

Abstract
Background
Although epidermal growth factor receptor (EGFR) and its truncated, autoactive mutant EGFR variant (v)III are bona fide drivers of tumorigenesis in some gliomas, therapeutic antibodies developed to neutralize this axis have not improved patient survival in a limited number of trials. Previous studies using cells transduced to exogenously express EGFRvIII may have compromised mechanistic studies of anti-EGFR therapeutics. Therefore, we re-assessed the activity of clinical EGFR antibodies in patient-derived gliomaspheres that endogenously express EGFRvIII.
Methods
The antitumor efficacy of antibodies was assessed using in vitro proliferation assays and intracranial orthografts. Receptor activation status, antibody engagement, oncogenic signaling, and mechanism of action after antibody treatment were analyzed by immunoprecipitation and western blotting. Tracking of antibody receptor complexes was conducted using immunofluorescence.
Results
The EGFR domain III–targeting antibodies cetuximab, necitumumab, nimotuzumab, and matuzumab did not neutralize EGFRvIII activation. Chimeric monoclonal antibody 806 (ch806) neutralized EGFRvIII, but not wild-type (wt)EGFR activation. Panitumumab was the only antibody that neutralized both EGFRvIII and wtEGFR, leading to reduction of p-S6 signaling and superior in vitro and in vivo antitumor activity. Mechanistically, panitumumab induced recycling of receptor but not degradation as previously described. Panitumumab, via its unique avidity, stably cross-linked EGFRvIII to prevent its activation, while ch806 induced a marked reduction in the active EGFRvIII disulphide-bonded dimer.
Conclusions
We discovered a previously unknown major resistance mechanism in glioma in that most EGFR domain III–targeting antibodies do not neutralize EGFRvIII. The superior in vitro and in vivo antitumor activity of panitumumab supports further clinical testing of this antibody against EGFRvIII-stratified glioma.
Keywords: antibody, EGFRvIII, glioma, neutralization, resistance
Key Points.
Most clinically approved antibodies targeting EGFR domain III do not neutralize EGFRvIII.
Panitumumab is the only antibody to neutralize both wtEGFR and EGFRvIII activation.
Panitumumab is an effective therapeutic in glioma orthograft models.
Importance of the Study.
EGFR and its autoactive, truncated mutant EGFRvIII are central to driving gliomagenesis, but therapeutic antibodies targeting this axis have failed clinically for unknown reasons. Our reassessment of the efficacy and mechanism of these antibodies in clinically relevant EGFRvIII-expressing gliomaspheres has yielded a previously undiscovered mechanism of resistance. Domain III–targeting EGFR antibodies, except for panitumumab, do not simultaneously neutralize EGFRvIII and wtEGFR activation, with panitumumab proving to be a superior antitumor agent in vivo. Furthermore, panitumumab induced receptor recycling but not degradation, in contrast to previous studies in transfected cells. This reappraisal challenges the central notion that EGFR antibodies induce wtEGFR and EGFRvIII degradation and deactivation. We also define 2 distinct and previously undescribed mechanisms of action of how panitumumab and ch806 neutralize EGFRvIII.
High-grade glioma (HGG) is the most common and deadly form of brain cancer.1 Epidermal growth factor receptor (EGFR) is a single-pass transmembrane receptor tyrosine kinase. The extracellular region consists of 4 domains—I, II, III, and IV. Ligands that engage EGFR bind domains I and III, resulting in non-covalent receptor dimerization and activation. EGFR is mutated, rearranged, alternatively spliced, and/or focally amplified in 57% of HGG tumors,2 making EGFR a prime target for glioma therapy. The autoactive deletion mutant, EGFR variant III (EGFRvIII), is among the most common alterations in HGG2; the deletion removes domain I (leaving the rest of the receptor intact) and liberates a free cysteine that enables interreceptor covalent dimerization and autoactivation.3 Despite the integral role of EGFR in HGG pathogenesis, attempts to therapeutically target EGFR with tyrosine kinase inhibitors in patients have largely failed,4 even at high intratumoral drug concentrations.5
Numerous EGFR-targeting antibodies have been clinically approved for the treatment of a range of cancers.6 Although it was initially assumed that EGFR antibodies would be ineffective for HGG treatment due to restricted tumor access through the blood–brain barrier, several studies have subsequently shown that this is not the case. In particular, the antibody–drug conjugate depatuxizumab mafodotin, which targets overexpressed and mutated EGFR, has shown encouraging activity.7 The initial concerns have limited the number of clinical studies using naked ligand-blocking EGFR-targeting antibodies in HGG.4 Indeed, most reports are restricted to 2 antibodies, cetuximab and nimotuzumab, and describe trials that were conducted without selection for patients most likely to benefit. Furthermore, the activity of the different antibodies against EGFRvIII, which occurs in 30% of HGG patients, has not been systematically analyzed preclinically in patient-derived gliomaspheres and orthografts.
Many therapeutic antibodies, such as the ErbB2-targeting trastuzumab in breast cancer, have entered the clinic with an incomplete understanding of their mechanisms of action.8 We recently showed that rilotumumab, an antibody designed to prevent hepatocyte growth factor (HGF) ligand binding to the c-Met receptor, which failed in phase III clinical trials, didn’t completely block HGF•c-Met interaction at the cell surface, allowing partial c-Met activation, propagation of oncogenic signaling, and continued cancer cell growth.9 The incomplete characterization of the mechanisms of action of therapeutic antibodies, particularly anti-EGFR therapeutics in glioma, is an important problem that has the potential to compromise the quality of clinical trials and the interpretation of outcomes. In this study, we examined the antitumor activity of all clinically approved EGFR antibodies and chimeric monoclonal antibody 806 (ch806), the antibody backbone used in depatuxizumab mafodotin, against a panel of patient-derived gliomaspheres, some of which express EGFRvIII. We also extended this work to an intracranial orthograft model. Importantly, for the first time, we investigated the mechanisms by which these antibodies bind and block EGFRvIII activity.
Materials and Methods
Cell Lines
The patient-derived gliomasphere lines GBML1, GBML2, GBML3, GBM4, BAH1, and Pr1.1 were obtained as previously described.10 GBM6 and GBM39 were obtained from Prof Paul Mischel (Ludwig Institute of Cancer Research). GBM9 was kindly donated by Prof Amyn Habib (UT Southwestern). HK296 and HK277 were generated and provided by Prof Harley Kornblum (UCLA). All cell lines were cultured in neural stem cell serum free medium (NSC SFM) as previously described.10 The known genotypes and phenotypes of the gliomaspheres used in this paper have been previously described.10
Antibodies and Reagents
Panitumumab was supplied by Amgen. Nimotuzumab was kindly donated by Dr Normando E. Iznaga Escobar at Innokeys. Ch806 and monoclonal antibody (mAb) 806 were obtained as previously described.11 Cetuximab was purchased from the Monash Medical Centre pharmacy. Matuzumab was purchased from Absolute Antibody. Necitumumab, pantiumumab mutants, ch806 antigen-binding fragment (Fab), and panitumumab Fab were generated as described below. The following antibodies were supplied by Cell Signaling: pY1068-EGFR (#3777), pY1045-EGFR (#2237), pan-AKT (#2920), pSer473-AKT (#4060), total ERK1/2 (#4696), pThr202/Tyr204 ERK1/2 (#4370), total S6 ribosomal protein (#2317), p-Ser235/236 S6 ribosomal protein (#4858), and total c-Cbl (#8447). Supplied by Santa Cruz Biotechnology were protein A/G beads, C-terminal EGFR mouse mAb conjugated to beads (#sc-373746 AC), and anti-pTyr antibody (#sc-7020). Total anti–pan-actin antibody (#MA5-11869) was sourced from Thermo Fisher Scientific. Anti-mouse Alexa Fluor (AF)680 and anti-rabbit AF800 conjugated secondary antibodies were from Life Technologies. Rapamycin was supplied by Selleckchem. ViaLight proliferation assays were from Lonza.
Assembly of Panitumumab, Necitumumab, and EGFR Expression Vectors
Codon-optimized DNA templates encoding the full-length heavy chain (HC) and light chain (LC) of panitumumab or necitumumab were synthesized by Genscript and subcloned into the mammalian expression vector, pCAGGS. A singular panitumumab mutant—HCmut:Y35H or LCmut:Y32H—and a panitumumab mutant containing both mutations (Dmut) were also synthesized and subcloned. To assemble a vector for the mammalian expression of the panitumumab Fab, splice-overlap PCR was performed to assemble a single open reading frame comprising the LC, the 2A peptide from Thosea asigna virus together with a furin cleavage site and glycine spacer,12 and the HC V region and CH1 domain terminating at residue 207. The resulting construct was subcloned into pCAGGS. Full-length (residues 25–645 from the mature N-terminus) and truncated (residues 274–645) EGFR ectodomain coding region fragments incorporating C-terminal Flag tags (DYKDDDDK) were cloned downstream of the mouse interleukin-3 signal peptide (MVLASSTTSIHTMLLLLLMLFHLGLQ) and the first 4 amino acids (AsisChem) from the mature N-terminus in pCAGGS.
Transient Transfection and Purification of Recombinant Proteins
Suspension-adapted cultures of FreeStyle 293 cells (Life Technologies) were grown in Freestyle 293 Expression Medium (Life Technologies) supplemented with Glutamax-I. Scale-up transient transfection was performed on 200 mL cultures as previously described.13 Soluble, recombinant EGFR was by anti-Flag immunoaffinity chromatography.14 Wild-type and mutant panitumumab were purified on a HiTrap MabSelect SuRe column (GE Healthcare), while panitumumab Fab was purified on a HiTrap Protein L column (GE Healthcare).
Preparation of Chimeric 806 Fab
For the generation of Fab from ch806, ch806 was digested at a previously determined optimal ratio with activated papain (Sigma-Aldrich), the reactions quenched with iodoacetamide and passed through a ProSep-vA column (Millipore). The flow-through was concentrated and then injected onto a Superdex 200 size exclusion column (GE Healthcare) equilibrated in 1× phosphate buffered saline to recover the ch806 Fab.
Proliferation Assays
Proliferation assays were conducted as previously described.10
Western Blotting
Reducing and non-reducing sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE) and western blotting were conducted as previously described.15
Immunofluorescent Antibody Internalization Assays
GBM6 and GBM39 cells were plated in 8-well iBidi chamber slides and starved of EGF for 36 h. Subsequently, panitumumab was conjugated to AF488 using a labeling kit (Life Technologies), diluted to 10 µg/mL in ice cold EGF-deficient NSC SFM and added to cells on ice for 2 h. Preheated 37°C EGF-deficient NSC SFM was added to cells after washing to internalize AF488-panitumumab•receptor complexes and slides incubated at 37°C, 5% CO2 for the specified times. At each timepoint, wells were fixed, mounted, and imaged as previously described.10,15
Blue Native Polyacrylamide Gel Electrophoresis and Native Western Blotting
Blue native (BN)-PAGE was performed as previously described16 using 50 µg of cell isolate solubilized in 0.2% (w/v) n-dodecyl-β-D-maltoside. Native proteins were transferred to polyvinylidene difluoride membrane using a semi-dry method and then probed and scanned as previously described.10
Surface Plasmon Resonance
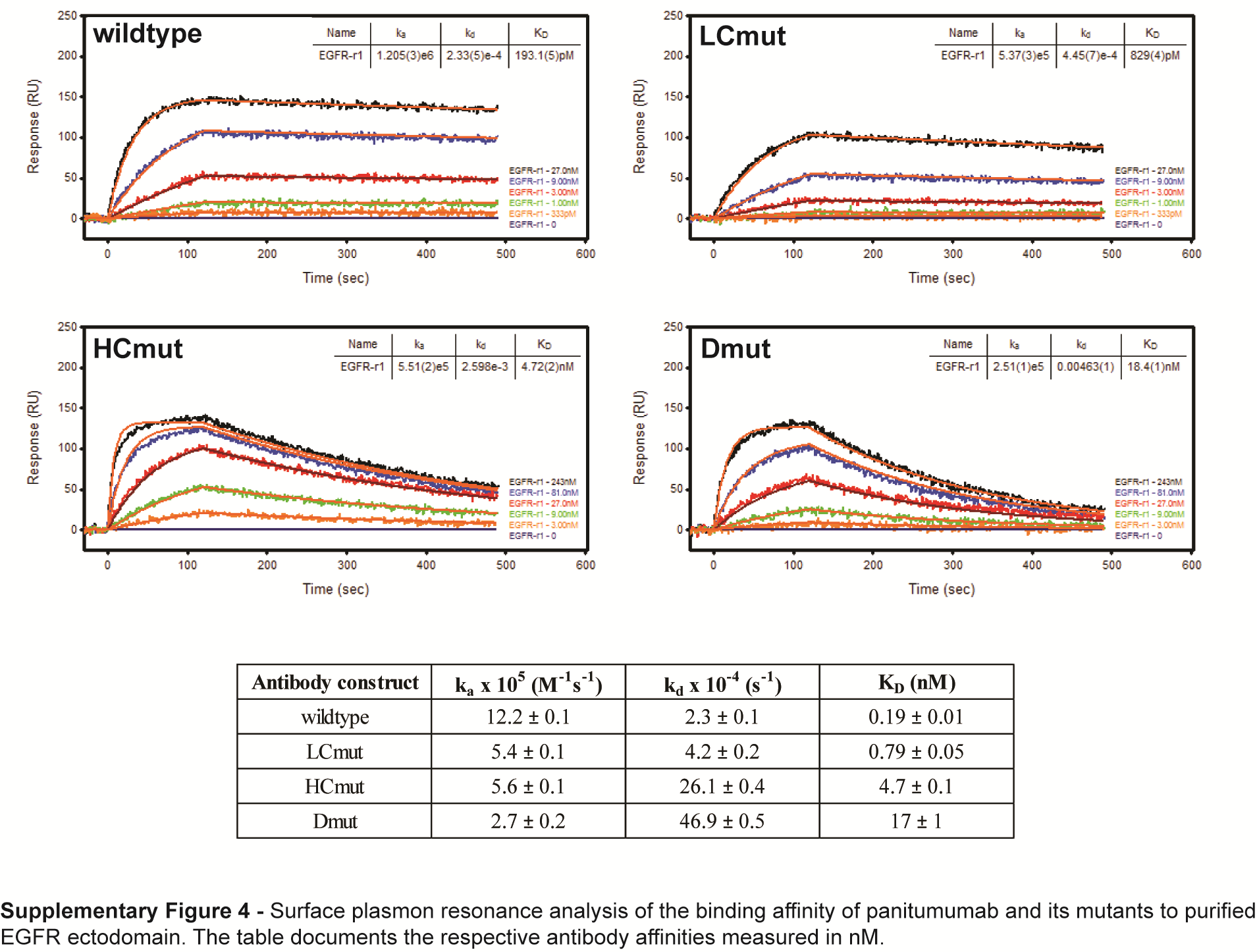
Using a ProteOn XPR36 Protein Interaction Array System, GLC sensor chips coated with anti-human immunoglobulin (Ig)G–fragment crystallizable (Fc) gamma-specific antibodies were used to capture panitumumab and mutant panitumumab, diluted to 1 µg/mL, for 90 s at a flow rate of 30 μL/min. Serial dilutions of soluble EGFR-621 ectodomain were then injected at a flow rate of 30 µL/min with an association time of 2 min and a dissociation time of up to 15 min. All binding experiments were conducted at 25°C in binding buffer (10 mM HEPES pH 7.5, 150 mM NaCl, 3 mM EDTA, 0.01% [v/v] Tween-20, 1 mg/mL bovine serum albumin) and were analyzed as described.14
In Vivo Orthotopic Trials
All animal experiments were approved by the Animal Ethics Committee of the Telethon Kids Institute and performed in accordance with the Australian Code for the Care and Use of Animals for Scientific Purposes. Intracranial GBM6 orthografts were established and monitored as recently described.17 Single agent EGFR antibodies were injected intraperitoneally at a dose of 1 mg every second day for 1 week, for a total of 3 injections. One group was treated with the combination of ch806 and cetuximab, where 0.5 mg of each antibody (a total of 1 mg) per injection was administered.
Results
Panitumumab Demonstrates Superior In Vitro and In Vivo Antitumor Efficacy Against EGFRvIII-Expressing Gliomaspheres
To reassess the anti-proliferative and antitumor efficacy of both clinical and preclinical antibodies targeting EGFR, we tested panitumumab, cetuximab, necitumumab, matuzumab, nimotuzumab (all domain III–binding antibodies), and ch806 (a domain II–binding antibody specific for binding EGFRvIII and overexpressed wild-type [wt]EGFR) against gliomaspheres expressing endogenous EGFRvIII (Fig. 1A), or wtEGFR or ins_773PH774 EGFR (Fig. 1B). Out of 5 EGFRvIII-expressing gliomaspheres, GBM9 was the only one resistant to anti-EGFR treatment. For GBM6, GBM39, and HK296, panitumumab was superior over the titration course, with anti-proliferative activity initiated at much lower concentrations. At no point with these gliomaspheres did cetuximab or necitumumab show equivalent anti-proliferative activity, even at antibody concentrations of 10 µg/mL. All domain III–targeting antibodies bound cell-surface EGFR identically by flow cytometry (Supplementary Fig. 1). Panitumumab, cetuximab, and necitumumab were equally effective at inhibiting the growth of BAH1 cells (Fig. 1A). Matuzumab, nimotuzumab, and ch806 generally had a poorer anti-proliferative effect against EGFRvIII-expressing gliomaspheres (Fig. 1A), with the exception of ch806 with GBM39 and matuzumab with BAH1, where anti-proliferative activity was observed.
Fig. 1.

Antitumor capabilities of EGFR antibodies in EGFRvIII and wtEGFR-expressing gliomasphere lines. (A, B) Representative plots for the in vitro inhibition of proliferation in gliomaspheres expressing EGFRvIII (A) or wtEGFR/EGFR mutants (B) after 7 days of treatment with increasing concentrations of anti-EGFR antibody as measured by Vialight assay. Data are presented as percentage of cell viability vs vehicle controls ± s.e.m. Horizontal dashed line = half-maximal inhibitory concentration demarcation. (C) Representative plots for the in vitro inhibition of proliferation in gliomaspheres expressing EGFRvIII after 7 days of treatment with 10 µg/mL of singular antibodies or domain III–targeting antibodies in combination with ch806 measured by Vialight assay. Data are presented as percentage of cell viability vs vehicle controls ± s.e.m. *P < 0.05 significant differences between combination treatments vs singular antibody treatment controls by one-way ANOVA. n.s. = not significant. (D) Kaplan–Meier survival curves demonstrating panitumumab single agent antitumor activity against GBM6 orthografts in nude mice. Data are presented as the percentage survival of mice in each group over time. A significant (P < 0.0001) increase in median survival in panitumumab-treated animals was observed vs all other groups after log-rank (Mantel–Cox) analysis.
In gliomaspheres expressing wtEGFR or ins_773PH774 EGFR (Fig. 1B), panitumumab was again more effective at inhibiting proliferation across a range of antibody concentrations. However, unlike EGFRvIII-expressing gliomaspheres, the anti-proliferative capacity of panitumumab, cetuximab, and necitumumab began to merge at the higher concentrations and was generally equal at the highest dose of 10 µg/mL. Matuzumab displayed some activity against GBML1 and HK277, while nimotuzumab and ch806 displayed no activity against any wtEGFR gliomaspheres (Fig. 1B). We are currently investigating why GBM9 and GBML3 do not respond to treatment, but our initial analyses after single cell isolation of clones containing a G13D Ras-activating mutation from GBML1 (which contains this mutation at a low allelic frequency) suggest that this mutation results in resistance to panitumumab treatment compared with wtRas clones (Supplementary Fig. 2).
To see if we could improve the anti-proliferative activity of the domain III–targeting antibodies panitumumab, cetuximab, and necitumumab against EGFRvIII-expressing gliomaspheres, we combined these antibodies with the domain II antibody ch806, which preferentially targets EGFRvIII and overexpressed wtEGFR (Fig. 1C). This approach generally did not improve anti-proliferative activity compared with antibody alone. Where a small but significant improvement was observed, such as in GBM6 (for cetuximab + ch806 and necitumumab + ch806) and HK296 (for necitumumab + ch806), it did not match the efficacy of panitumumab alone (Fig. 1C).
To verify these observations in vivo, we orthotopically engrafted NSG mice with GBM6 cells and treated groups with vehicle, panitumumab alone, cetuximab alone, nimotuzumab alone, ch806 alone, and cetuximab + ch806 at suboptimal doses and monitored their response (Figure 1D). The in vivo activity of cetuximab, nimotuzumab, and ch806 was low, with a small but significant 5–7 day (P < 0.05) improvement in survival versus vehicle controls observed. Antitumor activity was not improved by the combination of cetuximab + ch806 compared with single antibody treatments. Panitumumab displayed significant in vivo antitumor activity and improvement in survival (29 days vs vehicle, P < 0.0001 and 24 days vs other groups, P < 0.0001). Cumulatively, these results mirror our in vitro data, verify that anti-EGFR antibodies can pass the blood–brain barrier, and confirm that panitumumab is a superior in vivo antitumor agent over other EGFR antibodies.
Panitumumab Is the Only Antibody Capable of Neutralizing Both wtEGFR and EGFRvIII Activation
The lack of ability of other antibodies to achieve the same degree of cell growth inhibition as panitumumab in EGFRvIII-expressing gliomaspheres as well as with tumor orthografts led us to believe that there may be differences in each antibody’s ability to neutralize this receptor. After 24 h of antibody treatment of 5 EGFRvIII-expressing gliomaspheres, we found that EGFRvIII activity in whole cell lysates (measured by the phosphorylation of the Y1068 C-terminal domain residue) was only decreased by panitumumab and ch806 (Fig. 2A), with some minor decreases also observed for nimotuzumab in GBM9 but no other cell lines. We also observed an increase in total wtEGFR in GBM6, GBM39, and GBM9 following treatment with panitumumab, cetuximab, necitumumab, matuzumab, and to a lesser extent nimotuzumab. This was most likely due to these antibodies blocking the EGF in gliomasphere media from binding wtEGFR, thus preventing receptor activation, internalization, and subsequent degradation. In contrast to GBM6, GBM39, and GBM9, the most active receptor in BAH1 and HK296 appeared to be wtEGFR, which was effectively neutralized by panitumumab, cetuximab, necitumumab, and matuzumab but only partially so by nimotuzumab and not at all by ch806 (Fig. 2A). In HK296, effective neutralization of wtEGFR appeared to result in an upregulation of total and active EGFRvIII in cells treated with cetuximab, necitumumab, and matuzumab (Fig. 2A). Hence, only panitumumab effectively reduced the activity of both EGFRvIII and wtEGFR. This was also reflected in downstream oncogenic signaling analyses, with only panitumumab capable of robustly reducing p-S6 in GBM6, GBM39, BAH1, and HK296 (Fig. 2B). No real changes of note occurred for p-AKT or phosphorylated extracellular signal-regulated kinase 1 or 2 (p-ERK1/2) after antibody treatment (Fig. 2B). Importantly, in the resistant GBM9 cell line, p-S6 remained active after panitumumab treatment (Fig. 2B). Coadministration of panitumumab with rapamycin, which targets mammalian target of rapamycin (mTOR) complex 1, resulted in additive anti-proliferative activity in GBM9 cells (Supplementary Fig. 3), indicating that p-S6 may be a marker of response to EGFR antibodies.
Fig. 2.

Characterization of receptor activation and oncogenic signaling after antibody administration in EGFRvIII-expressing gliomaspheres. (A, B) Reducing SDS-PAGE and western analyses of (A) pY1068 wtEGFR/EGFRvIII and total EGFR status (* denotes wtEGFR) and (B) activation status of the major oncogenic signaling pathways AKT, ERK1/2, and S6 ribosomal protein in whole cell lysates isolated following antibody treatment. (C) Reducing SDS-PAGE and western analyses of the total phosphotyrosine and total EGFR status of immunoprecipitates following protein A/G or C-terminal EGFR-agarose bead immunoprecipitation of whole cell lysates after antibody treatment. * denotes wtEGFR.
To further clarify the status of wtEGFR and EGFRvIII activation following direct engagement by antibody, we treated cells with antibody for 24 h and then did immunoprecipitation (IP) on cell lysates using protein A/G only (which would enrich for receptor-bound antibody complexes) or C-terminal EGFR antibody conjugated to beads (which would enrich the entire EGFR population). We also probed IPs with a pan-phosphotyrosine antibody (PY99) that, in contrast to only interrogating the pY1068 site, yields the global phosphotyrosine status as an output of total receptor activation. Protein A/G IP confirmed that EGFRvIII was neutralized while bound by panitumumab and ch806, whereas EGFRvIII was active when engaged by either cetuximab or necitumumab, with increased wtEGFR activity also observed (Fig. 2C). As expected, panitumumab, cetuximab, and necitumumab bound both wtEGFR and EGFRvIII, whereas ch806 engaged EGFRvIII only. Analysis of the global EGFR activation status after antibody treatment, using the pan-phosphotyrosine probe, confirmed our previous finding that panitumumab was the only antibody capable of reducing both wtEGFR and EGFRvIII activity.
EGFRvIII and wtEGFR Are Recycled, Not Degraded, Following Anti-EGFR Antibody Treatment
The prevailing view of antibody-driven neutralization of EGFRvIII is that binding of antibody results in EGFRvIII internalization and degradation.18–21 To interrogate this further, we cultured GBM6 and GBM39 either with EGF in the medium (to maintain normal culture conditions) or starved of EGF overnight (to increase the expression of endogenous wtEGFR) and then treated the cells with antibody for 32 h to induce (any) potential degradation (Fig. 3A, B). Consistently, under both conditions, there was no notable reduction of EGFRvIII expression compared with vehicle. Starving cells of EGF (Fig. 3B) also confirmed that there was no reduction of wtEGFR after antibody treatment compared with vehicle. These results may be explained if, in response to antibody-induced receptor degradation, cells replace the receptors by either a transcriptional or a translational upregulation. As quantitative real time PCR of both wtEGFR and EGFRvIII expression showed no increase in gene expression in response to antibody (data not shown), we cultured cells in the presence of the translation inhibitor, cycloheximide (CHX), and treated them with each antibody for 32 h (Fig. 3A, B). No reduction of total wtEGFR and EGFRvIII was observed over vehicle controls, ruling out translational upregulation of receptor in response to antibody treatment. Receptor levels for both species in CHX experiments were reduced compared with untreated cells due to basal receptor turnover only. The presence of EGF made no difference to the ability of panitumumab to neutralize EGFRvIII, demonstrating that the mechanism is ligand independent. Hence, anti-EGFR antibody treatment of gliomasphere cultures do not induce receptor degradation to initiate neutralization.
Fig. 3.

Analysis of receptor degradation, trafficking, and recycling following EGFR antibody treatment. (A, B) Reducing SDS-PAGE and western analyses of total and pY1068 wtEGFR and EGFRvIII following antibody treatment of cells cultured in EGF-containing media (A) or EGF-deficient media (B) ±10 µM of the translation inhibitor cycloheximide [CHX]). (C) Reducing SDS-PAGE and western analyses of total and pY1045 wtEGFR and EGFRvIII following antibody treatment. (D) Immunoprecipitation enrichment of wtEGFR and EGFRvIII and western analyses for pY1045 EGFR, total EGFR, and c-Cbl from whole cell lysates after engagement of EGF-starved GBM6 cells with 10 µg/mL panitumumab in EGF-deficient NSC SFM at 4°C for 2 h followed by induction of antibody internalization by addition of 37°C EGF-deficient NSC SFM for the indicated timepoints. For EGF tests, 100 ng/mL of ligand was added to EGF-deficient NSC SFM and incubated with cells at 37°C for the indicated timepoints. (E) Immunofluorescence tracking of AF488-labeled panitumumab over time following engagement of EGF-starved cells at 4°C for 2 h and then internalization at 37°C over the indicated timepoints. Nuclei were stained using 4′,6′-diamidino-2-phenylindole (cyan), and AF488-panitumumab is depicted in gray. Red arrows = cell surface AF488 panitumumab•EGFR(vIII) signal concentrated in membrane ruffles. Yellow arrows = internalized AF488 panitumumab•EGFR(vIII) clusters. Scale bar = 10 µm.
Internalization of ligand-stimulated wtEGFR is often linked to the increase of pY1045, which is hypophosphorylated in EGFRvIII, followed by Casitas B-lineage lymphoma (c-Cbl) recruitment, engagement, and internalization and degradation.22,23 We analyzed the status of pY1045 after antibody treatment, confirming that EGFRvIII pY1045 remains hypophosphorylated after antibody engagement and does not increase (Fig. 3C). Time-course experiments with panitumumab engagement followed by C-terminal EGFR IP in EGF-starved GBM6 cells demonstrated that both total EGFRvIII, wtEGFR, and pY1045 remained constant over time following incubation at 4°C with panitumumab and subsequent 37°C media incubation (Fig. 3D). As a control, EGF stimulation resulted in pY1045 upregulation at 10 min followed by wtEGFR, but not EGFRvIII, degradation at 4 hours. Casitas B-lineage lymphoma was constitutively bound to EGFRvIII in vehicle controls—addition of EGF or panitumumab over time did not alter the association of c-Cbl with receptor, confirming that the mechanism of action of anti-EGFR antibodies appears to be independent of this pathway.
After ruling out receptor degradation in response to antibody treatment, we focused on 2 other remaining aspects—that either the antibody•receptor complex was not being internalized efficiently or it was being recycled back to the cell surface. To address these questions, we labeled panitumumab with AF488 (pani-488), bound it to GBM6 and GBM39 cells at 4°C, and initiated antibody•receptor internalization with 37°C media to track the progress of the antibody•receptor complex. Our data showed that both mechanisms are apparent (Fig. 3E). Over 180 min, we observed robust early internalization of pani-488 in GBM6 cells, followed by accumulation in perinuclear punctate clusters, which then dissociated to return to a peripheral cell surface staining at later stages. In GBM39, a peripheral cell surface pattern of pani-488 staining was maintained over the entire 180 min with some minor internalization of pani-488 into diffuse punctate clusters observed. This indicates that, in certain cases, panitumumab is not internalized after cell surface receptor engagement. When panitumumab is internalized, it appears that it is not degraded but shuttled back to the cell surface, consistent with receptor recycling.
Panitumumab and ch806 Display Two Distinct and Unique Mechanisms of EGFRvIII Inhibition
At this stage, we had ruled out receptor degradation as the reason for the neutralization of EGFRvIII by panitumumab. We next hypothesized that the antibody may be directly affecting the formation of the EGFRvIII disulphide-bonded dimer, the major active species of this receptor in U87MG.vIII cells.15 Using non-reducing SDS-PAGE and western analysis, we demonstrated the presence of the active EGFRvIII disulphide-bonded dimer in GBM6, GBM39, and GBM9 gliomaspheres (Fig. 4A). The addition of panitumumab did not affect total EGFRvIII disulphide-bonded dimer levels (Fig. 4A). Ch806 binding resulted in a reproducible and robust decrease in the amount of total EGFRvIII disulphide-bonded dimer (Fig. 4A), along with a loss of activity of EGFRvIII. Despite EGFRvIII disulphide-bonded dimer levels remaining intact after treatment, panitumumab was still able to neutralize EGFRvIII activity, suggesting that panitumumab does not block EGFRvIII activity by preventing the formation of the disulphide-bonded dimer (Fig. 4A). Cetuximab and necitumumab had no effect on either total or active EGFRvIII disulphide-bonded dimer (Fig. 4A).
Fig. 4.

Interrogation of the mechanism of action of how panitumumab and ch806 are able to neutralize EGFRvIII. (A) Non-reducing SDS-PAGE and western analyses of total and pY1068 wtEGFR and EGFRvIII in 3 EGFRvIII-expressing gliomasphere cell lines following treatment. (B) Non-reducing SDS-PAGE and western analyses of total and pY1068 wtEGFR and EGFRvIII in 3 EGFRvIII-expressing gliomasphere cell lines following treatment with panitumumab or its mutants. (C) Representative plots for the in vitro inhibition of proliferation in gliomaspheres expressing EGFRvIII after 7 days of treatment with increasing concentrations of panitumumab or its mutants as measured by Vialight assay. Data are presented as percentage of cell viability vs vehicle controls ± s.e.m. Horizontal dashed line = half-maximal inhibitory concentration demarcation. (D) BN-PAGE and native western analyses of total EGFR status in 3 EGFRvIII-expressing gliomasphere cell lines following treatment. Molecular weights in kDa are indicated to the left of panel. (E) BN-PAGE and native western analyses of total EGFR status in 3 EGFRvIII-expressing gliomasphere cell lines following treatment. Molecular weights in kDa are indicated to the left of panel. † = Fab•EGFRvIII complex. (F) Non-reducing SDS-PAGE and western analyses of total and pY1068 wtEGFR and EGFRvIII from the same tests as (E). (G) Reducing SDS-PAGE and western analyses of total and pY1068 wtEGFR and EGFRvIII from the same tests as (E). All treatments for western analyses were conducted for 24 h with 10 µg/mL antibody or molar equivalent Fab (6.6 µg/mL) and actin loading controls included. D = EGFRvIII disulphide-bonded dimer, M = EGFRvIII monomer, * = wtEGFR.
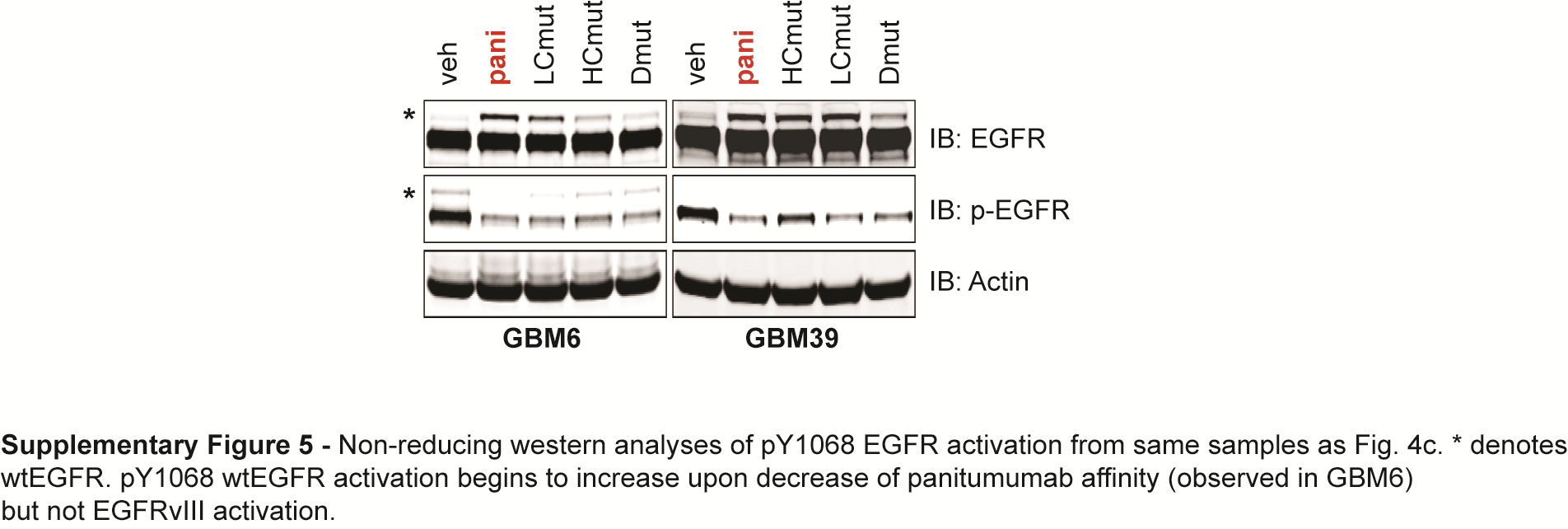
To see if the superior affinity of panitumumab over cetuximab and necitumumab was responsible for its ability to neutralize EGFRvIII activity, we introduced single point mutations into the VH domain (HCmut), VL domain (LCmut), or both domains (Dmut) of panitumumab to sequentially decrease its affinity from approximately 193 pM to 17 nM for purified wtEGFR, as measured by surface plasmon resonance (Supplementary Fig. 4). Non-reducing SDS-PAGE and western blot analyses of cells treated with these mutants showed that the loss of affinity had no effect on the ability of panitumumab to neutralize active EGFRvIII disulphide-bonded dimers (Fig. 4B) but did affect its ability to neutralize EGF-induced wtEGFR activation (Supplementary Fig. 5) and sequentially retarded its anti-proliferative activity (Fig. 4C). Hence, the high affinity of panitumumab governs its ability to neutralize ligand-induced wtEGFR activation, but not EGFRvIII activation, and wtEGFR activation is a major driver of glioma proliferation in these cells.
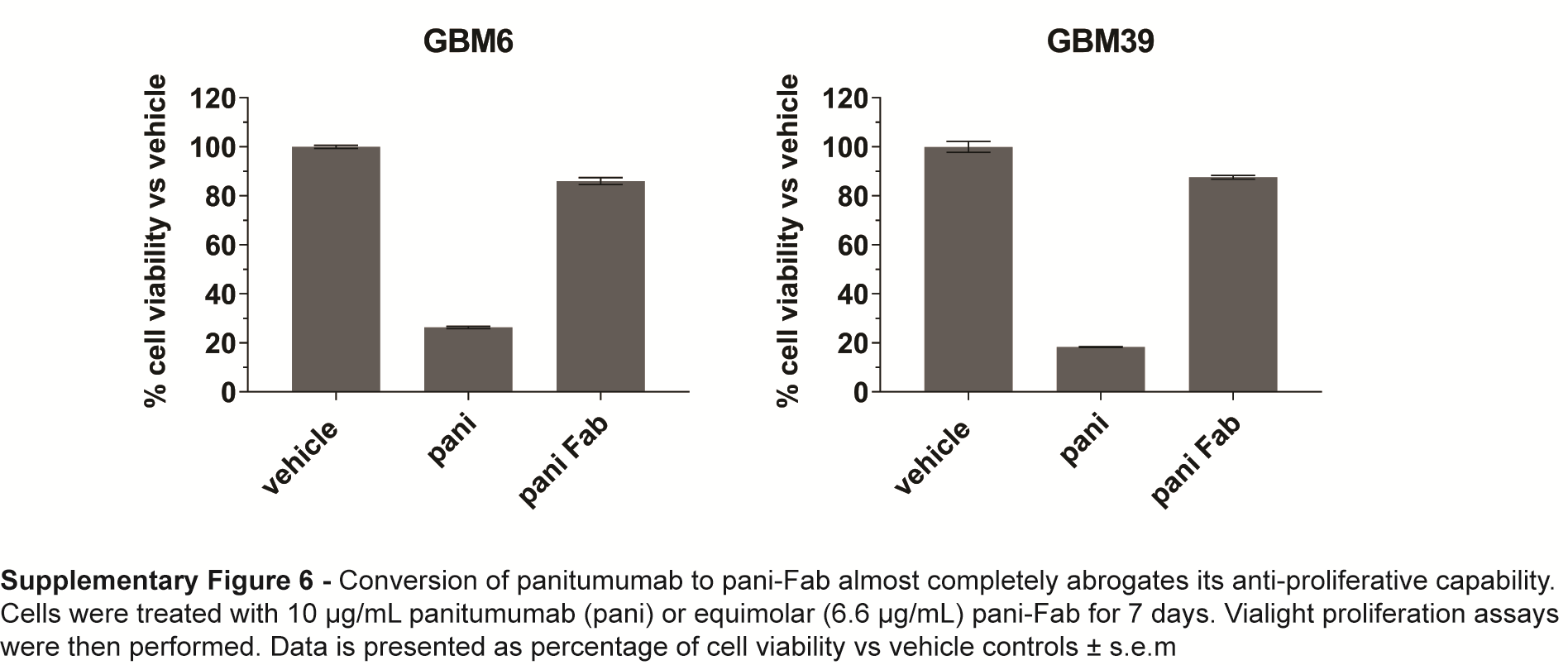
Another possibility was that panitumumab was binding EGFRvIII and locking it into inactive higher-molecular-weight clusters in order to prevent its activation. To interrogate this, we treated GBM6, GBM39, and GBM9 with antibody for 24 h followed by BN-PAGE and native western analyses to detect the presence of both covalent and non-covalent protein complexes. This approach revealed that EGFRvIII basally self-associates (either covalently or non-covalently) in a dimeric form of approximately 280 kDa (Fig. 4D). Treatment with panitumumab and ch806, but not cetuximab and necitumumab, resulted in the formation of stable, high-molecular-weight (~800 kDa) EGFRvIII•antibody complexes, indicating that the antibodies which neutralize EGFRvIII can stably cross-link the receptor (Fig. 4D). To investigate if the cross-linking abilities of panitumumab and ch806 were responsible for their anti-EGFRvIII activity, we created monovalent Fab forms of each antibody and treated cells with equimolar concentrations. BN-PAGE and native western analyses demonstrated that conversion to a monovalent Fab form for each antibody abrogated their ability to form stable EGFRvIII•antibody high-molecular-weight complexes (Fig. 4E). The Fabs retained their ability to bind (most likely monomeric) EGFRvIII, indicated by a small size shift observed (Fig. 4E). Non-reducing SDS-PAGE and western analyses on whole cell lysates derived from the same treated cell pool demonstrated that ch806 and its Fab were still able to reduce the total level of EGFRvIII disulphide-bonded dimer, resulting in an elimination of EGFRvIII activity (Fig. 4F). However, the conversion of panitumumab to monovalent Fab, while not affecting the total amount of EGFRvIII disulphide-bonded dimer, completely prevented its ability to neutralize EGFRvIII activity (Fig. 4F). Reducing SDS-PAGE and western analyses on the same lysates demonstrated that panitumumab and its Fab were able to rescue EGF-mediated wtEGFR degradation equally and neutralize wtEGFR activity, confirming that the Fab was fully active in blocking EGF binding to domain III (Fig. 4G). As expected, ch806 Fab did not affect total or active wtEGFR. Finally, conversion of panitumumab to pani-Fab almost completely abrogates its anti-proliferative activity against GBM6 and GBM39 (Supplementary Fig. 6). These results demonstrate 2 distinct mechanisms for panitumumab and ch806 in their ability to neutralize EGFRvIII.
Discussion
Our data uncover a previously unknown mechanism of resistance to EGFR antibody therapy, whereby EGFRvIII is not neutralized and remains active while bound by cetuximab and necitumumab. In contrast, our findings reveal that EGFR species are neutralized/internalized after panitumumab binding and then recycled back to the cell surface, resulting in no net loss of EGFRvIII or wtEGFR after antibody treatment. This directly challenges the prevailing view, derived primarily from transfected glioma models exogenously expressing EGFRvIII,18–21 that anti-EGFR antibodies bind EGFRvIII, resulting in its degradation and inactivation. Importantly, the efficacy of these antibodies in vitro is recapitulated in our in vivo intracranial models of EGFRvIII-expressing glioma, strengthening the translational relevance of our findings. Our studies again highlight the importance of utilizing appropriate cellular models for proper in vitro and in vivo assessment of antibody action, as we have found for HGF-targeting antibodies previously.9
Our studies here have also highlighted that, following EGFRvIII neutralization by panitumumab, p-AKT and p-ERK1/2 are unaffected, whereas p-S6 is strongly inhibited. The disconnect between lack of AKT inhibition and suppression of mTOR/p-S6 following EGFR and EGFRvIII inhibition observed here has been recorded before in glioma where EGFR is able to activate mTOR and p-S6 independently of AKT via protein kinase C activation. AKT activation has no effect on the proliferative capacity of glioma cells,24 agreeing with our data here. The continued propagation of p-AKT and p-ERK1/2 after panitumumab treatment may be due to the expression and coactivation of other apex receptor tyrosine kinases,25 which are all capable of redundantly sustaining AKT and ERK signaling after EGFR inhibition and may be more important for survival of these cells instead of a proliferative phenotype driven by AKT-independent p-S6 phosphorylation. The continued propagation of p-S6, through yet unidentified auxiliary pathways, is linked to panitumumab resistance in GBM9 (Fig. 2B) and can be counteracted in combination with mTOR inhibitors such as rapamycin (Supplementary Fig. 3). Furthermore, our other initial investigations have now verified that the abundant expression of a G13D Ras-activating mutation in isolated single cell clones directly leads to resistance to panitumumab as well as dacomitinib (Supplementary Fig. 2) in agreement with its role in resisting panitumumab treatment of colon cancers. Interestingly, the panitumumab-resistant GBML3 gliomasphere line (Fig. 1B) expresses G13D Ras at a higher allelic frequency than GBML1, and this may be the cause of the lack of response observed. We are now investigating this aspect.
We describe 2 new mechanisms of action by which ch806 and panitumumab abrogate EGFRvIII activation. Ch806 was able to potently reduce the levels of the active EGFRvIII disulphide-bonded dimer, most likely through stoichiometric interference, as a single domain Fab was just as effective as the bivalent antibody in preventing activation of the receptor, and its efficaciousness was not reliant on receptor cross-linking. We have not yet determined whether ch806 prevents the formation of nascent EGFRvIII disulphide-bonded dimers or binds and disrupts preexisting EGFRvIII disulphide-bonded dimers. On the other hand, the ability of panitumumab to neutralize EGFRvIII was independent of its affinity but entirely reliant on its bivalence/avidity, which promoted cross-linking of EGFRvIII into stable inactive complexes. Our data suggest that these complexes do not prevent EGFRvIII disulphide-bonded dimer formation, as this is unaffected by panitumumab treatment (Figure 4A), but interfere with the establishment of an active conformation. Cetuximab and necitumumab are unable to form stable complexes to cross-link EGFRvIII, and this appears to be the reason why these antibodies fail to neutralize the receptor. It is unlikely that affinity is responsible for this difference as (i) the panitumumab LCmut has equipotent affinity to cetuximab and necitumumab but is still able to neutralize EGFRvIII and (ii) ch806 has a low nM affinity,11 yet is still able to neutralize EGFRvIII. Another consideration that requires further investigation is whether panitumumab, an IgG2 isotype, has an altered flexibility to cross-link the receptor while bound to its epitope compared with all other domain III antibodies, which are of the IgG1 isotype.
The elucidation of the mechanisms of action of these antibodies may help to explain their therapeutic differences and the complex dynamic interplay between EGFRvIII and wtEGFR to sustain tumor proliferation. Our experiments utilizing panitumumab affinity mutants highlight the importance of the contribution of ligand-induced wtEGFR activation to glioma cell proliferation—a sequential loss of affinity results in gradual increases in wtEGFR activation, but not EGFRvIII activation, after which their anti-proliferative capability is compromised. This suggests that ligand-activated wtEGFR is a major driver of proliferation in the glioma models tested. Additionally, wtEGFR also remains partially active when directly engaged with cetuximab and necitumumab—as both of these antibodies completely neutralize ligand-induced wtEGFR activation, the activity observed is almost certainly EGFRvIII-mediated transactivation, a well-documented phenomenon.26 This may allow the continued perpetuation of wtEGFR activation in the presence of EGFR ligand blockade to sustain glioma growth, possibly explaining why cetuximab, nimotuzumab, and ch806 (which does not neutralize ligand-activated wtEGFR) fail to produce more than a small improvement in survival in our in vivo trials. Panitumumab, which neutralizes both axes simultaneously, is vastly superior in improving survival, highlighting the absolute importance of eliminating both axes of activation. These findings are also consistent with known biomarkers of response to cetuximab garnered from clinical trials, which reveal that glioma patients with EGFR amplification but lacking EGFRvIII expression have a significantly better progression-free and overall survival following treatment.27 As such, the failure of cetuximab and nimotuzumab to completely neutralize both EGFRvIII and wtEGFR activation may be an important contributing factor to the disappointing outcomes of the phase II cetuximab28,29 and phase III nimotuzumab30 clinical trials in glioma.
Cumulatively, our investigations reveal that only one anti-EGFR antibody, panitumumab, is capable of neutralizing EGFRvIII and wtEGFR and defines the mechanism by which EGFR antibodies affect the expression of EGFR species, receptor recycling, and receptor neutralization. Our data suggest that panitumumab treatment of patients stratified for EGFRvIII expression in combination with standard of care for nascent and/or recurrent glioma may significantly improve the outlook for those with this currently intractable disease.
Funding
This work was supported by the National Health and Medical Research Council, Cure Brain Cancer Foundation (TGJ and KLM), Victorian Government Infrastructure Program, and Therapeutic Innovation Australia. We acknowledge the support of the Ethan Davies Fellowship (SAG). The National Biologics Facility of the Commonwealth Scientific and Industrial Research Organisation (CSIRO) received financial support from Therapeutic Innovation Australia. TGJ is a Principal Research Fellow with the National Health and Medical Research Council of Australia.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Acknowledgments
We would like to thank staff of the National Biologics Facility, CSIRO, for performing scale-up cell culture and transfections. We thank Dr Peter Dallas for helping edit the manuscript.
Conflict of interest statement. TGJ has previously received funding from Amgen, the owner of panitumumab.
Authorship statement. Conception and intellectual direction of study: SAG, TEA, and TGJ. Experimental work: SAG, MM, ES, OD, LP, JB, MK, and SCC. Resource provision and funding: KLM, HIK, RE, TEA, and TGJ. Original manuscript drafting and Fig. construction: SAG. Manuscript editing and completion: SAG, TEA, RE, and TGJ.
References
- 1. Furnari FB, Fenton T, Bachoo RM, et al. . Malignant astrocytic glioma: genetics, biology, and paths to treatment. Genes Dev. 2007;21(21):2683–2710. [DOI] [PubMed] [Google Scholar]
- 2. Brennan CW, Verhaak RG, McKenna A, et al. ; TCGA Research Network The somatic genomic landscape of glioblastoma. Cell. 2013;155(2):462–477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Ymer SI, Greenall SA, Cvrljevic A, et al. . Glioma specific extracellular missense mutations in the first cysteine rich region of epidermal growth factor receptor (EGFR) initiate ligand independent activation. Cancers (Basel). 2011;3(2):2032–2049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Reardon DA, Wen PY, Mellinghoff IK. Targeted molecular therapies against epidermal growth factor receptor: past experiences and challenges. Neuro Oncol. 2014; 16(Suppl 8):viii7–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Hegi ME, Diserens AC, Bady P, et al. . Pathway analysis of glioblastoma tissue after preoperative treatment with the EGFR tyrosine kinase inhibitor gefitinib–a phase II trial. Mol Cancer Ther. 2011;10(6):1102–1112. [DOI] [PubMed] [Google Scholar]
- 6. Tebbutt N, Pedersen MW, Johns TG. Targeting the ERBB family in cancer: couples therapy. Nat Rev Cancer. 2013;13(9):663–673. [DOI] [PubMed] [Google Scholar]
- 7. Van Den Bent MJ, French P, Eoli M, et al. . Updated results of the INTELLANCE 2/EORTC trial 1410 randomized phase II study on Depatux –M alone, Depatux-M in combination with temozolomide (TMZ) and either TMZ or lomustine (LOM) in recurrent EGFR amplified glioblastoma (NCT02343406). J Clin Oncol. 2018;36(suppl; abstr 2023). [Google Scholar]
- 8. Vu T, Claret FX. Trastuzumab: updated mechanisms of action and resistance in breast cancer. Front Oncol. 2012;2:62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Greenall SA, Adams TE, Johns TG. Incomplete target neutralization by the anti-cancer antibody rilotumumab. MAbs. 2016;8(2):246–252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Greenall SA, Lim YC, Mitchell CB, et al. . Cyclin-dependent kinase 7 is a therapeutic target in high-grade glioma. Oncogenesis. 2017;6(5):e336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Panousis C, Rayzman VM, Johns TG, et al. . Engineering and characterisation of chimeric monoclonal antibody 806 (ch806) for targeted immunotherapy of tumours expressing de2-7 EGFR or amplified EGFR. Br J Cancer. 2005;92(6):1069–1077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Chng J, Wang T, Nian R, et al. . Cleavage efficient 2A peptides for high level monoclonal antibody expression in CHO cells. MAbs. 2015;7(2):403–412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Tom R, Bisson L, Durocher Y. Transfection of HEK293-EBNA1 cells in suspension with linear PEI for production of recombinant proteins. CSH Protoc. 2008; 2008:1–4. [DOI] [PubMed] [Google Scholar]
- 14. Madej MP, Coia G, Williams CC, et al. . Engineering of an anti-epidermal growth factor receptor antibody to single chain format and labeling by Sortase A-mediated protein ligation. Biotechnol Bioeng. 2012;109(6):1461–1470. [DOI] [PubMed] [Google Scholar]
- 15. Greenall SA, Donoghue JF, Van Sinderen M, et al. . EGFRvIII-mediated transactivation of receptor tyrosine kinases in glioma: mechanism and therapeutic implications. Oncogene. 2015;34(41):5277–5287. [DOI] [PubMed] [Google Scholar]
- 16. McKenzie M, Lazarou M, Ryan MT. Chapter 18 analysis of respiratory chain complex assembly with radiolabeled nuclear- and mitochondrial-encoded subunits. In: Methods in Enzymology. Vol. 456 London, UK: Academic Press; 2009:321–339. [DOI] [PubMed] [Google Scholar]
- 17. Endersby R, Whitehouse J, Hii H, Greenall SA, Johns TG, Gottardo NG. A pre-clinical assessment of the pan-ERBB inhibitor dacomitinib in pediatric and adult brain tumors. Neoplasia. 2018;20(5):432–442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Fukai J, Nishio K, Itakura T, Koizumi F. Antitumor activity of cetuximab against malignant glioma cells overexpressing EGFR deletion mutant variant III. Cancer Sci. 2008;99(10):2062–2069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Jutten B, Dubois L, Li Y, et al. . Binding of cetuximab to the EGFRvIII deletion mutant and its biological consequences in malignant glioma cells. Radiother Oncol. 2009;92(3):393–398. [DOI] [PubMed] [Google Scholar]
- 20. Patel D, Lahiji A, Patel S, et al. . Monoclonal antibody cetuximab binds to and down-regulates constitutively activated epidermal growth factor receptor vIII on the cell surface. Anticancer Res. 2007;27(5A):3355–3366. [PubMed] [Google Scholar]
- 21. Pillay V, Allaf L, Wilding AL, et al. . The plasticity of oncogene addiction: implications for targeted therapies directed to receptor tyrosine kinases. Neoplasia. 2009; 11(5):448–458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Davies GC, Ryan PE, Rahman L, Zajac-Kaye M, Lipkowitz S. EGFRvIII undergoes activation-dependent downregulation mediated by the Cbl proteins. Oncogene. 2006;25(49):6497–6509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Han W, Zhang T, Yu H, Foulke JG, Tang CK. Hypophosphorylation of residue Y1045 leads to defective downregulation of EGFRvIII. Cancer Biol Ther. 2006;5(10):1361–1368. [DOI] [PubMed] [Google Scholar]
- 24. Fan QW, Cheng C, Knight ZA, et al. . EGFR signals to mTOR through PKC and independently of Akt in glioma. Sci Signal. 2009; 2(55):ra4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Stommel JM, Kimmelman AC, Ying H, et al. . Coactivation of receptor tyrosine kinases affects the response of tumor cells to targeted therapies. Science. 2007;318(5848):287–290. [DOI] [PubMed] [Google Scholar]
- 26. Luwor RB, Zhu HJ, Walker F, et al. . The tumor-specific de2-7 epidermal growth factor receptor (EGFR) promotes cells survival and heterodimerizes with the wild-type EGFR. Oncogene. 2004;23(36):6095–6104. [DOI] [PubMed] [Google Scholar]
- 27. Lv S, Teugels E, Sadones J, et al. . Correlation of EGFR, IDH1 and PTEN status with the outcome of patients with recurrent glioblastoma treated in a phase II clinical trial with the EGFR-blocking monoclonal antibody cetuximab. Int J Oncol. 2012;41(3):1029–1035. [DOI] [PubMed] [Google Scholar]
- 28. Hasselbalch B, Lassen U, Hansen S, et al. . Cetuximab, bevacizumab, and irinotecan for patients with primary glioblastoma and progression after radiation therapy and temozolomide: a phase II trial. Neuro Oncol. 2010;12(5):508–516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Neyns B, Sadones J, Joosens E, et al. . Stratified phase II trial of cetuximab in patients with recurrent high-grade glioma. Ann Oncol. 2009;20(9):1596–1603. [DOI] [PubMed] [Google Scholar]
- 30. Westphal M, Heese O, Steinbach JP, et al. . A randomised, open label phase III trial with nimotuzumab, an anti-epidermal growth factor receptor monoclonal antibody in the treatment of newly diagnosed adult glioblastoma. Eur J Cancer. 2015;51(4):522–532. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.