

Abstract
In 2016, the World Health Organization deemed antibiotic resistance one of the biggest threats to global health, food security, and development. The need for new methods to combat infections caused by antibiotic resistant pathogens will require a variety of approaches to identifying effective new therapeutic strategies. One approach is the identification of small molecule adjuvants that potentiate the activity of antibiotics of demonstrated utility, whose efficacy is abated by resistance, both acquired and intrinsic. To this end, we have identified compounds that enhance the efficacy of antibiotics normally ineffective against Gram-negative pathogens because of the outer membrane permeability barrier. We identified two adjuvant compounds that dramatically enhance sensitivity of Acinetobacter baumannii to macrolide and glycopeptide antibiotics, with reductions in minimum inhibitory concentrations as high as 256-fold, and we observed activity across a variety of clinical isolates. Mode of action studies indicate that these adjuvants likely work by modulating lipopolysaccharide synthesis or assembly. The adjuvants were active in vivo in a Galleria mellonella infection model, indicating potential for use in mammalian infections.
Keywords: antibiotic adjuvant, antibiotic resistance, Acinetobacter baumannii, macrolide antibiotics, glycopeptide antibiotics, lipopolysaccharide
Graphical Abstract
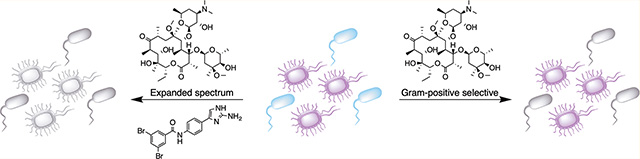
The increasing prevalence of multidrug-resistant (MDR) bacteria is a serious national and global threat.1 Antibiotics have been used as the primary treatment for bacterial infections over the last 70 years, but as new drugs are deployed in the clinic, bacteria unfailingly develop resistance. Resistance development is of particular concern with Gram-negative bacteria since the last novel class of Gram-negative acting antibiotics to be introduced into the clinic was the fluoroquinolone class in the 1960s,2 and the current development pipeline of antibiotics that are active against Gram-negative bacteria remains underpopulated.
One Gram-negative bacterium of particular concern is Acinetobacter baumannii one of the most insidious nosocomial pathogens known in military and civilian settings.3 A. baumannii causes life-threatening infections of multiple tissues and has evolved over the last 40 years from being a low-virulence pathogen of modest significance to being a leading cause of antibiotic-resistant infections worldwide.4 The bacterium is a scourge of intensive care units and is responsible for about a million infections per year globally,5 with clinical mortality rates as high as 70% reported.6
Due to its frequent multidrug resistance, current treatment options for A. baumannii infections are limited.β-Lactam antibiotics, especially carbapenems, represent the treatment of choice for susceptible infections. However, carbapenem resistant A. baumannii is becoming increasingly common, and for such infections, there is no consensus on the optimal alternative treatment.6 Because resistance has heretofore been relatively uncommon, colistin has become a favored treatment in spite of deleterious side effects.7 However, resistance to colistin in A. baumannii is becoming more frequent with the recent dissemination of plasmid-borne colistin resistance genes (mcr-1–7, etc.) into healthcare facilities.8
Given the discordance between the rising incidence of MDR bacterial infections and lack of new Gram-negative antibiotics in development, novel alternative approaches to develop new therapies are critical. There are several classes of clinically available antibiotics, including macrolides, glycopeptides, lipopeptides, and oxazolidinones that are considered selective for Gram-positive bacteria. The lack of activity of these antibiotic classes against Gram-negative bacteria is due not to the absence of the antibiotic target but rather to the inability of the antibiotic to access the target due to the impermeable nature of the Gram-negative outer membrane (in the case of the first three classes) or efflux of the antibiotic from the cell (in the case of oxazolidinones).9–14 Overcoming these intrinsic resistance mechanisms represents an attractive therapeutic approach, which would considerably expand treatment options for infections caused by Gram-negative bacteria. In addition to the Gram-positive selective antibiotics currently in use, these approaches also have the potential to boost the drug discovery pipeline by broadening the spectrum of novel otherwise Gram-positive selective antibiotics as they are identified. Previously reported examples of the potentiation of Gram-positive selective antibiotics against Gram-negative bacteria typically rely on disruption of the outer membrane. The truncated polymyxin, polymyxin B nonapeptide (PMBN), which lacks antibiotic activity, increases the susceptibility of several species of Gram-negative bacteria, including Klebsiella pneumoniae and Pseudomonas aeruginosa to erythromycin, novobiocin, and fusidic acid, while second-generation analogues that exhibit reduced renal toxicity have been reported to potentiate the activity of several antibiotics against Gram-negative species including Escherichia coli, K. pneumoniae, and A. baumannii.l5–17 Recently, the antiprotozoal drug pentamidine has been identified as having the ability to potentiate the activity of several Gram-positive selective antibiotics including rifampicin, erythromycin, and novobiocin (but not vancomycin) against E. coli and A. baumannii by a mechanism posited to involve disruption of lateral interactions between molecules of the lipid A component of the Gram-negative outer membrane.18
Both pentamidine and PMBN are examples of antibiotic adjuvants, compounds that undermine antibiotic resistance mechanisms. Our laboratories have been heavily involved in a concerted effort to identify such adjuvants,9–23 these molecules typically have little standalone microbicidal activity, but when paired with the appropriate antibiotic they provide a powerful combination therapy that is efficacious against MDR bacteria that would otherwise resist the antibiotic. The classic adjuvant example is Augmentin, a combination of the β-lactamase inhibitor clavulanic acid and amoxicillin. Augmentin represents one of the most powerful and successful antimicrobials, and it generated ca. $2 billion in annual sales between 2000 and 2004 (ranked as the top antimicrobial).24 Despite their potential, the only FDA-approved antibiotic adjuvants, like clavulanic acid, all target a single activity involving serine β-lactamase enzymes. However, there are many other antibiotic resistance mechanisms, including efflux, remodeling of the cell wall, permeation, stress responses, and biofilm formation, that could also be targeted to expand both the breadth of antibiotic classes and resistance mechanisms for which adjuvants could be designed.25
In this report, we disclose the discovery of small molecule adjuvants that potentiate the activity of macrolide and glycopeptide antibiotics against A. baumannii. Both of these antibiotic classes are typically limited to treating Gram-positive infections due to their inability to penetrate the Gram-negative cell envelope.26,27 These adjuvants are effective against all members of a panel of primary clinical A. baumannii isolates that encompass nearly all clinically relevant A. baumannii clades.28,29 Combination of one of the adjuvants with the macrolide clarithromycin is also effective in a Galleria mellonella model of infection. Therefore, pairing such adjuvants or their derivatives with macrolides or glycopeptides could provide an effective combination therapy for treatment of MDR A. baumannii infections, including pan-resistant variants.
RESULTS AND DISCUSSION
Our interest in pursuing adjuvants for macrolide antibiotics arose from analyzing the resistance profile of the primary clinical A. baumannii isolate AB5075, a highly virulent MDR strain isolated from an ultimately fatal bone infection of a soldier wounded in Iraq.30 Using standard minimum inhibitory concentration (MIC) measurements under Clinical and Laboratory Standards Institute (CLSI) microdilution protocols,31 we found that AB5075 was highly resistant to nearly every antibiotic class (Table S.1). Exceptionally, however, AB5075 displayed only modest resistance to macrolides (MICs 32–64 μg/mL). Screening of other MDR and non-MDR strains of A. baumannii revealed similar modest resistance to macrolides. Although there is no defined breakpoint for macrolide resistance in A. baumannii (i.e., the minimum in vitro MIC concentration that predicts clinical failure), the clinical breakpoint for the few Gram-negative species where macrolide antibiotics are employed for treatment falls between 1 and 8 μg/mL.32 Therefore, we reasoned that if we could discover an adjuvant that enhanced macrolide activity at least 8-fold, the combination could potentially allow macrolide therapy to become efficacious against A. baumannii.
We initiated this study by screening a diverse selection of small molecules from our internal library (Table S.2), which is based upon nitrogen dense marine alkaloid scaffolds, for potentiation of erythromycin, clarithromycin, and azithromycin activity against AB5075. Two concurrent assays were run in this screen: the first consisted of determining the MIC of each of the three antibiotics in the presence of molecules from the library at a concentration that mirrors the upper end at which the prototypical adjuvant (clavulanic acid) is typically viewed as useful (30 μM). The second assay consisted of determining the MIC of each compound alone against AB5075 to ensure that 30 μM is not overtly inherently toxic. The goal of this initial screen was to identify molecules that, with minimum stand-alone toxicity, enhance macrolide activity. From these initial experiments, we identified compounds 1 and 2 (Figure 1),20 which are both relatively nontoxic to the bacterium alone (MICs 100 μM), but at 30 μM potentiated the activity of all three macrolides (Table 1). Compounds 1 and 2, which have previously been reported to suppress resistance to β-lactam antibiotics in A. baumannii and P. aeruginosa,20 both reduced the erythromycin MIC from 32 μg/mL to 4 μg/mL, the azithromycin MIC from 64 μg/mL to 8 μg/mL, and the clarithromycin MIC from 32 μg/mL to 0.25 μg/mL.
Figure 1.

Structures of compounds identified from initial screen for potentiation of macrolide activity against AB5075.
Table 1.
Activity of Compounds Identified from Initial Screen for Potentiation of Macrolide Antibiotics against AB5075 (MIC fold reductions in parentheses)
| Compound | MIC (μM) | Cone Tested (μM) | Erythromydn MIC (μg/mL) | Clarithromydn MIC (μg/mL) | Azithromycin MIC (μg/mL) |
|---|---|---|---|---|---|
| 32 | 32 | 64 | |||
| 1 | 100 | 30 | 4 (8) | 0.25 (128) | 8 (4) |
| 2 | 100 | 30 | 4 (8) | 0.25 (128) | 8 (4) |
The activity of compounds 1 and 2 with several additional macrolide antibiotics, troleandomycin, josamycin, spiramycin, and oleandomycin (which are either not FDA approved or are used primarily in veterinary clinics), was also explored to determine whether adjuvant activity broadly affects this antibiotic class. Activity was observed with all four of the additional macrolides, with the highest level of activity observed with josamycin, the MIC of which was reduced 16-and 32-fold by compounds 1 and 2, respectively (Table S.3).
To further define the spectrum of potentiation activity of these adjuvants, a modified Biolog phenotypic microarray screen with AB5075 in the presence of either adjuvant 1 or 2 was conducted. A Biolog screen is a simple assay that employs antimicrobial/microbicidal agents that have been arrayed in 96-well format to rapidly measure the susceptibility of a given bacterium.33 In a typical Biolog experiment, bacteria of interest are added and growth kinetics evaluated in each well. Antibiotic/microbicidal agents to which the bacteria are susceptible reduce or eliminate growth compared to bacteria grown in media alone. In our modified Biolog screen, we compared the results from a standard Biolog experiment to those from a variation in which the bacteria were grown in the additional presence of adjuvants 1 or 2. Specifically, we were screening for additional antibiotics to which the adjuvants rendered the bacteria susceptible. Remarkably, in addition to macrolides, both adjuvants increased the sensitivity of AB5075 to the glycopeptide vancomycin, another important Gram-positive selective antibiotic. In follow-up assays, we found that compounds 1 and 2 reduced the MIC of vancomycin a remarkable 256-fold (from 256 μg/mL to 1 μg/mL). The presence of the adjuvants thus brought the MIC for vancomycin below the CLSI breakpoints for Staphylococci (≤2 μg/mL) and Enterococci (≤4 μg/mL). We also tested both adjuvants for their ability to potentiate aminoglycoside antibiotics; however, neither adjuvant was active toward AB5075 in combination with a range of aminoglycosides (Table S.4).
We next examined whether the adjuvant activity of compounds 1 and 2 with vancomycin and clarithromycin was exhibited against a diverse panel of A. baumannii isolates obtained from the Walter Reed Army Institute of Research (WRAIR). The panel includes strains that represent all major clades and many of the minor clades that are clinically relevant.28,29 Of the 26 strains tested (Table S.5), clarithromycin MICs ranged from 16 to 64 μg/mL. In the presence of either 30 μM adjuvant 1 or adjuvant 2, the MICs were suppressed to between 0.5 and ≤0.125 μg/mL or between 1 and ≤0.125 μg/mL, respectively. When analyzing the same strains for vancomycin activity, it was observed that all strains exhibited MICs of 256 >512 μg/mL. In the presence of 30 μM of adjuvant 1 or adjuvant 2, the vancomycin MIC was reduced to 32–0.5 μg/mL or 128–1 μg/mL, respectively. These results show that both adjuvants were highly active against diverse A. baumannii strains with both clarithromycin and vancomycin. Neither compound, however, was able to significantly potentiate the activity of clarithromycin against P. aeruginosa, K. pneumoniae, E. coli, or Salmonella typhimurium.
To quantify if these adjuvants display early growth toxicity, time-kill curves were constructed in the presence of compound 1 (chosen as the representative compound for this assay). AB5075 was grown in the presence of compound 1 at either 20 μM or 30 μM, and samples were plated at 2, 4, 6, 8, and 24-h time points. Although growth inhibition was observed at early times (until the 4-h time point for 20μM and 8-h time point for 30 μM), the viable count caught up with the control culture after 6 h for 20 μM and 24 h for 30 μM (Figure S.1).
Due to the early growth toxicity exhibited by compound 1 at 30 μM, the effect of lower concentrations of compounds 1 and 2 on macrolide and vancomycin MICs was examined (Table 2). We observed that decreasing compound concentration from 30 μM to 20 μM only reduces macrolide potentiation by 2-fold, and at 10 μM, significant activity (16-fold MIC reduction) is still observed for both compounds with clarithromycin. At 5 μM, activity becomes negligible with only 2- or 4-fold MIC reductions observed for either compound. Contrarily, the activity with vancomycin decreases more rapidly when the concentration is lowered, and by 10 μM there is no detectable activity.
Table 2.
Dose Dependent Resistance Suppression in AB5075 by Adjuvants 1 and 2
| Cone Tested (μM) | Erythromycin MIC (μg/mL) | Clarithromycin MIC (μg/mL) | Azithromycin MIC (μg/mL) | Vancomycin MIC (μg/mL) | |
|---|---|---|---|---|---|
| 32 | 32 | 64 | 256 | ||
| 1 | 30 | 4 | 0.2S | 8 | 1 |
| 20 | 4 | 0.5 | 16 | 8 | |
| 15 | 8 | 0.5 | 16 | 64 | |
| 10 | 8 | 2 | 16 | 256 | |
| 5 | 16 | 16 | 32 | 256 | |
| 2 | 30 | 4 | 0.2S | 8 | 1 |
| 20 | 4 | 0.2S | 16 | 4 | |
| 15 | 8 | 0.5 | 16 | 32 | |
| 10 | 8 | 2 | 32 | 128 | |
| 5 | 16 | 8 | 64 | 256 |
Next, we explored the potential in vivo relevance of this adjuvant approach for enhancing clarithromycin efficacy by examining activity in a Galleria mellonella model of AB5075 infection. This model has been previously established by researchers at WRAIR to predict activity in murine models of A. baumannii infection,30 and thus it represents a first step toward evaluating in vivo activity. We again chose compound 1 as our representative compound for this study. Compound 1 has been previously shown to exhibit no hemolytic activity up to 400 μM (highest concentration tested) and to exhibit a TCID50 of 387 μM against HaCaT keratinocyte cells as determined by a methylthiazolyldiphenyl-tetrazolium (MTT) assay.20 To this end, we tested adjuvant 1 alone, clarithromycin alone, and the combination of compound 1 and clarithromycin. Rifampin was used as a positive control, which, at a single dose of 30 mg/kg, afforded 70% survival of infected worms (vs 3% survival for untreated worms). Treatment of infected worms with only compound 1 at 100 mg/kg or clarithromycin at 25 mg/kg provided no improvement in survival in comparison to untreated controls. However, treatment with a combination of 1 and clarithromycin at those same concentrations afforded 42% worm survival from a single dose (Figure 2).
Figure 2.

Kaplan—Meier curve showing treatment of A. baumannii infection in G. mellonella model using combination therapy of compound 1 and clarithromycin. Results are an average of seven trials each containing ten larvae. Blue, A. baumannii only; Red, Clarithromycin at 25 mg/kg; Green, compound 1 at 100 mg/kg; Purple, Clarithromycin and compound 1 combination; Orange, Rifampin at 30 mg/kg.
Having established that both adjuvants 1 and 2 are highly active when paired with either clarithromycin or vancomycin, we compared the adjuvant activity of compounds 1 and 2 to the adjuvant activity of pentamidine (Table 3), which is known to potentiate macrolide activity by perturbing the outer membrane.18 This disruption is thought to be driven by direct physical interaction between the polycationic character of the compound and the negatively charged outer membrane of Gram-negative bacteria. We elected to measure the effect at two concentrations: 100 μg/mL (168 μM, ca. 5X higher than either 1 or 2), the concentration that established pentamidine’s adjuvant activity, to ensure that the compound was acting in our hands as had previously been reported,18 and 30 μM to directly compare with our adjuvants. At 100 μg/mL (168 μM), pentamidine was inactive in cation adjusted Mueller Hinton broth (CAMHB), the standard broth used to record MIC values, but it was active in LB medium, where it suppressed the MIC of clarithromycin to 0.25 μg/mL and the vancomycin MIC to 32 μg/mL. At 30 μM however, pentamidine was completely inactive in combination with clarithromycin or vancomycin in both media.
Table 3.
Comparison of Adjuvant Activity between Compound 1 and Pentamidine
| Cone Tested | Clarithromycin MIC (μg/mL) in CAMHB | Clarithromycin MIC (μg/mL) in LB | Vancomycin MIC (μg/mL) in CAMHB | Vancomycin MIC (μg/mL) in LB | |
|---|---|---|---|---|---|
| Control | N/A | 32 | 32 | 256 | 256 |
| Pentamidine | 100 [μg/mL] (168 μM) | 32 | 0.25 | 256 | 32 |
| 30 μM | 32 | 32 | 256 | 256 | |
| Compound 1 | 30 μM | 0.25 | 0.25 | 1 | 1 |
| 10 μM | 2 | 2 | 256 | 256 |
In efforts to characterize the mode of action of adjuvants 1 and 2, we first evaluated the impact of compound 1 on efflux in AB5075. The effect on efflux was studied by measuring the efflux of bisBenzimide H33342 (an established fluorescent efflux substrate) in the presence and absence of 1, compared to the known efflux inhibitor carbonyl cyanide-m-chlorophenyl hydrazone (CCCP).34 Cells treated with compound 1 exhibited fluorescence comparable to untreated controls, in contrast to a greater than 2-fold increase for cells treated with CCCP, indicating that compound 1 does not directly inhibit efflux. To determine whether prior exposure to compound 1 affects efflux rate, e.g., by affecting expression of efflux pump genes, bacteria were cultured in the presence of compound 1 prior to measuring efflux. After treatment, bacteria were collected, washed, and suspended in PBS before addition of bisBenzimide H33342 and assayed for efflux as described above. Again, treated cells exhibited comparable fluorescence intensity to untreated controls, thus establishing that prior exposure to 1 did not reduce efflux. Colony counts were performed after exposure to confirm that compound 1 did not affect bacterial growth under these conditions. Finally, we tested the ability of both compounds 1 and 2 to potentiate linezolid, as the mechanism of resistance to this antibiotic in A. baumannii involves efflux.35,36 Neither adjuvant potentiated linezolid activity, again indicating that they did not act by inhibiting efflux.
Next, we analyzed how compounds 1 and 2 affect membrane permeability and further compared the effect to that caused by pentamidine. At 10 μM of compounds 1 and 2, which is the lowest concentration that still elicited significant macrolide potentiation activity (i.e., a 16-fold reduction in clarithromycin MIC); we observed an increase in membrane permeability of 39 and 44%, respectively, compared to an untreated control, as determined by the Baclight assay.37 Interestingly, pentamidine induced a similar increase in membrane permeability, 27.1% at 30 μM, but as previously stated does not exhibit any potentiation activity at that concentration. With this knowledge, we analyzed the effect on membrane permeability of pentamidine at 100 μg/mL (168 μM), which is the concentration where activity was previously noted. At 168 μM, pentamidine induced a 96% increase in membrane permeability. As expected, when we analyzed compounds 1 and 2 at 30 μM membrane permeability increased by a similar amount (83 and 87% respectively), while when the concentration was lowered to 20 μM, the increase in membrane permeability was reduced to 78% and 83%, respectively. To further probe this apparent dichotomy, we studied the activity of control compound 3 (Figure 3) in the context of both adjuvant activity and effect on membrane permeability (Table S.6). At 60 μM and 30 μM, compound 3 induces a 72.9% and 60% increase in membrane permeability respectively and yet is devoid of adjuvant activity at both concentrations. Additionally, we performed a growth curve of AB5075 in the presence of 60 μM compound 3 to determine whether there was early toxicity similar to that observed with compound 1 (Figure S.2). Despite the similar degree of membrane permeability, compound 3 does not exhibit early toxicity. Despite the lack of toxicity displayed by compound 3, its effect upon membrane permeability indicates that membrane permeability appears decoupled from adjuvant activity for this class of compounds, and the modes of action of pentamidine and our lead compounds seem to differ (Figure 4).
Figure 3.

Structure of inactive analogue 3.
Figure 4.

Relationship of reduction in clarithromycin MIC and increase in membrane permeability between pentamidine and compounds 1 and 3.
As the adjuvant activity of compounds 1 and 2 appears decoupled from their effect on membrane permeability and neither compound appeared to inhibit efflux, we next studied the effect of compounds 1 and 2 on the MIC of colistin, as the microbicidal activity of colistin is driven by membrane disruption. We compared these results to the effect of pentamidine upon colistin activity. At 100 μg/mL (168 μM), pentamidine acted in a synergistic manner with colistin, reducing the colistin MIC > 4-fold. In contrast, the presence of 30 μM compound 1 or compound 2 increased the colistin MIC from 1 μg/mL to 4 μg/mL, which according to CLSI breakpoints for A. baumannii represents a shift from susceptible to resistant.38
As colistin is dependent upon direct interactions with the lipid A portion of lipopolysaccharide (LPS) to disrupt the outer membrane and cause bacterial death,39 a molecule such as pentamidine that also physically disrupts membranes should synergize with colistin, as we observed. To further evaluate the magnitude of antagonism between 1 and 2 and colistin, we performed a checkerboard assay, which generated ΣFIC values of 17 for compound 1 and 33 for compound 2 (Figures S.3 and S.4). These values indicate that these compounds are highly antagonistic with colistin as an ΣFIC ≥ 2 is considered antagonistic.40 Since adjuvants 1 and 2 antagonize colistin action, we hypothesized that their adjuvant activity with macrolide and glycopeptide antibiotics could be a result of inducing an alteration in LPS that leads to a reduced colistin interaction.
The direct necessity of LPS (as opposed to previously undiscovered ancillary resistance factors) for adjuvant activity was first established by generating a LPS deficient mutant (lpx−) of A. baumannii according to previously published conditions.41 The MIC of vancomycin and the macrolide antibiotics was then determined against this mutant in the absence and presence of adjuvant 1. The LPS deficient mutant was highly sensitive to all four antibiotics, and the presence of adjuvant 1 rendered no change in MIC, providing additional evidence that disruption of LPS biosynthesis is directly involved in the mechanism of action of the adjuvants (Table S.7).
LPS is a key factor in the limited permeation of several antibiotics through the Gram-negative outer membrane, so following our observations with the LPS deficient mutant, we next explored whether these compounds are effecting changes in the A. baumannii LPS. We first analyzed the composition of the lipid A component of LPS upon treatment with adjuvant 1 using gas chromatography (GC). AB5075 was incubated for 16 h with or without compound 1, and subsequent analysis of LPS revealed that treatment with compound 1 resulted in a shift toward palimitoylated lipid A species and an overall lesser amount of lipid A hydroxylation (Figure 5 and Figure S.5).
Figure 5.

Qpantification of LPS fatty acid content in untreated vs compound 1 treated AB5075.
To further evaluate the effects of adjuvant treatment, we analyzed LPS composition using gel electrophoresis. (Note: A. baumannii does not contain O-antigen in its LPS, which is thus sometimes referred to as lipooligosaccharide (LOS).)42 AB5075 was incubated for 4 and 16 h with and without compound 1 at 30 μM. Samples were then extracted by the hot phenol extraction protocol43 and run in a tricine gel with a 10–20% gradient using discontinuous buffering. The gel was then imaged using the Tsai and Frasch silver stain method.44 (Figure 6), while lipid A bands were quantified using ImageJ software (FigureS.6). The major band is LOS, and the three smaller bands underneath correspond to lipid A (highlighted). After 4 h of treatment, we noted a reduction in one of the lipid A bands, and after 16 h of treatment, this band is clearly absent with a significant difference between treated and untreated (P = 0.0192)45 samples, further indicating that compound treatment induces a change in structure by loss of lipid A components.
Figure 6.

LPS gel of AB5075 samples grown either in the absence or in the presence of compound 1. All lanes contain 20 μg of sample. A: protein ladder, B: AB5075 treated for 16 h with compound 1, C: 16 h treatment replicate, D: untreated control, E: untreated control replicate.
CONCLUSION
In conclusion, we describe the identification and mechanistic analysis of two adjuvants that considerably potentiate the activity of several macrolide antibiotics and a glycopeptide antibiotic against AB5075, with reductions in MIC as high as 256-fold. Additionally, the adjuvants display activity against a diverse panel of A. baumannii clinical isolates. Mode of action studies show that compounds 1 and 2 do not act through disruption of efflux or through increasing cell membrane permeability via physical disruption; however, compounds 1 and 2 strongly antagonize colistin action, suggesting that they may alter LPS biosynthesis or structure. GC analysis of Lipid A indicates that compound 1 decreases C14 hydroxylated species and increases palmitoylation of lipid A at Cl6. Additional analysis of LPS composition by gel electrophoresis indicates a structural alteration of the LPS. A model organism infection study was performed using G. mellonella and found that compound 1 dramatically enhanced clarithromycin activity in vivo. These compounds may provide a basis for combination therapy to broaden the antibiotic spectrum of antibiotics typically regarded as specific for Gram-positive infections to the treatment of MDR Gram-negative infections.
Supplementary Material
ACKNOWLEDGMENTS
The authors would like to thank the National Institutes of Health (AI136904 to C. Melander, C. Manoil, and R. Ernst, GM055769 to C. Melander). The authors would also like to acknowledge Dr. Larry Gallagher from the University of Washington for his work on the Biolog study.
ABBREVIATIONS
- MDR
multidrug resistant
- AB5075
Acinetobacter baumannii strain 5075
- FDA
Food and Drug Administration
- mcr
mobilized colistin resistance
- MIC
minimum inhibitory concentration
- CLSI
Clinical and Laboratory Standards Institute
- CCCP
carbonyl cyanide-m-chlorophenyl hydrazone
- PBS
phosphate buffered saline
- LPS
lipopolysaccharide
- LPX
LPS deficient mutant
- LOS
lipooligosaccharide
- CFU
colony forming units
- mg
milligram
- g
gram
- h
hours
- DMSO
dimethyl sulfoxide
- HB
Mueller Hinton Broth
- CAMHB
cation adjusted Mueller Hinton Broth
- HCl
hydrochloric acid
- LB
luria-bertani medium
Footnotes
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsinfec-dis.9b00067.
Description of materials used and protocols outlined for: MIC determination, antibiotic potentiation determination, biolog studies, time kill curves, Galleria mellonella screening, efflux and BacLight assays, checkerboard assay, LPX mutant generation, gas chromatography analysis, and lipid A analysis by gel electrophoresis. Table containing data regarding the antibiotic resistance profile of AB5075, table with the representative compounds screened for potentiation of erythromycin, tables containing the activity of lead compounds at 30 μM with a range of macrolide and aminoglycoside antibiotics against AB5075, table containing potentiation data of clarithromycin and vancomycin using compounds 1 and 2 against A. baumannii isolates, the growth curve of AB5075 in the presence of compound 1, the repotentiation and Baclight data for inactive adjuvant 3, and heat maps from the checkerboard assay used to determine FIC values for colistin and compounds 1 and 2 (PDF)
The authors declare the following competing financial interest(s): Dr. Melander is co-founder of Agile Sciences, a biotechnology company seeking to commercialize antibiotic adjuvants.
REFERENCES
- (1).Brown ED, and Wright GD (2016) Antibacterial drug discovery in the resistance era. Nature 529 (7586), 336–343. [DOI] [PubMed] [Google Scholar]
- (2).Lewis K (2013) Platforms for antibiotic discovery. Nat Rev. Drug Discovery 12 (5), 371–387. [DOI] [PubMed] [Google Scholar]
- (3).Alsan M, and Klompas M (2010) Acinetobacter baumannii: An Emerging and Important Pathogen. J. Clin Outcomes Manag 17 (8), 363–369. [PMC free article] [PubMed] [Google Scholar]
- (4).Boucher HW, Talbot GH, Bradley JS, Edwards JE, Gilbert D, Rice LB, and Bartlett J (2009) Bad bugs, no drugs: no ESKAPE! An update from the Infectious Diseases Society of America. Clin. Infect. Dis 48 (1), 1–12. [DOI] [PubMed] [Google Scholar]
- (5).Wong D, Nielsen TB, Bonomo RA, Pantapalangkoor P, Luna B, and Spellberg B (2017) Clinical and Pathophysiological Overview of Acinetobacter Infections: a Century of Challenges. Clin. Microbiol. Rev 30 (1), 409–447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Spellberg B, and Bonomo RA (2014) The deadly impact of extreme drug resistance in Acinetobacter baumannii. Crit. Care Med 42 (5), 1289–1291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Cheah SE, Li J, Tsuji BT, Forrest A, Bulitta JB, and Nation RL (2016) Colistin and Polymyxin B Dosage Regimens against Acinetobacter baumannii: Differences in Activity and the Emergence of Resistance. Antimicrob. Agents Chemother 60 (7), 3921–3933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Wang R, van Dorp L, Shaw LP, Bradley P, Wang Q, Wang X, and Balloux F (2018) The global distribution and spread of the mobilized colistin resistance gene mcr-1. Nat. Commun 9 (1), 1179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Nikaido H (2003) Molecular basis of bacterial outer membrane permeability revisited. Microbiol Mol. Biol. Rev 67 (4), 593–656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Mollmann U, Heinisch L, Bauernfeind A, Kohler T, and Ankel-Fuchs D (2009) Siderophores as drug delivery agents: application of the “Trojan Horse” strategy. BioMetals 22 (4), 615–624. [DOI] [PubMed] [Google Scholar]
- (11).Lambert PA (2002) Cellular impermeability and uptake of biocides and antibiotics in gram-positive bacteria and mycobacteria. J. Appl. Microbiol 31, 46S–54S. [PubMed] [Google Scholar]
- (12).Livermore DM (1990) Antibiotic uptake and transport by bacteria. Scand J. Infect Dis Suppl 74, 15–22. [PubMed] [Google Scholar]
- (13).Li XZ, Plesiat P, and Nikaido H (2015) The challenge of efflux-mediated antibiotic resistance in Gram-negative bacteria. Clin. Microbiol. Rev 28 (2), 337–418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Leclercq R (2002) Mechanisms of Resistance to Macrolides and Lincosamides: Nature of the Resistance Elements and Their Clinical Implications. Clin. Infect. Dis 34, 482–492. [DOI] [PubMed] [Google Scholar]
- (15).Viljanen P, and Vaara M (1984) Susceptibility of gram-negative bacteria to polymyxin B nonapeptide. Antimicrob. Agents Chemother 25 (6), 701–705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Ofek I, Cohen S, Rahmani R, Kabha K, Tamarkin D, Herzig Y, and Rubinstein E (1994) Antibacterial synergism of polymyxin B nonapeptide and hydrophobic antibiotics in experimental gram-negative infections in mice. Antimicrob. Agents Chemother 38 (2), 374–377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Zabawa TP, Pucci MJ, Parr TR Jr., and Lister T (2016) Treatment of Gram-negative bacterial infections by potentiation of antibiotics. Curr. Opin. Microbiol 33, 7–12. [DOI] [PubMed] [Google Scholar]
- (18).Stokes JM, MacNair CR, Ilyas B, French S, Cote JP, Bouwman C, and Brown ED (2017) Pentamidine sensitizes Gram-negative pathogens to antibiotics and overcomes acquired colistin resistance. Nat. Microbiol 2, 17028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Nguyen TV, Blackledge MS, Lindsey EA, Minrovic BM, Ackart DF, Jeon AB, and Melander C (2017) The Discovery of 2-Aminobenzimidazoles That Sensitize Mycobacterium smegmatis and M. tuberculosis to beta-Lactam Antibiotics in a Pattern Distinct from beta-Lactamase Inhibitors. Angew. ChemInt. Ed 56 (14), 3940–3944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Brackett CM, Melander RJ, An IH, Krishnamurthy A, Thompson RJ, Cavanagh J, and Melander C (2014) Small-molecule suppression of beta-lactam resistance in multidrug-resistant gram-negative pathogens. J. Med. Chem 57 (17), 7450–7458. [DOI] [PubMed] [Google Scholar]
- (21).Huggins WM, Barker WT, Baker JT, Hahn NA, Melander RJ, and Melander C (2018) Meridianin D Analogues Display Antibiofilm Activity against MRSA and Increase Colistin Efficacy in Gram-Negative Bacteria. ACS Med. Chem. Lett 9 (7), 702–707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Martin SE, Nguyen CM, Basaraba RJ, and Melander C (2018) Analogue synthesis reveals decoupling of antibiofilm and beta-lactam potentiation activities of a lead 2-aminoimidazole adjuvant against Mycobacterium smegmatis. Chem. Biol. Drug Des 92 (2), 1403–1408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Harris TL, Worthington RJ, and Melander C(2012) Potent Small-Molecule Suppression of Oxacillin Resistance in Methicillin-Resistant Staphylococcus aureus. Angew. Chem., Int. Ed 51 (45), 11254–11257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Renfrey S, and Featherstone J (2002) Structural proteomics. Nat. Rev. Drug Discovery 1 (3), 175–176. [DOI] [PubMed] [Google Scholar]
- (25).Melander RJ, and Melander C (2017) The Challenge of Overcoming Antibiotic Resistance: An Adjuvant Approach? ACS Infect. Dis 3 (8), 559–563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Munita JM, and Arias CA (2016) Mechanisms of Antibiotic Resistance. Microbiol Spectr, 4 (2), 481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Delcour AH (2009) Outer membrane permeability and antibiotic resistance. Biochim. Biophys. Acta, Proteins Proteomics 1794 (5), 808–816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Taitt CR, Leski TA, Stockelman MG, Craft DW, Zurawski DV, Kirkup BC, and Vora GJ (2014) Antimicrobial resistance determinants in Acinetobacter baumannii isolates taken from military treatment facilities. Antimicrob. Agents Chemother 58 (2), 767–781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Zurawski DV, Thompson MG, McQueary CN, Matalka MN, Sahl JW, Craft DW, and Rasko DA (2012) Genome sequences of four divergent multidrug-resistant Acinetobacter baumannii strains isolated from patients with sepsis or osteomyelitis. J. Bacteriol 194(6), 1619–1620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Jacobs AC, Thompson MG, Black CC, Kessler JL, Clark LP, McQpeary CN, and Zurawski DV (2014) AB5075, a Highly Virulent Isolate of Acinetobacter baumannii, as a Model Strain for the Evaluation of Pathogenesis and Antimicrobial Treatments. mBio 5 (3), e01076–01014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Institute, C. a. L. S.(2012) Performance Standards for Antimicrobial Susceptibility Testing; Twenty-Second Informational Supplement M100–S22, 32(3). [Google Scholar]
- (32).Testing, T. E. C. o. A. S.(2018) Breakpoint tables for interpretation of MICs and zone diameters, Version 8.1, http://www.eucast.org.
- (33).Bochner BR (2009) Global phenotypic characterization of bacteria. FEMS Microbiol Rev 33 (1), 191–205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Richmond GE, Chua KL, and Piddock LJ (2013) Efflux in Acinetobacter baumannii can be determined by measuring accumulation of H33342 (bis-benzamide). J. Antimicrob. Chemother 68 (7), 1594–1600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Schumacher A, Trittler R, Bohnert JA, Kummerer K, Pages JM, and Kern WV (2007) Intracellular accumulation of linezolid in Escherichia coli, Citrobacter freundii and Enterobacter aerogenes: role of enhanced efflux pump activity and inactivation. J. Antimicrob. Chemother 59 (6), 1261–1264. [DOI] [PubMed] [Google Scholar]
- (36).Vila J, Marti S, and Sanchez-Cespedes J (2007) Porins, efflux pumps and multidrug resistance in Acinetobacter baumannii. J Antimicrob. Chemother 59 (6), 1210–1215. [DOI] [PubMed] [Google Scholar]
- (37).Hilliard JJ, Goldschmidt RM, Licata L, Baum EZ, and Bush K (1999) Multiple mechanisms of action for inhibitors of histidine protein kinases from bacterial two-component systems. Antimicrob. Agents Chemother 43 (7), 1693–1699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).CLSI. (2012) Performance Standards for Antimicrobial Susceptibility Testing; Twenty-Second Informational Supplement, from Clinical and Laboratory Standards Institute. [Google Scholar]
- (39).Hancock RE (1997) Peptide antibiotics. Lancet 349 (9049), 418–422. [DOI] [PubMed] [Google Scholar]
- (40).Orhan G, Bayram A, Zer Y, and Balci I (2005) Synergy tests by E test and checkerboard methods of antimicrobial combinations against Brucella melitensis. J. Clin Microbiol 43 (1), 140–143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Moffatt JH, Harper M, Harrison P, Hale JD, Vinogradov E, Seemann T, and Boyce JD (2010) Colistin resistance in Acinetobacter baumannii is mediated by complete loss of lip-opolysaccharide production. Antimicrob. Agents Chemother 54 (12), 4971–4977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Weber BS, Harding CM, and Feldman MF (2016) Pathogenic Acinetobacter: from the Cell Surface to Infinity and Beyond. J Bacteriol 198 (6), 880–887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Haseley SR, Holst O, and Brade H (1997) Structural studies of the O-antigenic polysaccharide of the lipopolysaccharide from Acinetobacter (DNA group 11) strain 94 containing 3-amino-3,6-dideoxy-D-galactose substituted by the previously unknown amide-linked L-2-acetoxypropionic acid or L-2-hydroxypropionic acid. Eur. J. Biochem 247 (3), 815–819. [DOI] [PubMed] [Google Scholar]
- (44).Tsai CM, and Frasch CE (1982) A sensitive silver stain for detecting lipopolysaccharides in polyacrylamide gels. Anal. Biochem 119 (1), 115–119. [DOI] [PubMed] [Google Scholar]
- (45).Davis MR Jr., and Goldberg JB (2012) Purification and visualization of lipopolysaccharide from Gram-negative bacteria by hot aqueous-phenol extraction. J. Visualized Exp (63), DOI: 10.3791/3916. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.