

Abstract
Speckle-type POZ protein (SPOP) plays an important role in maintaining genome stability. Disability or mutation of the SPOP gene has been reported to contribute to prostate cancer incidence and prognosis. However, the functions of SPOP in lung cancer remain poorly understood, especially in lung adenocarcinoma (LUAD). Here, we found that SPOP affects the LUAD cell response to radiation by regulating the DNA damage response (DDR) pathway. SPOP is widely expressed in lung cancer cell lines, and SPOP protein levels are upregulated when cells experience DNA damage. SPOP knockdown affects DDR repair kinetics, apoptosis and cell cycle checkpoints that are induced by IR (ionizing radiation). Furthermore, we found that SPOP positively regulates the expression of DDR factors Rad51 and Ku80. Taken together, these data indicate the essential roles of SPOP in the DDR signaling pathways and LUAD cell response to radiation.
Keywords: SPOP, lung adenocarcinoma, DNA damage response, radiosensitivity, radiotherapy
Introduction
Lung cancer is one of the most common causes of cancer-related mortality. Non-small cell lung carcinoma (NSCLC) constitutes the main type of lung cancer, accounting for 85% of all cases [1]. There are several subtypes of NSCLC, among which lung adenocarcinoma (LUAD), lung squamous cell carcinoma (LUSC) and large cell lung carcinoma (LCLC) are the most common. LUSC exhibits a faster progression rate than LUAD, but it is sensitive to radiotherapy/chemotherapy and has a good response to surgical treatment. Although LCLC accounts for 10-15% of lung cancer and lacks specific differentiation [1,2], current treatments have greatly improved these patients’ survival. LUAD, however, comprises up to 40% of lung cancer cases and has a relatively poor treatment efficacy and prognosis for radiotherapy/chemotherapy. In clinical practice, patients with inoperable stage I or II lung cancer or who have postoperative residual tumor are recommended to undergo radical radiotherapy or postoperative radiotherapy. Meanwhile, chemoradiotherapy is commonly used to treat stage III and IV lung cancer patients. These situations emphasize that radiation therapy is an important regimen for various stages of lung cancer [3]. However, because of the intrinsic radiotherapy resistance of LUAD, conventional radiotherapy has a relatively poor therapeutic efficacy in LUAD patients [4].
Currently, many studies have shown that the DNA damage response (DDR) signaling pathway is involved in the resistance of tumors to radiotherapy. The DDR comprises four sub pathways-DNA repair, DNA damage checkpoints, transcriptional response and apoptosis-and is a genome surveillance system that repairs DNA lesions caused by cellular metabolites or exogenous DNA-damaging agents (such as IR and chemotherapeutics) [5,6]. Among the different types of DNA lesions, DNA double-strand breaks (DSB) are the most lethal forms and they cause the principal cytotoxic impact of ionizing radiation/radiotherapy [7]. In mammalian cells, DSBs are primarily repaired by nonhomologous end joining (NHEJ) and homologous recombination (HR), which are mainly regulated by the DNA-PK complex and Rad51-family [8]. Delaying or arresting DNA damage checkpoints can provide time and material conditions for the DNA repair process. If fatal DNA lesions cannot be repaired, the cell will initiate apoptosis programs and eliminate itself. This is how radiotherapy works. Defects in any part of these pathways may cause genomic instability and lead to carcinogenesis of normal cells, while abnormal activation of the DDR in tumor cells will weaken the treatment effect of IR, which is how the resistance of radiotherapy works [9,10]. Due to the key significance of DDR system components, they have been widely studied and used as therapeutic targets in cancer radiotherapy [10-12].
Speckle-type poxvirus and zinc finger protein (SPOP) was first reported in 1997, contains 374 amino acids and is distributed as scattered points within the nucleus under normal conditions [13]. SPOP serves as an adaptor of Cullin 3-based ubiquitin ligase and is responsible for the degradation of many nuclear proteins. The substrates of SPOP include the apoptosis factor DAXX [14], breast cancer metastasis suppressor BRMS1 [15], Hedgehog signaling transcription factors Gli2 and Gli3 [16], steroid receptor coactivator protein SRC-3 [17], and so on. Moussay E et al. found that SPOP is involved in the resistance of chronic lymphocytic leukemia to fludarabine treatment [18]. Kim MS et al. found that loss of SPOP expression was common in prostate, gastric and colorectal cancers [19]. A recent study revealed that SPOP acts as a novel participant in the DDR in cervical cancer and prostate cancer cells [20,21]. These works together highlighted a critical role of SPOP in maintaining genome stability and DNA damage response (DDR) integrity, further indicating its potential in increasing the treatment effect of DNA-damage-based therapeutics.
However, the above conjecture needs further elucidation. Therefore, in this study, we analyzed the expression level of SPOP in different lung cancer cell lines and uncovered the hidden mechanism by which SPOP increases the radiosensitivity of LUAD cells. Finally, our results indicate that SPOP is a potential therapeutic target for radiotherapy among LUAD patients.
Materials and methods
Cell cultures, antibodies and shRNA oligos
The NSCLC cell lines H226, H1703, H838, H1299, H1437, H1975, H1563, H460, H661 and H446 were cultured in RPMI-1640 Medium. A549 was cultured in F-12K Medium. These mediums were supplemented with 10% fetal bovine serum (FBS, HyClone), 100 U/ml penicillin and 100 μg/ml streptomycin. Cultures were grown in a 5% CO2 incubator at 37°C. In the case of camptothecin treatments, cells were incubated with CPT (500 nM) for 24 h. Drugs were purchased from Sigma. Two types of SPOP antibodies were used in this study. The SPOP antibody purchased from Santa Cruz Biotechnology (Dallas, TX) was used for immunofluorescence and western blotting at a dilution of 1:1000, whereas the SPOP antibody purchased from Proteintech (Wuhan, China) was used for immunohistochemistry at a dilution of 1:100. The γ-H2AX antibody, Ku80 antibody and Rad51 antibody were purchased from Cell Signaling Technology (Danvers, MA) and used at a dilution of 1:1000. The corresponding secondary antibody and fluorescent-conjugated secondary antibody were purchased from Cell Signaling Technology (Danvers, MA). The shSPOP plasmids were purchased from Genechem Technology (Shanghai, China), and the sequences of the SPOP shRNAs were 5’-CAAACGCCTGAAGCAATCCTA-3’, 5’-GAGGTGAGTGTTGTGCAAGAT-3’, and 5’-CACAGATCAAGGTAGTGAAAT-3’. Lentiviral particles containing shNC (negative control shRNA) or shSPOP were prepared according to the standard viral packaging protocol.
Bioinformatics analysis
CCLE (Cancer Cell Line Encyclopedia, Available online: https://portals.broadinstitute.org/ccle) is a large genetic and pharmacologic database of human cancer models. It integrates computational analyses by linking distinct pharmacologic vulnerabilities to genomic patterns for over 1100 cell lines [22]. The mRNA expression levels of SPOP in different pathological types of lung cancer were visualized using CCLE. GEPIA (Gene Expression Profiling Interactive Analysis, Available online: http://gepia.cancer-pku.cn/index.html) is an online tool that provides customizable functions such as tumor/normal differential expression analysis, profiling according to cancer types or pathological stages, and other important information [23]. We analyzed the expression of SPOP in LUAD and adjacent tissues with this website. Oncomine (Available online: https://www.oncomine.org) is a cancer microarray database and integrated data-mining platform by which we can interrogate genome-wide gene expression profiles and compare the profiles of major types of cancer to respective normal tissues [24]. We selected differentially expressed genes (DEGs) from lung adenocarcinomas and normal cervical mucosa tissues using this tool. The STRING database (Available online: http://string-db.org) collects the information of known and predicted protein interactions as well as physical and indirect functional associations [25]. The protein-protein interaction (PPI) network of how SPOP affects significantly enriched DEGs in LUAD and with which genes in the DNA double-strand break response pathway were constructed by STRING.
Immunohistochemistry and immunofluorescence
The tissue chip product of LUAD was purchased from Outdo Biotech (Shanghai, China) and included 75 pairs of lung adenocarcinoma tissues and matched paraneoplastic tissues (37 cases in stage I, 18 cases in stage II, 16 cases in stage III and 4 cases in stage IV). Immunohistochemistry and staining were performed according to the standard protocol. The tissue chip was incubated with primary antibody against SPOP overnight at 4°C followed by secondary antibody for 1 h at room temperature. Then, images were captured using microscopy and analyzed. The cells were plated on glass slides and divided into four groups: shNC, shSPOP, IR, and shSPOP combined with IR. The processing steps for the shSPOP combined with IR group were as follows: cells were incubated with shSPOP lentiviral particles for 12 h and then grown in exchanged fresh medium for 48 h. We cultured the cells for an additional 48 h in the presence of puromycin, eliminating the noninfected cells. After exposure to 6 Gy IR, the medium containing puromycin was removed, and the cells were maintained in normal medium. Twenty-four hours later, the cells were fixed with 4% PFA at 37°C for 20 min, treated with 0.02% Triton X-100 for 10 min, and then blocked at room temperature for 1 h. Cells were incubated with primary antibody overnight at 4°C followed by fluorescent-conjugated secondary antibody for 2 h at room temperature, and then the nuclei were stained with DAPI for 15 min. The images of the cells were obtained by fluorescence microscopy (Olympus Corporation, Japan).
Western blotting and real-time PCR
The cells were lysed in RIPA lysis buffer (Pioneer Biotechnology, China) containing a protease inhibitor and phosphatase inhibitor cocktail. Equal amounts of protein were separated by SDS-PAGE and transferred to polyvinylidene difluoride membranes. The membranes were blocked with 5% nonfat milk diluted with TBST (Tris-buffered saline with 0.05% Tween 20) for 2 h and then incubated with primary antibodies at 4°C overnight and secondary antibodies at 37°C for 2 h. The membranes were washed with TBST 3 times and Tris-buffered saline once. An enhanced chemiluminescence detection kit (Millipore, MA) was used to develop the signals of immunoblotted proteins, which were detected on a JS-380A automatic gel imaging system (Quantity One Quantitation software, Bio-Rad Laboratories, CA). RNA was isolated from cells using RNAfast 200 (Pioneer Biotechnology, China). The SPOP primer/probe sets (F: 5’-ATGCTTCGGATGTCTTGGAG-3’, R: 5’-GATTGCTTCAGGCGTTTGC-3’), Ku80 primer/probe sets (F: 5’-ATTTGCTGGAGGACATTGAAAG-3’, R: 5’-CTGAATCGGCTGCTGAGG-3’), Rad51 primer/probe sets (F: 5’-CAACACAGACCACCAGACC-3’, R: 5’-AGAAGCATCCGCAGAAACC-3’), GAPDH primer/probe sets (F: 5’-AAGGCTGTGGGCAAGGTCATC-3’, R: 5’-GCGTCAAAGGTGGAGGAGTGG-3’), PrimeScript™ RT Master Mix and SYBR® Premix Ex Taq™ were purchased from Takara Biotechnology (Dalian, China). All real-time PCR experiments were performed in triplicate.
Comet assay, cell cycle and apoptosis analysis
The cells were divided into four treatment groups: shNC, shSPOP, IR, and shSPOP combined with IR. Comet Assay kits (Trevigen, MD) were used in this study. Twenty-four hours after IR, the cells were harvested and mixed with agarose for electrophoresis. The nuclear DNA was stained with SYBR green dye for 10 min, and the images were then obtained by fluorescence microscopy. The olive tail moment was measured using CASP software. The cells were seeded in 6 well plates at a density of 2*105 cells/well and divided into four groups as before. We collected cells for subsequent cell cycle and apoptosis analyses 24 h after IR. Cells were fixed with 70% ethanol (2 h, 4°C) and treated with propidium iodide and RNase A (30 min, 37°C) for cell cycle analysis. Apoptosis was quantified with a KGI biotechnology apoptosis kit (Nanjing, China). Both cell cycle and apoptosis analyses were performed using flow cytometry (BD Biosciences, CA).
Methylthiazoltetrazolium (MTT) and colony forming assay
Cells were seeded in 96-well plates at a density of 4000 cells/well in 200 µL of culture medium and divided into four groups: shNC, shSPOP, IR, and shSPOP combined with IR. Cells were incubated for 24 h, 48 h and 72 h after IR, then 20 µL of 3-(4, 5-dimethylthiazol-2-y1)-2, 5-diphenytetrazolium bromide was added to each well and incubated for 4 h. The colored solution was quantified by a spectrophotometer at an absorbance of 490 nm. The relative cell proliferation of each group was calculated. The cells were incubated in 10 cm2 flasks, infected with shNC or shSPOP lentiviral particles and then exposed to IR at the indicated doses. The cell density of each group was 2000 cells/well. After 10-14 days, the cells were fixed and stained with crystal violet. Colonies containing >50 cells were counted. The cell survival curve was calculated.
Statistics
Data from immunofluorescence, real-time PCR, comet Assay, and cell cycle et al., are presented as the mean ± SD. One-way ANOVA with LSD’s post test was used to calculate the significance difference for each group. Analyses were performed with IBM SPSS Statistics, version 22 and GraphPad Prism, version 6 (GraphPad Software Inc.). P<0.05 was considered statistically significant.
Results
SPOP is widely expressed in different lung cancer cell lines
According to pathological classification, lung cancer can be divided mainly into 4 categories: LUSC, LUAD, LCLC and SCLC. To address the role of SPOP in LUAD, we started with profiling the mRNA expression of SPOP in different histopathological lung cancer cell lines with the CCLE online database. The results showed that SPOP was expressed in a broad range of tissue types and cell lines (Figure 1A). Western blotting results also show the protein level among these cell lines (Figure 1B). We further inquired the mRNA expression of SPOP in LUAD tissues (T) and adjacent normal tissues (N) by GEPIA and found no significant difference between these two types of tissues (Figure 1C). The LUAD tissue chip, which includes 75 pairs of cancer tissues (CT) and matched adjacent tissues (AT), was investigated for SPOP expression by immunohistochemistry, and similar results were obtained as GEPIA. Figure 1D shows representative images of one pair of cancer and adjacent tissues from the tissue chip. Next, we selected differentially expressed genes (DEGs) in lung adenocarcinomas and normal lung mucosa tissues from two GEO datasets, GSE19188 [26] and GSE31210 [27] (GSE19188 contains 45 LUAD and 65 adjacent normal lung samples; GSE31210 contains 226 LUAD and 20 normal lung tissues). The DEGs ranked by p-values from low to high are listed in Table 1. Then, by using another online database, STRING, we found that some of these DEGs were associated with SPOP to some extent (Figure 1E). Although the SPOP expression levels between LUAD and normal tissues had little difference, these findings still emphasize an important role of SPOP in LUAD.
Figure 1.
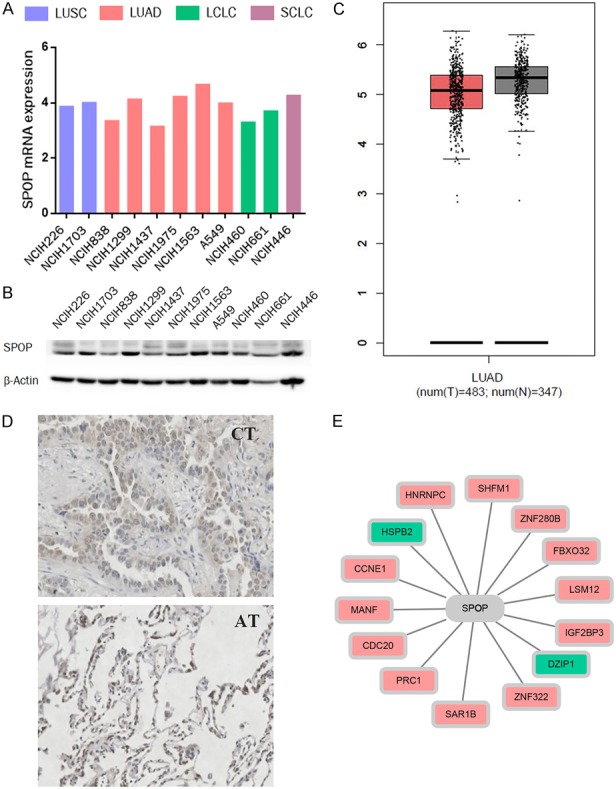
SPOP was widely expressed in different lung cancer cell lines. A. The mRNA expression of SPOP in different histopathological lung cancer cell lines from CCLE. B. Western blotting result showing the expression of SPOP across different lung cancer cell lines. β-Actin was probed as a loading control. C. The expression of SPOP in lung tumor tissues (T) and adjacent normal tissues (N) from GEPIA. D. SPOP immunohistochemical staining for a cancer tissue (CT) and matched adjacent tissue (AT), pair in the LUAD tissue chip. E. The relationship between SPOP and the 14 DEGs of LUAD (green represents the downregulated genes, and red represents upregulated genes).
Table 1.
| DEGs | Gene Name |
|---|---|
| Upregulated | TMEM106B, CCNB1, HIST1H2BD, HN1, EPRS, SRD5A1, GPR89B, GOLM1, GINS1, MDK, GPR89A, DNMT3A, HMGB3, MELK, SGPL1, COL11A1, PDIA4, KDM5B, SRPK1, ERO1L, SLC2A1, RALGPS2, UBE2C, DNAH14, TXNDC17, MARCH6, PDZD11, KIF2C, GFPT1, CDCA7, GJB2, BUB1B, SFXN1, PGM2L1, MTA3, MCTS1, TNPO1, CNPY2, MLF1IP, ZWINT, H2AFV, EIF2AK1, HIST2H2AA3, GORASP2, SLC35B2, MRPS24, FAM83A, FAM199X, TOP2A, GPT2 |
| Downregulated | ADAMTS8, AGER, GPM6A, ADH1B, IGSF10, GDF10, TNXB, ADAMTSL3, KCNT2, FHL5, ASPA, SPG20, CLIC5, FAM107A, TNNC1, CDH19, TGFBR3, PKNOX2, KCNK3, SYNE1, SLIT3, FXYD1, SPTBN1, FGF2, ITIH5, SHROOM4, NPR1, ROBO4, TAL1, FAM189A2, CBFA2T3, CELF2, MAMDC2, C19orf59, EMCN, HIGD1B, TMOD1, MYCT1, FHL1, CFD, SCN4B, TCF21, SFTPC, PELO, PEAR1, QKI, GKN2, GPM6B, RYR2, FABP4 |
SPOP knockdown leads to DNA DSB repair defects and DSB accumulation in H1299 cells
To characterize SPOP’s functions in the DDR signaling pathways, we started by examining whether the expression level of SPOP was changed after DNA damage. In unperturbed H1299 cells, SPOP expression was relatively weak. However, 24 h after the cells treated with 6 Gy IR, we observed increasing expression of SPOP protein and mRNA (Figure 2A, 2D and 2E) (P<0.05). These observations strongly indicated that SPOP was upregulated in response to DNA damage. To confirm this, we constructed a SPOP knockdown system using shRNA plasmids. All three independent shSPOP plasmids suppressed SPOP expression at the transcriptional level, but plasmid-2 and plasmid-3 were more effective (Figure 2F). Because lentiviral particles can be more efficient in infecting cells, we further constructed shRNA lentiviral particles corresponding to the sequences in plasmid-2 and plasmid-3. Lentivirus-1 (packed with the plasmid-2 sequence) and lentivirus-2 (packed with the plasmid-3 sequence) were both worked. Figure 2B, 2C and 2G, 2H show the mRNA and protein levels of SPOP that were knocked down by shSPOP lentivirus-1 (shSPOP-v1) and lentivirus-2 (shSPOP-v2) in H1299 and A549 cell lines. We chose these vectors to perform the following tests.
Figure 2.
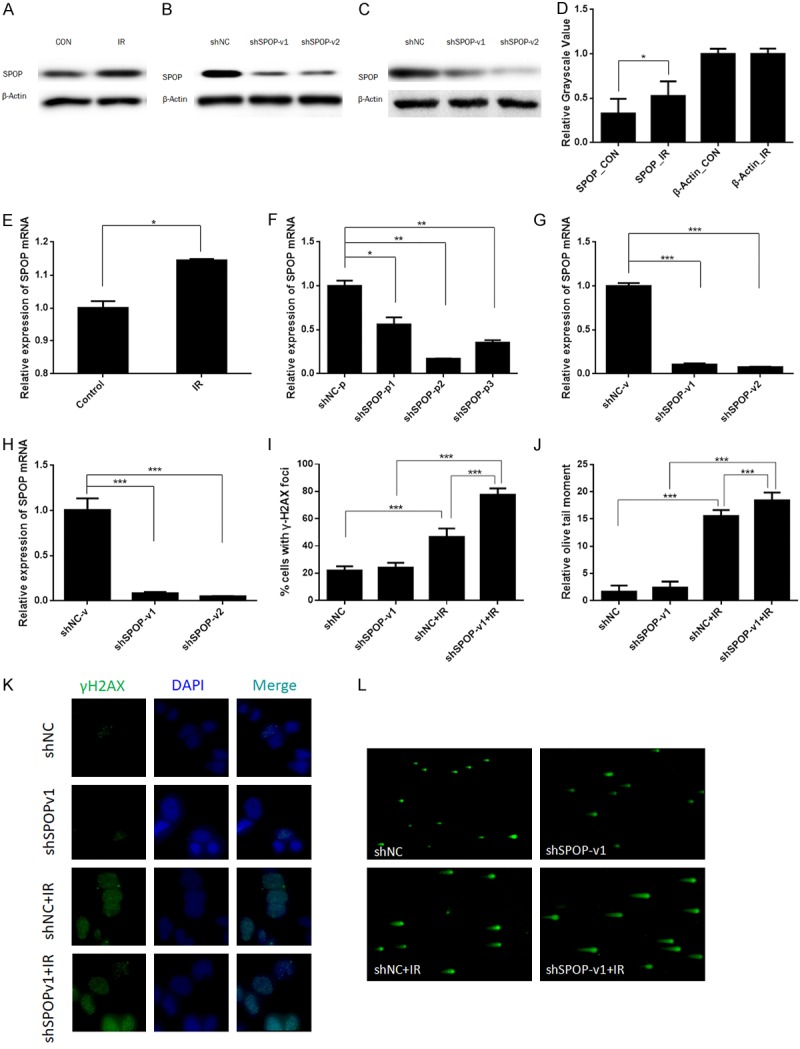
SPOP knockdown led to DNA DSB repair defects and DSB accumulation in H1299 cells. (A) Expression level of SPOP in H1299 control cells and cells treated by IR. β-Actin was probed as a loading control. (B) Expression level of SPOP in H1299 cells infected with shNC, lentivirus-1 or lentivirus-2. β-Actin was probed as a loading control. (C) Expression level of SPOP in A549 cells infected with shNC, lentivirus-1 or lentivirus-2. β-Actin was probed as a loading control. (D) Relative grayscale value of (A) (*P<0.05). (E) Relative mRNA levels of SPOP in control cells and cells treated with IR. Total RNA was extracted from H1299 cells, and mRNA expression was examined by RT-PCR. Relative levels of SPOP mRNA were normalized to those of GAPDH (internal control) (*P<0.05). (F) Three independent shSPOP plasmids suppressed SPOP expression at the transcriptional level (*P<0.05, **P<0.01, ***P<0.001). (G) Two independent shSPOP lentiviral particles suppressed SPOP expression in the H1299 cell line at the transcriptional level (***P<0.001). (H) Two independent shSPOP lentiviral particles suppressed SPOP expression in the A549 cell line at the transcriptional level (***P<0.001). (I) Quantitation of γ-H2AX foci in (K) (***P<0.001). (J) Quantitation of the olive tail moment in (L) (***P<0.001). (K) Immunofluorescence of the γ-H2AX foci. (L) The neutral single-cell gel electrophoresis assay (comet assay) for the olive tail moment.
It is widely accepted that γ-H2AX foci, a phosphorylated form of histone variant H2AX, are a marker of DNA DSBs [28]. Consequently, we monitored the DNA DSB repair capacity of H1299 cells by measuring γ-H2AX foci immunofluorescence. Four groups of H1299 cells, shNC, shSPOP-v1, IR, and shSPOP-v1 combined with IR, were fixed for immunofluorescence staining, following their respective treatments. The number of positive γ-H2AX foci in each group was counted by microscopy. Statistical comparison between shNC and IR told that the IR could induce gamma-H2AX foci formation (P<0.001). The degree of foci changes between non-IR stimulated and IR stimulated in SPOP knockdown cell lines showed a significant enhancement in the number of foci (P<0.001) (Figure 2I and 2K) Comparing these two increased foci levels, we could see the DNA DSB repair defects in SPOP knockdown cell line. We further performed a neutral single cell gel electrophoresis assay (comet assay), a method that exclusively visualizes cellular DSB, and confirmed the SPOP knockdown-mediated IR-induced accumulation of DSB. The higher mean comet tail moment in the shSPOP combined with IR group indicates that SPOP inhibition caused more DSB accumulation in H1299 cells after IR (P<0.001) (Figure 2J and 2L).
SPOP knockdown affects cell cycle checkpoints and apoptosis in LUAD
Because of the synergistic effect of the DDR system (including DNA repair, DNA damage checkpoints and apoptosis), we further investigated whether SPOP knockdown would affect these biological processes. H1299 cells were divided into four treatment groups: shNC, shSPOP-v1, IR, and shSPOP-v1 combined with IR. Twenty-four hours after IR treatment, we collected each group of cells for cell cycle and apoptosis analyses. As shown in Figure 3A and 3G, the number of cells in G2/M phase was significantly higher in the IR group than in the shNC group, indicating that activation of the G2/M checkpoint resulted from IR (P<0.001). Likewise, the shSPOP-v1 combined with IR group also showed increased activation of the G2/M checkpoint when compared with the shSPOP group (P<0.001). Figure 3B and 3H show similar trends in the A549 cell line. These findings reveal the effects of SPOP knockdown on cell cycle checkpoints. Next, we performed flow cytometry to measure the effect of the treatments on apoptosis. Comparison between shNC and IR groups showed that IR treatment increased cell apoptosis (P<0.001). The apoptosis level between non-IR stimulated and IR stimulated in SPOP knockdown cell lines also showed a significant enhancement (P<0.001). Comparing these two increased levels, the results indicated that IR-induced apoptosis was enhanced by SPOP knockdown in H1299 cells (Figure 3C and 3F). Camptothecin (CPT) is a classic DNA damage agent and is most commonly used to study DNA damage mechanisms. Here, we also used CPT to assess the sensitization effect of SPOP on lung cancer cell lines. Consistently, SPOP knockdown was found to increase the apoptosis ratio in both H1299 (Figure 3D and 3I) and A549 (Figure 3E and 3J) cell lines under DNA damage conditions.
Figure 3.

SPOP knockdown affected cell cycle checkpoints and apoptosis in H1299 cells. A and G. Cell cycle analysis of the H1299 cell line from four treatment groups (shNC, shSPOP, IR, shSPOP combined with IR) by flow cytometry (***P<0.001). B and H. Cell cycle analysis of the A549 cell line from four treatment groups (shNC, shSPOP, IR, shSPOP combined with IR) by flow cytometry (***P<0.001). C and F. Apoptosis analysis of H1299 cells from four treatment groups (shNC, shSPOP, IR, shSPOP combined with IR) by flow cytometry (**P<0.01, ***P<0.001). D and I. Apoptosis analysis of H1299 cells from six treatment groups (shNC, shSPOP-v1/2, shNC+CPT, shSPOP-v1/2+IR) by flow cytometry (*P<0.05, **P<0.01, ***P<0.001). E and J. Apoptosis analysis of A549 cells from six treatment groups (shNC, shSPOP-v1/2, shNC+CPT, shSPOP-v1/2+IR) by flow cytometry (*P<0.05, **P<0.01, ***P<0.001).
SPOP knockdown modulates the expression of the DSB repair protein Rad51
DNA DSBs are primarily repaired by nonhomologous end joining (NHEJ) and homologous recombination (HR). As SPOP knockdown led to DNA DSB repair defects and DSB accumulation in H1299 cells, we further tested whether SPOP affected the NHEJ and HR pathways. We downloaded the genes that participate in DNA double-strand break response pathways from the PathCards database (Available online: http://pathcards.genecards.org/). Then, a PPI network of SPOP with these genes was constructed by STRING. We found that SPOP was potentially connected to 9 related genes: MAPK8, TP53, UBA52, UBC, LIG4, RPS27A, UBB, RAD51 and XRCC5 (Ku80) (Figure 4A). These findings suggest that SPOP might participate in the DNA double-strand break response. Because of the key functions of Rad51 and Ku80 in HR and NHEJ, respectively, we examined their expression levels by western blotting and further found that SPOP knockdown affected the expression of the DSB repair proteins Rad51 and Ku80 in the H1299 cell line (Figure 4B and 4C). Next, we examined their mRNA levels by q-PCR and found that the mRNA level of Rad51 was upregulated under SPOP knockdown conditions (P<0.05), while there was no significant change in Ku80 levels (Figure 4D-G).
Figure 4.

SPOP knockdown mediated the expression of the DSB repair protein Rad51. A. The relationship between SPOP and 9 genes in the DNA double-strand break response pathways. B and C. The effect of knocking down SPOP (by shSPOP-v1 or shSPOP-v2) on Rad51 and Ku80 expression in H1299 cells. β-Actin was probed as a loading control. D-G. Relative mRNA levels of Rad51 and Ku80 in four groups (shNC, shSPOP, shNC+IR, shSPOP combined with IR) of H1299 cells. Relative levels of Rad51 and Ku80 mRNA were normalized to those of GAPDH (internal control) (*P<0.05).
SPOP knockdown sensitizes LUAD cells to irradiation
It has been reported that knockdown or mutation of SPOP may increase the response to DNA-damaging therapeutics in cervical cancer and prostate cancer cells, but there is no relative research reporting the radiosensitive effect of SPOP on LUAD cells. We divided the cancer cells into four groups as described above and incubated them 24 h, 48 h and 72 h after 6 Gy IR for MTT-based cell viability analysis or 10-14 days after 0 Gy to 6 Gy IR for colony forming assay. The MTT results showed that shSPOP combined with IR significantly inhibited cell proliferation compared to that of the IR group (Figure 5A and 5B). The clonogenic assay results (Figure 5D) and their quantification (Figure 5C) are also showed that SPOP knockdown significantly improved the radiosensitivity of H1299 and A549 cells. Thus, these results indicated not only that SPOP knockdown sensitized LUAD cells to irradiation but also that SPOP could be a potential therapeutic target in radiotherapy.
Figure 5.
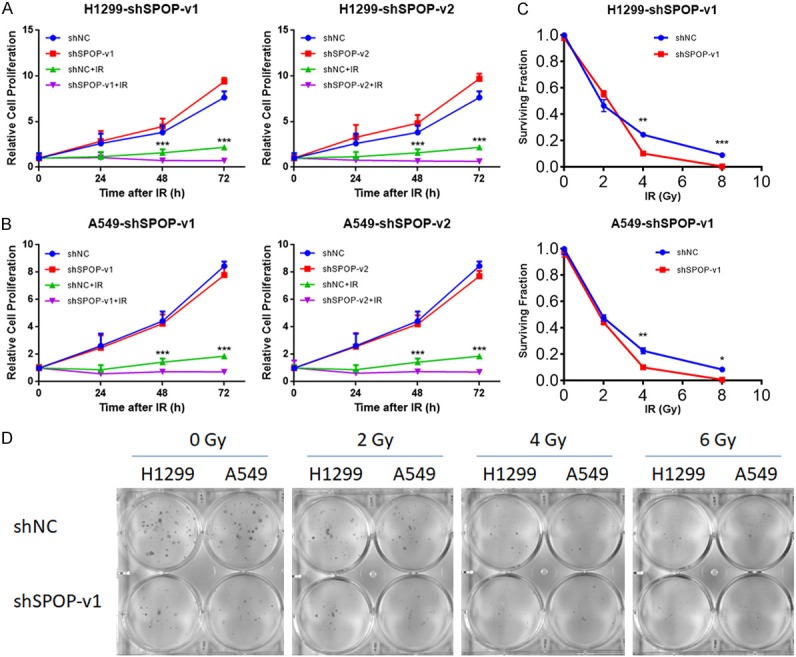
SPOP knockdown sensitized LUAD cells to irradiation. (A) MTT-based cell viability analysis of H1299 cells from four groups (shNC, shSPOP, shNC+IR, shSPOP+IR) (*P<0.05). (B) MTT-based cell viability analysis of A549 cells from four groups (shNC, shSPOP, shNC+IR, shSPOP+IR) (*P<0.05). (C) Clonogenic assay of H1299 and A549 cells under the indicated treatments (*P<0.05). (D) Photographs of the petridishes in the clonogenic assay corresponding to (C).
Discussion
LUAD is the most common type of lung cancer and radiotherapy is an important therapeutic strategy for inoperable and locally advanced LUAD [29,30]; however, patients’ treatment response to radiotherapy is largely limited by the cancer’s intrinsic radioresistance. The major mechanisms underlying radiotherapy resistance are increased DDR activity and DNA DSB repair efficiency in tumor cells. DNA carrying the basic information of the human genome is constantly subject to multiple endogenous or exogenous insults and manifests different patterns of damage (e.g., deamination, pyrimidine dimer, mismatches, interstrand crosslinking, DNA SSBs and DSBs) [31,32]. On the one hand, the DDR signaling system that evolved in eukaryotic organisms can repair these DNA damages and keep cells from undergoing mutation, carcinogenesis and even death. On the other hand, the unusual activation of DDR is also an important contributor to radioresistance in tumor cells because it tends to ignore or over repair the DNA DSBs, which is the principal cytotoxic effect of radiotherapy. Currently, DDR has been associated with radioresistance in many kinds of tumors, including LUAD [33-35]. Multiple works focusing on the inhibition of proteins involved in DDR signaling, such as ATM, ATR, Cyclin B, CHK1/2 and so on, have touted this approach as a viable strategy to increase the effectiveness of tumor radiotherapy [12,36-41].
Speckle-type POZ protein (SPOP), which has been reported in many tumor cells, plays an important role in maintaining genome stability and DDR activity [42-47]. However, the functions of SPOP and the possibilities for targeting this protein to increase the sensitivity of LUAD radiotherapy have not yet been reported. In this study, we found that SPOP was not only evenly and widely expressed in lung cancer cell lines but also related to some important DEGs in LUAD. Furthermore, we observed that SPOP protein was upregulated after DNA damage in H1299 cells, and SPOP knockdown led to DSB repair defects as well as DSB accumulation. Such intervention also affected the cell cycle checkpoints and apoptosis pathway after IR. Then, we investigated the underlying molecular mechanisms of DDR defects caused by SPOP knockdown and found that inhibiting the expression of SPOP could upregulate the DDR factor Rad51. Finally, we demonstrated that SPOP knockdown increased the radiation sensitivity of LUAD cell lines and identified SPOP as a potential therapeutic target for radiotherapy among LUAD patients.
The DDR signaling system mainly contains four subpathways as described above: DNA repair, DNA damage checkpoints, transcriptional response and apoptosis. Our research found that IR-induced DNA damage was sustained and accumulated under SPOP-inhibited conditions. These results indicated that conventional DNA repair function was disturbed by SPOP knockdown. Since IR-induced DSBs have been reported to be primarily repaired by nonhomologous end joining (NHEJ) and homologous recombination (HR), we further investigated the function of SPOP in these two repair modes. By using the PathCards database, we observed that multiple key proteins (MAPK8, TP53, UBC, UBA52, LIG4, RPS27A, UBB, RAD51 and XRCC5 (Ku80)) participating in DSB repair pathways showed a direct or indirect connection with SPOP. Among these genes, Rad51 is a central factor involved in HR-mediated DSB repair. It has been established as a radiosensitization target in multiple cancers [48,49]. King HO et al. found that Rad51 causes significant radiosensitization in glioblastoma stem cells [50], and Balbous A et al. showed that Rad51 inhibition can be a therapeutic strategy to help increase the efficacy of glioblastoma radiotherapy [51]. Similar conclusions were also observed in esophageal cancer and breast cancer [52,53]. In our study, Rad51 was markedly increased by shSPOP intervention. This result not only corresponds to previous conclusions and publications [21] that SPOP has a specific role in maintaining DNA damage repair but also details that SPOP may participate in the HR process by working with Rad51. This research illustrates the close relationship between SPOP and Rad51 and emphasizes the importance of SPOP in improving the radiotherapy efficacy in lung cancer patients. However, it seems confusing that Rad51 inhibition has been shown to increase the radiosensitization in previous reports, while Rad51 upregulation mediated by SPOP increases cell radiosensitization according to our research. To explain this, we tend to attribute this discrepancy to the E3 ubiquitin enzyme function of SPOP, whose inhibition upregulates Rad51 expression and possibly affects other HR-related proteins, even the whole HR signal pathway. We also believe this is related with the SPOP-related DNA repair impairment and DSB accumulation. Further experimental argumentation is required to prove this interference.
Taken together, the data from our study reveal that SPOP influences the DNA damage response by affecting cell cycle checkpoints, apoptosis progress and the HR repair pathway. Meanwhile, we identified SPOP as a potential target to increase the radiosensitivity of LUAD. In the future, in order to improve the prognosis of LAUD patients and the feasibility of clinical application of SPOP, it is necessary to further study the specific mechanisms behind these phenomena.
Acknowledgements
I would like to express my gratitude to all those who helped me during writing this thesis. Thank you to ChengYL for the encouragement. Thank you to Prof. Qing Zhu for the generous guidance on this research. Thank you to Joey Yung and Shanshan Deng for giving me strong spiritual support. This work was supported by the [National Natural Science Foundation of China] under Grant [81272488, 81472795, 81602802] to S.H., [81874223] to Q.Z.; [81602680] to D.Z.; [Innovation Capacity Support Plan of Shaanxi Province]under Grant [2018TD-002] to S.H.; and [China Postdoctoral Science Foundation]under Grant [2015M582677] to D.Z.
Disclosure of conflict of interest
None.
References
- 1.Denisenko TV, Budkevich IN, Zhivotovsky B. Cell death-based treatment of lung adenocarcinoma. Cell Death Dis. 2018;9:117. doi: 10.1038/s41419-017-0063-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Chen M, Liu X, Du J, Wang XJ, Xia L. Differentiated regulation of immune-response related genes between LUAD and LUSC subtypes of lung cancers. Oncotarget. 2017;8:133–144. doi: 10.18632/oncotarget.13346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Tyldesley S, Boyd C, Schulze K, Walker H, Mackillop WJ. Estimating the need for radiotherapy for lung cancer: an evidence-based, epidemiologic approach. Int J Radiat Oncol Biol Phys. 2001;49:973–985. doi: 10.1016/s0360-3016(00)01401-2. [DOI] [PubMed] [Google Scholar]
- 4.Willers H, Azzoli CG, Santivasi WL, Xia F. Basic mechanisms of therapeutic resistance to radiation and chemotherapy in lung cancer. Cancer J. 2013;19:200–207. doi: 10.1097/PPO.0b013e318292e4e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sancar A, Lindsey-Boltz LA, Unsal-Kacmaz K, Linn S. Molecular mechanisms of mammalian DNA repair and the DNA damage checkpoints. Annu Rev Biochem. 2004;73:39–85. doi: 10.1146/annurev.biochem.73.011303.073723. [DOI] [PubMed] [Google Scholar]
- 6.Helleday T, Eshtad S, Nik-Zainal S. Mechanisms underlying mutational signatures in human cancers. Nat Rev Genet. 2014;15:585–598. doi: 10.1038/nrg3729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Vignard J, Mirey G, Salles B. Ionizing-radiation induced DNA double-strand breaks: a direct and indirect lighting up. Radiother Oncol. 2013;108:362–369. doi: 10.1016/j.radonc.2013.06.013. [DOI] [PubMed] [Google Scholar]
- 8.Seluanov A, Mao Z, Gorbunova V. Analysis of DNA double-strand break (DSB) repair in mammalian cells. J Vis Exp. 2010 doi: 10.3791/2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Davies H, Glodzik D, Morganella S, Yates LR, Staaf J, Zou X, Ramakrishna M, Martin S, Boyault S, Sieuwerts AM, Simpson PT, King TA, Raine K, Eyfjord JE, Kong G, Borg A, Birney E, Stunnenberg HG, van de Vijver MJ, Borresen-Dale AL, Martens JW, Span PN, Lakhani SR, Vincent-Salomon A, Sotiriou C, Tutt A, Thompson AM, Van Laere S, Richardson AL, Viari A, Campbell PJ, Stratton MR, Nik-Zainal S. HRDetect is a predictor of BRCA1 and BRCA2 deficiency based on mutational signatures. Nat Med. 2017;23:517–525. doi: 10.1038/nm.4292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Srivastava M, Raghavan SC. DNA double-strand break repair inhibitors as cancer therapeutics. Chem Biol. 2015;22:17–29. doi: 10.1016/j.chembiol.2014.11.013. [DOI] [PubMed] [Google Scholar]
- 11.Sears CR, Cooney SA, Chin-Sinex H, Mendonca MS, Turchi JJ. DNA damage response (DDR) pathway engagement in cisplatin radiosensitization of non-small cell lung cancer. DNA Repair (Amst) 2016;40:35–46. doi: 10.1016/j.dnarep.2016.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Oike T, Ogiwara H, Torikai K, Nakano T, Yokota J, Kohno T. Garcinol, a histone acetyltransferase inhibitor, radiosensitizes cancer cells by inhibiting non-homologous end joining. Int J Radiat Oncol Biol Phys. 2012;84:815–821. doi: 10.1016/j.ijrobp.2012.01.017. [DOI] [PubMed] [Google Scholar]
- 13.Nagai Y, Kojima T, Muro Y, Hachiya T, Nishizawa Y, Wakabayashi T, Hagiwara M. Identification of a novel nuclear speckle-type protein, SPOP. FEBS Lett. 1997;418:23–26. doi: 10.1016/s0014-5793(97)01340-9. [DOI] [PubMed] [Google Scholar]
- 14.Kwon JE, La M, Oh KH, Oh YM, Kim GR, Seol JH, Baek SH, Chiba T, Tanaka K, Bang OS, Joe CO, Chung CH. BTB domain-containing speckle-type POZ protein (SPOP) serves as an adaptor of Daxx for ubiquitination by Cul3-based ubiquitin ligase. J Biol Chem. 2006;281:12664–12672. doi: 10.1074/jbc.M600204200. [DOI] [PubMed] [Google Scholar]
- 15.Kim B, Nam HJ, Pyo KE, Jang MJ, Kim IS, Kim D, Boo K, Lee SH, Yoon JB, Baek SH, Kim JH. Breast cancer metastasis suppressor 1 (BRMS1) is destabilized by the Cul3-SPOP E3 ubiquitin ligase complex. Biochem Biophys Res Commun. 2011;415:720–726. doi: 10.1016/j.bbrc.2011.10.154. [DOI] [PubMed] [Google Scholar]
- 16.Wang C, Pan Y, Wang B. Suppressor of fused and Spop regulate the stability, processing and function of Gli2 and Gli3 full-length activators but not their repressors. Development. 2010;137:2001–2009. doi: 10.1242/dev.052126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Geng C, He B, Xu L, Barbieri CE, Eedunuri VK, Chew SA, Zimmermann M, Bond R, Shou J, Li C, Blattner M, Lonard DM, Demichelis F, Coarfa C, Rubin MA, Zhou P, O’Malley BW, Mitsiades N. Prostate cancer-associated mutations in speckle-type POZ protein (SPOP) regulate steroid receptor coactivator 3 protein turnover. Proc Natl Acad Sci U S A. 2013;110:6997–7002. doi: 10.1073/pnas.1304502110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Moussay E, Palissot V, Vallar L, Poirel HA, Wenner T, El Khoury V, Aouali N, Van Moer K, Leners B, Bernardin F, Muller A, Cornillet-Lefebvre P, Delmer A, Duhem C, Ries F, van Dyck E, Berchem G. Determination of genes and microRNAs involved in the resistance to fludarabine in vivo in chronic lymphocytic leukemia. Mol Cancer. 2010;9:115. doi: 10.1186/1476-4598-9-115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kim MS, Je EM, Oh JE, Yoo NJ, Lee SH. Mutational and expressional analyses of SPOP, a candidate tumor suppressor gene, in prostate, gastric and colorectal cancers. Apmis. 2013;121:626–633. doi: 10.1111/apm.12030. [DOI] [PubMed] [Google Scholar]
- 20.Zhang D, Wang H, Sun M, Yang J, Zhang W, Han S, Xu B. Speckle-type POZ protein, SPOP, is involved in the DNA damage response. Carcinogenesis. 2014;35:1691–1697. doi: 10.1093/carcin/bgu022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Boysen G, Barbieri CE, Prandi D, Blattner M, Chae SS, Dahija A, Nataraj S, Huang D, Marotz C, Xu L, Huang J, Lecca P, Chhangawala S, Liu D, Zhou P, Sboner A, de Bono JS, Demichelis F, Houvras Y, Rubin MA. SPOP mutation leads to genomic instability in prostate cancer. Elife. 2015:4. doi: 10.7554/eLife.09207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Barretina J, Caponigro G, Stransky N, Venkatesan K, Margolin AA, Kim S, Wilson CJ, Lehar J, Kryukov GV, Sonkin D, Reddy A, Liu M, Murray L, Berger MF, Monahan JE, Morais P, Meltzer J, Korejwa A, Jane-Valbuena J, Mapa FA, Thibault J, Bric-Furlong E, Raman P, Shipway A, Engels IH, Cheng J, Yu GK, Yu J, Aspesi P Jr, de Silva M, Jagtap K, Jones MD, Wang L, Hatton C, Palescandolo E, Gupta S, Mahan S, Sougnez C, Onofrio RC, Liefeld T, MacConaill L, Winckler W, Reich M, Li N, Mesirov JP, Gabriel SB, Getz G, Ardlie K, Chan V, Myer VE, Weber BL, Porter J, Warmuth M, Finan P, Harris JL, Meyerson M, Golub TR, Morrissey MP, Sellers WR, Schlegel R, Garraway LA. The cancer cell line encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature. 2012;483:603–607. doi: 10.1038/nature11003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tang Z, Li C, Kang B, Gao G, Li C, Zhang Z. GEPIA: a web server for cancer and normal gene expression profiling and interactive analyses. Nucleic Acids Res. 2017;45:W98–W102. doi: 10.1093/nar/gkx247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rhodes DR, Yu J, Shanker K, Deshpande N, Varambally R, Ghosh D, Barrette T, Pandey A, Chinnaiyan AM. ONCOMINE: a cancer microarray database and integrated data-mining platform. Neoplasia. 2004;6:1–6. doi: 10.1016/s1476-5586(04)80047-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Szklarczyk D, Franceschini A, Wyder S, Forslund K, Heller D, Huerta-Cepas J, Simonovic M, Roth A, Santos A, Tsafou KP, Kuhn M, Bork P, Jensen LJ, von Mering C. STRING v10: protein-protein interaction networks, integrated over the tree of life. Nucleic Acids Res. 2015;43:D447–452. doi: 10.1093/nar/gku1003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hou J, Aerts J, den Hamer B, van Ijcken W, den Bakker M, Riegman P, van der Leest C, van der Spek P, Foekens JA, Hoogsteden HC, Grosveld F, Philipsen S. Gene expression-based classification of non-small cell lung carcinomas and survival prediction. PLoS One. 2010;5:e10312. doi: 10.1371/journal.pone.0010312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Okayama H, Kohno T, Ishii Y, Shimada Y, Shiraishi K, Iwakawa R, Furuta K, Tsuta K, Shibata T, Yamamoto S, Watanabe S, Sakamoto H, Kumamoto K, Takenoshita S, Gotoh N, Mizuno H, Sarai A, Kawano S, Yamaguchi R, Miyano S, Yokota J. Identification of genes upregulated in ALK-positive and EGFR/KRAS/ALK-negative lung adenocarcinomas. Cancer Res. 2012;72:100–111. doi: 10.1158/0008-5472.CAN-11-1403. [DOI] [PubMed] [Google Scholar]
- 28.Wang H, Adhikari S, Butler BE, Pandita TK, Mitra S, Hegde ML. A perspective on chromosomal double strand break markers in mammalian cells. Jacobs J Radiat Oncol. 2014;1 [PMC free article] [PubMed] [Google Scholar]
- 29.Wang C, Su H, Yang L, Huang K. Integrative analysis for lung adenocarcinoma predicts morphological features associated with genetic variations. Pac Symp Biocomput. 2017;22:82–93. doi: 10.1142/9789813207813_0009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Revannasiddaiah S, Thakur P, Bhardwaj B, Susheela SP, Madabhavi I. Pulmonary adenocarcinoma: implications of the recent advances in molecular biology, treatment and the IASLC/ATS/ERS classification. J Thorac Dis. 2014;6:S502–525. doi: 10.3978/j.issn.2072-1439.2014.05.19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Travers A, Muskhelishvili G. DNA structure and function. FEBS J. 2015;282:2279–2295. doi: 10.1111/febs.13307. [DOI] [PubMed] [Google Scholar]
- 32.Lakin ND, Jackson SP. Regulation of p53 in response to DNA damage. Oncogene. 1999;18:7644–7655. doi: 10.1038/sj.onc.1203015. [DOI] [PubMed] [Google Scholar]
- 33.Ali R, Rakha EA, Madhusudan S, Bryant HE. DNA damage repair in breast cancer and its therapeutic implications. Pathology. 2017;49:156–165. doi: 10.1016/j.pathol.2016.11.002. [DOI] [PubMed] [Google Scholar]
- 34.Tian H, Gao Z, Li H, Zhang B, Wang G, Zhang Q, Pei D, Zheng J. DNA damage response--a double-edged sword in cancer prevention and cancer therapy. Cancer Lett. 2015;358:8–16. doi: 10.1016/j.canlet.2014.12.038. [DOI] [PubMed] [Google Scholar]
- 35.Khanna A. DNA damage in cancer therapeutics: a boon or a curse? Cancer Res. 2015;75:2133–2138. doi: 10.1158/0008-5472.CAN-14-3247. [DOI] [PubMed] [Google Scholar]
- 36.Wang FZ, Fei HR, Cui YJ, Sun YK, Li ZM, Wang XY, Yang XY, Zhang JG, Sun BL. The checkpoint 1 kinase inhibitor LY2603618 induces cell cycle arrest, DNA damage response and autophagy in cancer cells. Apoptosis. 2014;19:1389–1398. doi: 10.1007/s10495-014-1010-3. [DOI] [PubMed] [Google Scholar]
- 37.Kim KS, Heo JI, Choi KJ, Bae S. Enhancement of cellular radiation sensitivity through degradation of Chk1 by the XIAP-XAF1 complex. Cancer Biol Ther. 2014;15:1622–1634. doi: 10.4161/15384047.2014.962305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Cui Y, Palii SS, Innes CL, Paules RS. Depletion of ATR selectively sensitizes ATM-deficient human mammary epithelial cells to ionizing radiation and DNA-damaging agents. Cell Cycle. 2014;13:3541–3550. doi: 10.4161/15384101.2014.960729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ayars M, Eshleman J, Goggins M. Susceptibility of ATM-deficient pancreatic cancer cells to radiation. Cell Cycle. 2017;16:991–998. doi: 10.1080/15384101.2017.1312236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zheng R, Liu Y, Zhang X, Zhao P, Deng Q. miRNA-200c enhances radiosensitivity of esophageal cancer by cell cycle arrest and targeting P21. Biomed Pharmacother. 2017;90:517–523. doi: 10.1016/j.biopha.2017.04.006. [DOI] [PubMed] [Google Scholar]
- 41.Wang W, Guo M, Xia X, Zhang C, Zeng Y, Wu S. XRRA1 targets ATM/CHK1/2-mediated DNA repair in colorectal cancer. Biomed Res Int. 2017;2017:5718968. doi: 10.1155/2017/5718968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Janouskova H, El Tekle G, Bellini E, Udeshi ND, Rinaldi A, Ulbricht A, Bernasocchi T, Civenni G, Losa M, Svinkina T, Bielski CM, Kryukov GV, Cascione L, Napoli S, Enchev RI, Mutch DG, Carney ME, Berchuck A, Winterhoff BJN, Broaddus RR, Schraml P, Moch H, Bertoni F, Catapano CV, Peter M, Carr SA, Garraway LA, Wild PJ, Theurillat JP. Opposing effects of cancer-type-specific SPOP mutants on BET protein degradation and sensitivity to BET inhibitors. Nat Med. 2017;23:1046–1054. doi: 10.1038/nm.4372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Dai X, Gan W, Li X, Wang S, Zhang W, Huang L, Liu S, Zhong Q, Guo J, Zhang J, Chen T, Shimizu K, Beca F, Blattner M, Vasudevan D, Buckley DL, Qi J, Buser L, Liu P, Inuzuka H, Beck AH, Wang L, Wild PJ, Garraway LA, Rubin MA, Barbieri CE, Wong KK, Muthuswamy SK, Huang J, Chen Y, Bradner JE, Wei W. Prostate cancer-associated SPOP mutations confer resistance to BET inhibitors through stabilization of BRD4. Nat Med. 2017;23:1063–1071. doi: 10.1038/nm.4378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wu F, Dai X, Gan W, Wan L, Li M, Mitsiades N, Wei W, Ding Q, Zhang J. Prostate cancer-associated mutation in SPOP impairs its ability to target Cdc20 for poly-ubiquitination and degradation. Cancer Lett. 2017;385:207–214. doi: 10.1016/j.canlet.2016.10.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Luo J, Bao YC, Ji XX, Chen B, Deng QF, Zhou SW. SPOP promotes SIRT2 degradation and suppresses non-small cell lung cancer cell growth. Biochem Biophys Res Commun. 2017;483:880–884. doi: 10.1016/j.bbrc.2017.01.027. [DOI] [PubMed] [Google Scholar]
- 46.Gao K, Jin X, Tang Y, Ma J, Peng J, Yu L, Zhang P, Wang C. Tumor suppressor SPOP mediates the proteasomal degradation of progesterone receptors (PRs) in breast cancer cells. Am J Cancer Res. 2015;5:3210–3220. [PMC free article] [PubMed] [Google Scholar]
- 47.Guo ZQ, Zheng T, Chen B, Luo C, Ouyang S, Gong S, Li J, Mao LL, Lian F, Yang Y, Huang Y, Li L, Lu J, Zhang B, Zhou L, Ding H, Gao Z, Zhou L, Li G, Zhou R, Chen K, Liu J, Wen Y, Gong L, Ke Y, Yang SD, Qiu XB, Zhang N, Ren J, Zhong D, Yang CG, Liu J, Jiang H. Small-molecule targeting of E3 ligase adaptor SPOP in kidney cancer. Cancer Cell. 2016;30:474–484. doi: 10.1016/j.ccell.2016.08.003. [DOI] [PubMed] [Google Scholar]
- 48.Chen X, Wong P, Radany EH, Stark JM, Laulier C, Wong JY. Suberoylanilide hydroxamic acid as a radiosensitizer through modulation of RAD51 protein and inhibition of homology-directed repair in multiple myeloma. Mol Cancer Res. 2012;10:1052–1064. doi: 10.1158/1541-7786.MCR-11-0587. [DOI] [PubMed] [Google Scholar]
- 49.Kobashigawa S, Morikawa K, Mori H, Kashino G. Gemcitabine induces radiosensitization through inhibition of RAD51-dependent repair for DNA double-strand breaks. Anticancer Res. 2015;35:2731–2737. [PubMed] [Google Scholar]
- 50.King HO, Brend T, Payne HL, Wright A, Ward TA, Patel K, Egnuni T, Stead LF, Patel A, Wurdak H, Short SC. RAD51 is a selective DNA repair target to radiosensitize glioma stem cells. Stem Cell Reports. 2017;8:125–139. doi: 10.1016/j.stemcr.2016.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Balbous A, Cortes U, Guilloteau K, Rivet P, Pinel B, Duchesne M, Godet J, Boissonnade O, Wager M, Bensadoun RJ, Chomel JC, Karayan-Tapon L. A radiosensitizing effect of RAD51 inhibition in glioblastoma stem-like cells. BMC Cancer. 2016;16:604. doi: 10.1186/s12885-016-2647-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Liu Q, Jiang H, Liu Z, Wang Y, Zhao M, Hao C, Feng S, Guo H, Xu B, Yang Q, Gong Y, Shao C. Berberine radiosensitizes human esophageal cancer cells by downregulating homologous recombination repair protein RAD51. PLoS One. 2011;6:e23427. doi: 10.1371/journal.pone.0023427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Gasparini P, Lovat F, Fassan M, Casadei L, Cascione L, Jacob NK, Carasi S, Palmieri D, Costinean S, Shapiro CL, Huebner K, Croce CM. Protective role of miR-155 in breast cancer through RAD51 targeting impairs homologous recombination after irradiation. Proc Natl Acad Sci U S A. 2014;111:4536–4541. doi: 10.1073/pnas.1402604111. [DOI] [PMC free article] [PubMed] [Google Scholar]