

Abstract
Proteolytic cleavage regulates numerous processes in health and disease. One key player is the ubiquitously expressed serine protease furin, which cleaves a plethora of proteins at polybasic recognition motifs. Mammalian substrates of furin include cytokines, hormones, growth factors and receptors. Thus, it is not surprising that aberrant furin activity is associated with a variety of disorders including cancer. Furthermore, the enzymatic activity of furin is exploited by numerous viral and bacterial pathogens, thereby enhancing their virulence and spread. In this review, we describe the physiological and pathophysiological substrates of furin and discuss how dysregulation of a simple proteolytic cleavage event may promote infectious diseases and cancer. One major focus is the role of furin in viral glycoprotein maturation and pathogenicity. We also outline cellular mechanisms regulating the expression and activation of furin and summarise current approaches that target this protease for therapeutic intervention.
Keywords: bacterial toxins, cancer, furin, guanylate‐binding proteins, proprotein convertases, viral glycoproteins
Introduction
The human genome encodes more than 550 proteases. These molecular scissors play important roles in essentially all physiological processes. They digest the proteins in our food, degrade misfolded or unwanted proteins and regulate the trafficking and activity of numerous cellular factors. Proteolytic cleavage is certainly one of the most important post‐translational modifications, generating a plethora of bioactive proteins and peptides with key roles in cell proliferation, immunity and inflammation. Not surprisingly, mutations in proteases and/or aberrant protease activity are associated with numerous pathological processes including cancer, cardiovascular disorders and autoimmune diseases.1 Intriguingly, also many viral pathogens exploit cellular proteases for the proteolytic processing and maturation of their own proteins. Similarly, activation of bacterial toxins frequently requires cleavage by proteases of the infected or intoxicated host.
In recent years, modulation of protease activity has therefore emerged as a potential therapeutic approach in a variety of infectious and noninfectious diseases. One particularly promising target for therapeutic intervention is the cellular protease furin. This protease most likely cleaves and activates more than 150 mammalian, viral and bacterial substrates.2 Among them are viral envelope glycoproteins and bacterial toxins, as well as cellular factors that promote tumor development and growth if they are hyperactivated (Figure 1). In this review, we summarise our current knowledge of furin‐mediated protein processing in health and disease with a focus on the role of furin in viral protein processing. Furthermore, we describe cellular mechanisms regulating furin activity at the transcriptional and post‐transcriptional level. Finally, we present approaches that aim at modulating furin activity for therapeutic purposes and discuss their suitability for clinical application.
Figure 1.
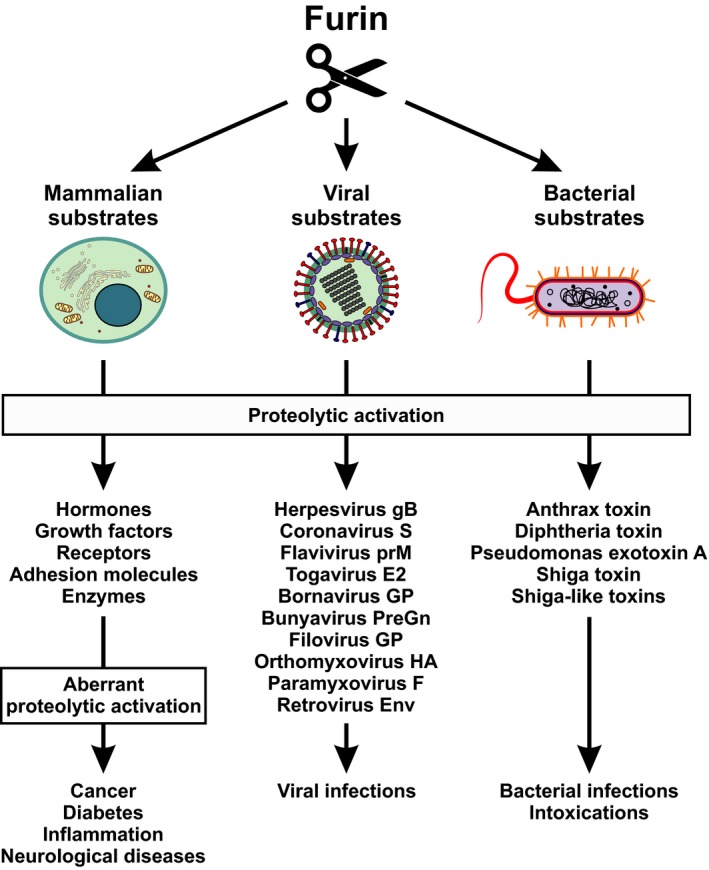
Furin cleaves and activates a variety of mammalian, viral and bacterial substrates. The serine protease furin is essential for proper activation of a variety of cellular precursor proteins. As a consequence, aberrant expression or activity of furin may result in a variety of disorders (left panel). Furthermore, numerous viruses exploit furin for the activation of their glycoproteins (central panel) and several bacterial exotoxins are activated by furin‐mediated cleavage (right panel). E2, glycoprotein E2; Env, envelope; F, fusion protein; gB, glycoprotein B; GP, glycoprotein; HA, hemagglutinin; PreGn, precursor glycoprotein Gn; prM, premembrane protein; S, spike protein.
The family of proprotein convertases
Furin is a member of the evolutionarily ancient family of proprotein convertases. Their similarity with bacterial subtilisin and yeast kexin proteases has coined the abbreviation PCSK (proprotein convertase subtilisin/kexin type). Humans encode nine members of this protease family (PCSK1–9), with PCSK3 representing furin (Table 1). PCSKs are well known for their ability to activate other cellular proteins. The proteolytic conversion of inactive precursor proteins into bioactive molecules has already been described in the 1960s.3 However, it took more than 20 years until furin was identified as the first mammalian proprotein convertase.4, 5 To date, more than 200 cellular substrates of PCSKs have been described, including hormones, receptors, growth factors and adhesion molecules.
Table 1.
The family of proprotein convertases
| PCSK nomenclature | Alternative names | trans‐cleavage | Target sequence |
|---|---|---|---|
| PCSK1 | PC1, PC3, PC1/3, SPC3, NEC1 | Yes | K/R‐Xn‐K/R↓ |
| PCSK2 | PC2, SPC2 | Yes | K/R‐Xn‐K/R↓ |
| PCSK3 | Furin, PACE, SPC1 | Yes | R‐X‐K/R‐R↓ |
| PCSK4 | PC4, SPC5 | Yes | K/R‐Xn‐K/R↓ |
| PCSK5 | PC5(A/B), PC6, PC5/6, SPC7 | Yes | K/R‐Xn‐K/R↓ |
| PCSK6 | PACE4, SPC4 | Yes | K/R‐Xn‐K/R↓ |
| PCSK7 | PC7, PC8, LPC, SPC7 | Yes | K/R‐Xn‐K/R↓ |
| PCSK8 | SKI‐1, S1P, MBTPS1 | Yes | R‐X‐L/V/I‐X↓ |
| PCSK9 | PC9, NARC‐1 | No | Internal V‐F‐A‐Q152↓ |
LPC, lymphoma proprotein convertase; MBTPS1, membrane‐bound transcription factor peptidase site 1; NARC‐1, neural apoptosis‐regulated convertase 1; NEC1, neuroendocrine convertase 1; PACE, paired basic amino acid cleaving enzyme; PC, proprotein convertase; S1P, site‐1 protease; SKI‐1, subtilisin/kexin isoenzyme 1; SPC, subtilisin proprotein convertase; Xn, 0‐, 2‐, 4‐ or 6‐amino acid spacer; X represents any amino acid; ↓, cleavage site.
Although PCSKs are frequently coexpressed in the same cell and may cleave the same substrates, there is no complete redundancy and the inactivation of individual PCSKs results in specific knock‐out phenotypes in mice.6 PCSK1–7 cleave their substrates after basic residues, with the typical recognition motif K/R‐Xn‐K/R↓6 (Table 1). In contrast, PCSK8 cleaves after nonbasic residues and is best known for its regulation of cholesterol and lipid metabolism by activating sterol regulatory element‐binding protein (SREBP) transcription factors.6, 7 Like PCSK8, PCSK9 also plays a key role in cholesterol metabolism as it regulates low‐density lipoprotein (LDL) particle levels in the blood. However, this effect does not involve proteolytic cleavage of a specific substrate, but is mediated by a direct binding of PCSK9 to LDL receptors.8 With two FDA‐approved inhibitors for the treatment of hypercholesterolaemia, PCSK9 is also the prime example of a protease that is successfully targeted for therapy.6
The ‘life cycle’ of furin
The prototypical and best‐characterised member of the PCSK family is furin/PCSK3. Since it cleaves basic amino acid motifs, it has also been termed PACE (paired basic amino acid cleaving enzyme). Furin is expressed by the FUR (FES upstream region) gene on chromosome 15. Although furin is ubiquitously expressed, its mRNA and protein levels vary depending on the cell type and tissue. High levels can be found in salivary glands, liver and bone marrow, whereas muscle cells express relatively low amounts of furin.9 Three promoters (P1, P1A and P1B), each harbouring an alternative transcription start site, have been described (Figure 2). However, the respective transcripts differ only in the first untranslated exon and are therefore predicted to express the same protein.10 While the P1A and P1B promoters resemble those of constitutively expressed housekeeping genes, the P1 promoter binds the transcription factor C/EBPβ and can be trans‐activated upon cytokine stimulation.10 In line with this, IFNγ, TGFβ, IL‐12 and PMA induce furin expression.11, 12, 13, 14
Figure 2.

Maturation of the cellular protease furin. Furin expression is driven by three different promoters, sharing characteristics of either cytokine‐activated (P1) or housekeeping gene (P1A and P1B) promoters. During translation, furin is integrated into ER membranes and glycosylated. After the N‐terminal signal peptide (red) is removed, an autocatalytic cleavage event occurs, generating a short propeptide (light blue). This propeptide remains associated with furin and acts as an intramolecular chaperone and inhibitor. After transit to the Golgi complex, the propeptide is removed and glycans are trimmed before furin gains its proteolytic activity. Furin accumulates in the trans‐Golgi network (TGN), but can also traffic to the plasma membrane and cycle between these two compartments via endosomes. Proteolytic cleavage at the C terminus of furin separates the transmembrane domain (orange) from the catalytically active domain. As a result, furin can be shed into the extracellular space as an active enzyme.
Upon translation of the mRNA, furin enters the secretory pathway as an inactive proenzyme and is integrated into the ER membrane via its C‐terminal transmembrane domain (Figure 2). Like most type I transmembrane proteins, it harbours a short N‐terminal signal peptide that is cleaved off cotranslationally. Similar to other proprotein convertases, furin contains an inhibitory N‐terminal 83‐amino acid propeptide, whose chaperone function is required for correct folding of the catalytic domain.15 During the transition of furin from the ER to the trans‐Golgi network (TGN), the inhibitory propeptide is removed in a two‐step autoproteolytic process and furin gains its enzymatic activity.16 At the same time, N‐linked oligosaccharides are added and trimmed. Although furin accumulates in the TGN, it can be further transported to the cell surface and back via the endosomal pathway.17, 18 Finally, furin can also be shed and released into the extracellular space upon proteolytic separation of the catalytic domain from the membrane‐bound C terminus.19 Whether this cleavage step is mediated by furin itself or another protease remains to be determined.20 The presence of furin in the TGN and endosomal compartments, at the cell surface and in the extracellular space, may explain its ability to process a large variety of intra‐ and extracellular substrates.
Mammalian substrates of furin
The canonical furin cleavage site is frequently described as R‐X‐K/R‐R↓. However, variations of this motif may also be recognised and a stretch of 20 amino acids surrounding the cleavage site as well as post‐translational modifications determine interaction with the furin binding pocket.21 Bioinformatic analyses and functional studies uncovered more than 100 furin cleavage sites in mammalian proteins. These comprise growth factors and cytokines (e.g. IGF1, IGF2, TGFβ, PDGFα, PDGFβ, VEGF‐C, NGF, CXCL10), hormones (e.g. PTH, TRH, GHRH), adhesion molecules (e.g. integrins, vitronectin), collagens, metalloproteinases, coagulation factors, receptors, membrane channels and albumin.2 While most of these target factors are activated upon furin‐mediated cleavage, furin also exerts inactivating cleavage steps. For example, the furin paralogue PCSK9 and endothelial lipase can be inactivated by furin.22, 23
The physiological importance of furin is reflected by furin knock‐out mice, which die at embryonic day 11 because of cardial ventral closure defects and hemodynamic insufficiency.24 Similarly, endothelial cell‐specific knock‐out of furin results in cardiac malformation and death shortly after birth.25 Even mutations in the cleavage site of a single furin target protein may have detrimental effects and result in genetic disorders such as haemophilia B or X‐linked hypohidrotic ectodermal dysplasia.26, 27 Because of its pleiotropic effects, variations in furin expression levels and/or its enzymatic activity may have detrimental effects and promote the pathogenesis of a variety of disorders, including rheumatoid arthritis, amyloid dementia and cancer.17
Role of furin in tumor development and progression
Furin has been termed a ‘master switch of tumor growth and progression’28, 29 as its aberrant expression or activation can promote the formation and progression of various malignancies including colon carcinoma, rhabdomyosarcoma, head and neck cancers, lung, skin and brain tumors.30 In some cases, furin levels positively correlate with aggressiveness, and increased furin expression has been proposed as prognostic marker for advanced cancers.30
The oncogenic and prometastatic activity of furin has been ascribed to its ability to activate proteins that promote cell proliferation, angiogenesis, migration and tissue invasion. For example, furin cleaves and activates growth factors such as IGFs, PDGFs or NGF that enhance cell proliferation and consequently tumor growth. Similarly, angiogenic and lymphangiogenic factors such as VEGF‐C and VEGF‐D may be hyperactivated and promote the vascularisation and growth of solid tumors.30 Notably, furin expression is induced by hypoxia, as all three FUR promoters harbour binding sites for the hypoxia‐inducible factor‐1 (HIF‐1).31 Thus, furin‐mediated vascularisation may preferentially occur in otherwise growth‐restricted hypoxic tumors. Interestingly, hypoxia also results in subcellular relocalisation of furin to the cell surface, which may further enhance processing of growth factors and other extracellular tumorigenic precursor proteins.32
Besides effects on tumor growth, furin can also promote migration and extravasation of malignant cells as it processes adhesion molecules mediating cell–cell and cell–matrix interactions. The cleavage of integrins may be particularly relevant as they not only mediate adhesion of cells to the extracellular matrix, but also act as signal transducers regulating cell growth, division and survival.33 In addition, furin activates matrix metalloproteinases (e.g. MMP14) that facilitate metastasis by degrading components of the extracellular matrix.30
Finally, increased furin activity may also promote cancer development by suppressing protective antitumor mechanisms. For example, increased furin‐mediated activation of TGFβ reduces immune surveillance by promoting the development of suppressive Treg cells and inhibiting effector T‐cell functions.34 The key role of furin in immunity is highlighted by T‐cell‐specific furin knock‐out mice, which harbour inherently over‐reactive effector T cells that secrete reduced levels of active TGFβ.35
Notably, positive feedback loops can further enhance the oncogenic potential of furin. For example, the furin substrate TGFβ not only increases furin mRNA expression, but also enhances its proteolytic activity by an unknown mechanism.36 Similarly, furin enhances the secretion of IFNγ, which in turn activates the FUR promoter.11, 14 This mutual enhancement seems particularly important given the key role of IFNγ in tumor development and progression.37 On the one hand, furin‐driven IFNγ release may have beneficial effects as it boosts the tumorlytic activity of natural killer cells and cytotoxic T lymphocytes. Furthermore, IFNγ may act as an antiangiogenic factor and directly inhibit tumor cell proliferation by inducing the expression of tumor suppressors such as p21 or p27. On the other hand, however, recent evidence suggests that IFNγ may also exert tumor‐promoting effects, for example by selecting for immune evasive phenotypes and promoting an immunosuppressive tumor microenvironment.37 Intriguingly, a study on laryngeal cancer patients suggests that the IFNγ‐furin feedback loop may be further boosted iatrogenically since radiotherapy increased furin expression in some patients.38
In summary, aberrant furin activation promotes several steps of cancer development, including cell proliferation, vascularisation, metastasis and antitumor immunity. However, the relative contribution of individual furin substrates to tumor progression and the role of other proprotein convertases remain largely unclear.
Role of furin in the activation of bacterial exotoxins
Furin may not only promote disease upon aberrant expression, but also by activating a variety of pathogen‐derived proteins. One example for cleavage of unwanted proteins is the proteolytic activation of bacterial toxins. Particularly, the group of AB toxins comprises several well‐described furin substrates. These exotoxins are secreted by bacteria and exert their effect in the cytoplasm of the target cell. They usually consist of an enzymatically active A subunit and a B subunit that mediates membrane binding and translocation. To exert its toxic effect, the A subunit has to be separated from the membrane‐associated B subunit by proteolytic cleavage.39
In case of diphtheria toxin and Pseudomonas exotoxin A, furin cleaves R‐V‐R‐R↓ and R‐Q‐P‐R↓ target sequences, respectively.40, 41 Cleavage most likely occurs in endosomes before the A subunit translocates into the nucleus where it inhibits protein synthesis by inhibiting the elongation factor eEF2.42 In line with an important role of furin in bacterial virulence, toxicity is highest if an optimal furin cleavage site is present.43 Similarly, furin cleaves Shiga and Shiga‐like toxins expressed by certain Shigella spp. and Escherichia coli strains and enhances their ability to halt protein synthesis. Although a furin target sequence is conserved among all Shiga‐like toxins, mutational analyses suggested that furin‐mediated cleavage augments toxin activity, but is not essential.39 Another well‐characterised example is anthrax toxin, a three‐protein exotoxin consisting of the receptor binding protective antigen (PA) and the enzymatically active components oedema factor (EF) and lethal factor (LF). Upon binding to its receptor, PA is cleaved by furin at the cell surface. This cleavage step triggers the oligomerisation of PA into a prepore that binds EF and LF. Subsequently, this toxin complex is endocytosed and PA forms a channel that allows the translocation of EF and LF into the cytoplasm.39 Although PA can be activated by different proprotein convertase family members, furin seems to be the major protease activating anthrax toxin.44
These examples illustrate that several bacterial pathogens exploit furin and related convertases for the activation of their exotoxins. Strictly speaking, however, some toxins produced by bacteria (e.g. diphtheria toxin and Shiga toxins) represent viral gene products as they are encoded by bacteriophages.45 In these cases, the term ‘viral exotoxin’ may be more appropriate. This strongly suggests that furin‐mediated toxin activation confers a selection advantage to both, the bacterium and its phage. For example, induction of cell death by furin‐activated toxins may promote tissue invasion, increase transmission rates (e.g. by causing diarrhoea) or suppress cellular immune responses. Without the proteolytic activation of exotoxins, diseases such as dysentery or diphtheria would not occur.
Role of furin in viral protein processing and pathogenicity
Like bacterial and viral exotoxins, most viral envelope glycoproteins need to be proteolytically cleaved before they can mediate viral entry into host cells. In many cases, viruses exploit cellular trypsin‐ or subtilisin‐like endoproteases for this purpose. While subtilisin‐like proteases such as furin require polybasic cleavage sites, trypsin‐like proteases also recognise monobasic motifs and cleave after single arginine or lysine residues.46
Notably, the dependency on specific proteases can also be an important determinant of tissue tropism and viral spread in an infected organism. For example, avirulent Newcastle disease virus (NDV) strains harbour a monobasic cleavage site in their Fusion (F) protein and result only in local infections (mainly in the respiratory tract) since expression of the respective host proteases is limited to a few cell types. In contrast, the F proteins of virulent NDV strains can be cleaved by furin or related proprotein convertases that are ubiquitously expressed. Consequently, these viruses are able to spread systemically and cause high rates of mortality in infected birds.47 Another well‐described example is the cleavage of influenza A virus hemagglutinin (HA). In contrast to low pathogenic avian influenza A viruses, their highly pathogenic counterparts harbour a polybasic furin cleavage site in the HA protein.48 Thus, the ability of viruses to exploit furin may have drastic effects on their pathogenicity.
To date, furin‐mediated cleavage has been described for envelope glycoproteins encoded by numerous evolutionarily diverse virus families, including Herpes‐, Corona‐, Flavi‐, Toga‐, Borna‐, Bunya‐, Filo‐, Orthomyxo‐, Paramyxo‐, Pneumo‐ and Retroviridae (Table 2). Although viral furin substrates generally harbour the canonical polybasic cleavage site, timing and subcellular localisation of furin‐mediated activation may differ substantially between virus families. Since furin and viral glycoproteins both enter the secretory pathway, proteolytic activation can occur at different steps of the viral replication cycle. While the envelope proteins of some viruses are cleaved in the producer cell, others are processed in the extracellular space or during entry into their target cells (Figure 3). The following sections highlight the characteristics of a few selected viral glycoproteins, their processing by furin and the importance of furin‐mediated cleavage for infection and pathogenicity.
Table 2.
Viral substrates of furin
| Taxonomy | Virus | Protein | Exemplary cleavage site | Reference |
|---|---|---|---|---|
| dsDNA | Herpesviridae | |||
| Human cytomegalovirus | gB | THRTRR↓ST | 121 | |
| Varicella‐zoster virus | gB | NTRSRR↓SV | 122 | |
| Epstein–Barr virus | gB | LRRRRR↓DA | 123 | |
| Papillomaviridae | ||||
| Human papillomavirus type 16 | L2 | AKRTKR↓AS | 79 | |
| (+) ssRNA | Coronaviridae | |||
| Infectious bronchitis virus | S | TRRFRR↓SI | 124 | |
| Mouse hepatitis virus | S | SRRARR↓SV | 125 | |
| Flaviviridae | ||||
| Yellow fever virus | prM | SGRSRR↓SV | 126 | |
| Tick‐borne encephalitis virus | prM | GSRTRR↓SV | 127 | |
| Dengue virus type 2 | prM | HRREKR↓SV | 75 | |
| Togaviridae | ||||
| Sindbis virus | E2 | SGRSKR↓SV | 128 | |
| Semliki forest virus | E2 | GTRHRR↓TV | 129 | |
| (−) ssRNA | Bornaviridae | |||
| Borna disease virus | GP | LKRRRR↓DT | 130, 131 | |
| Bunyaviridae | ||||
| Crimean‐Congo haemorrhagic fever orthonairovirus | PreGn | TNRSKR↓NL | 132 | |
| Filoviridae | ||||
| Ebola virus | GP | GRRTRR↓EA | 63 | |
| Reston virus | GP | TRKQKR↓SV | 63 | |
| Marburg virus | GP | YFRRKR↓SI | 64 | |
| Orthomyxoviridae | ||||
| Influenza A virus (H5) | HA | TRRQKR↓GL | 133 | |
| Influenza A virus (H7) | HA | KKREKR↓GL | 134 | |
| Influenza A virus (H9) | HA | PARSKR↓GL | 62 | |
| Paramyxoviridae | ||||
| Newcastle disease virus | F | GRRQRR↓FI | 135 | |
| Human parainfluenza virus 3 | F | DPRTKR↓FF | 136 | |
| Mumps virus | F | SRRHKR↓FA | 137 | |
| Measles virus | F | SRRHKR↓FA | 138 | |
| Simian virus 5 | F | TRRRRR↓FA | 139 | |
| Respiratory syncytial virus | F | KKRKRR↓FL | 140 | |
| Pneumoviridae | ||||
| Metapneumovirus | F | NPRQSR↓FV | 91 | |
| ssRNA‐RT | Retroviridae | |||
| Human immunodeficiency virus 1 | Env | VQREKR↓AV | 141 | |
| Rous sarcoma virus | Env | GIRRKR↓SV | 142 | |
| Murine leukaemia virus | Env | SNRHKR↓EP | 143 | |
| Feline foamy virus | Env | SSRRRR↓DI | 144 | |
| dsDNA‐RT | Hepadnaviridae | |||
| Hepatitis B virus | HBeAg | VRRRGR↓SP | 82, 83 | |
E2, glycoprotein E2; Env, envelope; F, fusion protein; gB, glycoprotein B; GP, glycoprotein; HA, hemagglutinin; HBeAg, hepatitis B external core antigen; L2, minor capsid protein; PreGn, precursor glycoprotein Gn; prM, premembrane protein; S, spike protein; ↓, cleavage site. Virus families and polybasic cleavage sites are highlighted in bold.
Figure 3.

Furin‐mediated processing of viral proteins can occur at several steps of the viral replication cycle. While some viral proteins are proteolytically processed during maturation or egress (a), others are cleaved during entry into new target cells (b). (a) In case of the human immunodeficiency virus (HIV) (left panel), the viral envelope (Env) glycoprotein precursor (green) migrates through the ER to the Golgi complex where it is cleaved by furin (pink scissors) into the functional mature Env glycoprotein (blue). Processed Env glycoproteins are transported to the cell surface and incorporated into assembling viral particles. In contrast, dengue viruses bud into the ER lumen and incorporate the uncleaved premembrane protein (prM) (right panel). During virus particle transit through the secretory pathway, virion‐associated prM proteins can be cleaved by furin (dark blue to light blue). (b) Some prM molecules escape furin‐mediated cleavage in the producer cells resulting in the release of immature or partially mature dengue virus particles. In this case, processing can also occur in endosomes of new target cells, upon receptor‐mediated endocytosis (left panel). During human papillomavirus (HPV) infection (right panel), attachment to heparan sulphate proteoglycans induces a conformational change that allows proteolytic processing of the minor capsid protein L2 (red) by furin, which is present at the cell surface. Furin processing induces a structural rearrangement that allows binding to a secondary receptor and subsequent receptor‐mediated endocytosis.
Human immunodeficiency viruses (HIV)
Retroviral glycoprotein trimers, such as those of human immunodeficiency, Rous sarcoma or murine leukaemia viruses, are proteolytically processed and activated in the producer cells (Figure 3a, left panel). In case of HIV‐1, the gp160 precursor of the viral envelope protein (Env) is cleaved into gp120 and membrane‐anchored gp41 that remain associated through noncovalent interactions. Cleavage occurs in intracellular compartments, before the assembly of virions at the plasma membrane. Notably, proteolytic processing of Env depends on correct N‐linked glycosylation as aberrant carbohydrate side chains may result in subcellular mistrafficking or sequestration of Env.49 Most likely, HIV‐1 takes advantage of the redundancy of several proprotein convertases recognising the polybasic cleavage motif in Env. Furin, PCSK5, PCSK6 and PCSK7 have all been shown to cleave gp160 in cells, albeit with different efficiencies.49 Notably, in vitro cleavage experiments using recombinant proteases did not always reflect cleavage efficiency in transfected cells and the relative contribution of individual PCSKs to HIV‐1 maturation in vivo remains unclear.49 Interestingly, HIV‐1 Env harbours a second polybasic cleavage site, about eight amino acid residues upstream of the major one. Although cleavage at this site does not result in fusiogenic Env species, about 15% of all gp160 molecules are cleaved at this position, at least in case of the cell‐culture‐adapted HIV‐1 clone LAI.50, 51
In some cases, gp160 escapes intracellular cleavage and may be incorporated as an unprocessed precursor into budding virions. Whether these Env molecules may be processed extracellularly by shed furin is unknown. In this context, it is noteworthy that membrane‐bound plasmin has been shown to convert extracellular gp160 into gp120 and gp41.52 However, it remains to be determined whether plasmin‐mediated cleavage results in fully infectious HIV‐1 particles.
Highly pathogenic avian influenza A viruses
Influenza A virus hemagglutinin (HA) can be cleaved and primed by a variety of cellular proteases. Even bacterial proteases may promote influenza virus spread by cleaving HA0 into HA1 and HA2. 53 Both subunits remain linked via disulphide bonds and form trimeric structures. While HA1 binds to the sialic acid receptor on viral target cells, HA2 harbours the fusion peptide that mediates fusion of viral and cellular endosomal membranes. HA cleavage can occur within producer cells, upon release of virions from infected cells or directly prior to entry into new target cells.54 As a general rule, hemagglutinins of mammalian and low pathogenic avian influenza A viruses cannot be cleaved by furin as they usually only harbour a mono‐ or dibasic cleavage site. Instead, they depend on trypsin‐like proteases such as transmembrane protease serine S1 member 2 (TMPRSS2) or human airway trypsin‐like protease (HAT).55 Expression of such trypsin‐like proteases is largely restricted to the respiratory and gastrointestinal tract.
In contrast, H5 and H7 hemagglutinins of a large number of highly pathogenic avian influenza A viruses (HPAIV) can be cleaved by furin or PCSK5, which are present in many cell types.56, 57 This is because they acquired a polybasic cleavage site upon insertion of additional lysine and/or arginine residues. Duplication of lysine and arginine residues in HA is facilitated by polymerase slippage as these amino acids are encoded by purine‐rich codons.58 Instead of the prototypical R‐X‐K/R‐R↓ motif, some HPAIVs harbour a suboptimal K‐X‐K/R‐R↓ cleavage motif. Proteolytic cleavage at this site is only efficient if additional positively charged amino acids upstream of this cleavage motif are present or if attachment sites for masking oligosaccharide chains are missing.59, 60 Notably, a subset of H9N2 lowly pathogenic avian influenza A virus strains also harbour R‐S‐K‐R↓ or R‐S‐R‐R↓ sites that are not only cleaved by trypsin‐like proteases, such as TMPRSS2 or HAT, but also by PCSKs.61 However, their cleavage is only efficient in the presence of very high amounts of furin or upon mutation of a glycosylation site in HA.62 Thus, the ability to exploit furin for efficient HA cleavage and the associated increase in pathogenicity are not only determined by the presence of a furin consensus target site, but also by adjacent residues and the absence of masking oligosaccharide chains.
Ebola and Marburg viruses
Furin cleaves the glycoproteins (GPs) of Marburg virus (MARV) and all five Ebolavirus species into a large N‐terminal subunit (GP1) that mediates receptor binding and a small membrane‐anchored C‐terminal part (GP2) that contains the fusion peptide.63, 64 MARV and human pathogenic Ebolavirus species harbour canonical furin cleavage sites (R‐X‐K/R‐R↓). In contrast, the GP of the closely related Reston virus, which causes asymptomatic infections in humans, is processed less efficiently by furin as it carries the suboptimal cleavage site K‐Q‐K‐R↓.63 Surprisingly, however, uncleavable GP mutants of highly pathogenic Ebola virus (EBOV) are able to mediate infection and furin‐mediated cleavage is not required for replication in cell culture.65 Furthermore, an EBOV mutant lacking the furin cleavage site replicated efficiently in nonhuman primates and showed no differences in disease progression or lethality compared to wild‐type viruses.66 Thus, the high conservation of the furin cleavage site among different Ebolavirus species is surprising and it remains to be determined whether furin‐mediated GP processing plays a role in the natural reservoir hosts of these viruses.65
Notably, the EBOV GP gene harbours an RNA editing site sequence and may not only express full‐length GP, but also a soluble form of the glycoprotein (pre‐sGP) that lacks the C‐terminal transmembrane domain.67 Intriguingly, pre‐sGP harbours another furin recognition site and is cleaved into mature sGP and a short so‐called Δ peptide. Both of them are ultimately released from infected cells.68 Although pre‐sGP is produced in higher amounts than GP, its role in viral replication is under debate. Among others, sGP has been suggested to serve as a decoy antigen, to act as a structural substitute for GP1 and to induce apoptosis of uninfected lymphocytes.69
Flaviviruses
Flavivirus RNA is translated into a single large polyprotein that is cleaved by cellular and viral proteases into all structural and nonstructural proteins of the virus. The structural proteins comprise the envelope proteins prM and E that are incorporated as prM/E heterodimers into budding virions.70 prM acts as a chaperone and facilitates correct folding of the E glycoprotein.71 Many flaviviruses bud into the lumen of the ER and enter the secretory pathway.70 In the acidic milieu of the trans‐Golgi network (TGN), a furin cleavage site is exposed and prM can be cleaved into mature pr and M proteins.72 Thus, furin‐mediated cleavage of the viral glycoprotein occurs only after its incorporation into newly formed virions (Figure 3a, right panel). This is in contrast to viruses such as HIV, whose envelope proteins are cleaved before assembly. The pr peptide remains associated with the E protein until the virion is released from the cell, thereby preventing premature unintended fusion with membranes of the producer cell.73 Furin‐mediated prM cleavage is essential for replication of flaviviruses such as tick‐borne encephalitis or dengue virus.74, 75
In some cases, prM molecules escape furin processing in the producer cell and result in the release of immature or partially mature viral particles. Partially mature virions are still infectious since low amounts of mature M are sufficient to mediate fusion with target cell membranes.70 Furthermore, uncleaved prM may participate in virion attachment to target cells and can be cleaved by furin during the entry process, in the acidic milieu of endosomes76, 77 (Figure 3b, left panel). The relative contribution of prM processing during viral entry into new target cells, however, remains to be determined.
The ratio of mature to immature prM depends on a variety of factors, including the producer cell type and the flavivirus species. For example, dengue viruses are known to release high amounts of immature or partially mature viruses, most likely because of a conserved acidic residue within the furin recognition site78 (Table 2). Importantly, the content of prM in viral particles also affects antibody recognition and consequently antibody‐dependent enhancement of dengue virus infection.77 Thus, furin‐mediated protein processing may once again markedly affect the outcome of infection.
Papillomaviruses
While furin plays a key role in activating envelope glycoproteins of a variety of viruses, its activity is also exploited for the cleavage of other viral proteins. One example is the cleavage of the L2 protein of papillomaviruses.79 Together with the major capsid protein L1, this minor capsid protein builds the viral capsid. The furin cleavage site is located close to the N terminus of L2 and highly conserved among different human papillomavirus (HPV) strains.80 Cleavage is not required for virus assembly or release, but essential for infection of new target cells. For example, LoVo and CHO cells lacking furin expression are completely resistant to infection with pseudoviruses of HPV16,79, 80 which is one of the high‐risk HPV types causing cervical cancer.
In contrast to flavivirus prM, which can be cleaved during egress and entry, papillomavirus L2 seems to be exclusively cleaved on target cells.80 A model has been proposed, in which attachment of papillomaviruses to heparan sulphate proteoglycans induces a conformational change in L2 that exposes the polybasic cleavage site.81 Upon proteolytic processing of L2, L1 may engage a secondary cellular receptor and mediate infection80 (Figure 3b, right panel). Furthermore, interaction of cleaved L2 with an unknown intracellular receptor may be required for escape of L2 from the endosomal compartment and its ability to escort viral DNA into the nucleus.80 This illustrates that also nonenveloped viruses have evolved the ability to exploit furin or related PCSKs for their own purposes.
Hepatitis B virus
Another nonenvelope protein that is cleaved by furin is the external core antigen (HBeAg) of hepatitis B virus (HBV).82, 83 Cleaved HBeAg is secreted from infected cells and exerts immunosuppressive effects.84 It has been suggested to act as a T‐cell tolerogen that prevents killing of infected hepatocytes by cytotoxic T lymphocytes.85 In contrast, uncleaved HBeAg may have the opposite effects as it is transported to the plasma membrane where it can trigger antiviral immune responses.86 Thus, furin‐mediated cleavage of HBeAg may affect the outcome of infection. Intriguingly, Han Chinese frequently harbour a single nucleotide polymorphism in the P1 promoter of the FUR gene that is associated with increased risk of developing persistent HBV infection with detectable amounts of HBeAg in the serum.87 This polymorphism increases the binding efficiency of the hepatic transcription factor NF‐E2, thereby most likely increasing furin expression.87 Whether the observed increase in HBV persistence is the result of increased HBeAg processing and/or other effects of furin remains to be determined.
Suppression of furin‐mediated processing of viral glycoproteins by par1, gbp2 and gbp5
Viral pathogens and their hosts are in a continuous arms race.88 Although viruses have evolved sophisticated strategies to exploit the metabolism, protein synthesis and trafficking pathways of an infected cell, the host is not defenceless. Besides innate and adaptive immune responses that directly target components of the virus, infected cells may also restrict viral spread by limiting the availability of cellular factors that are critical for viral replication, so‐called ‘virus dependency factors’. For example, the host protein SAMHD1 restricts replication of several viral pathogens by depleting cellular dNTP levels.89 Furthermore, IFI16 targets the cellular transcription factor Sp1 to suppress viral gene expression.90 Intriguingly, accumulating evidence suggests that inhibition of the virus dependency factor furin represents another efficient and broadly active mechanism of antiviral immunity.
In 2013, Aerts and colleagues found that protease‐activated receptor 1 (PAR1), a G‐protein‐coupled receptor, interferes with the expression of furin and furin‐mediated processing of the human metapneumovirus F protein.91 Follow‐up experiments revealed that PAR1 harbours an R41XXXXR46 motif that mediates interaction with several PCSKs.92 In line with this, soluble PC5A/PCSK5 and PCSK6 cleave PAR1 at R46↓ and abrogate its ability to induce calcium signalling upon thrombin‐mediated cleavage at the plasma membrane. Surprisingly, however, membrane‐bound PCSKs such as furin fail to cleave PAR1 at this position. Instead, furin traps PAR1 in the trans‐Golgi network and prevents its anterograde transport to the cell surface (Figure 4a). At the same time, PAR1 also blocks the proteolytic activity of furin, inhibiting for example the maturation of HIV‐1 Env. This inhibitory activity is not shared by its paralogue PAR2, which is efficiently cleaved by furin.93 Notably, expression of PAR1 is induced in proinflammatory environments such as the brain of HIV‐1 infected individuals suffering from HIV‐associated neurocognitive disorders (HAND).92 Thus, PAR1‐mediated furin inhibition may represent a mechanism of innate immunity limiting the spread of HIV‐1 and potentially additional furin‐dependent viral pathogens.
Figure 4.
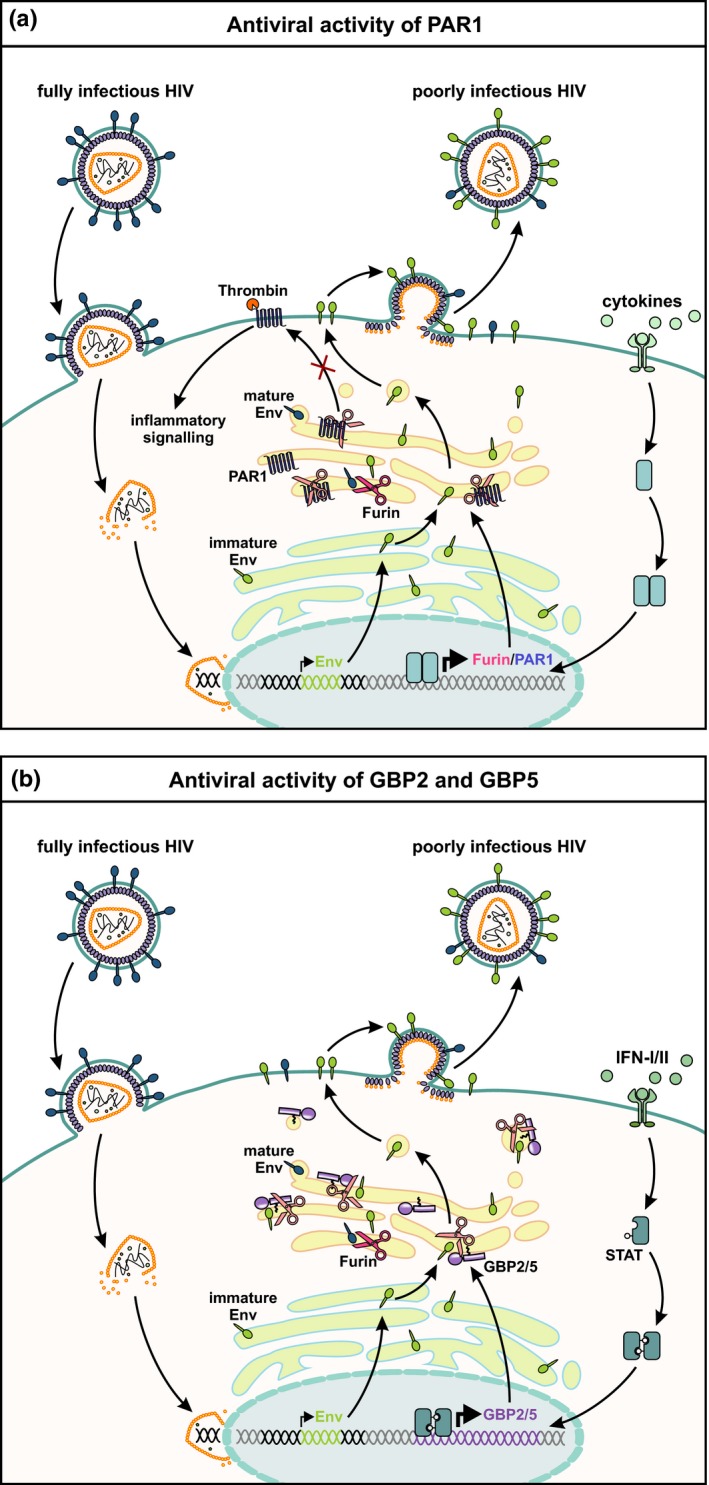
PAR1 as well as GBP2 and GBP5 reduces HIV particle infectivity by inhibiting furin‐mediated Env processing. Human immunodeficiency virus (HIV) particles containing functional mature envelope (Env) glycoproteins fuse with the plasma membrane of the target cell to release the capsid core into the host cell cytoplasm. Upon reverse transcription and integration of the retroviral genome, viral gene expression is initiated. (a) In a cytokine (e.g. IL‐1β)‐induced inflammatory state, furin and protease‐activated receptor 1 (PAR1) expression are induced. Furin and PAR1 interact with each other and are trapped as inactive proteins in the trans‐Golgi network (TGN). As a consequence, PAR1 cannot traffic to the cell surface, where it is usually cleaved by thrombin to induce inflammatory signalling pathways. Moreover, production of infectious HIV‐1 particles is impaired because of reduced furin‐mediated cleavage of HIV Env. (b) At the same time, cells of the infected host may induce the expression of interferon‐stimulated genes such as guanylate‐binding proteins 2 and 5 (GBP2 and GBP5). Both proteins colocalise with furin and inhibit its proteolytic activity. As a result, HIV Env maturation is impaired and newly forming viral particles are poorly infectious since they incorporate immature Env glycoproteins. IFN‐I/II, type I and type II interferons; STAT, signal transducers and activators of transcription.
A similar inhibitory activity was recently described for two IFNγ‐inducible GTPases, termed guanylate‐binding proteins 2 and 5 (GBP2 and GBP5). Initially, GBP5 was described in a screening for novel restriction factors of HIV and shown to interfere with the maturation of the retroviral Env protein.94, 95 Follow‐up experiments revealed that this antiviral activity is shared by its paralogue GBP2 and that both proteins reduce the proteolytically active amount of furin. As a result, cleavage of the Env precursor gp160 into mature gp120 and gp41 is reduced and newly formed virions are only poorly infectious (Figure 4b). Since many viral pathogens rely on furin or related PCSKs for the maturation of their own (glyco)proteins, GBP2 and GBP5 exert broad antiviral activity, inhibiting replication of highly pathogenic avian influenza A, measles and Zika viruses. In contrast, GBP2 and GBP5 do not decrease infectivity of virions carrying the glycoprotein of vesicular stomatitis virus, which does not require a proteolytic activation step. Notably, inhibition of furin in infected cells comes at a cost, since furin‐mediated processing of matrix metalloproteinases and other cellular substrates is also reduced in the presence of increased GBP2 or GBP5 levels.96 Future experiments will reveal whether PAR‐1‐ or GBP‐mediated inhibition of furin activity may also prevent the development or proliferation of certain cancers. Remarkably, increased GBP2/5 expression is associated with favorable outcome in patients suffering from melanoma or breast cancer.97, 98 Thus, a better understanding of cellular mechanisms regulating furin activity will help to understand the pathogenesis of infectious diseases and cancer and may uncover novel targets for therapeutic intervention.
Therapeutic inhibition of furin
Because of the key role of furin in the pathogenesis of cancer and infectious diseases, its suitability as a therapeutic target has raised significant interest for several years. Many laboratories have explored the possibility to limit tumor growth, viral replication or bacterial intoxication by reducing the amount or proteolytic activity of furin.
Initially, most studies focused on peptides or proteins that bind to the active site of furin and inhibit substrate binding in a competitive manner. For example, a variant of the naturally occurring serine protease inhibitor α‐1 antitrypsin was modified to harbour a consensus furin cleavage site. This variant, termed α‐1 antitrypsin Portland (α1‐PDX), inhibits furin and PCSK5 and has been shown to prevent the processing of HIV‐1 Env and measles virus F in vitro.99, 100 Similarly, peptides derived from the cleavage site of influenza A virus hemagglutinin and polyarginines compete with natural furin substrates.101, 102 Even exogenous addition of the autoinhibitory propeptide of furin has been shown to reduce its enzymatic activity, limiting for example the activation of MMP9 in breast cancer cells.103, 104 However, the therapeutic potential of the propeptide has never been evaluated in vivo and the inhibitory effects are most likely limited as it is known to dissociate from furin in the TGN.
Several approaches, including incorporation of D‐ instead of L‐amino acids, have been applied to increase the stability and hence efficacy of furin inhibitors. For example, hexa‐D‐arginine (D6R), one of the first furin inhibitors, exhibits good stability and prevents the cytotoxic effects of Pseudomonas exotoxin A in vitro and in vivo.105 Similarly, topical application of nona‐D‐arginine (D9R) has been shown to reduce corneal damage in mice infected with Pseudomonas aeruginosa.106 Interestingly, D9R also showed direct bactericidal activity, probably because of its polycationic nature.107 Besides D‐amino acids, incorporation of amino acid analogs such as decarboxylated arginine mimetics or 4‐amidinobenzylamide (Amba) has been used to increase the stability of peptide‐derived furin inhibitors.108 Furthermore, the addition of a chloromethyl ketone (CMK) moiety to the C terminus of a polybasic cleavage motif has proven useful as it results in the alkylation of the active site of furin that irreversibly blocks its enzymatic activity.109 Nevertheless, the cytotoxicity of CMK‐based inhibitors and the instability of the CMK moiety may limit their use to topical applications such as the treatment of HPV skin infections.110 Finally, the elucidation of the crystal structure of furin enabled the targeted modelling of nonpeptidic inhibitors such as streptamine‐based compounds. Upon addition of guanidine residues, streptamine derivatives mimic the cationic furin cleavage site and inhibit its enzymatic activity in the nanomolar range in vitro.111 Dahms and colleagues describe an interesting example of a 2,5‐dideoxystreptamine‐derived inhibitor, where two molecules of the inhibitor form a complex with furin.112 While the first inhibitor molecule directly interferes with the conformation of the catalytic triad, the second molecule binds to an adjacent planar peptide stretch.
Besides stability, the subcellular localisation of furin inhibitors is a key determinant of their efficacy in vivo. Notably, optimal localisation of the inhibitor strongly depends on the processing event targeted for therapeutic intervention. To inhibit activation of anthrax toxin, for example, the inhibitor does not need to enter the cells, as furin cleaves the toxin precursor at the cell surface. In contrast, penetration of the inhibitor into the cell is essential to prevent the maturation of HIV‐1 Env and other viral glycoproteins that are cleaved intracellularly. While some inhibitors (e.g. HA‐derived peptides) efficiently enter cells, others were modified to increase their intracellular availability. For example, addition of a decanoyl moiety to CMK inhibitors increases their ability to penetrate cells.113 Streptamine derivatives may be particularly promising for targeted therapy as the positioning of the guanidyl substituents determines the localisation of the inhibitor to distinct subcellular compartments such as endosomes or the Golgi complex.29
While many of the inhibitors described above potently reduce furin activity both in vitro and in vivo, most of them also inhibit other proprotein convertases recognising the same or similar polybasic cleavage sites. This limitation is inherent to competitive inhibitors that aim at mimicking the target sequence of furin and may be overcome by allosteric inhibitors that bind furin‐specific motifs outside the active site. One example is the nanobody Nb14, which binds to the C‐terminal P‐domain of furin, thereby blocking the access of larger substrates to the active site. Notably, Nb14 specifically binds to the P‐domain of furin and does not recognise other PCSKs.114
Instead of targeting furin at the protein level, therapeutic approaches may also aim at targeting its RNA. For example, the endogenous degradation of furin mRNA by Regnase‐1 (ZC3H12A) and/or Roquin (RC3H1)115 could be modulated to interfere with the expression and thus proteolytically active amount of furin. However, modulation of Regnase‐1 and Roquin will most likely have off‐target effects as both endoribonucleases also degrade additional mRNAs. A more selective RNA‐based approach is the silencing of furin via shRNA. In fact, shRNA‐mediated suppression of furin expression is currently the clinically most advanced therapy targeting this protease. In a phase III clinical trial, patients suffering from metastatic Ewing's sarcoma family of tumors (ESFT) are treated with an immunotherapy that involves the silencing of furin and simultaneous overexpression of GM‐CSF.116 More specifically, tumor cells are extracorporeally transfected and reintroduced as so‐called furin knock‐down and GM‐CSF augmented (FANG) cancer vaccine, also known as Vigil. While GM‐CSF boosts the antitumor response by dendritic cells and T cells, knock‐down of furin prevents the proteolytic activation of TGFβ, which may otherwise revert the beneficial effects of GM‐CSF.117, 118, 119 In a phase II clinical trial, the autologous FANG/Vigil vaccine has already proven successful as it increased relapse‐free survival of ovarian cancer patients from 481 to 826 days and showed only limited adverse effects.120
Conclusions and perspectives
The proprotein convertase furin has become an attractive target for the treatment of various infectious and noninfectious diseases as it regulates the activity of numerous mammalian, bacterial and viral proteins. In recent years, several peptidic and nonpeptidic inhibitors have been developed that block the activation of bacterial toxins, prevent the maturation of viral proteins and suppress tumor growth in vitro. Although some of them also yielded promising results in mouse models, there have been only a limited number of clinical trials in humans.
One major challenge is the redundancy of furin with related proprotein convertases that also recognise polybasic cleavage sites. On the one hand, selective inhibition of furin may be beneficial as it limits unwanted side effects due to inhibition of other PCSKs. On the other hand, treatment of some diseases may require the simultaneous inhibition of several PCSKs to efficiently block pathological substrate conversion. To advance current approaches, a better understanding of the relative contribution of individual PCSKs to (nonphysiological) proteolytic protein processing is urgently needed. Therapeutic intervention needs to specifically target the convertase(s) that drive disease progression. Currently, the most promising approaches for selective inhibition are shRNA‐mediated silencing and nanobodies as they show no or only little off‐target effects.
Even if selective inhibition of individual PCSKs can be achieved, systemic long‐term inhibition will most likely have detrimental effects, as PCSKs are required for the activation of hundreds of cellular substrates. Thus, local applications such as targeted treatment of tumors or topical treatment of bacterial and viral infections may be more feasible than systemic therapy. Finally, the ability of tumor cells or pathogens to evolve resistance or evasion mutations remains poorly investigated. For example, several substrates such as dengue virus prM harbour suboptimal furin target sequences and may optimise their cleavage sites upon therapy to enable sufficient cleavage in the presence of inhibitors.
Although the therapeutic application of furin inhibitors may be full of pitfalls, it is certainly a promising approach that should be further pursued. Future studies will elucidate the role of individual PCSKs and their substrates in disease progression and a better understanding of cellular pathways regulating furin activity may uncover additional targets for therapeutic intervention.
Conflict of interest
The authors declare no conflict of interest.
Author contributions
EB and DS performed literature research. EB drafted the figures, and DS wrote the initial version of the manuscript.
Acknowledgments
We thank Frank Kirchhoff and Dominik Hotter for their helpful comments. This work was funded by the DFG priority programme ‘Innate Sensing and Restriction of Retroviruses’ (SPP 1923) to DS; EB was supported by the International Graduate School in Molecular Medicine Ulm (IGradU).
References
- 1. Chakraborti S, Dhalla NS. Pathophysiological Aspects of Proteases. Berlin, Germany: Springer, 2017. [Google Scholar]
- 2. Tian S, Huang Q, Fang Y et al FurinDB: a database of 20‐residue furin cleavage site motifs, substrates and their associated drugs. Int J Mol Sci 2011; 12: 1060–1065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Steiner DF, Cunningham D, Spigelman L et al Insulin biosynthesis: evidence for a precursor. Science 1967; 157: 697–700. [DOI] [PubMed] [Google Scholar]
- 4. Roebroek AJ, Schalken JA, Leunissen JA et al Evolutionary conserved close linkage of the c‐fes/fps proto‐oncogene and genetic sequences encoding a receptor‐like protein. EMBO J 1986; 5: 2197–2202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. van de Ven WJ, Voorberg J, Fontijn R et al Furin is a subtilisin‐like proprotein processing enzyme in higher eukaryotes. Mol Biol Rep 1990; 14: 265–275. [DOI] [PubMed] [Google Scholar]
- 6. Seidah NG, Prat A. The biology and therapeutic targeting of the proprotein convertases. Nat Rev Drug Discov 2012; 11: 367–383. [DOI] [PubMed] [Google Scholar]
- 7. Cheng D, Espenshade PJ, Slaughter CA et al Secreted site‐1 protease cleaves peptides corresponding to luminal loop of sterol regulatory element‐binding proteins. J Biol Chem 1999; 274: 22805–22812. [DOI] [PubMed] [Google Scholar]
- 8. McNutt MC, Lagace TA, Horton JD. Catalytic activity is not required for secreted PCSK9 to reduce low density lipoprotein receptors in HepG2 cells. J Biol Chem 2007; 282: 20799–20803. [DOI] [PubMed] [Google Scholar]
- 9. Uhlén M, Fagerberg L, Hallström BM et al Tissue‐based map of the human proteome. Science 2015; 347: 1260419. [DOI] [PubMed] [Google Scholar]
- 10. Ayoubi TA, Creemers JW, Roebroek AJ et al Expression of the dibasic proprotein processing enzyme furin is directed by multiple promoters. J Biol Chem 1994; 269: 9298–9303. [PubMed] [Google Scholar]
- 11. Pesu M, Muul L, Kanno Y et al Proprotein convertase furin is preferentially expressed in T helper 1 cells and regulates interferon γ. Blood 2006; 108: 983–985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Blanchette F, Day R, Dong W et al TGFβ1 regulates gene expression of its own converting enzyme furin. J Clin Invest 1997; 99: 1974–1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Decroly E, Vandenbranden M, Ruysschaert JM et al The convertases furin and PC1 can both cleave the human immunodeficiency virus (HIV)‐1 envelope glycoprotein gp160 into gp120 (HIV‐1 SU) and gp41 (HIV‐I TM). J Biol Chem 1994; 269: 12240–12247. [PubMed] [Google Scholar]
- 14. Hipp MM, Shepherd D, Gileadi U et al Processing of human toll‐like receptor 7 by furin‐like proprotein convertases is required for its accumulation and activity in endosomes. Immunity 2013; 39: 711–721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Shinde U, Inouye M. Intramolecular chaperones and protein folding. Trends Biochem Sci 1993; 18: 442–446. [DOI] [PubMed] [Google Scholar]
- 16. Anderson ED, VanSlyke JK, Thulin CD et al Activation of the furin endoprotease is a multiple‐step process: requirements for acidification and internal propeptide cleavage. EMBO J 1997; 16: 1508–1518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Thomas G. Furin at the cutting edge: from protein traffic to embryogenesis and disease. Nat Rev Mol Cell Biol 2002; 3: 753–766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Molloy SS, Thomas L, VanSlyke JK et al Intracellular trafficking and activation of the furin proprotein convertase: localization to the TGN and recycling from the cell surface. EMBO J 1994; 13: 18–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Vey M, Schäfer W, Berghöfer S et al Maturation of the trans‐Golgi network protease furin: compartmentalization of propeptide removal, substrate cleavage, and COOH‐terminal truncation. J Cell Biol 1994; 127: 1829–1842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Plaimauer B, Mohr G, Wernhart W et al “Shed” furin: mapping of the cleavage determinants and identification of its C‐terminus. Biochem J 2001; 354: 689–695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Tian S. A 20 Residues motif delineates the furin cleavage site and its physical properties may influence viral fusion. Biochem Insights 2009; 2: BCI.S2049. [Google Scholar]
- 22. Jin W, Fuki IV, Seidah NG et al Proprotein convertases [corrected] are responsible for proteolysis and inactivation of endothelial lipase. J Biol Chem 2005; 280: 36551–36559. [DOI] [PubMed] [Google Scholar]
- 23. Essalmani R, Susan‐Resiga D, Chamberland A et al In vivo evidence that furin from hepatocytes inactivates PCSK9. J Biol Chem 2011; 286: 4257–4263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Roebroek AJ, Umans L, Pauli IG et al Failure of ventral closure and axial rotation in embryos lacking the proprotein convertase Furin. Development 1998; 125: 4863–4876. [DOI] [PubMed] [Google Scholar]
- 25. Kim W, Essalmani R, Szumska D et al Loss of endothelial furin leads to cardiac malformation and early postnatal death. Mol Cell Biol 2012; 32: 3382–3391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Chen Y, Molloy SS, Thomas L et al Mutations within a furin consensus sequence block proteolytic release of ectodysplasin‐A and cause X‐linked hypohidrotic ectodermal dysplasia. Proc Natl Acad Sci USA 2001; 98: 7218–7223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Giannelli F, Green PM, High KA et al Haemophilia B: database of point mutations and short additions and deletions. Nucleic Acids Res 1990; 18: 4053–4059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Bassi DE, Fu J, Lopez de Cicco R et al Proprotein convertases: “master switches” in the regulation of tumor growth and progression. Mol Carcinog 2005; 44: 151–161. [DOI] [PubMed] [Google Scholar]
- 29. Klein‐Szanto AJ, Bassi DE. Proprotein convertase inhibition: paralyzing the cell's master switches. Biochem Pharmacol 2017; 140: 8–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Jaaks P, Bernasconi M. The proprotein convertase furin in tumour progression: the PC furin in tumour progression. Int J Cancer 2017; 141: 654–663. [DOI] [PubMed] [Google Scholar]
- 31. McMahon S, Grondin F, McDonald PP et al Hypoxia‐enhanced expression of the proprotein convertase furin is mediated by hypoxia‐inducible factor‐1: impact on the bioactivation of proproteins. J Biol Chem 2005; 280: 6561–6569. [DOI] [PubMed] [Google Scholar]
- 32. Arsenault D, Lucien F, Dubois CM. Hypoxia enhances cancer cell invasion through relocalization of the proprotein convertase furin from the trans‐Golgi network to the cell surface. J Cell Physiol 2012; 227: 789–800. [DOI] [PubMed] [Google Scholar]
- 33. Stupack DG, Cheresh DA. Get a ligand, get a life: integrins, signaling and cell survival. J Cell Sci 2002; 115: 3729–3738. [DOI] [PubMed] [Google Scholar]
- 34. Dahmani A, Delisle J‐S. TGF‐β in T cell biology: implications for cancer immunotherapy. Cancers 2018; 10: 194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Pesu M, Watford WT, Wei L et al T cell‐expressed proprotein convertase furin is essential for maintenance of peripheral tolerance. Nature 2008; 455: 246–250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Bourne GL, Grainger DJ. Development and characterisation of an assay for furin activity. J Immunol Methods 2011; 364: 101–108. [DOI] [PubMed] [Google Scholar]
- 37. Mojic M, Takeda K, Hayakawa Y. The dark side of IFN‐γ: its role in promoting cancer immunoevasion. Int J Mol Sci 2017; 19: 89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Lee M, Ryu CH, Chang HW et al Radiotherapy‐associated furin expression and tumor invasiveness in recurrent laryngeal cancer. Anticancer Res 2016; 36: 5117–5125. [DOI] [PubMed] [Google Scholar]
- 39. Gordon VM, Leppla SH. Proteolytic activation of bacterial toxins: role of bacterial and host cell proteases. Infect Immun 1994; 62: 333–340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Tsuneoka M, Nakayama K, Hatsuzawa K et al Evidence for involvement of furin in cleavage and activation of diphtheria toxin. J Biol Chem 1993; 268: 26461–26465. [PubMed] [Google Scholar]
- 41. Ogata M, Fryling CM, Pastan I et al Cell‐mediated cleavage of Pseudomonas exotoxin between Arg279 and Gly280 generates the enzymatically active fragment which translocates to the cytosol. J Biol Chem 1992; 267: 25396–25401. [PubMed] [Google Scholar]
- 42. Collier RJ. Effect of diphtheria toxin on protein synthesis: inactivation of one of the transfer factors. J Mol Biol 1967; 25: 83–98. [DOI] [PubMed] [Google Scholar]
- 43. Williams DP, Wen Z, Watson RS et al Cellular processing of the interleukin‐2 fusion toxin DAB486‐IL‐2 and efficient delivery of diphtheria fragment A to the cytosol of target cells requires Arg194. J Biol Chem 1990; 265: 20673–20677. [PubMed] [Google Scholar]
- 44. Molloy SS, Bresnahan PA, Leppla SH et al Human furin is a calcium‐dependent serine endoprotease that recognizes the sequence Arg‐X‐X‐Arg and efficiently cleaves anthrax toxin protective antigen. J Biol Chem 1992; 267: 16396–16402. [PubMed] [Google Scholar]
- 45. Wagner PL, Waldor MK. Bacteriophage control of bacterial virulence. Infect Immun 2002; 70: 3985–3993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Klenk HD, Garten W. Host cell proteases controlling virus pathogenicity. Trends Microbiol 1994; 2: 39–43. [DOI] [PubMed] [Google Scholar]
- 47. Nagai Y, Hamaguchi M, Toyoda T. Molecular biology of Newcastle disease virus. Prog Vet Microbiol Immunol 1989; 5: 16–64. [PubMed] [Google Scholar]
- 48. Klenk HD, Rott R. The molecular biology of influenza virus pathogenicity. Adv Virus Res 1988; 34: 247–281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Moulard M, Decroly E. Maturation of HIV envelope glycoprotein precursors by cellular endoproteases. Biochim Biophys Acta 2000; 1469: 121–132. [DOI] [PubMed] [Google Scholar]
- 50. Fenouillet E, Gluckman JC. Immunological analysis of human immunodeficiency virus type 1 envelope glycoprotein proteolytic cleavage. Virology 1992; 187: 825–828. [DOI] [PubMed] [Google Scholar]
- 51. Kieny MP, Lathe R, Rivière Y et al Improved antigenicity of the HIV env protein by cleavage site removal. Protein Eng 1988; 2: 219–225. [DOI] [PubMed] [Google Scholar]
- 52. Okumura Y, Yano M, Murakami M et al The extracellular processing of HIV‐1 envelope glycoprotein gp160 by human plasmin. FEBS Lett 1999; 442: 39–42. [DOI] [PubMed] [Google Scholar]
- 53. Tashiro M, Ciborowski P, Klenk H‐D et al Role of Staphylococcus protease in the development of influenza pneumonia. Nature 1987; 325: 536. [DOI] [PubMed] [Google Scholar]
- 54. Garten W, Braden C, Arendt A et al Influenza virus activating host proteases: identification, localization and inhibitors as potential therapeutics. Eur J Cell Biol 2015; 94: 375–383. [DOI] [PubMed] [Google Scholar]
- 55. Böttcher E, Matrosovich T, Beyerle M et al Proteolytic activation of influenza viruses by serine proteases TMPRSS2 and HAT from human airway epithelium. J Virol 2006; 80: 9896–9898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Stieneke‐Gröber A, Vey M, Angliker H et al Influenza virus hemagglutinin with multibasic cleavage site is activated by furin, a subtilisin‐like endoprotease. EMBO J 1992; 11: 2407–2414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Horimoto T, Nakayama K, Smeekens SP et al Proprotein‐processing endoproteases PC6 and furin both activate hemagglutinin of virulent avian influenza viruses. J Virol 1994; 68: 6074–6078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Perdue ML, García M, Senne D et al Virulence‐associated sequence duplication at the hemagglutinin cleavage site of avian influenza viruses. Virus Res 1997; 49: 173–186. [DOI] [PubMed] [Google Scholar]
- 59. Röhm C, Süss J, Pohle V et al Different hemagglutinin cleavage site variants of H7N7 in an influenza outbreak in chickens in Leipzig, Germany. Virology 1996; 218: 253–257. [DOI] [PubMed] [Google Scholar]
- 60. Ohuchi M, Orlich M, Ohuchi R et al Mutations at the cleavage site of the hemagglutinin alter the pathogenicity of influenza virus a/chick/penn/83 (H5N2). Virology 1989; 168: 274–280. [DOI] [PubMed] [Google Scholar]
- 61. Baron J, Tarnow C, Mayoli‐Nüssle D et al Matriptase, HAT, and TMPRSS2 activate the hemagglutinin of H9N2 influenza A viruses. J Virol 2013; 87: 1811–1820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Tse LV, Hamilton AM, Friling T et al A novel activation mechanism of avian influenza virus H9N2 by furin. J Virol 2014; 88: 1673–1683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Volchkov VE, Feldmann H, Volchkova VA et al Processing of the Ebola virus glycoprotein by the proprotein convertase furin. Proc Natl Acad Sci USA 1998; 95: 5762–5767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Volchkov VE, Volchkova VA, Ströher U et al Proteolytic processing of Marburg virus glycoprotein. Virology 2000; 268: 1–6. [DOI] [PubMed] [Google Scholar]
- 65. Wool‐Lewis RJ, Bates P. Endoproteolytic processing of the Ebola virus envelope glycoprotein: cleavage is not required for function. J Virol 1999; 73: 1419–1426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Neumann G, Geisbert TW, Ebihara H et al Proteolytic processing of the Ebola virus glycoprotein is not critical for Ebola virus replication in nonhuman primates. J Virol 2007; 81: 2995–2998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Volchkov VE, Becker S, Volchkova VA et al GP mRNA of Ebola virus is edited by the Ebola virus polymerase and by T7 and vaccinia virus polymerases. Virology 1995; 214: 421–430. [DOI] [PubMed] [Google Scholar]
- 68. Volchkova VA, Klenk HD, Volchkov VE. ∆‐peptide is the carboxy‐terminal cleavage fragment of the nonstructural small glycoprotein sGP of Ebola virus. Virology 1999; 265: 164–171. [DOI] [PubMed] [Google Scholar]
- 69. de la Vega M‐A, Wong G, Kobinger GP et al The multiple roles of sGP in Ebola pathogenesis. Viral Immunol 2015; 28: 3–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Pierson TC, Diamond MS. Degrees of maturity: the complex structure and biology of flaviviruses. Curr Opin Virol 2012; 2: 168–175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Guirakhoo F, Heinz FX, Mandl CW et al Fusion activity of flaviviruses: comparison of mature and immature (prM‐containing) tick‐borne encephalitis virions. J Gen Virol 1991; 72: 1323–1329. [DOI] [PubMed] [Google Scholar]
- 72. Yu I‐M, Zhang W, Holdaway HA et al Structure of the immature dengue virus at low pH primes proteolytic maturation. Science 2008; 319: 1834–1837. [DOI] [PubMed] [Google Scholar]
- 73. Yu I‐M, Holdaway HA, Chipman PR et al Association of the pr peptides with dengue virus at acidic pH blocks membrane fusion. J Virol 2009; 83: 12101–12107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Elshuber S, Allison SL, Heinz FX et al Cleavage of protein prM is necessary for infection of BHK‐21 cells by tick‐borne encephalitis virus. J Gen Virol 2003; 84: 183–191. [DOI] [PubMed] [Google Scholar]
- 75. Zybert IA, van der Ende‐Metselaar H, Wilschut J et al Functional importance of dengue virus maturation: infectious properties of immature virions. J Gen Virol 2008; 89: 3047–3051. [DOI] [PubMed] [Google Scholar]
- 76. Davis CW, Nguyen H‐Y, Hanna SL et al West Nile virus discriminates between DC‐SIGN and DC‐SIGNR for cellular attachment and infection. J Virol 2006; 80: 1290–1301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Rodenhuis‐Zybert IA, van der Schaar HM, da Silva Voorham JM et al Immature dengue virus: a veiled pathogen? PLoS Pathog 2010; 6: e1000718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Junjhon J, Lausumpao M, Supasa S et al Differential modulation of prM cleavage, extracellular particle distribution, and virus infectivity by conserved residues at nonfurin consensus positions of the dengue virus pr‐M junction. J Virol 2008; 82: 10776–10791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Richards RM, Lowy DR, Schiller JT et al Cleavage of the papillomavirus minor capsid protein, L2, at a furin consensus site is necessary for infection. Proc Natl Acad Sci USA 2006; 103: 1522–1527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Day PM, Schiller JT. The role of furin in papillomavirus infection. Future Microbiol 2009; 4: 1255–1262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Day PM, Gambhira R, Roden RBS et al Mechanisms of human papillomavirus type 16 neutralization by l2 cross‐neutralizing and l1 type‐specific antibodies. J Virol 2008; 82: 4638–4646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Ito K, Kim K‐H, Lok AS‐F et al Characterization of genotype‐specific carboxyl‐terminal cleavage sites of hepatitis B virus e antigen precursor and identification of furin as the candidate enzyme. J Virol 2009; 83: 3507–3517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Messageot F, Salhi S, Eon P et al Proteolytic processing of the hepatitis B virus e antigen precursor. Cleavage at two furin consensus sequences. J Biol Chem 2003; 278: 891–895. [DOI] [PubMed] [Google Scholar]
- 84. Milich DR, Chen MK, Hughes JL et al The secreted hepatitis B precore antigen can modulate the immune response to the nucleocapsid: a mechanism for persistence. J Immunol 1998; 160: 2013–2021. [PubMed] [Google Scholar]
- 85. Chen MT, Billaud J‐N, Sällberg M et al A function of the hepatitis B virus precore protein is to regulate the immune response to the core antigen. Proc Natl Acad Sci USA 2004; 101: 14913–14918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Schlicht HJ, Schaller H. The secretory core protein of human hepatitis B virus is expressed on the cell surface. J Virol 1989; 63: 5399–5404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Lei RX, Shi H, Peng XM et al Influence of a single nucleotide polymorphism in the P1 promoter of the furin gene on transcription activity and hepatitis B virus infection. Hepatology 2009; 50: 763–771. [DOI] [PubMed] [Google Scholar]
- 88. Daugherty MD, Malik HS. Rules of engagement: molecular insights from host‐virus arms races. Annu Rev Genet 2012; 46: 677–700. [DOI] [PubMed] [Google Scholar]
- 89. Lahouassa H, Daddacha W, Hofmann H et al SAMHD1 restricts HIV‐1 by reducing the intracellular pool of deoxynucleotide triphosphates. Nat Immunol 2012; 13: 223–228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Gariano GR, Dell'Oste V, Bronzini M et al The intracellular DNA sensor IFI16 gene acts as restriction factor for human cytomegalovirus replication. PLoS Pathog 2012; 8: e1002498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Aerts L, Hamelin M‐È, Rhéaume C et al Modulation of protease activated receptor 1 influences human metapneumovirus disease severity in a mouse model. PLoS ONE 2013; 8: e72529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Kim W, Zekas E, Lodge R et al Neuroinflammation‐induced interactions between protease‐activated receptor 1 and proprotein convertases in HIV‐associated neurocognitive disorder. Mol Cell Biol 2015; 35: 3684–3700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Sachan V, Lodge R, Mihara K et al HIV‐induced neuroinflammation: impact of PAR1 and PAR2 processing by Furin. Cell Death Differ 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. McLaren PJ, Gawanbacht A, Pyndiah N et al Identification of potential HIV restriction factors by combining evolutionary genomic signatures with functional analyses. Retrovirology 2015; 12: 41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Krapp C, Hotter D, Gawanbacht A et al Guanylate Binding Protein (GBP) 5 is an interferon‐inducible inhibitor of HIV‐1 infectivity. Cell Host Microbe 2016; 19: 504–514. [DOI] [PubMed] [Google Scholar]
- 96. Braun E, Hotter D, Koepke L et al Guanylate‐Binding Proteins 2 and 5 exert broad antiviral activity by inhibiting furin‐mediated processing of viral envelope proteins. Cell Rep 2019; 27: 2092–2104. e10. [DOI] [PubMed] [Google Scholar]
- 97. Wang Q, Wang X, Liang Q et al Distinct prognostic value of mRNA expression of guanylate‐binding protein genes in skin cutaneous melanoma. Oncol Lett 2018; 15: 7914–7922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Godoy P, Cadenas C, Hellwig B et al Interferon‐inducible guanylate binding protein (GBP2) is associated with better prognosis in breast cancer and indicates an efficient T cell response. Breast Cancer 2014; 21: 491–499. [DOI] [PubMed] [Google Scholar]
- 99. Anderson ED, Thomas L, Hayflick JS et al Inhibition of HIV‐1 gp160‐dependent membrane fusion by a furin‐directed α 1‐antitrypsin variant. J Biol Chem 1993; 268: 24887–24891. [PubMed] [Google Scholar]
- 100. Watanabe M, Hirano A, Stenglein S et al Engineered serine protease inhibitor prevents furin‐catalyzed activation of the fusion glycoprotein and production of infectious measles virus. J Virol 1995; 69: 3206–3210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Strongin A, Lebl M, Day R. Targeting host proteinases as a therapeutic strategy against viral and bacterial pathogens. 2009.
- 102. Cameron A, Appel J, Houghten RA et al Polyarginines are potent furin inhibitors. J Biol Chem 2000; 275: 36741–36749. [DOI] [PubMed] [Google Scholar]
- 103. Zhong M, Munzer JS, Basak A et al The prosegments of furin and PC7 as potent inhibitors of proprotein convertases. In vitro and ex vivo assessment of their efficacy and selectivity. J Biol Chem 1999; 274: 33913–33920. [DOI] [PubMed] [Google Scholar]
- 104. Lapierre M, Siegfried G, Scamuffa N et al Opposing function of the proprotein convertases furin and PACE4 on breast cancer cells’ malignant phenotypes: role of tissue inhibitors of metalloproteinase‐1. Cancer Res 2007; 67: 9030–9034. [DOI] [PubMed] [Google Scholar]
- 105. Sarac MS, Cameron A, Lindberg I. The furin inhibitor hexa‐D‐arginine blocks the activation of Pseudomonas aeruginosa exotoxin A in vivo . Infect Immun 2002; 70: 7136–7139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Karicherla P, Hobden JA. Nona‐D‐arginine therapy for Pseudomonas aeruginosa keratitis. Invest Ophthalmol Vis Sci 2009; 50: 256–262. [DOI] [PubMed] [Google Scholar]
- 107. Karicherla P, Hobden JA. Nona‐D‐arginine amide for prophylaxis and treatment of experimental Pseudomonas aeruginosa keratitis. Curr Eye Res 2010; 35: 220–224. [DOI] [PubMed] [Google Scholar]
- 108. Becker GL, Sielaff F, Than ME et al Potent inhibitors of furin and furin‐like proprotein convertases containing decarboxylated P1 arginine mimetics. J Med Chem 2010; 53: 1067–1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Henrich S, Cameron A, Bourenkov GP et al The crystal structure of the proprotein processing proteinase furin explains its stringent specificity. Nat Struct Biol 2003; 10: 520–526. [DOI] [PubMed] [Google Scholar]
- 110. Couture F, Kwiatkowska A, Dory YL et al Therapeutic uses of furin and its inhibitors: a patent review. Expert Opin Ther Pat 2015; 25: 379–396. [DOI] [PubMed] [Google Scholar]
- 111. Jiao G‐S, Cregar L, Goldman ME et al Guanidinylated 2,5‐dideoxystreptamine derivatives as anthrax lethal factor inhibitors. Bioorg Med Chem Lett 2006; 16: 1527–1531. [DOI] [PubMed] [Google Scholar]
- 112. Dahms SO, Jiao G‐S, Than ME. Structural studies revealed active site distortions of human furin by a small molecule inhibitor. ACS Chem Biol 2017; 12: 1211–1216. [DOI] [PubMed] [Google Scholar]
- 113. Garten W, Stieneke A, Shaw E et al Inhibition of proteolytic activation of influenza virus hemagglutinin by specific peptidyl chloroalkyl ketones. Virology 1989; 172: 25–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Zhu J, Declercq J, Roucourt B et al Generation and characterization of non‐competitive furin‐inhibiting nanobodies. Biochem J 2012; 448: 73–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Cui X, Mino T, Yoshinaga M et al Regnase‐1 and roquin nonredundantly regulate th1 differentiation causing cardiac inflammation and fibrosis. J Immunol 2017; 199: 4066–4077. [DOI] [PubMed] [Google Scholar]
- 116. Vigil + Irinotecan and Temozolomide in Ewing's Sarcoma ‐ Full Text View ‐ ClinicalTrials.gov.
- 117. Inaba K, Inaba M, Romani N et al Generation of large numbers of dendritic cells from mouse bone marrow cultures supplemented with granulocyte/macrophage colony‐stimulating factor. J Exp Med 1992; 176: 1693–1702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Dubois CM, Laprise MH, Blanchette F et al Processing of transforming growth factor β 1 precursor by human furin convertase. J Biol Chem 1995; 270: 10618–10624. [DOI] [PubMed] [Google Scholar]
- 119. Yamaguchi Y, Tsumura H, Miwa M et al Contrasting effects of TGF‐β1 and TNF‐α on the development of dendritic cells from progenitors in mouse bone marrow. Stem Cells 1997; 15: 144–153. [DOI] [PubMed] [Google Scholar]
- 120. Oh J, Barve M, Matthews CM et al Phase II study of Vigil® DNA engineered immunotherapy as maintenance in advanced stage ovarian cancer. Gynecol Oncol 2016; 143: 504–510. [DOI] [PubMed] [Google Scholar]
- 121. Spaete RR, Thayer RM, Probert WS et al Human cytomegalovirus strain Towne glycoprotein B is processed by proteolytic cleavage. Virology 1988; 167: 207–225. [DOI] [PubMed] [Google Scholar]
- 122. Keller PM, Davison AJ, Lowe RS et al Identification and structure of the gene encoding gpII, a major glycoprotein of varicella‐zoster virus. Virology 1986; 152: 181–191. [DOI] [PubMed] [Google Scholar]
- 123. Gong M, Ooka T, Matsuo T et al Epstein‐Barr virus glycoprotein homologous to herpes simplex virus gB. J Virol 1987; 61: 499–508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124. Cavanagh D, Davis PJ, Pappin DJ et al Coronavirus IBV: partial amino terminal sequencing of spike polypeptide S2 identifies the sequence Arg‐Arg‐Phe‐Arg‐Arg at the cleavage site of the spike precursor propolypeptide of IBV strains Beaudette and M41. Virus Res 1986; 4: 133–143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125. de Haan CAM, Stadler K, Godeke G‐J et al Cleavage inhibition of the murine coronavirus spike protein by a furin‐like enzyme affects cell‐cell but not virus‐cell fusion. J Virol 2004; 78: 6048–6054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126. Rice CM, Lenches EM, Eddy SR et al Nucleotide sequence of yellow fever virus: implications for flavivirus gene expression and evolution. Science 1985; 229: 726–733. [DOI] [PubMed] [Google Scholar]
- 127. Stadler K, Allison SL, Schalich J et al Proteolytic activation of tick‐borne encephalitis virus by furin. J Virol 1997; 71: 8475–8481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128. Rice CM, Strauss JH. Nucleotide sequence of the 26S mRNA of Sindbis virus and deduced sequence of the encoded virus structural proteins. Proc Natl Acad Sci USA 1981; 78: 2062–2066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129. Zhang X, Fugère M, Day R et al Furin processing and proteolytic activation of Semliki Forest virus. J Virol 2003; 77: 2981–2989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130. Gonzalez‐Dunia D, Cubitt B, de la Torre JC. Mechanism of Borna disease virus entry into cells. J Virol 1998; 72: 783–788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131. Richt JA, Fürbringer T, Koch A et al Processing of the Borna disease virus glycoprotein gp94 by the subtilisin‐like endoprotease furin. J Virol 1998; 72: 4528–4533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132. Sanchez AJ, Vincent MJ, Erickson BR et al Crimean‐Congo hemorrhagic fever virus glycoprotein precursor is cleaved by Furin‐like and SKI‐1 proteases to generate a novel 38‐kilodalton glycoprotein. J Virol 2006; 80: 514–525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133. Kawaoka Y, Nestorowicz A, Alexander DJ et al Molecular analyses of the hemagglutinin genes of H5 influenza viruses: origin of a virulent turkey strain. Virology 1987; 158: 218–227. [DOI] [PubMed] [Google Scholar]
- 134. Porter AG, Barber C, Carey NH et al Complete nucleotide sequence of an influenza virus haemagglutinin gene from cloned DNA. Nature 1979; 282: 471–477. [DOI] [PubMed] [Google Scholar]
- 135. Toyoda T, Sakaguchi T, Imai K et al Structural comparison of the cleavage‐activation site of the fusion glycoprotein between virulent and avirulent strains of Newcastle disease virus. Virology 1987; 158: 242–247. [DOI] [PubMed] [Google Scholar]
- 136. Spriggs MK, Olmsted RA, Venkatesan S et al Fusion glycoprotein of human parainfluenza virus type 3: nucleotide sequence of the gene, direct identification of the cleavage‐activation site, and comparison with other paramyxoviruses. Virology 1986; 152: 241–251. [DOI] [PubMed] [Google Scholar]
- 137. Waxham MN, Server AC, Goodman HM et al Cloning and sequencing of the mumps virus fusion protein gene. Virology 1987; 159: 381–388. [DOI] [PubMed] [Google Scholar]
- 138. Richardson C, Hull D, Greer P et al The nucleotide sequence of the mRNA encoding the fusion protein of measles virus (Edmonston strain): a comparison of fusion proteins from several different paramyxoviruses. Virology 1986; 155: 508–523. [DOI] [PubMed] [Google Scholar]
- 139. Paterson RG, Harris TJ, Lamb RA. Fusion protein of the paramyxovirus simian virus 5: nucleotide sequence of mRNA predicts a highly hydrophobic glycoprotein. Proc Natl Acad Sci USA 1984; 81: 6706–6710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140. Collins PL, Huang YT, Wertz GW. Nucleotide sequence of the gene encoding the fusion (F) glycoprotein of human respiratory syncytial virus. Proc Natl Acad Sci USA 1984; 81: 7683–7687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141. McCune JM, Rabin LB, Feinberg MB et al Endoproteolytic cleavage of gp160 is required for the activation of human immunodeficiency virus. Cell 1988; 53: 55–67. [DOI] [PubMed] [Google Scholar]
- 142. Grandgenett D, Quinn T, Hippenmeyer PJ et al Structural characterization of the avian retrovirus reverse transcriptase and endonuclease domains. J Biol Chem 1985; 260: 8243–8249. [PubMed] [Google Scholar]
- 143. Freed EO, Risser R. The role of envelope glycoprotein processing in murine leukemia virus infection. J Virol 1987; 61: 2852–2856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144. Geiselhart V, Bastone P, Kempf T et al Furin‐mediated cleavage of the feline foamy virus Env leader protein. J Virol 2004; 78: 13573–13581. [DOI] [PMC free article] [PubMed] [Google Scholar]