

Abstract
Current immunotherapy has limited efficacy on metastatic castrate-resistant prostate cancer (mCRPC). We therefore sought to improve the antitumor ability of mCRPC patient-derived CD8+ T-cells by the endowment of specificity to prostate-specific membrane antigen (PSMA) and insensitivity to immunosuppressant molecule transforming growth factor-β (TGF-ß) under the control of herpes simplex virus-1 thymidine kinase. CD8+ T-cells were collected by leukapheresis and cultured in a Food and Drug Administration-approved Cell Processing Work Station. We developed a chimeric antigen receptor retroviral construct using an anti-PSMA chimeric immunoglobulin-T-cell receptor(ζ) gene (PZ1) and dominant negative TGF-ß type II receptor (TßRIIDN), that could induce CD8+ T-cells to be PSMA reactive and insensitive toTGF-ß. Cr51 release assay was performed on PC-3 and PC-3-PSMA. The further antitumor functions of PSMA-specific, TGF-ß insensitive CD8+ T-cells was evaluated using an immunodeficient RAG-1−/− mouse model. We found PSMA-specific, TGF-ß insensitive CD8+ T-cells from mCRPC were expanded with strong expression of PZ1 and thymidine kinase genes, and their growth was not suppressed by TGF-ß. The survival of these cells decreased sharply after treatment with ganciclovir. Treatment of PSMA-specific TGF-ß, insensitive CD8+ T-cells was associated with 61.58% specific lysis on PC-3-PSMA, and significantly suppressed PC3-PSMA tumor compared with the PC3 tumor. A large amount of tumor apoptosis and CD8+ T-cellinfiltration were found only in the PC3-PSMA tumor. This study verified that PSMA-specific, TGF-ß insensitive CD8+ T-cells derived from mCRPC patients could be successfully expanded and used to overcome the immunosuppressive effects of the tumor microenvironment to control PSMA-expressing PC in vitro and in vivo. This may provide a promising approach for men with mCRPC who fail androgen deprivation therapy.
Patient summary:
We investigated the role of a novel chimeric antigen receptor T-immunotherapy based on autologous metastatic castrate-resistant prostate cancer patient-derived prostate-specific membrane antigen (PSMA)-specific, transforming growth factor-ß insensitive CD8+ T-cells on PSMA-positive prostate cancer. We found that this chimeric antigen receptor T-cells could kill PSMA-positive prostate cancer specifically. The results suggest that this novel immunotherapy treatment is a potential new approach for men with metastatic castrate-resistant prostate cancer.
Keywords: PSMA, TGF-, CD8+ T-cells, Prostate cancer, mCRPC
Despite treatment with surgery or radiation, approximately 30% of men with prostate cancer will develop recurrent disease that develops into castration-resistant PC (mCRPC) requiring palliative androgen deprivation or chemotherapy [1]. Previous immunotherapies, including sipuleucel-T, have been based on activating antigen presenting cells and have been shown to improve overall survival [2], albeit in a subset of patients with lower prostate-specific antigen values and less aggressive disease [3,4]. While this type of treatment may ultimately not benefit the majority of patients with mCRPC, it provides a platform from which to develop novel immunotherapies.
Prostate-specific membrane antigen (PSMA) is an integral cell-surface membrane protein expressed by PC cells. Its expression progressively increases in higher-grade tumors, and correlates with hormone-refractory and metastatic disease like mCRPC, making PSMA an ideal immunotherapy target [5]. In this study, we developed a chimeric antigen T-cell receptor (CAR) retroviral construct TβRIIDN-herpes simplex virus-1 thymidine kinase-IRES-PZ1 (TβRIIDN-TK-IRES-PZ1; Fig. 1A) by using an anti-PSMA IgTCR(ζ) gene (PZ1) which encompasses a scFv derived from the J591 hybridoma, joining the VH and VL fragments and human CD8α hinge, and the transmembrane domain which links the scFv to the intracellular domain of human TCR CD3ζ thus mediating activation signals upon binding and results in major histocompatibility complex independent activation [6]. We also included a dominant negative transforming growth factor-β (TGF-ß) type II receptor (TβRIIDN) gene [7,8], that infers resistance to tumor-secreted TGF-ß, a molecule that normally suppresses cytotoxic T lymphocyte function, and has been implicated in different aspects of carcinogenesis including mCPRC cell proliferation and metastasis [9]. As a safety mechanism, the TK gene was also incorporated into the construct.
Fig. 1 -.
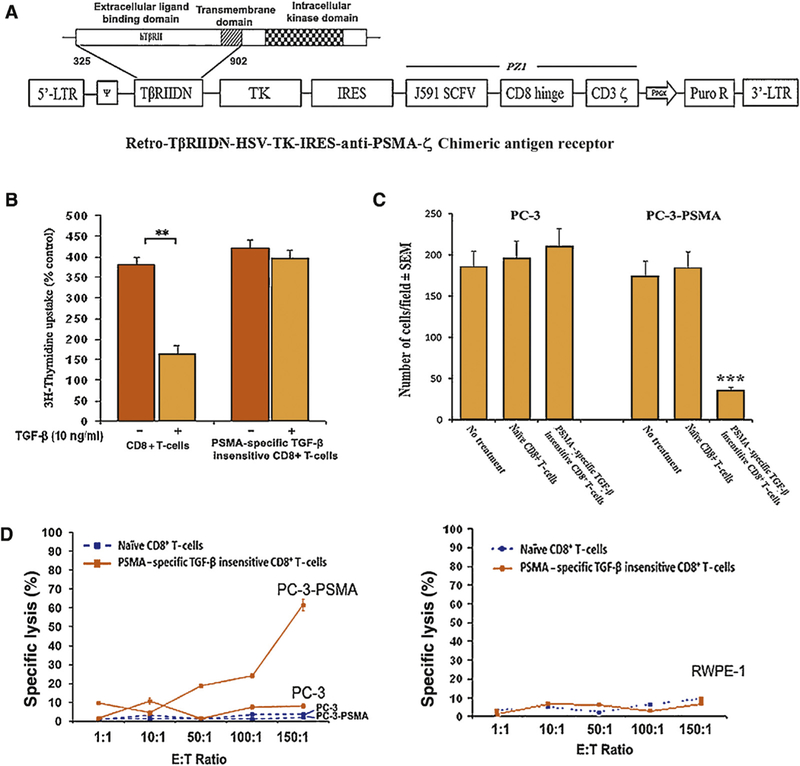
(A) Schematic diagram of the chimeric antigen T-cell receptor retroviral construct dominant negative transforming growth factor-ß type II receptor (TβRIIDN)-thymidine kinase (TK)-internal ribosomal entry sequence (IRES)-antiprostate-specific membrane antigen (PSMA) IgTCR(ζ; PZ1) retroviral construct. A truncated sequence of the human TpRIIDN, lacking the intracellular kinase signaling domain, and HSV-TK1 gene was cloned into the pMig-IRES vector following by PZ1 which is a PSMA-specific fusion receptor that encompasses an scFv derived from the J591 hybridoma, joining the VH and VL fragments through a serine/glycine linker and human CD8α hinge, and the transmembrane domain which links the scFv to the intracellular domain of human TCR CD3ζ thus mediating activation signals upon binding and results in major histocompatibility complex independent activation. Furthermore, we included the gene for TβRIIDN which can bind with TGF-β but will not transmit the signal into the cell, which reduces the sensitivity to TGF-ß, and for safety the herpes simplex virus-1 TK gene. (B) [3H]-Thymidine incorporation into naïve CD8+ T-cells or PSMA-specific TGF-β insensitive CD8+ T-cells before and after the treatment of with TGF-β1. Bars, standard deviation. PSMA-specific TGF-β insensitive CD8+ T-cells are insensitive to the treatment of TGF-β when naïve CD8+ T-cells were inhibited by 57.1%. (C) PC-3 and PC-3-PSMA possessed equal invasive capabilities. There were no significant changes in cell motility through a Matrigel-coated polycarbonate membrane when cocultured with naïve CD8+ T-cells. The invasion of PC-3-PSMA cells, but not PC-3 cells, could be inhibited by coculture with PSMA-specific TGF-β insensitive CD8+ T-cells. Corresponding numbers of invasive cells are listed. (D) In-vitro cytotoxic T-lymphocyte assay. Cytotoxic T-lymphocyte assay was done using the conventional 51Cr release assay (Supplementary data). Naïve CD8+ T-cells and PSMA-specific TGF-β insensitive CD8+ T-cells were cocultured with 51Cr-labeled targets at the specific E/T ratios. PC-3 and PC-3-PSMA cells were used as the targets in the top part of the figure and RWPE-1 cells were used as the targets in the bottom part of the figure. Point, average observations obtained from eight wells; bars, standard deviation. PSMA-specific TGF-p insensitive CD8+ T-cells generated 61.5% specific lysis against PC-3-PSMA but not PC-3 or RWEP-1 when there was lower than 10% lysis found by treating with naïve CD8+ T-cells.
After Institutional Review Board approval, peripheral blood CD8+ T-cells were collected by leukapheresis from a patient with confirmed mCRPC who was previously treated with docetaxel chemotherapy (Supplementary data). The CD8+ T-cells were expanded in an Food and Drug Administration-approved good manufacturing practices-compliant enclosed negative pressure Cell Processing Work Station (Panasonic, Kadoma, Osaka Prefecture, Japan). We redirected the mCRPC patient’s CD8+ T-cells by infecting the TβRIIDN-TK-IRES-PZ1 CAR construct into them, and generated PSMA-specific, TGF-β insensitive CD8+ T-cells which expressed high levels of PZ1 and TK (71.1% positive) and low levels of TGF-β signal (Supplementary Figs. 1 and 2A). These cells were expanded 23.4-fold in 21 d (Supplementary Fig. 2B), and their proliferation was not suppressed by TGF-β in contrast to control naïve CD8+ T-cells which were suppressed by 57.1% (Fig. 1B). Treatment with ganciclovir induced a marked decrease in the survival of PSMA-specific, TGF-β insensitive CD8+ T-cells to only 1.3% in 5 d (Supplementary Fig. 2C), representing an important safety feature to guard against unexpected off target toxicity.
In comparison to the control group, significantly higher numbers of memory type CD8+ T-cells were found in PSMA-specific, TGF-β insensitive CD8+ T-cells (16.72%; Supplementary Fig. 3A), suggesting that these cells have much stronger antitumor immune activation following tumor antigen stimulation. These cells could also induce significantly greater numbers of early- (32.0%, marked byAnnexin V) and late-apoptotic events (marked by 7aad; Supplementary Fig. 3B). Our results demonstrated the feasibility of exvivo expansion of mCRPC autologous PSMA-specific, TGF-β insensitive CD8+ T-cells which can overcome TGF-β immune suppression and induce tumor apoptosis.
We continued to elucidate the specific antitumor ability of these PSMA-specific, TGF-β insensitive CD8+ T-cells. Human PC PC-3-PSMA cell (PSMApositive) invasion, but not PC-3 cell (PSMAnegative) invasion, could be dramatically inhibited by coculture with PSMA-specific, TGF-ß-insensitive CD8+ T-cells (from 175.5/field to 36.7/field; Fig. 1C, Supplementary Fig. 3C and D). These PSMA-specific, TGF-β insensitive CD8+ T-cells showed greater specific lysis against PC-3-PSMA (61.5%) and another two PSMA-positive PC cell lines–LNCaP (53.7%) and VCaP (57.6%)–compared with PC-3 cells (8.1%) when naïve CD8+ T-cells maintained low tumor-killing activity or invasion suppression against both PC cells by Cr51 release assay (Fig. 1D, Supplementary Fig. 4A and B). An in-vitro study was performed by subcutaneous injection of xenotransplanted PC-3 and PC-3-PSMA cells (2 × 105 cells/each) infected with human simplex virus 1-TK-green fluorescent protein-luciferase reporter (SFG-nTGL) into the left and right flank region, respectively, in each of the 56 immunodeficient BALB/c-RAG-1−/− mice. These animals were provided 2-weekly adoptive transfer treatments (on days 7 and 14) with either naïve CD8+ T-cells or PSMA-specific, TGF-β insensitive CD8+ T-cells (2 × 106 cells/mouse), respectively, by intraperitoneal injection. Tumors were measured by in Vivo Luciferase Imaging Systems weekly. All mice were sacrificed 3 wk after the second adoptive transfer (day 35). Our results demonstrated that adoptive transfer of PSMA-specific, TGF-ß insensitive CD8+ T-cells is able to suppress the growth of PC-3-PSMA tumor in vivo within 2 wk, and eventually mediate complete regression of established solid PC-3-PSMA tumors in two of 20 (10%) mice with a significant reduction in total PC-3-PSMA tumor burden (> 82%) but not PC-3 tumors which were found in all the same animals. In comparison, treatment with naïve CD8+ T-cells did not suppress the growth of any tumors (Fig. 2A and B, Supplementary Fig. 4C and D). These results indicated that PSMA-specific, TGF-β insensitive CD8+ T-cells can specifically eliminate PSMA-positive PC and suppress their invasion dramatically in vitro and in vivo.
Fig. 2 -.
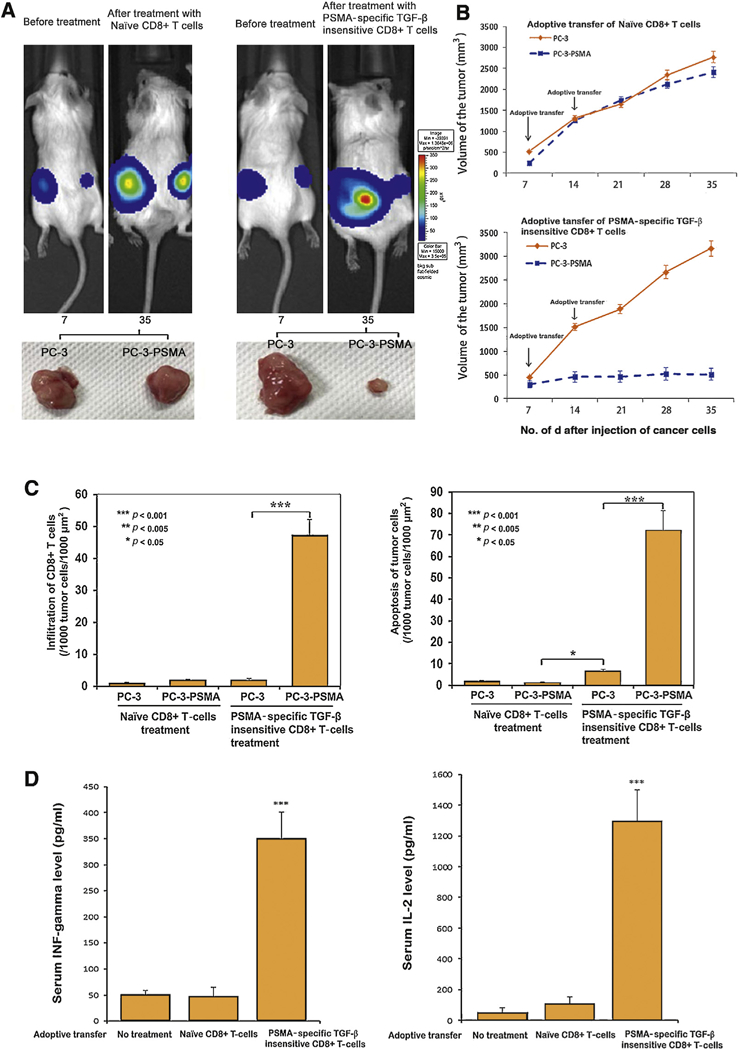
(A) IVIS 100 imaging system was used to monitor tumor growth in real-time. We found that PC-3-prostate-specific membrane antigen (PSMA) tumor (right side) growth is inhibited dramatically by with the treatment of adoptive transfer of PSMA-specific transforming growth factor (TGF)-β insensitive CD8+ T-cells compared with PC-3 tumor (left side) from the same animals after 35 d. Adoptive transfer of naïve CD8+ T-cells did not affect the growth of either PC-3 or PC-3-PSMA. (B) Twice adoptive transfer of PSMA-specific TGF-β insensitive CD8+ T-cells inhibits the PC-3-PSMA tumor growth in a time dependent manner. At the end of the 35-d treatment period, mice were sacrificed and tumors were isolated. The average volume of PC-3-PSMA tumor was 514.2 mm3 compared with 3165.2 mm3 in PC-3 tumors, respectively, in mice treated with PSMA-specific TGF-ß-insensitive CD8+ T-cells (p < 0.05). The corresponding average volume of PC-3-PSMA tumors and PC-3 tumors was 2411.9 mm3 and 2768.5 mm3, respectively, in the mice that received adoptive transfer of naïve CD8+ T-cells. (C) Immunofluorescent staining of nuclei, CD8+ T-cells, and apoptosis in tumor parenchyma. Representative tissue sections of tumors were simultaneously stained for cell nuclei (blue), CD8+ T-cells (red), and markers of apoptosis (green). The most prominent histologic feature of tumor tissue is the evidence of infiltration of CD8+ T-cells into the tumor parenchyma (left panel) and frequent tumor apoptosis in PC-3-PSMA but not PC-3 tumors treated with PSMA-specific TGF-ß-insensitive CD8+ T-cells (right panel). Although few CD8+ T-cells were found undergoing apoptosis (yellow), the majority of the apoptotic cells were derived from the tumor cells (green). Tumor cell apoptosis (72.5/1000 μm2) and CD8+ T-cell infiltration (45.5/1000 μm2) in PC3-PSMA tumor parenchyma was significantly higher compared with PC-3 tumors (6.7/1000 μm2 and 3.1/1000 μm2, respectively) which received adoptive transfer of PSMA-specific TGF-ß-insensitive CD8+ T-cells. In contrast, tumors in animals that received naïve CD8+ T-cells treatment did not exhibit significant infiltration of CD8+ T-cells or tumor cell apoptosis within either PC3 or PC3-PSMA. Magnification, ×40. (D) Serum specimens were collected at the time of the animals’ euthanasia, which was 3 wk after the second adoptive transfer of CD8+ T-cells. Circulating levels of interferon (IFN)-γ and interleukin-2 (IL-2) in experimental mice are shown in the left panel and right panel, respectively. Bars, standard deviation. * p < 0.001.
Evidence of nuclear fusion, fragmentation, necrosis (Supplementary Fig. 5), a large amount of infiltration of CD8+ T-cells into the tumor parenchyma (45.5/1000 μm2), and the presence of tumor apoptosis (72.5/1000 μm2; Fig. 2C) were found in PC-3-PSMA but not in PC-3 tumors, and there was a significant increase in serum interferon-γ (351 pg/ml) and interleukin-2 (1307 pg/ml) levels was observed among animals derived from mice that received the adoptive transfer of PSMA-specific, TGF-ß-insensitive CD8+ T-cells in comparison with the naïve CD8+ T-cell treatment group, suggesting that the presence of PSMA-specific, TGF-ß-insensitive CD8+ T-cells activated both local and systemic antitumor immunity (Fig. 2D).
Taken together, there are currently no PSMA-targeted immunotherapies that are routinely incorporated in the clinical setting for mCRPC. In this study (we summarized our strategy in a schematic diagram in Supplementary Fig. 6), mCRPC-derived, PSMA-specific, TGF-ß-insensitive CD8+ T-cells exhibited a 10-fold higher tumor killing ability and 75% decreased tumor-invasive abilites against PSMA-positive PC because of: (A) high recognition against PSMA antigen, (B) insensitive to immune suppression by tumor-derived TGF-ß, (C) capability of infiltration into PSMA-positive tumor parenchyma resulting in greater tumor apoptosis, (D) activating systemic level of interleukin-2 and interferon-γ and increasing central memory CD8+ T-cells which provide long-lasting cell-mediated systemic antitumor activity in the host [10], and (E) the safety of these cells are supported by patient autohomology and TK/ganciclovir system controllable. Our study provides proof of principle that this engineered CAR can improve the function of mCRPC patient-derived CD8+ T-cells by enhancing CD8+ T-cell expansion, providing a mechanism to overcome CD8+ cell tolerance, and promoting PSMA-specific cytolytic activity. Therefore, these CAR T-cells may offer a novel and promising therapeutic intervention for both men with recurrent androgen-sensitive PC as well as mCRPC.
Supplementary Material
Acknowledgments
Financial disclosures: Qiang Zhang certifies that all conflicts of interest, including specific financial interests and relationships and affiliations relevant to the subject matter or materials discussed in the manuscript (eg, employment/affiliation, grants or funding, consultancies, honoraria, stock ownership or options, expert testimony, royalties, or patents filed, received, or pending), are the following: this study was supported in part by grants from Walter S. and Lucienne Driskill Immunotherapy Research Program, Developmental Therapeutics Program of the Division of Hematology Oncology, Feinberg School of Medicine, Northwestern University, Matthews Center for regenerative medicine pilot grants; National Cancer Institute-SPORE in Prostate Cancer; Department of Defense (W81XWH-09–1-0311), NIH SPORE Grant (2 P50CA090386–06A2), gifts from Mr. Fred L. Turner and Lynn Sage Breast Cancer Research OncoSET Program, Robert H. Lurie Cancer Center.
Footnotes
Appendix A. Supplementary data
Supplementary data associated with this article can be found, in the online version, at https://doi.org/10.1016/j.eururo.2017.12.008.
References
- [1].Drake CG. Prostate cancer as a model for tumor immunotherapy. Nat Rev Immunol 2010;10:580–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Kantoff PW, Higano CS, Shore ND, et al. Sipuleucel-T immunotherapy for castration-resistant prostate cancer. N Engl J Med 2008;363:411–22. [DOI] [PubMed] [Google Scholar]
- [3].Gulley JL, Arlen PM, Madan RA, et al. Immunologic and prognostic factors associated with overall survival employing a poxviral-based PSA vaccine in metastatic castrate-resistant prostate cancer. Cancer Immunol Immunother 2010;59:663–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Schellhammer PF, Chodak G, Whitmore JB, Sims R, Frohlich MW, Kantoff PW. Lower baseline prostate-specific antigen is associated with a greater overall survival benefit from sipuleucel-T in the immunotherapy for prostate adenocarcinoma treatment (IMPACT) trial. Urology 2013;81:1297–302. [DOI] [PubMed] [Google Scholar]
- [5].Ananias HJK, van den Heuvel MC, Helfrich W, De Jong IJ. Expression of the gastrin-releasing peptide receptor, the prostate stem cell antigen and the prostate-specific membrane antigen in lymph node and bone metastases of prostate cancer. Prostate 2009;69:1101–8. [DOI] [PubMed] [Google Scholar]
- [6].Maher J, Brentjens RJ, Gunset G, Riviere I, Sadelain M. Human T-lymphocyte cytotoxicity and proliferation directed by a single chimeric TCRzeta/CD28 receptor. Nat Biotechnol 2002;20:70–5. [DOI] [PubMed] [Google Scholar]
- [7].Zhang Q, Yang XJ, Kundu SD, et al. Blockade of transforming growth factor-{beta} signaling in tumor-reactive CD8(+) T cells activates the antitumor immune response cycle. Mol Cancer Ther 2006; 5:1733–43. [DOI] [PubMed] [Google Scholar]
- [8].Zhang Q, Yang X, Pins M, et al. Adoptive transfer of tumor-reactive transforming growth factor-beta-insensitive CD8+ T cells: eradication of autologous mouse prostate cancer. Cancer Res 2005;65: 1761–9. [DOI] [PubMed] [Google Scholar]
- [9].Chen W, Ten Dijke P. Immunoregulation by members of the TGFβ superfamily. Nat Rev Immunol 2016;16:723–40. [DOI] [PubMed] [Google Scholar]
- [10].Klebanoff CA, Gattinoni L, Restifo NP. CD8+ T-cell memory in tumor immunology and immunotherapy. Immunol Rev 2006; 211:214–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.