scientific, Sayreville, NJ) in Tris–borate–ethylenediaminetetraacetic acid buffer 1× (Amresco, Solon, OH). Boiled agarose solution was cooled to 50 °C and then supplemented with SYBR Safe DNA gel stain (Invitrogen, Carlsbad, CA) according to the manufacturer’s instructions. After solidification of the cast gel, 5 μL from each reaction was mixed with 1 μL of gel loading dye (New England BioLabs, Ipswich, MA) and loaded into the wells. Electrophoresis was performed for 5 min at 60 V, followed by 40 min at 160 V, using a Wide Mini-Sub Horizontal Electrophoresis System and PowerPac HC High-Current Power Supply (Bio-Rad, Hercules, CA). Following the electrophoresis step, the gel was transferred to the ChemiDoc imaging system (Bio-Rad, Hercules, CA) and analyzed.
Comparing Chick Sexing in Ovo Using qPCR and the Magnetically Modulated FRET-Based Assay
Following successful determination of chick sex in ovo using PCR, we evaluated the capabilities of our modified FRET-based assay to determine the chick sex using qPCR and the MMB system. Genomic DNA samples extracted from 25 fertilized eggs were used. For each DNA sample, we prepared a reaction mixture containing 1 μL of DNA (50 ng/μL), 12.5 μL of JumpStart Taq ReadyMix (Sigma-Aldrich, MO), 0.5 μL (500 pmol) of forward and 0.5 μL (500 pmol) of reverse primers, 10 μL (10 pmol) of a modified double-quenched probe, and 0.5 μL of DNase/RNase-free ultrapure water.
For the qPCR analysis, the 25 PCR mixtures were subjected to 1 min at 94 °C, followed by 30 cycles of 10 s at 94 °C, 10 s at 54 °C, and 5 s at 68 °C. The total duration of each cycle was 25 s, and the total duration of the PCR phase (including heating, cooling, imaging, and analysis) was approximately 25 min.
For the MMB analysis, the 25 PCR mixtures were subjected to 1 min at 94 °C, followed by eight cycles of 10 s at 94 °C, 10 s at 54 °C, and 5 s at 68 °C. The total duration of each cycle was 25 s, and the total duration of the PCR phase (including heating and cooling) was approximately 6 min. Subsequently, the reaction tubes were removed from the thermocycler; then, their contents were supplemented with 75 μL of ultrapure water for a final volume of 100 μL and analyzed using the MMB system.
MMB System Testing
Prior to testing in the MMB system, each PCR was incubated under constant rotation (TR-1550, MRC, Israel, 40 rpm) for 5 min at room temperature with ∼100 000 streptavidin-coupled magnetic beads (M-280 Streptavidin, Thermo Fisher Scientific, Waltham, MA). Subsequently, the beads were collected and resuspended in 400 μL of phosphate-buffered saline 1× buffer (Biological Industries, Beit HaEmek, Israel) supplemented with 0.01% (v/v) Tween 20 (Sigma-Aldrich, MO). Then, a total of three borosilicate cuvettes (W2540, VitroCom, Mountain Lakes, NJ), each loaded with 100 μL of the final solution, were measured using the MMB system. To reduce their autofluorescence, prior to their use in the assay, the magnetic beads were photobleached for 18 h.34
MMB System Setup
The MMB system, shown in Figure 4, uses a 532 nm laser diode module (ThorLabs, CPS532), working at 0.25 mW. The laser beam is diverted by a dichroic mirror (Semrock, BrightLine Di02-R532) and focused by an objective lens (Newport, M-10X, 0.25 NA) to a 150 μm spot size on the borosilicate sample cell, which contains the magnetic beads. The emitted fluorescence is collected using the same objective lens, passed through the dichroic mirror and two emission filters (Semrock, FF03-575/25), and detected by a digital camera (FLIR, GS3-U3-23S6M-C). Two electromagnets, one on each side of the glass sample cell, apply an alternating magnetic field gradient at 1 Hz, causing the magnetic beads to aggregate and move in a periodic lateral motion inside the sample cell. As the bead aggregate passes in front of the optical laser beam, the fluorescence emitted from the assay complex creates a flashing signal that is distinguishable from the constant background of the sample matrix or from unbound fluorescent molecules. A sequence of 600 images are acquired over a period of 12 s. The gray value from the laser beam area of each image is integrated, and the peak-to-peak voltage differences over time are calculated and averaged. The MMB system is enclosed in a lightproof box, which allows for the detection of low analyte concentrations and greatly reduces the measurement fluctuations that would otherwise be introduced by intrusive exterior light.
Figure 4.

Magnetic modulation biosensing system setup. The 532 nm laser beam is reflected by a dichroic mirror and focused by an objective lens onto a borosilicate cuvette, which contains the sample of magnetic beads. The cuvette is positioned between two electromagnets that cause the beads to aggregate and move in a periodic motion, in and out of the laser beam. The laser excites the fluorophores in the sample, and the modulated emitted fluorescence is collected by the same objective lens, filtered by two emission filters, and detected by a camera.
Acknowledgments
The authors thank Shira Roth and Yehudit Michelson for their valuable discussions, and Meir Cohen for technical assistance. This research was supported by the Israel Science Foundation (Grant No. 1142/15) and the Israel Innovation Authority (Kamin, Grant No. 59042). James Ballard provided an editorial review of the manuscript.
The authors declare the following competing financial interest(s): A.D. has a financial interest in MagBiosense, Inc., which, however, did not financially support this work.
References
- Romero D.; Martínez-Salazar J.; Ortiz E.; Rodríguez C.; Valencia-Morales E. Repeated sequences in bacterial chromosomes and plasmids: a glimpse from sequenced genomes. Res. Microbiol. 1999, 150, 735–743. 10.1016/s0923-2508(99)00119-9. [DOI] [PubMed] [Google Scholar]
- Mehrotra S.; Goyal V. Repetitive sequences in plant nuclear DNA: types, distribution, evolution and function. Genomics, Proteomics Bioinf. 2014, 12, 164–171. 10.1016/j.gpb.2014.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biscotti M. A.; Olmo E.; Heslop-Harrison J. S. Repetitive DNA in eukaryotic genomes. Chromosome Res. 2015, 23, 415–420. 10.1007/s10577-015-9499-z. [DOI] [PubMed] [Google Scholar]
- Agrawal S.; Ganley A. R.. Complete Sequence Construction of the Highly Repetitive Ribosomal RNA Gene Repeats in Eukaryotes Using Whole Genome Sequence Data. In The Nucleolus; Németh A., Ed.; Methods in Molecular Biology; Humana Press: New York, NY, 2016; Vol. 1455, pp 161–181. [DOI] [PubMed] [Google Scholar]
- Treangen T. J.; Salzberg S. L. Repetitive DNA and next-generation sequencing: computational challenges and solutions. Nat. Rev. Genet. 2011, 13, 36–46. 10.1038/nrg3117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeffreys A. J.; Wilson V.; Thein S. L. Hypervariable Minisatellite Regions in Human DNA. Nature 1985, 314, 67–73. 10.1038/314067a0. [DOI] [PubMed] [Google Scholar]
- Rerkamnuaychoke B.; Chantratita W.; Jomsawat U.; Thanakitgosate J.; Rojanasunan P. Paternity testing by PCR-based STR analysis. J. Med. Assoc. Thai 2000, 83, S55–S62. [PubMed] [Google Scholar]
- Abbasi I.; Webster B. L.; King C. H.; Rollinson D.; Hamburger J. The substructure of three repetitive DNA regions of Schistosoma haematobium group species as a potential marker for species recognition and interbreeding detection. Parasites Vectors 2017, 10, 364 10.1186/s13071-017-2281-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sohn S. H.; Lee C. Y.; Ryu E. K.; Han J. Y.; Multani A. S.; Pathak S. Rapid sex identification of chicken by fluorescence in situ hybridization using a W chromosome-specific DNA probe. Asian-Australas. J. Anim. Sci. 2002, 15, 1531–1535. 10.5713/ajas.2002.1531. [DOI] [Google Scholar]
- Petitte J. N.; Kegelmeyer A. E. Rapid sex determination of chick embryos using the polymerase chain reaction. Anim. Biotechnol. 1995, 6, 119–130. 10.1080/10495399509525841. [DOI] [Google Scholar]
- Dawson D. A.; Darby S.; Hunter F. M.; Krupa A. P.; Jones I. L.; Burke T. A critique of avian CHD-based molecular sexing protocols illustrated by a Z-chromosome polymorphism detected in auklets. Mol. Ecol. Notes 2001, 1, 201–204. 10.1046/j.1471-8278.2001.00060.x. [DOI] [Google Scholar]
- Hosseinzadeh-Colagar A.; Haghighatnia M. J.; Amiri Z.; Mohadjerani M.; Tafrihi M. Microsatellite (SSR) amplification by PCR usually led to polymorphic bands: Evidence which shows replication slippage occurs in extend or nascent DNA strands. Mol. Biol. Res. Commun. 2016, 5, 167–174. [PMC free article] [PubMed] [Google Scholar]
- Hommelsheim C. M.; Frantzeskakis L.; Huang M.; Ulker B. PCR amplification of repetitive DNA: a limitation to genome editing technologies and many other applications. Sci. Rep. 2014, 4, 5052 10.1038/srep05052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holland P. M.; Abramson R. D.; Watson R.; Gelfand D. H. Detection of Specific Polymerase Chain-Reaction Product by Utilizing the 5′-]3′ Exonuclease Activity of Thermus-Aquaticus DNA-Polymerase. Proc. Natl. Acad. Sci. U.S.A. 1991, 88, 7276–7280. 10.1073/pnas.88.16.7276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Didenko V. V. DNA probes using fluorescence resonance energy transfer (FRET): designs and applications (vol 31, pg 1106, 2001). Biotechniques 2002, 32, 1012. 10.2144/01315rv02. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson P. M.; Labonte M. J.; Russell J.; Louie S.; Ghobrial A. A.; Ladner R. D. A novel fluorescence-based assay for the rapid detection and quantification of cellular deoxyribonucleoside triphosphates. Nucleic Acids Res. 2011, 39, e112 10.1093/nar/gkr350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pilotte N.; Papaiakovou M.; Grant J. R.; Bierwert L. A.; Llewellyn S.; McCarthy J. S.; Williams S. A. Improved PCR-Based Detection of Soil Transmitted Helminth Infections Using a Next-Generation Sequencing Approach to Assay Design. PLoS Neglected Trop. Dis. 2016, 10, e0004578 10.1371/journal.pntd.0004578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danielli A.; Arie A.; Porat N.; Ehrlich M. Detection of fluorescent-labeled probes at subpicomolar concentrations by magnetic modulation. Opt. Express 2008, 16, 19253–19259. 10.1364/OE.16.019253. [DOI] [PubMed] [Google Scholar]
- Danielli A.; Porat N.; Arie A.; Ehrlich M. Rapid homogenous detection of the Ibaraki virus NS3 cDNA at picomolar concentrations by magnetic modulation. Biosens. Bioelectron. 2009, 25, 858–863. 10.1016/j.bios.2009.08.047. [DOI] [PubMed] [Google Scholar]
- Griffiths R. Sex identification in birds. Semin. Avian Exot. Pet Med. 2000, 9, 14–26. 10.1016/S1055-937X(00)80012-2. [DOI] [Google Scholar]
- Krautwald-Junghanns M. E.; Cramer K.; Fischer B.; Forster A.; Galli R.; Kremer F.; Mapesa E. U.; Meissner S.; Preisinger R.; Preusse G.; Schnabel C.; Steiner G.; Bartels T. Current approaches to avoid the culling of day-old male chicks in the layer industry, with special reference to spectroscopic methods. Poult. Sci. 2018, 97, 749–757. 10.3382/ps/pex389. [DOI] [PubMed] [Google Scholar]
- Bruijnis M. R. N.; Blok V.; Stassen E. N.; Gremmen H. G. J. Moral “Lock-In” in Responsible Innovation: The Ethical and Social Aspects of Killing Day-Old Chicks and Its Alternatives. J. Agric. Environ. Ethics 2015, 28, 939–960. 10.1007/s10806-015-9566-7. [DOI] [Google Scholar]
- Stevens L. Sex chromosomes and sex determining mechanisms in birds. Sci. Prog. 1997, 80, 197–216. [PubMed] [Google Scholar]
- Kodama H.; Saitoh H.; Tone M.; Kuhara S.; Sakaki Y.; Mizuno S. Nucleotide sequences and unusual electrophoretic behavior of the W chromosome-specific repeating DNA units of the domestic fowl, Gallus gallus domesticus. Chromosoma 1987, 96, 18–25. 10.1007/BF00285878. [DOI] [PubMed] [Google Scholar]
- Saitoh Y.; Saitoh H.; Ohtomo K.; Mizuno S. Occupancy of the majority of DNA in the chicken W chromosome by bent-repetitive sequences. Chromosoma 1991, 101, 32–40. 10.1007/BF00360684. [DOI] [PubMed] [Google Scholar]
- Saitoh Y.; Mizuno S. Distribution of XhoI and EcoRI family repetitive DNA sequences into separate domains in the chicken W chromosome. Chromosoma 1992, 101, 474–477. 10.1007/BF00352469. [DOI] [PubMed] [Google Scholar]
- Granevitze Z.; Blum S.; Cheng H.; Vignal A.; Morisson M.; Ben-Ari G.; David L.; Feldman M. W.; Weigend S.; Hillel J. Female-specific DNA sequences in the chicken genome. J. Hered. 2007, 98, 238–242. 10.1093/jhered/esm010. [DOI] [PubMed] [Google Scholar]
- Thomas M. R.; Matsumoto S.; Cain P.; Scott N. S. Repetitive DNA of grapevine: classes present and sequences suitable for cultivar identification. Theor. Appl. Genet. 1993, 86, 173–180. 10.1007/BF00222076. [DOI] [PubMed] [Google Scholar]
- Hamburger J.; He N.; Abbasi I.; Ramzy R. M.; Jourdane J.; Ruppel A. Polymerase chain reaction assay based on a highly repeated sequence of Schistosoma haematobium: a potential tool for monitoring schistosome-infested water. Am. J. Trop. Med. Hyg. 2001, 65, 907–911. 10.4269/ajtmh.2001.65.907. [DOI] [PubMed] [Google Scholar]
- He L.; Martins P.; Huguenin J.; Van T. N.; Manso T.; Galindo T.; Gregoire F.; Catherinot L.; Molina F.; Espeut J. Simple, sensitive and robust chicken specific sexing assays, compliant with large scale analysis. PLoS One 2019, 14, e0213033 10.1371/journal.pone.0213033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haugland R. A.; Brinkman N.; Vesper S. J. Evaluation of rapid DNA extraction methods for the quantitative detection of fungi using real-time PCR analysis. J. Microbiol. Methods 2002, 50, 319–323. 10.1016/S0167-7012(02)00037-4. [DOI] [PubMed] [Google Scholar]
- Guha P.; Das A.; Dutta S.; Chaudhuri T. K. A rapid and efficient DNA extraction protocol from fresh and frozen human blood samples. J. Clin. Lab. Anal. 2018, 32, e22181 10.1002/jcla.22181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warren W. C.; Hillier L. W.; Tomlinson C.; Minx P.; Kremitzki M.; Graves T.; Markovic C.; Bouk N.; Pruitt K. D.; Thibaud-Nissen F.; Schneider V.; Mansour T. A.; Brown C. T.; Zimin A.; Hawken R.; Abrahamsen M.; Pyrkosz A. B.; Morisson M.; Fillon V.; Vignal A.; Chow W.; Howe K.; Fulton J. E.; Miller M. M.; Lovell P.; Mello C. V.; Wirthlin M.; Mason A. S.; Kuo R.; Burt D. W.; Dodgson J. B.; Cheng H. H. A New Chicken Genome Assembly Provides Insight into Avian Genome Structure. G3: Genes, Genomes, Genet. 2017, 7, 109–117. 10.1534/g3.116.035923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roth S.; Hadass O.; Cohen M.; Verbarg J.; Wilsey J.; Danielli A. Improving the Sensitivity of Fluorescence-Based Immunoassays by Photobleaching the Autofluorescence of Magnetic Beads. Small 2019, 15, 1803751 10.1002/smll.201803751. [DOI] [PubMed] [Google Scholar]