

ABSTRACT
Daratumumab is an anti-CD38 directed monoclonal antibody approved for the treatment of multiple myeloma (MM) and functions primarily via Fc-mediated effector mechanisms such as complement-dependent cytotoxicity (CDC), antibody-dependent cell cytotoxicity (ADCC), antibody-dependent cellular phagocytosis, and T-cell activation. However, not all patients respond to daratumumab therapy and management of MM remains challenging. Radioimmunotherapy with alpha particle-emitting radionuclides represents a promising approach to significantly enhance the potency of therapeutic antibodies in cancer treatment. Here we report the results of mechanistic and feasibility studies using daratumumab radiolabeled with an alpha-emitter 225Actinium for therapy of MM.
CD38-positivelymphoma Daudi cell line and MM cell lines KMS-28BM and KMS-28PE were treated in vitro with 225Ac-daratumumab. 225Ac-daratumumab Fc-functional properties were assessed with C1q binding and ADCC assays. The pharmacokinetics and tumor uptake of 111In-daratumumab in Daudi tumor-bearing severe combined immunodeficiency (SCID) mice were measured with microSPECT/CT. The therapeutic effects of 225Ac-daratumumab on Daudi and KSM28BM tumors in mice and treatment side effects were evaluated for 50 days posttreatment. The safety of 225Ac-labeled antimurine CD38 mAb in immunocompetent mice was also evaluated.
225Ac-daratumumab efficiently and specifically killed CD38-positive tumor cells in vitro, while its complement binding and ADCC functions remained unaltered. MicroSPECT/CT imaging demonstrated fast clearance of the radiolabeled daratumumab from the circulation and tissues, but prolonged retention in the tumor up to 10 days. Therapy and safety experiments with 225Ac-daratumumab showed a significant increase in the antitumor potency in comparison to naked antibody without any significant side effects.
Our results highlight the potential of targeting alpha-emitters to tumors as a therapeutic approach and suggest that 225Ac-daratumumab may be a promising therapeutic strategy for the treatment of hematologic malignancies.
KEYWORDS: Multiple myeloma, daratumumab, 225Actinium, microSPECT/CT, radioimmunotherapy
Introduction
Overall survival for patients suffering from multiple myeloma (MM) has improved considerably in recent years thanks to the introduction of many new therapeutic options, including immunomodulatory agents, proteasome inhibitors and, more recently, targeted monoclonal antibodies (mAbs).1–7 Daratumumab is a first-in-class CD38-targeting antibody, that has been approved in combination for the treatment of newly diagnosed MM patients who are ineligible for autologous stem cell transplant and as a single agent or in combination with standard of care in relapsed or refractory MM. Based on its favorable efficacy and safety profile, other CD38-specific antibodies such as isatuximab, MOR202, and TAK-079, are now being developed and tested in MM and other CD38 positive hematologic malignancies.1 CD38-therapeutic antibodies as a class, typically rely on classical Fc-dependent immune effector function as a primary mechanism of action, e.g. antibody-dependent cellular cytotoxicity (ADCC), antibody-dependent cellular phagocytosis, and complement-dependent cytotoxicity (CDC), although recent studies suggest that targeting of immune suppressor mechanisms may also contribute to the anti-tumor activity of certain CD38 antibodies.2,8 Despite the notable improvement in patient responses with anti-CD38 antibody single agent or combination therapy, not all patients respond and management of MM remains challenging. For MM, stem cell transplant remains the primary curative therapy. The American Cancer Society projects that about 30,770 new cases of MM will be diagnosed in the United States in 2018 (16,400 in men and 14,370 in women) and about 12,770 deaths will occur. One obstacle potentially hindering patient responses to CD38 antibody therapy is the dependence of this approach on tumors possessing sufficient CD38 expression levels before the start of daratumumab therapy to elicit the above-mentioned mechanisms of action. Therefore, patients with low or heterogeneous tumor CD38 expression may respond poorly to the antibody therapy. Options to increase the therapeutic index of the drug, such as antibody-drug conjugates, where a cytotoxic toxin or chemotherapeutic is conjugated to the antibody, are also largely dependent on high antigen density to deliver sufficient payload to enable potent tumor cell killing. Thus, therapies that could potentially work in the setting of low CD38 expression would be able to improve the outcomes of antibody-based therapies.
A potential solution to increase the potency and potential response to CD38-directed targeted therapy that is not dependent on high expression of CD38, is radioimmunotherapy (RIT). RIT has emerged as a useful antitumor therapy via the targeting of radionuclides with antibodies against tumor-associated antigens.9 RIT has historically proven successful in the treatment of primary, recurrent, and refractory non-Hodgkin’s lymphomas using anti-CD20 mAbs coupled to two β-emitters, Yttrium-90, or Iodine-131.10 In the last decade, the use of α-emitting radionuclides has gained momentum both in clinical trials and in preclinical studies, driven by the advantages of α-emitters over β-emitters. These advantages include very specific targeting of the diseased cells due to the α-particles’ short 50–80 µm tissue range, and increased killing efficiency due to high linear energy transfer.11,12 Actinium-225 (225Ac) is an α-particle emitting radionuclide that has potent cytotoxic activities over short distances (approx. 3–4 cell diameters), allowing for precise targeting of a lethal dose of radiation generating double strand DNA breaks in antigen positive tumor cells13 (Figure 1). Given its high potency, as little as a single hit from an alpha particle may be sufficient to kill a tumor cell.14 Further, via cross-fire effect, adjacent antigen negative tumor cells may also be within the radius of the 225Ac alpha particle path length, while sparing distal normal tissues. In this study, we demonstrate that 225Ac conjugation of the anti-CD38 mAb daratumumab significantly increases its antitumor potency while not impeding its ability mediate Fc-dependent effector functions.
Figure 1.
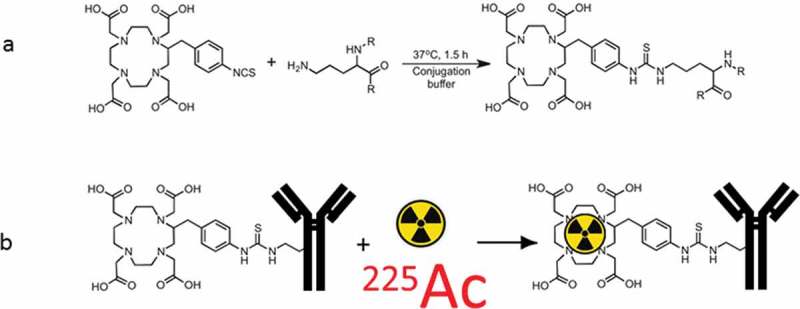
Schematic of radiolabeling daratumumab with 225Ac: (a) attachment of bifunctional chelator DOTA to daratumumab; (b) radiolabeling of DOTA-daratumumab conjugate with 225Ac.
Results
Conjugation of DOTA to daratumumab does not compromise its CD38 binding or therapeutic properties. We tested the ability of daratumumab-DOTA conjugate to bind CD38 on Daudi cells. The binding of the conjugate to Daudi cells, as assessed by flow cytometry, was identical to that of the naïve daratumumab, and no binding was observed for the secondary antibody alone (Figure 2(a)). Likewise, conjugation of DOTA did not affect binding of daratumumab to complement (C1q) (Figure 2(b)) nor did it inhibit the ability of daratumumab to initiate ADCC (Figure 2(c)).
Figure 2.
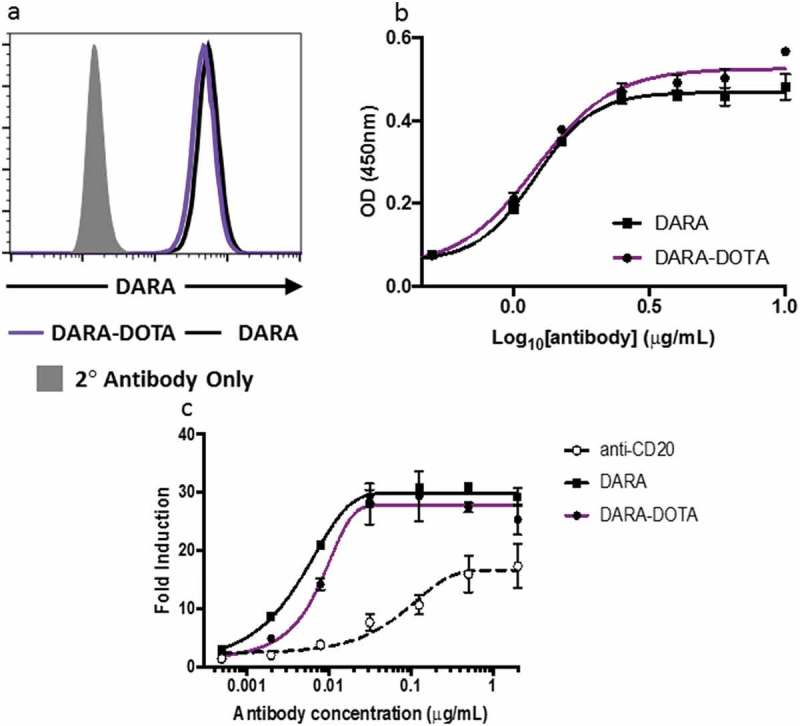
Binding of DOTA-daratumumab to CD38 on Daudi cells and associated immune mechanisms: (a) flow cytometry of naïve daratumumab and DOTA-daratumumab binding to Daudi cells. Secondary antibody alone was used as a negative control; (b) C1q Binding. Various concentrations of daratumumab and DOTA-daratumumab were immobilized on plastic and incubated with hC1q. The amount of C1q bound was assessed using anti-C1q-HRP as a probe; (c) ADCC activity. Various concentrations of daratumumab and DOTA-daratumumab were added to CD38 expressing target cells. Effector cells that express luciferase when activated through FcγR(III) were added to target cells and luminescence was measured after the addition of Bio-Glow™. Fold induction = RLU(induced-background)/RLU(no antibody control-background). Anti-CD20 antibody, which is known to activate ADCC, was used as a positive control.
225Ac-daratumumab specifically killed Daudi and multiple myeloma cells in vitro. The in vitro killing of Daudi cells with 225Ac-daratumumab showed that the maximum killing of the cells was reached with 60 nCi/0.06 μg at 72 h after the start of incubation (Figure 3). For MM cell lines KMS-28BM and KMS-28PE, the maximum killing of cells was observed at 96 h following the start of incubation. Tumor cell killing was shown to be CD38-specific as the isotype matching irrelevant control antibody labeled with the same 225Ac specific activity did not demonstrate any cytocidal effects on the cells (Figure 3). Notably, the unlabeled daratumumab in 0.02–0.06 μg amounts used in this experiment also did not have any effect on the cells, although PMBC were not included in the cytotoxicity assay.
Figure 3.
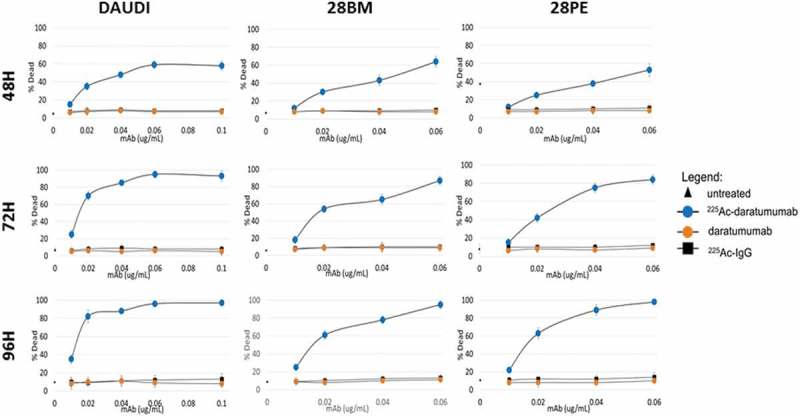
Cytotoxicity of 225Ac-daratumumab toward CD38-positive tumor cells. Titrations of 225Ac-daratumumab, daratumumab, and nonspecific control 225Ac-IgG were tested in three different cell lines (Daudi, KMS-28BM, KMS-28PE) and cellular viability was assessed by XTT and Trypan blue assays at 48, 72, and 96 h. 2 × 105 cells in 1 mL PBS per sample were treated with RIT.
Radiolabeled daratumumab localizes to and is extensively retained in Daudi tumors. We performed microSPECT/CT imaging of 111In-daratumumab in Daudi tumor-bearing mice to ascertain its specific uptake in the tumors. MicroSPECT/CT imaging demonstrated the noticeable localization of 111In-daratumumab in the tumors already at 24 h postadministration. 111In-daratumumab continued to concentrate in the tumor up to 10 days, while activity had cleared from the rest of the body (Figure 4). These imaging results provided impetus to perform RIT of Daudi tumors in mice with 225Ac-daratumumab.
Figure 4.
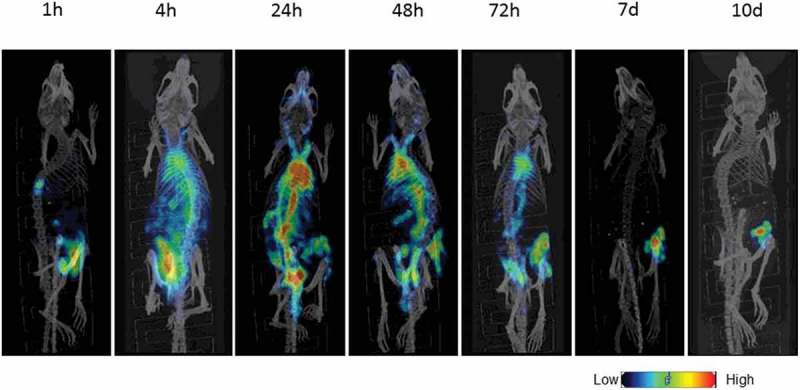
Biodistribution of 111In-daratumumab in Daudi tumor-bearing SCID mice. Mice were administered a single intraperitoneal (IP) injection of 111In-daratumumab (400 µCi) and imaged with microSPECT/CT at 1, 4, and 24 h postinjection followed by 2, 3, 7, and 10 days.
Conjugation of daratumumab to 225Ac significantly increases its anti-tumor potency against Daudi and KSM28BM tumors in vivo. At an antibody concentration of 0.3 μg, a single dose of naked unlabeled daratumumab was ineffective at controlling tumor growth and was not significantly different than vehicle control (Figure 5(a)). To the contrary, both 200 nCi/0.3 μg and 400 nCi/0.3 μg 225Ac-daratumumab were effective at stabilizing the growth of established tumors. Only at a 30-fold increase in antibody concentration (10 μg) was naked daratumumab able to effect a similar level of tumor growth inhibition. At Day 15 posttreatment, tumor growth control was statistically significant (p = .02) in the 400 nCi 225Ac-daratumumab group in comparison to both 200 nCi 225Ac-daratumumab and 10 μg daratumumab groups. Further, at the conclusion of the observation period, Day 55 posttreatment, 50% of mice in 400 nCi 225Ac-daratumumab were still alive versus 20% in 10 μg daratumumab group, demonstrating a survival benefit with the 225Ac-daratumumab at 400 nCi/0.3 μg (Figure 5(b)). A comparison of 225Ac-daratumumab to naked daratumumab in vivo was also performed in KSM28BM multiple myeloma tumor-bearing mice. In the multiple myeloma xenograft model, the results also demonstrated a significant antitumor effect (p < .01) of 400 nCi 225Ac-daratumumab on controlling KSM28BM tumor growth in comparison to unlabeled daratumumab and untreated controls (Figure 5(c)).
Figure 5.
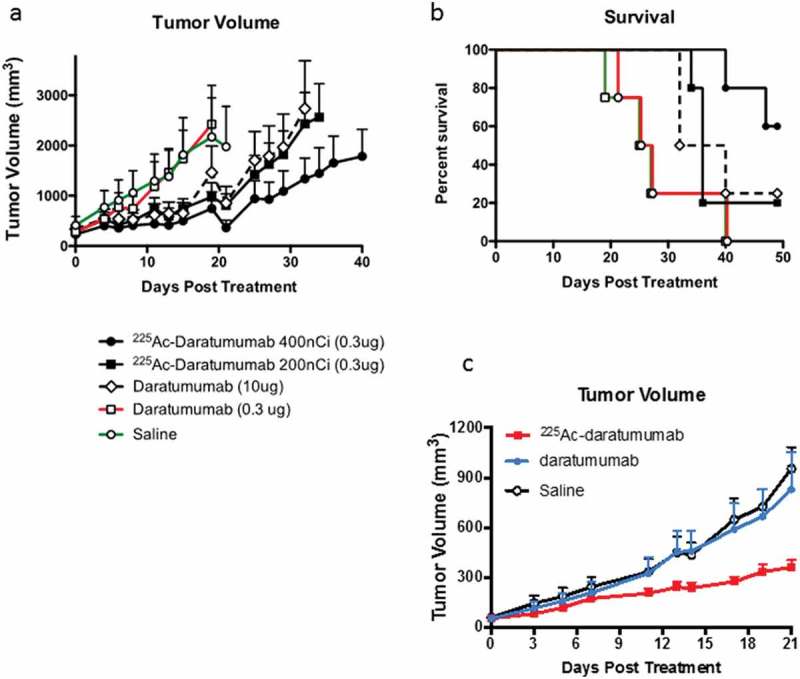
RIT of Daudi and KMS-28BM tumor-bearing SCID mice. (a) Daudi tumor volume post RIT; (b) Kaplan-Meier plot of Daudi tumor-bearing mice survival post RIT; (c) KMS-28BM tumor volume post RIT. SCID mice (five animals per group) were injected with CD38 positive Daudi or KMS-28BM cells into the right flank. When tumors reached ∼200 mm3 mice with Daudi tumors were treated with a single IP injection of 200 or 400 nCi 225Ac-daratumumab (0.3 µg), or an equivalent dose of 0.3 µg naked daratumumab or a 30-fold higher dose (10 µg) naked daratumumab, or saline vehicle. Mice with KMS-28BM tumors were treated IP with 400 nCi 225Ac-daratumumab (0.3 µg), or an equivalent dose of 0.3 µg naked daratumumab, or saline vehicle. Tumor volume was calculated using the formula V = 0.5(L×W2). Mice were sacrificed when the tumor volume reached 4,000 mm3.
RIT with 225Ac-daratumumab and 225Ac-labeled antimurine CD38 mAb was safe in Daudi tumor-bearing mice. We performed a comprehensive safety evaluation of 225Ac-daratumumab by monitoring the weight of the mice and assessing hematologic parameters and systemic toxicity (kidneys and liver). The weight of mice in the 200 nCi 225Ac-daratumumab group did not change throughout the experiment while mice in the 400 nCi group demonstrated only transient weight loss during the second week posttreatment, with rapid recovery noted in the third week posttreatment (Figure S1(a)). Of note, the experiment was conducted in SCID mice which are very sensitive to radiation due to a defect in DNA repair that causes their immunodeficiency. The evaluation of hematologic and toxicity parameters demonstrated that there was no statistically significant difference in any of the 225Ac-daratumumab versus naked daratumumab groups in any parameter tested (Figure S1(b)). This indicated that there was no impact of the presence of 225Ac conjugate on blood parameters or evidence of increased liver damage as measured by AST and ALT levels in treated mice. Further, there was no change in creatinine and BUN demonstrating the absence of any apparent kidney toxicity. Additional testing with 400 nCi 225Ac-labeled antimurine CD38 mAb produced very similar results – no observed weight loss and absence of signs of either hematologic or systemic toxicity (Figure S2).
Immunohistochemistry demonstrated significant tumor damage via apoptosis. Staining of the Daudi tumors from saline-treated mice with anti-CD38 Ab demonstrated abundant levels of CD38 in these tumors (Figure 6(a)). While hematoxylin and eosin (H&E)-stained tumors from saline-treated mice showed coherent sheets of tumor cells (Figure 6(b)), the tumors from the 225Ac-daratumumab group demonstrated disruption in tumor architecture and evidence of necrosis (Figure 6(c)). TUNEL staining for nicked DNA, which is characteristic of apoptosis did not show any significant apoptosis in the tumors in either saline or 0.3 μg daratumumab-treated groups (Figure 6(d,e), respectively) while approximately 80% of cells were apoptotic in 225Ac-daratumumab-treated tumors (Figure 6(f)). Ki67 staining was performed to determine the proliferation index of the tumor cells following treatment. The proliferation index was determined to be 95% for tumors in saline-treated group (Figure 6(g)), 94.7% in 0.3 μg daratumumab group (Figure 6(h)) and 70% – 225Ac-daratumumab group (Figure 6(i)). The tumors in patients with Burkitt lymphoma, which is the origin of Daudi cells, are known to have proliferation index of almost 100%.15
Figure 6.
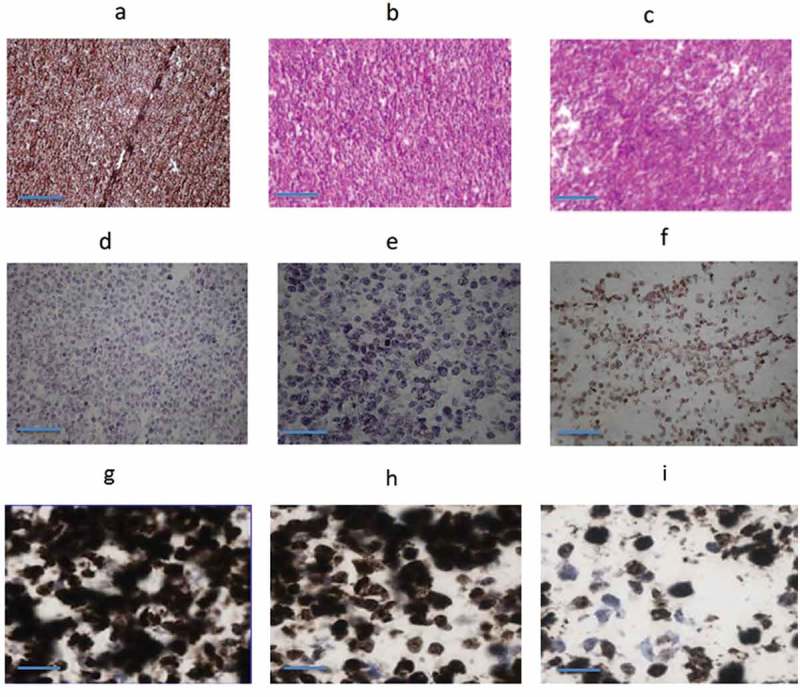
Immunohistochemistry of Daudi tumors: (a) CD38 expression; (b) H&E, untreated group; (c) H&E, 400 nCi 225Ac-daratumumab-treated group; (d) TUNEL, untreated group; (e) TUNEL, 0.3 ug cold daratumumab treated group; (f) TUNEL, 400 nCi 225Ac-daratumumab-treated group; (g) Ki67, untreated group; (h) Ki67, 0.3 ug cold daratumumab-treated group; and (i) Ki67, 400 nCi 225Ac-daratumumab-treated group. Bar length is 100 µm.
Discussion
Therapeutic monoclonal antibodies have become critically important in the treatment and control of cancer. Despite many approved therapeutic antibodies, such as the anti-CD38 antibody daratumumab, demonstrating improvements in response rate and outcome for patients, many patients fail to respond to treatment and durable responses are infrequent. The potential reasons for such failures are complex, and some of them such as pretreatment CD38 tumor expression level; relationship of CD38-expression to survival, disease stages, molecular entities, and high-risk definitions; and alternative splicing or lack of CD38-expression as potential mechanisms of upfront resistance are discussed by Seckinger et al.16 Improvements in antibody technology may be leveraged to enhance therapeutic potency, to expand the responding population of patients, without increasing toxicity risk. Engineered antibody conjugates have shown promise, recognizing the exquisite specificity of antibodies as delivery vehicles. For example, very recently B cell maturation antigen (BCMA) was identified as a potential therapeutic target in 778 newly diagnosed and relapsed myeloma patients, and IgG-based BCMA-T cell bispecific antibody (EM801) was shown to increase CD3+ T-cell/myeloma cell crosslinking, followed by CD4+/CD8+ T-cell activation, and secretion of interferon-γ, granzyme B, and perforin.17 Chemoconjugates, however, have had relatively few regulatory successes and often have narrow therapeutic windows. Increasingly, payloads with greater potency have been developed, particularly those focused on eliciting DNA damage, such as the pyrrolobenzodiazepine (PBD).18 However, with increased potency, toxicity risk may also increase.
Larson et al.9 and Kraeber-Bodéré et al.10 compared the advantages and disadvantages of beta- and alpha-emitting radionuclides for therapy of cancer. Alpha radioimmunotherapy, particularly with 225Ac, represents a promising approach to “arming” antibodies for the treatment of cancer. The 225Ac payload conjugated to a monoclonal antibody, antibody fragment, or small peptide ligand can deliver high energy alpha particles directly to the tumor site, generating lethal double strand DNA breaks without the necessitating significant payload accumulation within the tumor cell. Due to its short path length, the range of its high energy alpha particle emission is only a few cell diameters thick, thereby limiting damage to nearby normal tissues. Furthermore, 225Ac-antibody conjugates offer a crucial advantage over ADCs in that they can be potentially effective even in patients with low target antigen expressing tumors. This is because only one or two hits of α-particles emitted by 225Ac is sufficient to cause significant cytocidal effects.9–14 This means that only one antibody molecule decorated with one or two atoms of 225Ac is sufficient to kill a cancer cell. This is in contrast to beta-emitters where hundreds of antibody molecules are needed to bind to their respective antigens to deliver sufficient radioactive payload to exert an antitumor effect on a cancer cell.10,11 Other advantages of radioactive payload over drugs or toxins are: (1) the antibody used for radiation delivery does not need to be internalized to kill the cell; (2) not every cancer cell in the tumor needs to be targeted by the antibody because of the “cross-fire” effect of the radiation; (3) in contrast to ADCs, the radioisotope linked to the antibody is unlikely to elicit significant immune responses that would limit subsequent use; and (4) RIT is potentially less toxic since the chemistry of linking different radioisotopes to antibodies has been well developed, and the exceptional stability of radiolabeled mAbs in vitro and in vivo has been confirmed. Notably, a number of clinical studies are currently in progress evaluating the utility and potency of 225Ac radioimmunoconjugate therapy in diseases such as acute myeloid leukemia and prostate cancer.12,19 The goal of this study was to investigate the impact of conjugation of the α-particle emitting radionuclide 225Ac on the reactivity, potency and safety of the anti-CD38 antibody daratumumab on in vitro and in vivo models of CD38 positive cancer. The flow cytometry, CDC and ADCC experiments demonstrated that attachment of the bifunctional chelating agent DOTA to daratumumab, which enabled subsequent radiolabeling with 225Ac, did not interfere with daratumumab immunoreactivity or its primary immune mechanisms of action, indicating the potential for additive, or even synergistic, radiation, and immune effects of the 225Ac-radioimmunoconjugate daratumumab in treating CD38+ cancer. The most likely reason for the preservation of daratumumab CDC and ADCC capabilities is that the Fc region of the daratumumab was not affected by the DOTA conjugation. If this is the case, other immune mechanisms which involve Fc region of the daratumumab should not be affected either.
To investigate the effects of α-radiation delivered by daratumumab to CD38-positive cells, nanogram levels of antibody, well below those needed to effect pronounced CDC and ADCC in vitro were chosen for in vitro cytotoxicity analysis. In this regard, de Weers et al. who described the development of daratumumab and its mechanisms of action,20 observed the most pronounced lysis of cells via CDC at 1 μg/ml of daratumumab, and lysis via ADCC at 0.1 μg/ml. At titrations as much as two orders of magnitude below that used by de Weers et al.,20 0.01–0.06 μg/ml robust tumor cell killing was evident in in vitro cytotoxicity analysis (Figure 3).
Radiolabeled daratumumab localized in Daudi tumors in vivo, as shown by microSPECT/CT, and was retained in the tumors for at least 10 days. These results demonstrated that radiolabeling of daratumumab did not adversely impact the antibody’s ability to traffic and localize to the site of the C38+ tumor. The ability of daratumumab to internalize into tumor cells upon binding to CD38 likely contributed to its retention in the tumor. Such prolonged retention in the tumor could be beneficial for 225Ac with its 10-day physical half-life enabling not only delivery but also retention of tumoricidal doses of radiation within the tumor. The RIT experiments in vivo in Daudi and KMS28BM tumor-bearing mice, and additional safety evaluation with 225Ac-labeled anti-murine CD38 mAb, demonstrated that 225Ac-daratumumab conjugate was not only well tolerated, but also effected an increased in vivo potency of the daratumumab of at least 30-fold over naked daratumumab, resulting in a significant survival advantage in the high-dose 225Ac treatment groups.
In conclusion, this study showed that conjugating daratumumab to 225Ac increased its antitumor activity ~30-fold, without negatively impacting its inherent functional mechanistic properties. Due to the addition of the potent 225Ac payload, the alpha particle conjugation of daratumumab may be a promising approach to significantly expand the population of patients responsive to daratumumab treatment through its increased mechanistic potency, potentially enabling tumor growth control on low or heterogeneous CD38 positive tumors. Furthermore, with increased potency following 225Ac conjugation, there is a potential for reductions in dose level and scheduling in the amount of daratumumab needed clinically for the control of multiple myeloma. The results also highlight the potential of targeting α-emitters to tumors as a viable therapeutic approach and suggest that 225Ac-daratumumab may be a promising therapeutic strategy for the treatment of hematologic malignancies.
Materials and methods
Antibodies, reagents, and cell lines. Daratumumab manufactured by Janssen Biotech (USA) was purchased from the pharmacy at the Montefiore Medical Center, New York, USA. Isotype control human IgG1 was purchased from Creative Diagnotics (New York, USA). 225Ac in dry nitrate form was obtained from Oak Ridge National Laboratory, USA. Indium-111 (111In) in form of 111In chloride was purchased from MDS Nordion (Vancouver, BC, Canada). The CD38-positive lymphoma cell line Daudi was procured from ATCC (Manassas, USA), and CD38-positive multiple myeloma cell lines KMS-28BM and KMS-28PE from XenoTech (Japan). The Daudi, and KMS-28BM and KMS-28PE cell lines were grown in RPMI, 10% fetal bovine serum (FBS), and antimicrobial cocktail. Bifunctional chelating agent p-SCN-Bn-DOTA (DOTA) was purchased from Macrocyclics (Texas, USA).
Antibody conjugation and immunoreactivity determination. Daratumumab was first conjugated to bifunctional chelating agent DOTA (Figure 1(a)) to enable subsequent radiolabeling with 111In or 225Ac (Figure 1(b)). The conjugation reaction was performed with 5 molar excess of DOTA over daratumumab for 1.5 h at 37°C in sodium carbonate buffer at pH 8.5 followed by centrifugal filter purification into 0.15 M ammonium acetate buffer, pH 6.5. The isotype control human IgG1 was conjugated to DOTA in the same way. To test if the DOTA-conjugated daratumumab had preserved its immunoreactivity toward CD38, Daudi cells were incubated with daratumumab alone and daratumumab-DOTA and the amount of bound Ab was determined by flow cytometry using anti-hIgGPE to detect bound antibodies.
Radiolabeling of daratumumab-DOTA with 225Ac and 111In. For in vitro cell killing experiments DOTA-daratumumab at a concentration of 0.3 mg/mL in 0.15 M ammonium acetate buffer, pH 6.5, was radiolabeled with 225Ac in 0.01 M HCl with specific activity of 1 μCi/μg antibody for 60 min at 37°C. The labeling reactions were quenched DTPA and iTLC was performed immediately. ITLC strips were cut in half and counted in a gamma counter in the 95–105 keV 225Ac energy window to determine the labeling efficiency. If the labeling efficiency was below 99%, the samples were subsequently purified on disposable spin columns to 99 ± 1 radiochemical purity. For the treatment of tumor-bearing mice, daratumumab-DOTA conjugate was labeled with 225Ac at a ratio of 400 nCi to 0.3 μg and either used “as is” or 200 nCi was aliquoted and diluted with unlabeled daratumumab until the total amount of antibody per dose became 0.3 μg. For in vivo imaging, the daratumumab-DOTA conjugate was labeled with 111In which served as an imaging surrogate of 225Ac at a ratio of 400 μCi to 80 μg. 111In labeling was performed analogous to 225Ac labeling with a labeling efficiency of >95% without spin column purification.
In vitro killing of the Daudi and multiple myeloma CD38 positive cells with 225Ac-daratumumab. Daudi cells (2 × 105 in 1 mL PBS) were placed in BSA-blocked Eppendorf tubes and treated in triplicate with 0, 20, 40, 60, and 100 nCi/sample with 225Ac-daratumumab, or the matching activities of the control human IgG, or unlabeled daratumumab. The total amount of daratumumab per 1 mL sample was 0.02 μg for 20 nCi samples, 0.04 μg for 40 nCi samples, 0.06 μg for 60 nCi samples and for matching samples of unlabeled daratumumab. After 48, 72 and 96 h incubation, the effect of the radiolabeled antibodies on the cells was evaluated using the tetrazolium dye (2,3)-bis-(2-methoxy-4-nitro-5-sulphenyl)-(2H)-terazolium-5-carboxanilide (XTT) assay. Wells were washed, fresh media added along with 50 μl XTT (Sigma) at 1 mg/ml in PBS, and 4 μl menadione (Sigma) at 1 mM in acetone. Cells were incubated for another 3 h and the absorbance at 492 nm was read. All conditions were performed in triplicate. In parallel, the killing was evaluated with Trypan blue staining. In the follow-up studies, the MM CD38 expressing cell lines KMS-28BM and KMS-28PE were used under identical conditions.
Flow cytometry. The antigen-binding ability of daratumumab and DOTA conjugated daratumumab was determined by flow cytometry. Daudi cells were washed three times with FACS buffer (0.01 M azide, 2% FBS, PBS), incubated with FcR blocking antibody for 10 min at 4ºC, then antibodies were added and incubated for an additional 20 min at 4ºC. Cells were washed three times and incubated with phycoerythrin labeled anti-hIgG antibody (BD Pharrmingen, San Jose, CA) for 20 min. Stained cells were washed thrice with FACS buffer and analyzed with a CytoFLEX flow cytometer (Beckman Coulter, Mississauga, ON) and the data analyzed using FlowJo software (Tree Star Inc., Ashland, OR).
C1q binding assay. The binding of C1q to daratumumab and DOTA conjugated daratumumab was assessed using an ELISA binding assay. High binding 96-well plates (Corning) were coated overnight at 4°C with varying concentrations of antibody in coating buffer (100 mM sodium carbonate, pH 9.6). After each incubation, sample wells were washed three times with PBST (0.05% Tween 20/PBS, pH 7.4). After coating, plates were blocked for one hour at RT with blocking buffer (0.1% BSA/PBST) that was then replaced with 2 μg/mL human C1q in blocking buffer. After one hour, wells were washed three times and 100 μL 1 μg/mL anti-hC1qHRP in blocking buffer was added. After one hour wells were washed and 100 μL TMB substrate (Pierce, Rockford, IL) was added. After 15 min, the reaction was stopped with 100 μL 1 M HCl and the absorbance at 450 nm was read with a Spectra MAX 250 plate reader (Molecular Devices, San Jose, CA).
ADCC assay. The ability of daratumumab and DOTA conjugated daratumumab to mediate ADCC was assessed using the ADCC Reporter Bioassay, Complete Kit (Raji) purchased from Promega (Madison, WI) and used according to manufacturer’s protocol.
Daudi tumor model, microSPECT/CT imaging and RIT. Animal experiments were approved by the University of Saskatchewan’s Animal Research Ethics Board and adhered to the Canadian Council on Animal Care guidelines for humane animal use. For tumor induction, female SCID mice (CB17/Icr-Prkdcscid/IcrIcoCrl, procured from Charles River Laboratories, Canada) were injected subcutaneously with 5 × 106 Daudi cells in 50 μL of saline into the right flank. Tumor growth was measured with electronic calipers every 3 days, and tumor volume was calculated using the formula V = 0.5(L×W2), with width being the shorter tumor diameter. On day 14 posttumor cells inoculation, when tumors reached an average volume of ~200 mm3, mice were either imaged by microSPECT/CT or treated with RIT. For imaging, three mice were injected intraperitoneally (IP) with 400 μCi 111In-daratumumab and imaged on a microSPECT/CT (MI Labs, Netherlands) at 1, 4, and 24 h postinjection followed by 2, 3, 7, and 10 days. For RIT experiments, the tumor-bearing mice were randomized into the groups of five animals and injected IP with either: 100 μL saline, or unlabeled daratumumab at concentrations of 10 or 0.3 μg per mouse, or 225Ac-daratumumab at 400 nCi and 200 nCi per 0.3 μg of antibody. Mice were observed for tumor progression for 40 days and any mouse whose tumor reached 4,000 mm3 volume, or became necrotic, was humanely euthanized.
RIT of KMS28BM multiple myeloma tumor-bearing mice. Female SCID mice were injected subcutaneously with 2 × 106 KMS28BM cells in 50 μL of saline into the right flank. Ten days later, when tumors reached an average volume of ~100 mm3, mice were treated i.p. with 0.3 μg per mouse of unlabeled daratumumab, or with 400 nCi 225Ac-DARA per 0.3 μg of antibody or saline. Tumor volume measured as in the Daudi study.
Safety evaluation of RIT. To evaluate the side effects of RIT with 225Ac-daratumumab, the weight of the mice was recorded every 3 days. On Day 7 posttreatment the blood was taken from the mice in 0.3 μg daratumumab alone and in 400 nCi 225Ac-daratumumab groups and analyzed for white blood cells, platelet and red blood cells counts, as well as for aspartate aminotransferase (AST) and alanine transaminase (ALT), creatinine and blood urea nitrogen (BUN). In addition, we conducted a series of experiments with the mAb binding to murine CD38 to evaluate any possible off target effects of radiolabeled mAb binding to CD38 on normal tissues. Anti-mouse CD38 (clone 90) mAb was purchased from ThermoFisher (Waltham, MA), conjugated to DOTA and labeled with 225Ac in the same manner as daratumumab. Female C57Bl/6 mice were injected with either 400 nCi free 225Ac or 225Ac-antiCD38 mAb. Their weight was determined every 3 days. Blood was collected 33 days after the treatment and analyzed for blood cell counts and liver and kidney toxicity as above.
Immunohistochemistry. SCID mice bearing subcutaneous Daudi tumors were treated with 0.3 μg daratumumab, or with 225Ac-daratumumab at 400 nCi per 0.3 μg of daratumumab, or 100 μL saline. On day 7 posttreatment, the mice were sacrificed, their tumors removed, fixed in 10% formalin, and embedded into paraffin. The tumor blocks were cut into 4 μm sections, subjected to the standard deparaffinization/antigen retrieval protocols and were stained with: (1) H&E, (2) polyclonal rabbit anti-CD38 Ab antibody with 1:1,500 dilution (catalog HPA022132, Sigma, Saint Louis, MO) and visualized with EnVision+ System-HRP labelled polymer conjugated to goat anti-rabbit immunoglobulin (Code K4003, Dako, Glostrup, Denmark); (3) TUNEL (Click-iT™ TUNEL Colorimetric IHC Detection Kit, Cat. No. C10625, ThermoFisher Scientific, Waltham, MA, USA); and (4) Ki67 (MIB-1 Ab, Dako). To quantitate TUNEL and Ki67 staining results, all cells and positively stained cells per field of view were counted, and the percentage of positively stained cells was calculated by dividing the number of positively stained cells by the number of all cells per field of view and multiplying the result by 100.
Funding Statement
Funding for the study was provided by Actinium Pharmaceuticals.
Disclosure of Potential Conflicts of Interest
ED has received the research support from Actinium Pharmaceuticals; MSB and DLL are employees of Actinium Pharmaceuticals.
Supplementary material
Supplemental data for this article can be accessed on the publisher’s website.
References
- 1.D’Agostino M, Gazzera G, Cetani G, Bringhen S, Boccadoro M, Gay F.. Clinical and pharmacologic features of monoclonal antibodies and checkpoint blockade therapy in multiple myeloma. Curr Med Chem. 2018. May 13. doi: 10.2174/0929867325666180514114806. [DOI] [PubMed] [Google Scholar]
- 2.van de Donk NWCJ. Immunomodulatory effects of CD38-targeting antibodies. Immunol Lett. 2018. April 24;199:16–22. doi: 10.1016/j.imlet.2018.04.005. [DOI] [PubMed] [Google Scholar]
- 3.Busch L, Mougiakakos D, Büttner-Herold M, Müller MJ, Volmer DA, Bach C, Fabri M, Bittenbring JT, Neumann F, Boxhammer R, et al. Lenalidomide enhances MOR202-dependent macrophage-mediated effector functions via the vitamin D pathway. Leukemia. 2018. March 28;32:2445–2458 [Epub ahead of print]. doi: 10.1038/s41375-018-0114-0. [DOI] [PubMed] [Google Scholar]
- 4.Frerichs KA, Nagy NA, Lindenbergh PL, Bosman P, Marin Soto J, Broekmans M, Groen RWJ, Themeli M, Nieuwenhuis L, Stege C, et al. CD38-targeting antibodies in multiple myeloma: mechanisms of action and clinical experience. Expert Rev Clin Immunol. 2018. March;14(3):197–206. doi: 10.1080/1744666X.2018.1443809. [DOI] [PubMed] [Google Scholar]
- 5.Chim CS, Kumar SK, Orlowski RZ, Cook G, Richardson PG, Gertz MA, Giralt S, Mateos MV, Leleu X, Anderson KC. Management of relapsed and refractory multiple myeloma: novel agents, antibodies, immunotherapies and beyond. Leukemia. 2018. February;32(2):252–262. doi: 10.1038/leu.2017.329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.van de Donk NWCJ, Richardson PG, Malavasi F. CD38 antibodies in multiple myeloma: back to the future. Blood. 2018. January 4;131(1):13–29. doi: 10.1182/blood-2017-06-740944. [DOI] [PubMed] [Google Scholar]
- 7.van de Donk NW, Janmaat ML, Mutis T, Lammerts van Bueren JJ, Ahmadi T, Sasser AK, Lokhorst HM, Parren PW. Monoclonal antibodies targeting CD38 in hematological malignancies and beyond. Immunol Rev. 2016. March;270(1):95–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Adams HC 3rd, Stevenaert F, Krejcik J, Van der Borght K, Smets T, Bald J, Abraham Y, Ceulemans H, Chiu C, Vanhoof G, et al. High-parameter mass cytometry evaluation of relapsed/refractory multiple myeloma patients treated with daratumumab demonstrates immune modulation as a novel mechanism of action. Cytometry A. 2018. December 11 [Epub ahead of print]. doi: 10.1002/cyto.a.23693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Larson SM, Carrasquillo JA, Cheung NV, Press OW. Radioimmunotherapy of human tumours. Nature. 2015;15:347–360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kraeber-Bodéré F, Bodet-Milin C, Rousseau C, Eugène T, Pallardy A, Frampas E, Carlier T, Ferrer L, Gaschet J, Davodeau F, et al. Radioimmunoconjugates for the treatment of cancer. Semin Oncol. 2014;41:613–622. doi: 10.1053/j.seminoncol.2014.07.004. [DOI] [PubMed] [Google Scholar]
- 11.Baidoo KE, Yong K, Brechbiel MW. Molecular pathways: targeted α-particle radiation therapy. Clin Cancer Res. 2013;19:530–537. doi: 10.1158/1078-0432.CCR-12-0298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jurcic JG, Rosenblat TL. Targeted alpha-particle immunotherapy for acute myeloid leukemia. Am Soc Clin Oncol Educ Book. 2014;34:e126–131. doi: 10.14694/EdBook_AM.2014.34.e126. [DOI] [PubMed] [Google Scholar]
- 13.Scheinberg DA, McDevitt MR. Actinium-225 in targeted alpha-particle therapeutic applications. Curr Radiopharm. 2011. October;4(4):306–320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Nikula TK, McDevitt MR, Finn RD, Wu C, Kozak RW, Garmestani K, Brechbiel MW, Curcio MJ, Pippin CG, Tiffany-Jones L, et al. Alpha-emitting bismuth cyclohexylbenzyl DTPA constructs of recombinant humanized anti-CD33 antibodies: pharmacokinetics, bioactivity, toxicity and chemistry. J Nucl Med. 1999. January;40(1):166–176. [PubMed] [Google Scholar]
- 15.Chuang SS, Ye H, Du MQ, Lu CL, Dogan A, Hsieh PP, Huang WT, Jung YC. Histopathology and immunohistochemistry in distinguishing Burkitt lymphoma from diffuse large B-cell lymphoma with very high proliferation index and with or without a starry-sky pattern: a comparative study with EBER and FISH. Am J Clin Pathol. 2007;128(4):558–564. doi: 10.1309/EQJR3D3V0CCQGP04. [DOI] [PubMed] [Google Scholar]
- 16.Seckinger A, Hillengass J, Emde M, Beck S, Kimmich C, Dittrich T, Hundemer M, Jauch A, Hegenbart U, Raab MS, et al. CD38 as immunotherapeutic target in light chain amyloidosis and multiple myeloma-association with molecular entities, risk, survival, and mechanisms of upfront resistance. Front Immunol. 2018. July 20;9:1676. doi: 10.3389/fimmu.2018.01676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Seckinger A, Delgado JA, Moser S, Moreno L, Neuber B, Grab A, Lipp S, Merino J, Prosper F, Emde M, et al. Target expression, generation, preclinical activity, and pharmacokinetics of the BCMA-T cell bispecific antibody EM801 for multiple myeloma treatment. Cancer Cell. 2017. March 13;31(3):396–410. doi: 10.1016/j.ccell.2017.02.002. [DOI] [PubMed] [Google Scholar]
- 18.Mantaj J, Jackson PJM, Rahman KM, Thurston DE. From Anthramycin to Pyrrolobenzodiazepine (PBD)-Containing Antibody-Drug Conjugates (ADCs). Angew Chem Int Ed Engl. 2017;56:462–488. doi: 10.1002/anie.201510610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kratochwil C, Bruchertseifer F, Giesel FL, Weis M, Verburg FA, Mottaghy F, Kopka K, Apostolidis C, Haberkorn U, Morgenstern A. 225Ac-PSMA-617 for PSMA-targeted α-radiation therapy of metastatic castration-resistant prostate cancer. J Nucl Med. 2016. December;57(12):1941–1944. doi: 10.2967/jnumed.116.178673. [DOI] [PubMed] [Google Scholar]
- 20.de Weers M, Tai Y-T, van der Veer MS, Bakker JM, Vink T, Jacobs DCH, Oomen LA, Peipp M, Valerius T, Slootstra JW, et al. Daratumumab, a novel therapeutic human CD38 monoclonal antibody, induces killing of multiple myeloma and other hematological tumors. J Immunol. 2011;186:1840–1848. doi: 10.4049/jimmunol.1003032. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.