

Abstract
Background
Neurofibromatosis type 1 (NF1) is a tumor-predisposition disorder caused by germline mutations in NF1. NF1 patients have an 8–16% lifetime risk of developing a malignant peripheral nerve sheath tumor (MPNST), a highly aggressive soft-tissue sarcoma, often arising from preexisting benign plexiform neurofibromas (PNs) and atypical neurofibromas (ANFs). ANFs are distinct from both PN and MPNST, representing an intermediate step in malignant transformation.
Methods
In the first comprehensive genomic analysis of ANF originating from multiple patients, we performed tumor/normal whole-exome sequencing (WES) of 16 ANFs. In addition, we conducted WES of 3 MPNSTs, copy-number meta-analysis of 26 ANFs and 28 MPNSTs, and whole transcriptome sequencing analysis of 5 ANFs and 5 MPNSTs.
Results
We identified a low number of mutations (median 1, range 0–5) in the exomes of ANFs (only NF1 somatic mutations were recurrent), and frequent deletions of CDKN2A/B (69%) and SMARCA2 (42%). We determined that polycomb repressor complex 2 (PRC2) genes EED and SUZ12 were frequently mutated, deleted, or downregulated in MPNSTs but not in ANFs. Our pilot gene expression study revealed upregulated NRAS, MDM2, CCND1/2/3, and CDK4/6 in ANFs and MPNSTs, and overexpression of EZH2 in MPNSTs only.
Conclusions
The PN-ANF transition is primarily driven by the deletion of CDKN2A/B. Further progression from ANF to MPNST likely involves broad chromosomal rearrangements and frequent inactivation of the PRC2 genes, loss of the DNA repair genes, and copy-number increase of signal transduction and cell-cycle and pluripotency self-renewal genes.
Keywords: atypical neurofibromas, benign-to-malignant transformation, malignant peripheral nerve sheath tumor, neurofibromatosis type 1, plexiform neurofibromas
Key Points
1. ANFs are relatively chromosomally stable tumors with low somatic mutation burden.
2. Frequent inactivation of NF1, CDKN2A/B, and SMARCA2 genetically defines ANF.
3. Unlike MPNST, we detect no recurrent PRC2 gene inactivation in ANF.
Importance of the Study
NF1-associated ANFs are rare premalignant lesions with a high risk of transformation to MPNST, a soft-tissue sarcoma with poor prognosis. Early detection and treatment of ANF may prevent MPNST, for which surgery remains the only therapeutic option. At present, the genetics of ANF development and transformation to MPNST are not fully understood. Here we present the first multisample/multipatient comprehensive genomic study of ANF. We show that somatic mutation burden and genomic instability in ANF is relatively low, with only NF1, CDKN2A/B, and, to a lesser extent, SMARCA2 mutated in the tumors. SUZ12, EED, and TP53, which are frequently inactivated in MPNST, are intact in ANF. We conclude that in ANF, loss of CDKN2A/B is the main genetic event that in addition to NF1 inactivation leads to premalignancy. The transition to MPNST coincides with a dramatic rise in genomic instability, inactivation of PRC2, and copy-number gains of cell-cycle and pluripotency genes.
Neurofibromatosis type 1 (NF1) is a common (~1/2000–1/3500 worldwide1) autosomal dominant, archetypal tumor-predisposition disorder secondary to mutations in the tumor suppressor NF1. Phenotypically, NF1 is associated with neurocutaneous abnormalities, including a variety of benign and malignant tumors.2 Life expectancy is reduced by 8–15 years in both men and women, primarily due to malignancy and cardiovascular disease.3,4 A significant proportion of the excess mortality in NF1 is attributable to malignant peripheral nerve sheath tumors (MPNSTs), an aggressive soft-tissue sarcoma with limited therapeutic options.4 The relative risk of MPNST in NF1 is enormously increased (~2000-fold), with a lifetime risk of 8–16%.4,5
MPNSTs typically arise from a preexisting plexiform neurofibroma (PN), a histologically benign congenital neurofibroma affecting up to 50% of people with NF16 and readily identified on whole-body MRI.7 More widespread use of whole-body MRI has prompted the identification of “distinct nodular lesions” (DNLs), frequently within PNs. These lesions are >3 cm in longest diameter, well demarcated, and distinct from surrounding tissue and lack the “central dot” sign of PN. Distinct nodular lesions appear after early childhood and typically grow faster than the surrounding or adjacent PN and may be precursors of an MPNST.8 Some DNLs are biopsy-proven atypical neurofibromas (ANFs).9 ANFs are pathologically defined lesions that have increased variable cellularity, cytological atypia, and more pronounced fascicular growth patterns, but lack the widespread atypia and fascicular growth mitotic activity and necrosis seen in MPNST. In one study, 15/16 ANFs harbored a deletion of chromosome 9p21.3, a region that includes cell-cycle regulators cyclin-dependent kinase inhibitor 2A and 2B (CDKN2A/B),10 both frequently deleted somatically in MPNSTs.11–14
ANF is hypothesized to be a premalignant lesion, with CDKN2A/B deletion the first step in the progression to MPNST. Whole-exome sequencing (WES) and copy-number analysis of PNs showed remarkably low somatic mutation rates, stable chromosomal architecture, and intact CDKN2A/B and revealed the primacy of NF1 inactivation in its pathogenesis.15 WES of NF1-associated MPNST shows that biallelic loss of NF1 and mutation in polycomb repressive complex 2 (PRC2) genes are essential to its pathogenesis.16–18 It remains unclear what other genes and pathways play a role in PN transformation into a premalignant state. Resection of ANF may prevent MPNST9; therefore, identification of genetic biomarkers for ANF is important for the disease management. A better understanding of the PN-to-MPNST transformation is a key recommendation from a recent international consensus meeting on research priorities in MPNST.19,20 To investigate this, we characterized ANFs and a small set of MPNSTs using WES, whole transcriptome sequencing, and copy-number determination.
Materials and Methods
Sample Collection and Clinical Information
Clinical information and tumor samples with matching normal DNA were collected at the NCI in Bethesda, Maryland, the University of Leuven in Belgium, and the University of Florida in Gainesville. The diagnosis of all ANF cases was confirmed by one pathologist at NCI and one pathologist at the University of Leuven. All protocols were approved by the appropriate investigational review board, and subjects and/or their parents or guardians provided written, informed consent. Detailed description of the materials and methods used in this study can be found in the Supplementary material.
Whole-Exome Sequencing of Matching Pairs of Tumor and Normal DNA
Capture of the exome and library preparation was done using the SeqCap EZ Exome plus UTR Library kit (Roche, #06740308001). Sequencing was done according to the manufacturer’s instructions. Among sequenced exomes, the average breadth of coverage was 89% of targeted bases (range 88–91%), and the average depth of coverage was 59x (range 44–82x) for both tumor and normal samples.
Whole-Exome Sequencing Analysis
Sequencing reads were aligned to the National Center for Biotechnology Information’s Build GRCh37 (hg19) using Novoalign v2.08.02 (for ANF1 through ANF7) and v3.02.07 (for ANF8 through ANF15). Point mutations and small indels were called for all tumor/normal pairs with Mutect (v1.1.4),21 SomaticSniper (v1.0.5),22 and Shimmer (v0.1.1).23 Mutect and Shimmer used default parameters; filtering of SomaticSniper variants was as per program authors. MPNST samples were analyzed in the same manner, except that an additional somatic caller, Strelka v1.0.14,24 was added to the pipeline. Coding variants identified as somatic mutations by at least one caller were further filtered by comparing them with 1000 Genomes (internationalgenome.org), ExAC (exac.broadinstitute.org), ESP6500 (evs.gs.washington.edu), and ClinSeq (https://www.ncbi.nlm.nih.gov/projects/gap/cgi-bin/study.cgi?study_id=phs000971.v1.p1, Accessed March 1, 2019) databases; variants with minor allele frequency >1% were removed from further consideration. Variants in known false-positive genes25 were excluded from further consideration.
Somatic Mutation Verification and Deep NF1 Mutation Detection by AmpliSeq/Ion Torrent
Multiplex PCR primers for somatic mutation verification were designed using the Ion AmpliSeq Designer tool (v3.0.1, Life Technologies). Multiplex PCR amplification, library preparation, and sequencing on the Ion Proton sequencer (Life Technologies) were performed as per the manufacturer’s instructions.
Copy-Number Variation and Loss of Heterozygosity
Single nucleotide polymorphism (SNP) genotyping was performed using HumanOmniExomeExpress BeadChip kits (Illumina, #20004207) as per the manufacturer’s instructions. Analysis by allele-specific copy-number analysis of tumors (ASCAT, v2.1) was performed as previously described.26 For samples ANF1‒ANF7, SNP-array data were not available. To analyze somatic copy-number alterations in the NF1 and CDKN2A/B loci in these samples, we used ExomeCNV (v1.4)27 with the WES data. A copy-number variation (CNV) meta-analysis using previously published data10 was performed. Paired CNV and loss-of-heterozygosity analysis of tumor and matching normal DNA was performed by using Nexus v6.1 software (BioDiscovery) as described previously.28 Analysis by Genomic Identification of Significant Targets in Cancer (GISTIC) (https://software.broadinstitute.org/software/cprg/?q=node/31, Accessed March 1, 2019) was performed using the GISTIC module in Nexus v6.1 software.
Whole Transcriptome RNA Sequencing and Analysis of the Data
Total RNA isolation, library construction, and sequencing on the Illumina platform was done as previously described.15 Expression data were analyzed with Gene Set Enrichment Analysis (GSEA) v2 (Broad Institute; software.broadinstitute.org/gsea) according to the developer’s recommendations.
Immunohistochemical Staining of Tumors
All immunohistochemical (IHC) stains were done on formalin-fixed paraffin embedded 5-micron tissue sections mounted on charged microscopic slides.
Results
Clinical Characteristics of Patients and Tumors
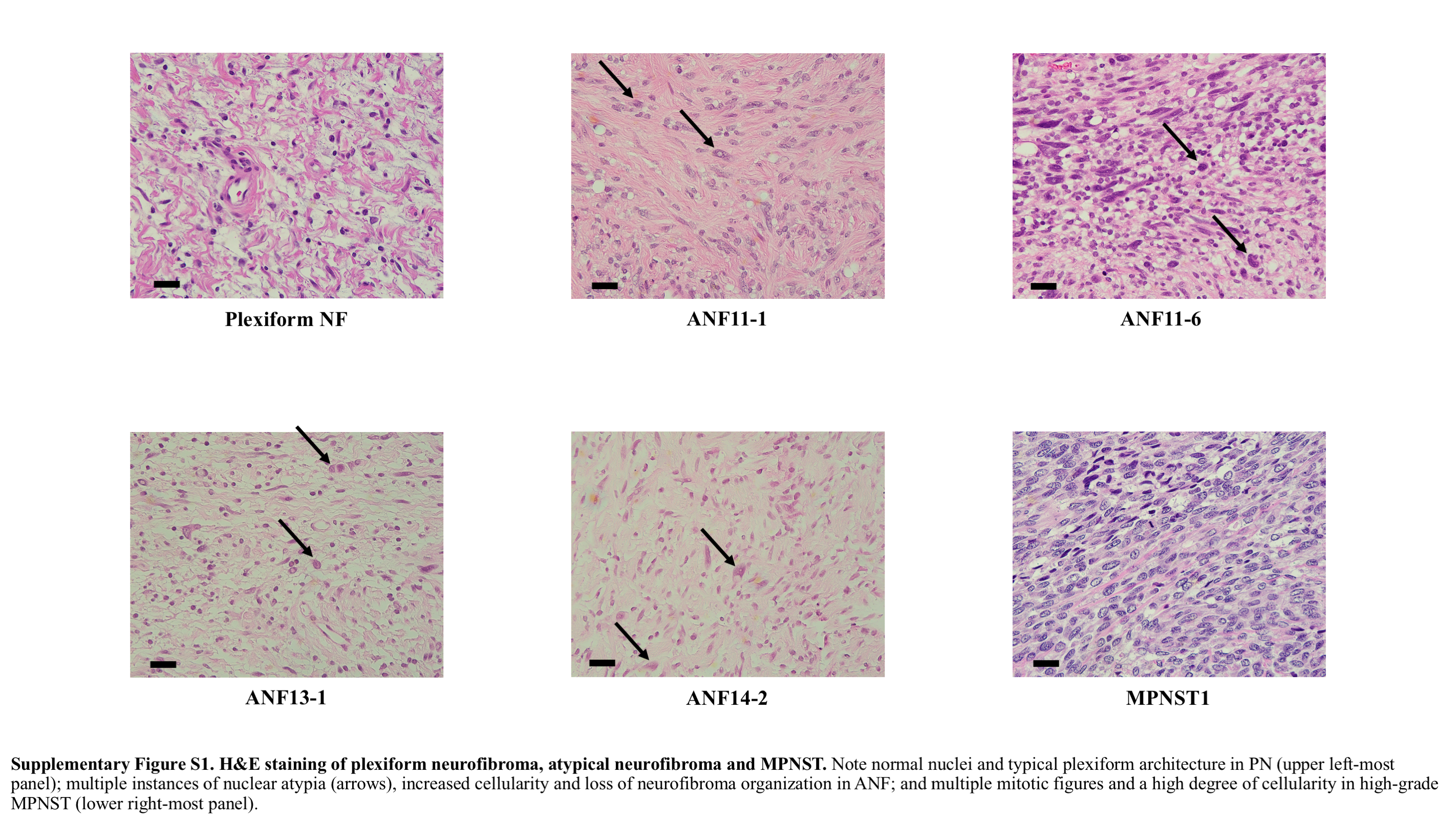
Clinical information for the tumors and NF1 patients is summarized in Table 1 and a recent publication.9 The pathologic classification of ANF was done prior to the development of the new proposed term “atypical neurofibromatous neoplasms of uncertain biologic potential.”20 Hematoxylin and eosin preps of select atypical neurofibromas are shown in comparison to PN and MPNST in Supplementary Figure 1. While normal nuclei and typical plexiform architecture could be observed in PN, multiple instances of nuclear atypia, increased cellularity, and loss of neurofibroma organization are prominent in the ANFs. On the other hand, in high-grade MPNST, multiple mitotic figures and a high degree of cellularity are well distinguished.
Table 1.
Clinical information for patients and tumors
| Tumor Sample ID | Tumor ID in Higham et al, 2018 | Tumor Type | Age at Diagnosis, y | Sex | Inheritance | Family History of MPNST | Personal History of MPNST | Location | Reason for Biopsy/ Resection | Additional Concerning Lesions | Isolated or within Plexiform |
|---|---|---|---|---|---|---|---|---|---|---|---|
| A15_ANF1 | BEL-13-1 | ANF | 26.9 | M | Unknown | NA | No | Neck | G | Yes | Isolated |
| ANF2 | BEL-17 | ANF | 34.4 | F | de novo | No | Yes | Neck | P, G | No | Isolated |
| A13_ANF3 | BEL-11 | ANF | 58.8 | F | Unknown | NA | No | Neck | P, G, E | No | Isolated |
| A4_ANF4 | BEL-4 | ANF | 28.3 | M | Familial-maternal | NA | Yes | Abdomen/ Pelvis | E | No | Unknown |
| A7_ANF5 | N/A | ANF | 18.8 | F | de novo | No | No | Chest | G, E | No | Within PN |
| ANF6 | BEL-16 | ANF | 32.7 | F | Unknown | NA | No | Lower extremity | P | No | Within PN |
| A11_ANF7 | N/A | ANF | 26.7 | F | Familial-maternal | No | Yes | Neck | P | Yes | Unknown |
| ANF8 | BEL-14 | ANF | 28.3 | F | Familial-maternal | NA | No | Chest | P, G | No | Unknown |
| ANF9 | N/A | ANF | 26.4 | M | Familial-maternal | NA | No | Lower extremity | P | No | Unknown |
| ANF10 | BEL-18 | ANF | 31.8 | F | Unknown | NA | No | Chest | G, E | No | Isolated |
| ANF11-1a | NCI-14-3 | ANF | 28.3 | M | Familial-paternal | No | No | Abdomen/ pelvis | G, E | Yes | Within PN |
| ANF11-2a | NCI-14-3 | ANF | 28.3 | M | Familial-paternal | No | No | Abdomen/ pelvis | G, E | Yes | Within PN |
| ANF11-6 | NCI-14-4 | ANF | 30.4 | M | Familial-paternal | No | No | Lower extremity | E | Yes | Isolated |
| ANF11-7 | NCI-14-1 | ANF | 25.4 | M | Familial-paternal | No | No | Abdomen/ pelvis | G, E | Yes | Within PN |
| ANF13-1 | NCI-15 | ANF | 26 | F | Familial-paternal | Yes | Yes | Lower extremity | G, E | Yes | Isolated |
| ANF14-1b | N/A | NF/ ANF& | 9.4 | M | de novo | No | No | Abdomen/ pelvis | G, E | No | Within PN |
| ANF14-2b | NCI-2 | ANF | 9.4 | M | de novo | No | No | Abdomen/ pelvis | G, E | No | Within PN |
| ANF15 | NCI-3 | ANF | 13.7 | M | Familial-maternal | Yes | No | Lower extremity | G, E | Yes | Isolated |
| MPNST1 | N/A | MPNST | 17.1 | M | Familial-maternal | No | Yesd | Abdomen/ pelvis | P, G, E | No | Within PN |
| MPNST2 | N/A | MPNST | 47 | M | de novo | No | Yes | Upper extremity | P, G | No | Within PN |
| MPNST3-1c | N/A | MPNST | 23.6 | M | de novo | No | Yesd | Abdomen/ pelvis | P, G, E | Yes | Within PN |
| MPNST3-2c | N/A | MPNST | 23.6 | M | de novo | No | Yesd | Abdomen/ pelvis | P, G, E | Yes | Within PN |
| MPNST4 | N/A | MPNST | 28 | M | de novo | No | Yes | Lower extremity | P, G | No | Within PN |
| MPNST5 | N/A | MPNST | 39 | M | de novo | No | Yes | Brain metastasise | Neurological symptoms | No | Brain metastasis |
Abbreviations: NA = not available, N/A = not applicable; P = pain associated with tumor; G = tumor growth; E = elevated standard uptake value (SUV) on fluorodeoxyglucose-positron emission tomography (FDG-PET); F = female; M = male. Superscripts: afragments of a larger tumor; b2 distinct nodular lesions within the same PN; cfragments of a larger tumor; dlesions included in this study; eprimary MPNST in a leg; &tumor ANF14-1 was initially classified as neurofibroma (NF) by a pathologist; however, based on clinical (fast growth and elevated SUV on FDG-PET scan) and genetic (deletion of the CDKN2A/B locus) evaluations in this study, this tumor was reclassified as ANF. IDs for ANF investigated in Higham et al, 2018 (Neuro-Oncology, Vol. 20, pp. 818–825) are shown in the second column.
NF1, CDKN2A/B, and PRC2 Mutational and Copy-Number Status in ANFs and MPNSTs
Information for the analyses performed on the tumor samples is available in Supplementary Table 1. We first sought mutations or deletions in NF1 and CDKN2A/B (Table 2). We identified 14/14 (100%) and 13/16 (81%) NF1 germline and somatic mutations, respectively, in the 16 ANFs. We identified no point mutations or small indels in CDKN2A or CDKN2B; however, SNP-array and ExomeCNV analyses revealed hetero- or homozygous loss of the CDKN2A/2B locus (9p21.3) in 12/16 (75%) tumors. We sought, but did not find, damaging somatic variation in PRC2 genes (embryonic ectoderm development [EED], suppressor of zeste 12 homolog [SUZ12], EZH1/2, RBBP4/7, AEBP2, JARID2) or in TP53.
Table 2.
Mutation and CNV in NF1, CDKN2A/B, and PRC2 genes in ANF and MPNST
| Sample ID | NF1, Germline | NF1, Somatic | CDKN2A/2B | PRC2 Genes |
|---|---|---|---|---|
| A15_ANF1 | Frameshift, p.M991Ifs*2 | Gains with multiple breakpointsb | Not detected | Not detected |
| ANF2 a | Missense, p.A706F | Not detected | Not detected | Not detected |
| A13_ANF3 | Splicing, c.3113+1G>A | Frameshift, p.E2624Rfs*33 | Het loss | Not detected |
| A4_ANF4 | Type I microdeletion | Nonsense, p.Q1801X | Not detected | Not detected |
| A7_ANF5 | Frameshift, p.F199Lfs*5 | Large deletion | Homozygous loss | Not detected |
| ANF6 a | Exons 2-28 deletion | Not detected | Not detected | Not detected |
| ANF7 | Splicing, c.3113+1G>A | Frameshift, p.D2614Nfs*7 | Homozygous loss | Not detected |
| ANF8 | Missense, p.L2317P | Large deletionb | Het loss | Not detected |
| ANF9 | Missense, p.Y575C NM_001128147 | Missense, p.C845Y | Partial gain, with a breakpoint in CDKN2A | Not detected |
| ANF10 | Splicing, c.1261-1G>A | Frameshift, p.V224fs*0 | Homozygous loss | Not detected |
| ANF11-1 | Frameshift, p.N1582Kfs*19 | Copy-neutral LOH | Het loss | Not detected |
| ANF11-2 | Same as in ANF11-1 | Copy-neutral LOH | Het loss | Not detected |
| ANF13 | Nonsense, p.R416X | Not detected | Het loss | Not detected |
| ANF14-1 | Splicing, c.288+2T>G | Frameshift, p.I1381Mfs*3 | Het loss | Not detected |
| ANF14-2 | Same as in ANF14-1 | Same as in ANF14-1 | Het loss | Not detected |
| ANF15 | Frameshift, p.T2264Tfs*4 | Frameshift, p.K2643Rfs*14 | Partial gain, with a breakpoint in CDKN2A | Not detected |
| MPNST1 | Nonsense, p.R440X | Frameshift, p.L996Sfs*15 | Homozygous loss | Not available |
| MPNST2 | Nonsense, p.R1276X | Copy-neutral LOH | Homozygous loss CDKN2A, Het loss CDKN2B | Not detected, but SUZ12 expression is sharply decreased |
| MPNST3-1 | Frameshift, p.Q2050Hfs*10 | Large deletion | Het loss | SUZ12, p.T415Gfs*4, homozygous |
| MPNST4 | Frameshift, p.N1229Mfs*10 | Copy-neutral LOH | Homozygous loss | EED, p.S241R, homozygous |
| Detection rate in ANF | 100% (14/14) | 81% (13/16) | 75% (12/16) | 0% (0/16) |
| Detection rate in MPNST | 100% (4/4) | 100% (4/4) | 100% (4/4) | 67% (2/3) |
Abbreviations: Het = heterozygous; LOH = loss of heterozygosity. Superscripts: aonly ExomeCNV (v1.4) analysis of the WES data for the NF1 and CDKN2A/B loci was performed; bcopy-number changes detected in WES data only.
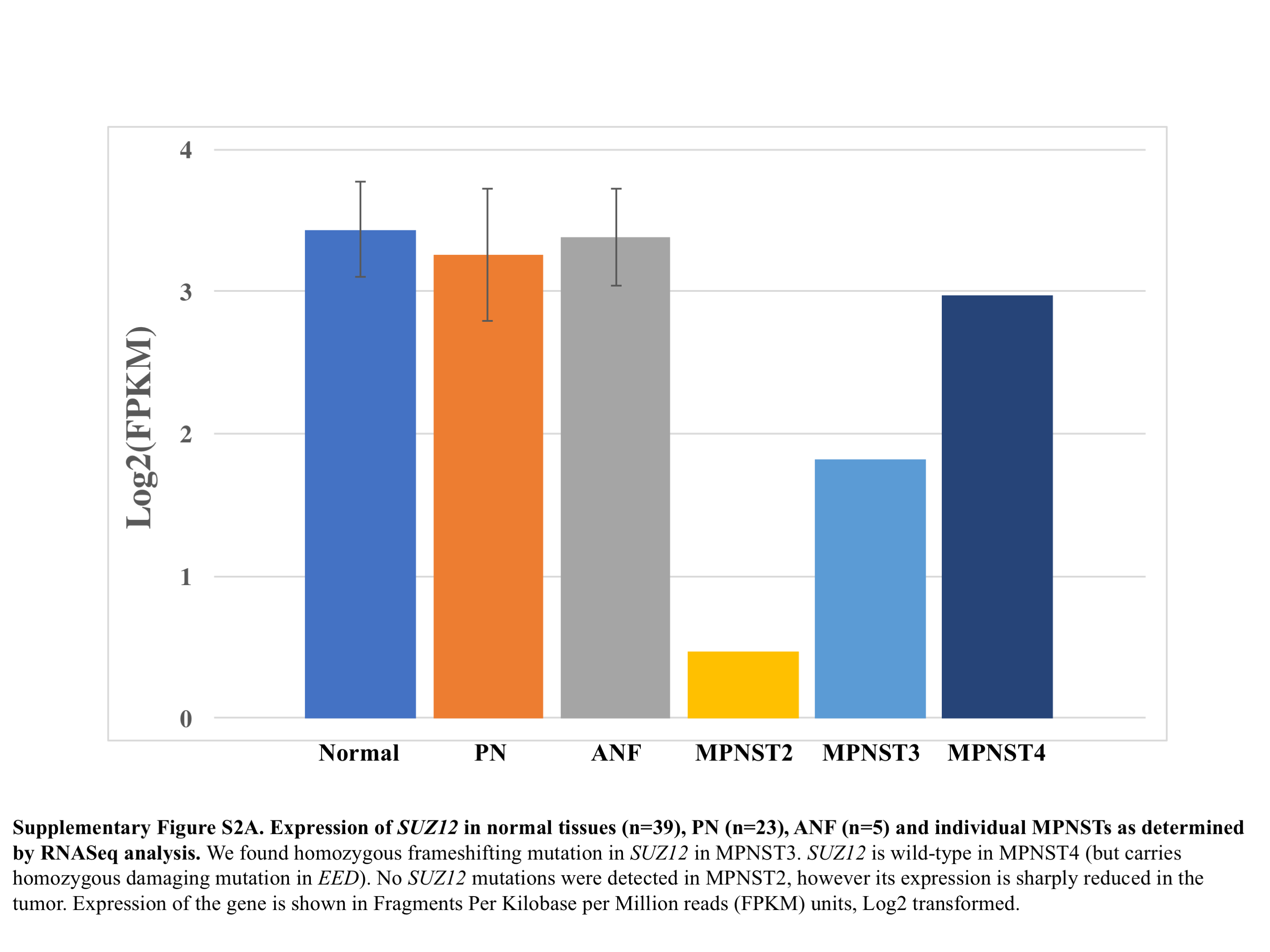
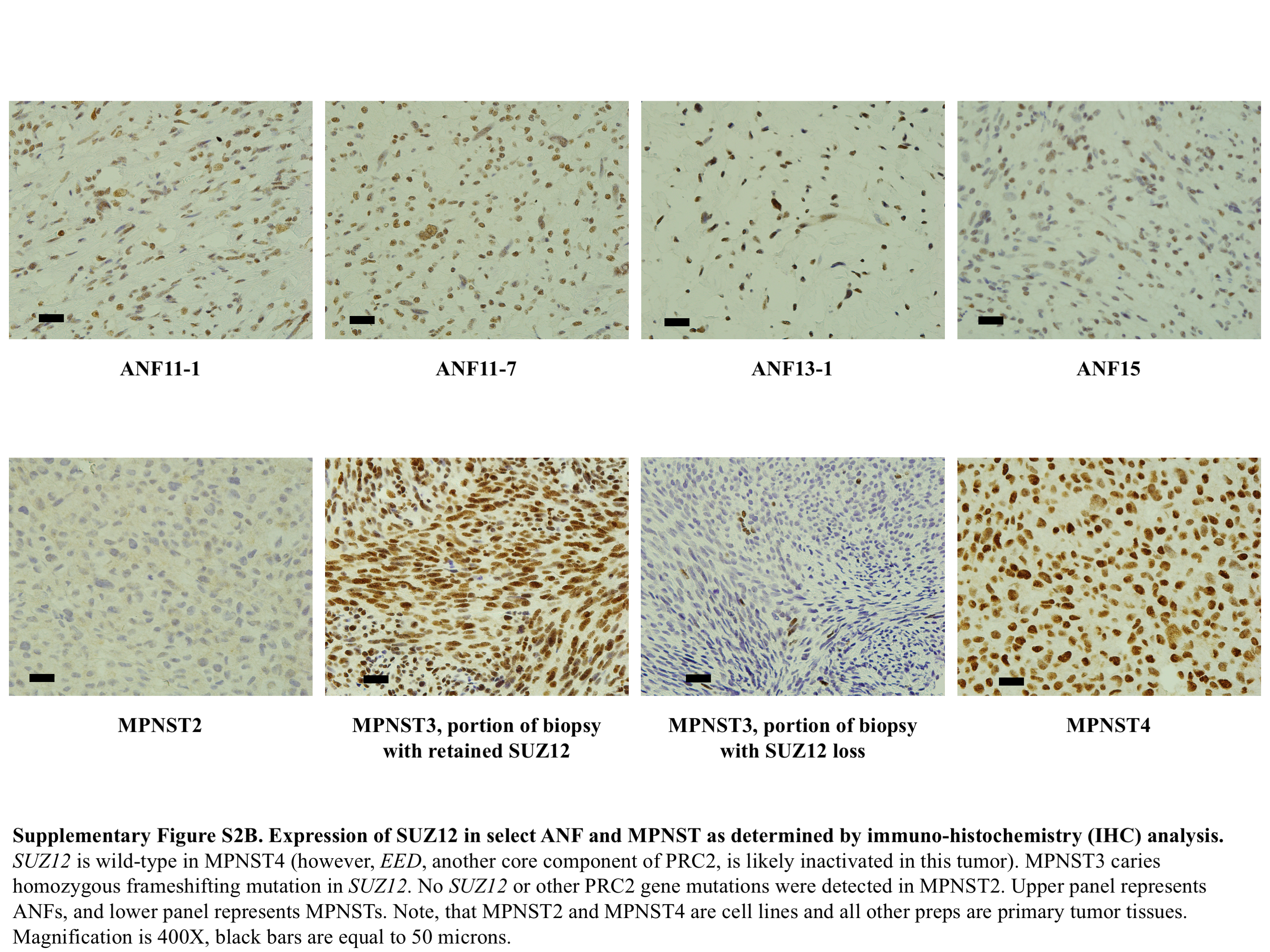
In MPNSTs, we found pathogenic NF1 germline and somatic mutations/deletions in 4/4 (100%) samples. There was heterozygous or homozygous loss of the CDKN2A/B locus in 4/4 (100%) MPNSTs. Like ANFs, we did not identify point mutations or small indels at this locus. We identified a potentially deleterious homozygous missense mutation in EED (but intact SUZ12) in MPNST4 and a frame-shifting homozygous indel in SUZ12 in MPNST3-1. In MPNST2, no PRC2 genes were mutated; however, expression of SUZ12, as demonstrated by RNA sequencing (RNAseq) analysis, was sharply reduced in this tumor, implying that in some cases epigenetic inactivation of PRC2 genes could be at play (Supplementary Figure 2A). The RNAseq findings for SUZ12 expression in the MPNSTs were confirmed by IHC staining with anti-SUZ12 antibodies: We observed robust SUZ12 expression in MPNST4 and sharply reduced levels of the protein in MPNST2 (Supplementary Figure 2B). Interestingly, in MPNST3, in which SUZ12 carried a homozygous frameshifting indel and in which the SUZ12 RNA expression was at ~50% of normal (Supplementary Figure 2A), we observed substantial tumor heterogeneity, with distinct areas in the tumor biopsy exhibiting drastically varying levels of SUZ12 expression (Supplementary Figure 2B). It appears that in some MPNST3 cells the mutant protein is still expressed; however, it is likely that its function is affected by this truncating mutation.
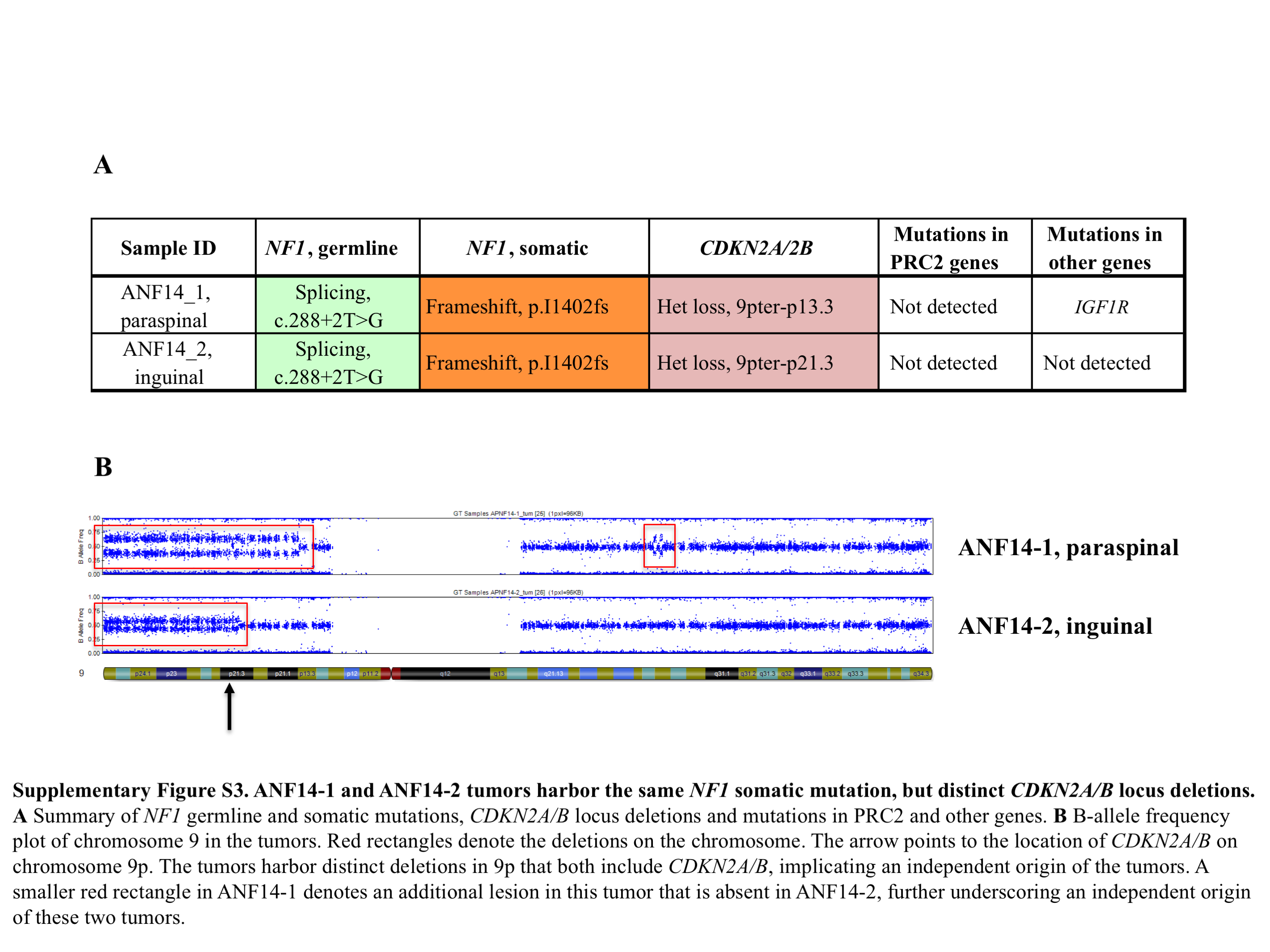
We observed a particularly informative case of a 9-year-old NF1 patient with a PN involving the right inguinal area, pelvis, and thigh (Figure 1). MRI evaluation at NCI revealed 2 DNLs in the inguinal and paraspinal areas. Core biopsies of the inguinal and paraspinal lesions were classified as an ANF and neurofibroma, respectively. The inguinal lesion was later resected and the paraspinal tumor was observed. Genetic analysis of the paraspinal (ANF14-1) and inguinal nodular lesions (ANF14-2) confirmed that they originated within the same PN, since they shared the same second hit in NF1. However, ANF14-1 and ANF14-2 harbor distinct deletions of chromosome 9p, which includes CDKN2A/B (Supplementary Figure 3). These observations suggest that ANF14-1 and ANF14-2 arose within the PN and that deletion of CDKN2A/B is a predominant driving event in this transformation.
Fig. 1.

MRI and fluorodeoxyglucose (FDG) PET evaluation of 2 distinct nodular lesions within a single large PN. (A, B) A 9-year-old boy with NF1 and newly diagnosed PN involving the right retroperitoneum, pelvis, and thigh. MRI evaluation at the NCI revealed 2 distinct nodular lesions, one in the right inguinal area (A; sample ANF14-2), and one in the right paraspinal area (B; sample ANF14-1). (C‒E) Volumetric MRI analysis demonstrated faster growth rates for the nodular lesions (black contour) compared with the PN (green contour). (F, G) FDG-PET demonstrated FDG avidity of the nodular lesions with minimal uptake in the surrounding PN (F and G, inguinal and paraspinal lesions, respectively). Core biopsy of the inguinal lesion showed ANF; core biopsy of the paraspinal lesion showed neurofibroma. The inguinal lesion was resected, and pathology confirmed atypical neurofibroma. Follow-up with MRI continues to demonstrate more rapid growth of the remaining paraspinal lesion compared with the PN (C, D).
Small-Scale Somatic Mutations in ANF and MPNST Exomes
We identified 13 somatic mutations in 13 genes in 9 ANFs (Supplementary Table 2A). Of the 13 mutated genes, one (FOXP1) belonged to the cancer genes in the census of COSMIC (Catalogue of Somatic Mutations in Cancer). One tumor (ANF8) harbored 5 mutations in 5 different genes, 3 of which were predicted to be damaging. All mutations in ANFs were heterozygous, with a median variant allele frequency (VAF) of 0.21 (range 0.18–0.34); NF1 was the only recurrently mutated gene.
In contrast to ANFs, we observed 70 somatic mutations in 3 MPNSTs, 34 of which were potentially deleterious (Supplementary Table 2B). Unlike ANF mutations, 19/70 mutations in MPNSTs were homozygous (including EED and SUZ12), and the median VAF was 0.51 (range 0.15–1.00). One gene, KAT7, was mutated twice in a single tumor (MPNST2), but both mutations were in cis. NF1 and PRC2 genes (EED and SUZ12 in MPNST4 and MPNST3, respectively) were the only genes found recurrently mutated in the MPNSTs.
Chromosomal Landscape of ANF and MPNST as Revealed by ASCAT Analysis
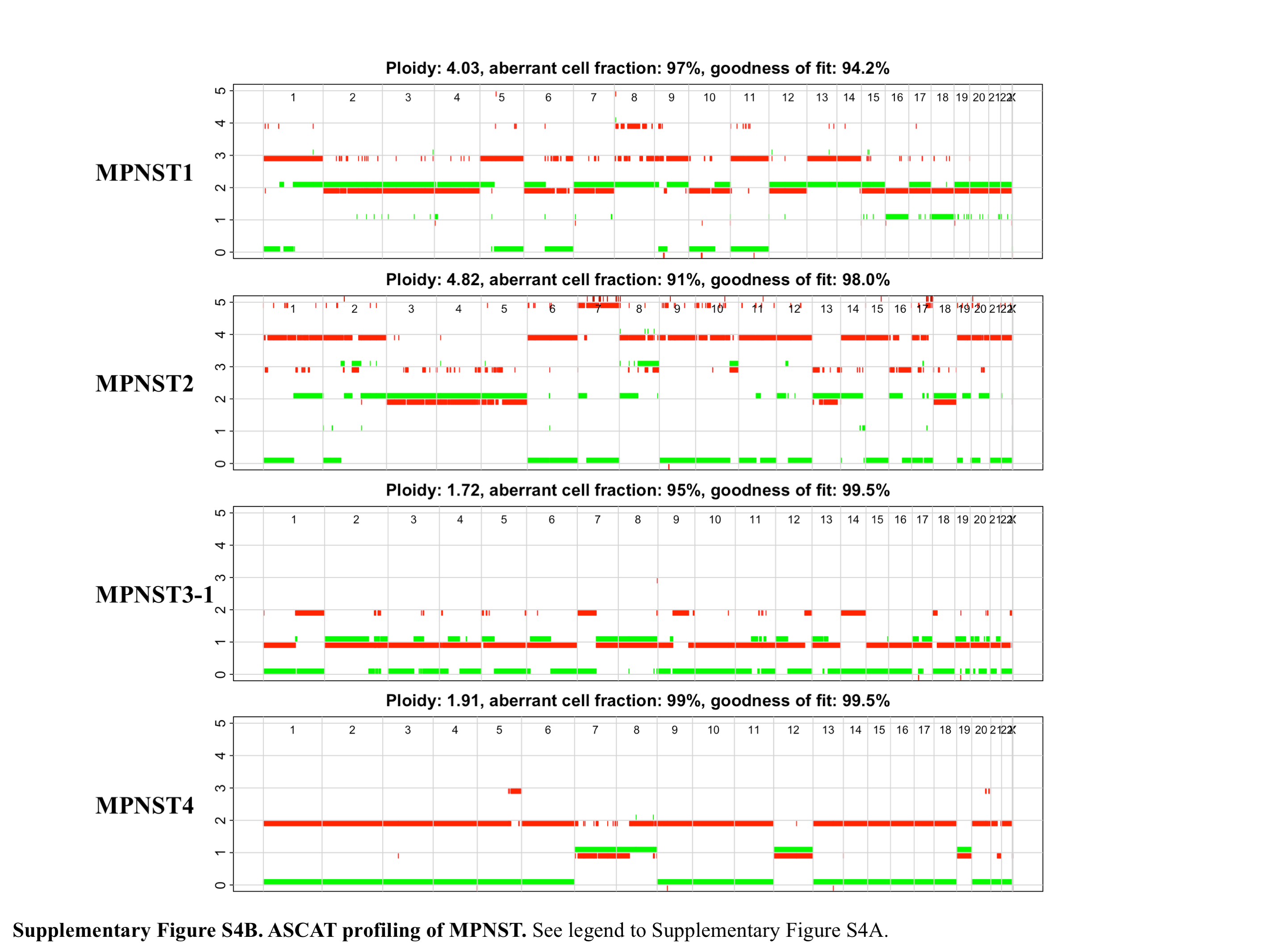
Next, we used ASCAT to determine allele-specific copy number in 10 ANFs and 4 MPNSTs. Most ANFs were diploid, except for a single tumor (ANF13), which was nearly tetraploid. The median value of the fraction of atypical cells containing CNV in the ANFs was 40.5% (range 22–100%) (Supplementary Figure 4A). Two of the MPNSTs (MPNST1 and MPNST2) were polyploid (4.03 and 4.82, respectively), while MPNST3 and MPNST4 were nearly diploid (1.72 and 1.91). The median proportion of aberrant cells in the MPNSTs was 96% (range 91–99%) (Supplementary Figure 4B).
CNV Meta-Analysis of the Combined ANF Set
Next, we characterized individual CNVs in the combined set of 26 ANFs using Nexus and GISTIC analyses (Figure 2). In 17/26 ANFs (65%), the most frequently affected locus was in 9p21.3, which harbors CDKN2A/B (Figure 2B and Supplementary Figure 5A). Most deletions were heterozygous. One deletion in one sample (A2) was called homozygous by the Nexus software, and an additional 6 samples (A7_ANF5, A9, A12, A14, A16, and ANF10) had probe median log2R ratio values considerably lower than the rest of the samples with the deletion (<−0.7), suggesting that the CDKN2A/B deletion in these samples was homozygous as well, a finding masked by heterogeneity of the tumors. We found several other regions adjacent to the CDKN2A/B locus on 9p that were deleted in more than one-third of the samples, but most of these regions were part of the larger deletion spanning CDKN2A/B, implying that the majority of genes within these CNVs might simply be passengers (Figure 2B). We identified one exception: Switching-defective/sucrose nonfermentable (SWI/SNF) related, matrix associated, actin dependent regulator of chromatin, subfamily A2 (SMARCA2) was heterozygously deleted in 42% of samples. In ~12% of ANFs (A11_ANF7, A12, ANF11-7), the deletion was clearly independent from CDKN2A/B locus loss (Supplementary Figure 2B), suggesting a causative role of SWI/SNF complex disruption in tumor progression.
Fig. 2.

CNV in ANF and MPNST. (A) GISTIC analysis of 26 ANFs (upper panel) and 28 MPNSTs (lower panel). Gains and losses are shown in blue and red, respectively. Statistically significant CNVs (false discovery rate <0.25) are denoted with gray vertical bars. Chromosome numbers are shown on top of each panel. (B) CNV in chromosome 9p in 26 ANFs (left panel) and 28 MPNSTs (right panel). Gains and losses are shown in blue and red, respectively. Homozygous losses are shown in dark red and thick bars. Vertical rectangles in the center denote minimally overlapped regions in ANF and MPNST that both include the CDKN2A and CDKN2B genes. Left vertical rectangle in the ANF panel denotes SMARCA2. Arrows show samples (A11_ANF7, A12, ANF11-7) in which deletion of SMARCA2 is clearly independent from CDKN2A/B.
Among all ANFs, we identified 253 genes that were affected in at least 4/26 samples. Eleven of these 253 genes, including CDKN2A, were cancer genes (Supplementary Table 3A–C). One of them, FANCG, is a DNA repair protein implicated in maintenance of chromosome architecture (www.uniprot.org), Accessed March 1, 2019 frequent deletion of this gene could increase genomic instability, a common feature in MPNST. Another gene, PTPRD, is a tumor suppressor and a negative regulator of the STAT3 oncogene.29 A frequent loss of PTPRD or FANCG in ANF warrants further investigation. The role of other genes (eg, JAK2, PDCD1LG2, PSIP1, FCGR2B) that are predominantly deleted in ANF but are known as oncogenes or fusion genes is more challenging to define. It appears these genes are more likely to be passengers (Supplementary Table 3B).
CNV Meta-Analysis of the Combined MPNST Set
We observed a highly rearranged genomic architecture in the MPNSTs (Figure 2, Supplementary Figure 5B). Like the ANFs, the CDKN2A/B locus was one of the most frequently deleted loci (20/28 [71%]) (Figure 2B). Moreover, we observed a homozygous deletion of this locus in 14/28 (50%) samples, which was the most frequently observed homozygous loss in these tumors (Figure 2B).
Across all samples, there were 23 229 genes (including non-coding RNA genes) affected by gains or losses in the regions that overlapped in at least 4/28 samples; 700 were cancer genes (Supplementary Table 3D–F). Out of these 700 genes, we identified 178 oncogenes that were at increased copy-number state, and 144 tumor suppressor genes (TSGs) that were hetero- or homozygously deleted in at least 4/28 samples (Supplementary Table 3E, F). We observed copy-number gains in receptor tyrosine kinase genes that control cellular circuitry, including Ras–mitogen-activated protein kinase (MAPK), phosphatidylinositol-3 kinase–Akt, phospholipase C gamma–protein kinase C, and Janus kinase/signal transducers and activators of transcription pathways (eg, EGFR, ERBB2/3/4); signal transduction kinases/phosphatases (eg, BRAF, AKT2/3); G1-S phase transition genes (eg, CDK6, CCND1/3); negative regulators of TP53 (PPM1D, MDM2/4), and telomerase reverse transcriptase, among others. We detected gains and amplifications in MYC in 71% of samples and the transcriptional activator of MYC, TRRAP, was gained in 46% of samples. In addition to MYC, we identified increased copy number of KLF4 and SOX2, the genes involved in maintaining “stemness” and pluripotency. EZH2, the methyltransferase subunit of PRC2, was found at elevated copy-number state in 50% of MPNSTs.
In addition to multiple gains in oncogenes, we observed a number of losses in TSGs, including DNA repair/recombination genes (eg, ATM, PALB2); chromatin modification genes, including PRC2 EED and SUZ12 and subunits of chromatin remodeling complex SWI/SNF (ARID1A, PBRM1), and genes controlling pluripotency of stem cells (ZFHX3, JAK1, BMPR1A, AXIN1, TCF3, TBX3).
GISTIC Analysis of ANF and MPNST
We identified statistically significant CNVs (false discovery rate <0.25) in 26 ANFs and 28 MPNSTs by GISTIC (Figure 2A, Supplementary Table 3G). Frequent loss of the CDKN2A/B locus was confirmed in both types of tumors. In MPNST, we confirmed frequent gain of oncogenes MDM2 and PDGFRA and frequent loss of PRC2 genes EED and SUZ12.
Significant Increase in Genomic Instability in Transition from ANF to MPNST
We compared the median number of CNVs, median CNV size, median number of bases in all CNVs combined in a tumor and the affected portion of the genome in ANF and MPNST (Table 3). There was a ~7-fold increase in the median number of CNV, a ~4-fold increase of the median size of CNV, and a 33-fold increase of the affected portion of the genome (expressed either as a number of bases or percent of the genome) in MPNST versus ANF. P-values for all comparisons were <10−5 (Mann–Whitney test), indicating a substantially and significantly increased level of genomic instability in the transition from premalignant to malignant state.
Table 3.
Summary of CNVs in ANF and MPNST
| Type of Tumor (number of samples) | Median Number of CNVs per Tumor (range) | Median Size of CNV in Kb (range) | Median Number of Bases in All CNVs in Mb (range) | Percent of the Affected Part of the Genome per Tumor (range) |
|---|---|---|---|---|
| ANF (26) | 48 (4–555) | 70 (0.6–25,188) | 37 (0.5–160) | 0.62 (0.01- 2.66) |
| MPNST (28) | 323 (73–4946) | 275 (0.7–138,465) | 1234 (152–3017) | 20.6 (2.5–50.3) |
Analysis of Genome-Wide Expression RNAseq Data by GSEA
We analyzed differential expression in ANFs and MPNSTs versus normal tissues using GSEA. Significant gene sets upregulated in ANF related to the immune response, signal transduction, and processes affected in various types of cancer. Downregulated gene sets in ANF were associated with oxidative phosphorylation and cellular respiration. Upregulated gene sets in MPNST were associated with cell cycle, DNA replication/repair, and chromosomal organization, while downregulated gene sets often included genes that were underexpressed in tumors with activated KRAS (Supplementary Table 4). There was a minimal overlap between significant gene sets in ANF and MPNST (data not shown).
Next, we identified up- and downregulated genes in ANF and MPNST that contributed to the enrichment score by performing leading edge analysis. There were 2075 and 3711 upregulated, and 396 and 1738 downregulated genes in ANF and MPNST, respectively. Overlapping analysis between ANF and MPNST revealed 787 up- and 127 downregulated genes in common. Both overlaps were highly statistically significant by the hypergeometric test. There were 30 known cancer genes among the genes in these overlaps (Supplementary Table 5). CCND1, CDK6, NRAS, MDM2, and MET were among the overexpressed genes, and AXIN2 was among the downregulated genes.
Differential Expression of Genes Frequently Affected by CNV/Mutations, Genes Identified by GSEA Leading Edge Analysis, and Select Biologically Relevant Genes
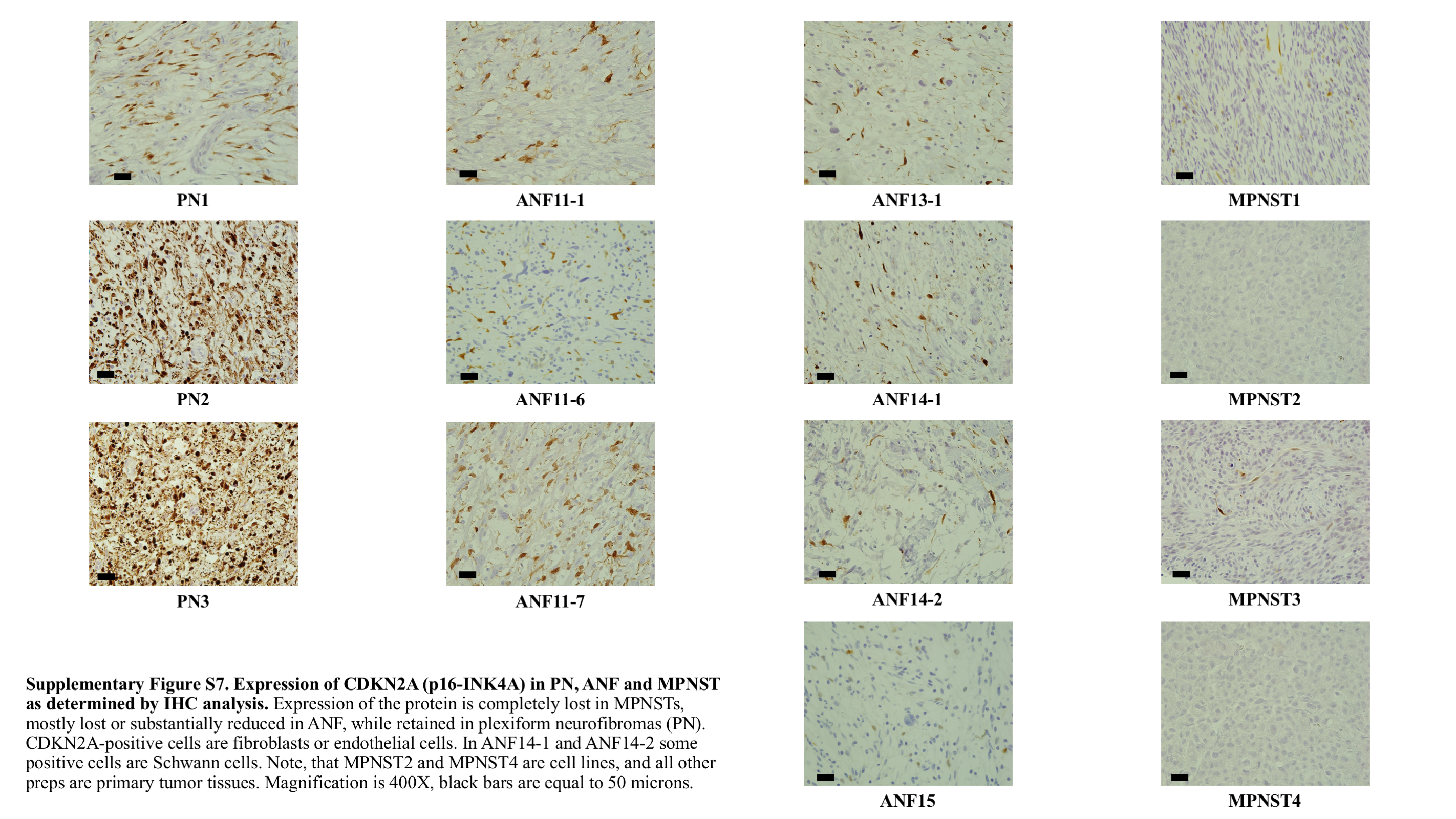
To better understand the transition of expression profiles from normal tissues to a premalignant state in ANF, we have included RNAseq data for 23 primary Schwann cell cultures established from NF1-associated PNs and enriched for NF1−/− cells. First, we compared expression of 12 genes, including NF1, CDKN2A/B, SMARCA2, NRAS, PRC2 genes, CCND1, CDK6, MDM2, TP53 in PN, ANF, MPNST, and normal tissues (Figure 3). In addition, we explored differential expression of genes associated with these 12 genes through participation in protein complexes (eg, SWI/SNF, PRC1/2) or signaling pathways (eg, cell cycle, TP53) (Supplementary Figure 6). We observed highly statistically significant overexpression of NRAS (but not K- and HRAS) in all 3 types of tumors: PN, ANF, and MPNST. Cell-cycle inhibitors CDKN2A and CDKN2B were expressed at very low levels in normal tissues, as expected. Sharply increased expression of these 2 genes in PN is likely due to inactivation of NF1 and subsequent activation of RAS, which in turn mediates oncogene-induced senescence (see Discussion). In ANF and MPNST, where frequent deletion of the CDKN2A/B locus was observed, expression of these genes was predictably lower than in PN, but still higher than in normal tissues. We confirmed these observations by performing IHC analysis of PN, ANF, and MPNST using anti–p16-INK4A antibodies. One can observe a robust expression of CDKN2A in most PNs and substantially lower levels of this protein in ANF and MPNST (Supplementary Figure 7). However, we observed a wide spectrum of intensities of p16 staining in the tumors, thus p16-INK4A IHC should be used in combination with other clinical and molecular analyses. Expression of cyclins D1 and D2 (CCND1/2) and CDK4/6, which control G1/S transition, was significantly higher in all 3 types of tumors compared with normal tissues (Supplementary Figure 6A). We observed elevated expression of MDM2 in all types of tumors as well; however, expression of TP53, which is the target of MDM2 suppression, was mainly unchanged in the tumors (Supplementary Figure 6B). We found that expression of SUZ12 and EED was not significantly affected in the tumors; however, EZH2, the catalytic subunit of PRC2, was highly overexpressed in MPNST. In contrast, in ANF, expression of EZH2 was similar to the normal controls (Supplementary Figure 6C). Expression of SMARCA2 in ANF was essentially the same as in normal tissues (Figure 3, Supplementary Figure 6E), despite the observation that the gene was heterozygously deleted in 42% of the tumors. However, examination of the samples, which were used for the expression analysis, has shown that only 1 of 5 samples carried heterozygous deletion of SMARCA2. In addition, ANFs are heterogeneous tumors with a substantial proportion of nontumor cells, which further challenges accurate estimation of gene expression in these tumors. A modest but statistically significant downregulation of SMARCA2 in the PNs (Figure 3) warrants further investigation.
Fig. 3.

Expression of genes frequently affected by CNV/mutations, genes identified by GSEA leading edge analysis, and select biologically relevant genes in normal tissues (n = 39), PN (n = 23), ANF (n = 5), and MPNST (n = 5) as determined by RNAseq analysis. Expression values are shown in fragments per kilobase per million (FPKM) reads, log2 transformed, log2(FPKM) units. Blue, red, gray, and orange bars correspond to normal tissues, PN, ANF, and MPNST, respectively. Asterisks indicate statistically significant difference (by 2-tailed t-tests) between normal and PN, ANF, or MPNST expression values at P < 0.0001.
Discussion
This report is the first to describe comprehensive multiplatform genomic analyses of NF1-associated atypical neurofibromas from multiple patients. We show that NF1 and CDKN2A/B loss are the primary genetic drivers in the development of ANF. In ANF, we observed SMARCA2 loss in 42% of samples; we did not observe mutation or copy-number changes in TP53 or the PRC2 complex. The overall somatic mutation burden in the ANFs was low and similar to that observed in PN.15 However, unlike in PN, where chromosomal architecture is essentially normal,15 we detected frequent (69%) deletions in chromosome 9p, which included CDKN2A/B. Overall, we observed a relatively low level of genomic instability in ANF and a profound, significantly increased perturbation of chromosomal architecture in MPNST, resulting in frequent gains of 178 oncogenes and in frequent losses in 144 TSGs. Gene expression analysis with RNAseq revealed upregulated NRAS, MDM2, CCND1/2/3, and CDK4/6 in both ANF and MPNST, but CCNA/E/B, CDK2/1, and EZH2 as well as the genes controlling mitosis were overexpressed in MPNST only. We provide a case report of 2 genomically distinct ANFs (both harboring 9p [CDKN2A/B] deletions) that arose as radiographically distinct nodular lesions from the same PN in a child with NF1.
Nielsen and colleagues14 observed that benign neurofibromas expressed p16, whereas the MPNSTs were essentially p16 negative. They concluded that inactivation of CDKN2A is associated with malignant transformation of neurofibromas. The most comprehensive study of ANF to date found one highly recurrent (15/16) deletion of the CDKN2A/B locus.10 A recent study has demonstrated that there is normal status of CDKN2A/B in PN but a deletion of this locus in 2 ANFs resected from the same patient and that, importantly, a copy-number status of CDKN2A/B correlated with a degree of histological atypia in these ANFs.30 Consistent with the previous studies, we identified hetero- or homozygous deletion of the CDKN2A/B locus as the most frequent genetic aberration in ANF tumors and one of the most frequent in MPNST.
In addition to frequent loss of CDKN2A/B, we found deletion of SMARCA2 in 42% of ANF; moreover, in at least 3 samples the SMARCA2 deletion was clearly separate from the CDKN2A/B loss, pointing to a possible causative role of SWI/SNF complex disruption in the tumors. SMARCA2 is an integral part of the ATP-dependent chromatin remodeling and transcriptional activator complex SWI/SNF, which in many instances acts as an antagonist of PRC1 and PRC2.31 Given that the CDKN2A/B locus is itself a target for PRC1/2 inactivation,32 heterozygous deletion of SMARCA2 may lead to a partial inactivation of the SWI/SNF complex, which in turn may lower activity of p16-INK4a/b and p14-ARF by establishing a more repressive chromatin structure by PRC1/2 at this locus. It will be important to further investigate what role inactivation of SMARCA2 and other SWI/SNF genes might play in clinical severity of ANF.
In a first, we conducted RNAseq on ANFs. We acknowledge that the number of samples in our study was modest. Consistent with clinical observations that demonstrated continuous growth of ANF,8,9,33 we detected elevated expression of NRAS, CCND1/2/3, and CDK4/6 in these tumors. We detected elevated levels of CDKN2A/B in ANF compared with normal tissues; however, this is consistent with findings that an activated Ras-MAPK pathway (which in PNs is caused by NF1 inactivation) induces senescence by stimulating these cell-cycle inhibitors.34 The frequent deletions of the CDKN2A/B locus that we observed in ANF may aid in overcoming p16-, p15-, and p14-mediated inhibition of the cell cycle in these tumors and, in turn, activate MDM2, which was also one of the genes with frequent gains in the MPNST and overexpressed in both types of tumors. Interestingly, in a recent study35 it was demonstrated that MDM2 directly binds to EZH2 and modifies methylation of histones, thus promoting stemness and enabling cancer cell survival independently of p53. In light of these observations, inhibition of MDM2 and/or EZH2 in MPNST might be an appealing therapeutic strategy.
At present, the diagnosis of ANF is difficult and primarily is based on pathological examination of the tumors. A new term, “atypical neurofibromatous neoplasms of uncertain biologic potential (ANNUBP),” and diagnostic criteria have been recently proposed.20 In this study, we evaluated 2 distinct ANFs (ANF14-1 and ANF14-2) from an NF1 patient and arising from the same PN. Although clinically both lesions were worrisome, the pathology examination classified only one tumor as an ANF, while deeming the other as benign neurofibroma. Subsequent genomic analysis of the tumors revealed that both lesions had concerning CDKN2A/B locus deletion. Careful reexamination of the tumors by the pathologist left the diagnoses unchanged; however, based on clinical and genetic evidence, we believe that both tumors should be treated as ANF. This example underscores the importance of genetic information in clinical decision making and illustrates that PN-ANF transformation could be relatively frequent, at least in some patients.
In conclusion, in our data, in addition to PN-related NF1 inactivation, transition from benign PN to premalignant ANF frequently proceeds through inactivation of CDKN2A/B. CDKN2A/B appears to be the primary driver of this transition, but perhaps other genetic events (eg, deletion of SMARCA2) are also involved. Upon further transformation to the malignant state the level of genomic instability rises dramatically, accelerating complete loss of function of key gatekeepers via loss of heterozygosity (eg, EED, SUZ12) and by affecting copy number of multiple oncogenes and TSGs.
The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services, nor does mention of trade names, commercial products, or organizations imply endorsement by the US government.
Funding
This work was supported by the Intramural Research Program of the Division of Cancer Epidemiology and Genetics and the Center for Cancer Research of the National Cancer Institute, Bethesda, Maryland, the Intramural Research Program of the National Human Genome Research Institute, Bethesda, Maryland; NIH grant R29 #NS31550 (to MRW); Department of Defense DAMD 179810689 (to MRW) and Children’s Tumor Foundation Neurofibroma Therapeutics Acceleration Award (to MRW).
Conflict of interest statement. The authors declare no conflicts of interest.
Authorship statement. AP—performed experiments, analyzed data, wrote and prepared manuscript for publication, submitted manuscript for publication; NFH, SS, RP, BZ—analyzed high-throughput data, wrote portion of the manuscript; CSH—collected and analyzed clinical data, wrote portion of the manuscript; ED—analyzed clinical data, wrote portion of the manuscript; MMM—performed pathology analysis of tumors; PF—prepared tumor tissues for pathology analysis; HB—collected surgical material, isolated DNA from blood and tumors, collected and analyzed clinical data; SC—performed SNP-microarray experiments, wrote portion of the manuscript; KJ—performed high-throughput experiments, wrote portion of the manuscript; JSW—wrote portion of the manuscript; JCM—contributed to experimental design and its implementation; JK—contributed to experimental design, its implementation, and interpretation of the data; EL, MRW—provided biospecimens, contributed to experimental design and interpretation of the data; BCW—provided biospecimens, contributed to experimental design and its implementation, wrote portion of the manuscript; DRS—contributed to experimental design, its implementation and interpretation of the data, wrote portion of the manuscript.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
References
- 1. Uusitalo E, Leppävirta J, Koffert A, et al. . Incidence and mortality of neurofibromatosis: a total population study in Finland. J Invest Dermatol. 2015;135(3):904–906. [DOI] [PubMed] [Google Scholar]
- 2. Friedman JM. GeneReviews®. Pagon RAet al. , eds. Seattle: University of Washington; 1993. [Google Scholar]
- 3. Evans DG, O’Hara C, Wilding A, et al. . Mortality in neurofibromatosis 1: in North West England: an assessment of actuarial survival in a region of the UK since 1989. Eur J Hum Genet. 2011;19(11):1187–1191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Uusitalo E, Rantanen M, Kallionpää RA, et al. . Distinctive cancer associations in patients with neurofibromatosis type 1. J Clin Oncol. 2016;34(17):1978–1986. [DOI] [PubMed] [Google Scholar]
- 5. Evans DG, Baser ME, McGaughran J, Sharif S, Howard E, Moran A. Malignant peripheral nerve sheath tumours in neurofibromatosis 1. J Med Genet. 2002;39(5):311–314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Mautner VF, Asuagbor FA, Dombi E, et al. . Assessment of benign tumor burden by whole-body MRI in patients with neurofibromatosis 1. Neuro Oncol. 2008;10(4):593–598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Kim A, Stewart DR, Reilly KM, Viskochil D, Miettinen MM, Widemann BC. Malignant peripheral nerve sheath tumors state of the science: leveraging clinical and biological insights into effective therapies. Sarcoma. 2017;2017:7429697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Meany H, Dombi E, Reynolds J, et al. . 18-fluorodeoxyglucose-positron emission tomography (FDG-PET) evaluation of nodular lesions in patients with Neurofibromatosis type 1 and plexiform neurofibromas (PN) or malignant peripheral nerve sheath tumors (MPNST). Pediatr Blood Cancer. 2013;60(1):59–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Higham CS, Dombi E, Rogiers A, et al. . The characteristics of 76 atypical neurofibromas as precursors to neurofibromatosis 1 associated malignant peripheral nerve sheath tumors. Neuro Oncol. 2018;20(6):818–825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Beert E, Brems H, Daniëls B, et al. . Atypical neurofibromas in neurofibromatosis type 1 are premalignant tumors. Genes Chromosomes Cancer. 2011;50(12):1021–1032. [DOI] [PubMed] [Google Scholar]
- 11. Berner JM, Sørlie T, Mertens F, et al. . Chromosome band 9p21 is frequently altered in malignant peripheral nerve sheath tumors: studies of CDKN2A and other genes of the pRB pathway. Genes Chromosomes Cancer. 1999;26(2):151–160. [PubMed] [Google Scholar]
- 12. Kourea HP, Orlow I, Scheithauer BW, Cordon-Cardo C, Woodruff JM. Deletions of the INK4A gene occur in malignant peripheral nerve sheath tumors but not in neurofibromas. Am J Pathol. 1999;155(6):1855–1860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Mantripragada KK, Spurlock G, Kluwe L, et al. . High-resolution DNA copy number profiling of malignant peripheral nerve sheath tumors using targeted microarray-based comparative genomic hybridization. Clin Cancer Res. 2008;14(4):1015–1024. [DOI] [PubMed] [Google Scholar]
- 14. Nielsen GP, Stemmer-Rachamimov AO, Ino Y, Moller MB, Rosenberg AE, Louis DN. Malignant transformation of neurofibromas in neurofibromatosis 1 is associated with CDKN2A/p16 inactivation. Am J Pathol. 1999;155(6):1879–1884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Pemov A, Li H, Patidar R, et al. ; NISC Comparative Sequencing Program; NCI DCEG Cancer Genomics Research Laboratory The primacy of NF1 loss as the driver of tumorigenesis in neurofibromatosis type 1-associated plexiform neurofibromas. Oncogene. 2017;36(22):3168–3177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Lee W, Teckie S, Wiesner T, et al. . PRC2 is recurrently inactivated through EED or SUZ12 loss in malignant peripheral nerve sheath tumors. Nat Genet. 2014;46(11):1227–1232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Zhang M, Wang Y, Jones S, et al. . Somatic mutations of SUZ12 in malignant peripheral nerve sheath tumors. Nat Genet. 2014;46(11):1170–1172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. De Raedt T, Beert E, Pasmant E, et al. . PRC2 loss amplifies Ras-driven transcription and confers sensitivity to BRD4-based therapies. Nature. 2014;514(7521):247–251. [DOI] [PubMed] [Google Scholar]
- 19. Reilly KM, Kim A, Blakely J, et al. . Neurofibromatosis type 1-associated MPNST state of the science: outlining a research agenda for the future. J Natl Cancer Inst. 2017;109(8):djx124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Miettinen MM, Antonescu CR, Fletcher CDM, et al. . Histopathologic evaluation of atypical neurofibromatous tumors and their transformation into malignant peripheral nerve sheath tumor in patients with neurofibromatosis 1—a consensus overview. Hum Pathol. 2017;67:1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Cibulskis K, Lawrence MS, Carter SL, et al. . Sensitive detection of somatic point mutations in impure and heterogeneous cancer samples. Nat Biotechnol. 2013;31(3):213–219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Larson DE, Harris CC, Chen K, et al. . SomaticSniper: identification of somatic point mutations in whole genome sequencing data. Bioinformatics. 2012;28(3):311–317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Hansen NF, Gartner JJ, Mei L, Samuels Y, Mullikin JC. Shimmer: detection of genetic alterations in tumors using next-generation sequence data. Bioinformatics. 2013;29(12):1498–1503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Saunders CT, Wong WS, Swamy S, Becq J, Murray LJ, Cheetham RK. Strelka: accurate somatic small-variant calling from sequenced tumor-normal sample pairs. Bioinformatics. 2012;28(14):1811–1817. [DOI] [PubMed] [Google Scholar]
- 25. Fuentes Fajardo KV, Adams D, Mason CE, et al. ; NISC Comparative Sequencing Program Detecting false-positive signals in exome sequencing. Hum Mutat. 2012;33(4):609–613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Van Loo P, Nilsen G, Nordgard SH, et al. . Analyzing cancer samples with SNP arrays. Methods Mol Biol. 2012;802:57–72. [DOI] [PubMed] [Google Scholar]
- 27. Sathirapongsasuti JF, Lee H, Horst BA, et al. . Exome sequencing-based copy-number variation and loss of heterozygosity detection: ExomeCNV. Bioinformatics. 2011;27(19):2648–2654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Dewan R, Pemov A, Kim HJ, et al. . Evidence of polyclonality in neurofibromatosis type 2-associated multilobulated vestibular schwannomas. Neuro Oncol. 2015;17(4):566–573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Kim M, Morales LD, Jang IS, Cho YY, Kim DJ. Protein tyrosine phosphatases as potential regulators of STAT3 signaling. Int J Mol Sci 2018;19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Carrió M, Gel B, Terribas E, et al. . Analysis of intratumor heterogeneity in Neurofibromatosis type 1 plexiform neurofibromas and neurofibromas with atypical features: correlating histological and genomic findings. Hum Mutat. 2018;39(8):1112–1125. [DOI] [PubMed] [Google Scholar]
- 31. Kadoch C, Copeland RA, Keilhack H. PRC2 and SWI/SNF chromatin remodeling complexes in health and disease. Biochemistry. 2016;55(11):1600–1614. [DOI] [PubMed] [Google Scholar]
- 32. Gil J, Peters G. Regulation of the INK4b-ARF-INK4a tumour suppressor locus: all for one or one for all. Nat Rev Mol Cell Biol. 2006;7(9):667–677. [DOI] [PubMed] [Google Scholar]
- 33. Ferner RE, Golding JF, Smith M, et al. . [18F]2-fluoro-2-deoxy-D-glucose positron emission tomography (FDG PET) as a diagnostic tool for neurofibromatosis 1 (NF1) associated malignant peripheral nerve sheath tumours (MPNSTs): a long-term clinical study. Ann Oncol. 2008;19(2):390–394. [DOI] [PubMed] [Google Scholar]
- 34. DeNicola GM, Tuveson DA. RAS in cellular transformation and senescence. Eur J Cancer. 2009;45(Suppl 1):211–216. [DOI] [PubMed] [Google Scholar]
- 35. Wienken M, Dickmanns A, Nemajerova A, et al. . MDM2 associates with polycomb repressor complex 2 and enhances stemness-promoting chromatin modifications independent of p53. Mol Cell. 2016;61(1):68–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.