

Abstract
Gap junctions contain intercellular channels that enable intercellular communication of small molecules while also serving as a signaling scaffold. Connexins, the proteins that form gap junctions in vertebrates, are highly regulated and typically have short (<2 h) half-lives. Connexin43 (Cx43), the predominate connexin in the myocardium and epithelial tissues, is phosphorylated on more than a dozen serine residues and interacts with a variety of protein kinases. These interactions regulate Cx43 and gap junction formation and stability. Casein kinase 1 (CK1)–mediated phosphorylation of Cx43 promotes gap junction assembly. Using murine knock-in technology and quantitative PCR, immunoblotting, and immunoprecipitation assays, we show here that mutation of the CK1 phosphorylation sites in Cx43 reduces the levels of total Cx43 in the myocardium and increases Cx43 phosphorylation on sites phosphorylated by extracellular signal–regulated kinase (ERK). In aged myocardium, we found that, compared with WT Cx43, mutant Cx43 expression increases ERK activation, phosphorylation of Akt substrates, and protection from ischemia-induced injury. Our findings also uncovered that Cx43 interacts with the hypoxia-inducible protein N-Myc downstream-regulated gene 1 protein (NDRG1) and that Cx43 phosphorylation status controls this interaction and dramatically affects NDRG1 stability. We propose that, in addition to altering gap junction stability, Cx43 phosphorylation directly and dynamically regulates cellular signaling through ERK and Akt in response to ischemic injury. We conclude that gap junction–dependent NDRG1 regulation might explain some cellular responses to hypoxia.
Keywords: gap junction, connexin, cell signaling, phosphorylation, cell junction, hypoxia, casein kinase 1, myocardium, N-Myc downstream-regulated gene 1 (NDRG1)
Introduction
Gap junctions are specialized membrane domains that contain channels that allow exchange of small molecules (<1000 Da), including ions, metabolites, and second messengers (e.g. Ca2+ and inositol 1,4,5-trisphosphate) between neighboring cells (1–3). Connexins, like other junctional proteins, also play critical signaling and scaffolding roles that can be fully or partially independent of channel function. Cx43, expressed in over 34 tissues and 46 cell types (4), is phosphorylated at multiple (>12) serine residues found in the cytoplasmic C-terminal region (5–10). Cx43 phosphorylation regulates Cx43 trafficking, localization, and gap junction function (11, 12). Cx43 phosphorylation by casein kinase 1 (CK1) at serines 325, 328, and/or 330 increases the efficiency of gap junction assembly, whereas mutation at these residues causes cell–cell coupling to develop more slowly and with a severely decreased frequency of fully opened channels (13, 14). In contrast, Cx43 phosphorylation by extracellular signal–regulated kinase (ERK),3 protein kinase C (PKC) is typically increased during gap junction remodeling and disassembly (15–18). Phosphorylation by Akt is dynamically regulated and can increase gap junction size (17). ERK, Akt, and PKC are well known to modulate cell survival, particularly in the myocardium, where they can regulate propagation and protect from ischemia reperfusion injury (IRI) (19–21). Gap junction remodeling and phosphorylation of Cx43 by PKC and Akt occur in response to short-term ischemia in the myocardium (17, 22), and phosphorylation on Ser-262, a potential ERK site (12), has been suggested to promote cell survival and protection of the myocardium from IRI (23, 24). Myocardial preconditioning, where short bouts of ischemia result in protection from IRI, also results in altered gap junction remodeling and phosphorylation of Cx43 by these very same kinases (17, 24, 25). Thus, interaction between Cx43 and these kinases may play an important role in cellular responses to injury.
Previous studies have shown that mutation of the Cx43 sites phosphorylated by CK1 to alanine in a knock-in mouse model (hereafter called Cx43CK1) results in animals that develop normally but have diminished expression of Cx43 in many tissues, including the heart (13, 26–28). It has also been shown that these animals are sensitive to induced arrhythmia (27). We were interested in understanding the mechanism driving diminished Cx43 protein levels in tissue, and given the overlapping abilities of Cx43 and CK1, ERK, and Akt to affect cellular and tissue function, we hypothesized that modulation of specific phosphorylation sites on Cx43 could alter the ability of cardiac tissue to respond to damage.
In this study, we examined the effect of the mutation of the CK1 sites in Cx43 and discovered an interplay between phosphorylation of Cx43 by CK1, ERK, and Akt that affected ERK kinase activity and the ability of Akt to phosphorylate the hypoxia-responsive protein NDRG1. We found that mutation of the CK1 sites resulted in increased ERK activation and that these sites were involved in ERK binding to Cx43, indicating a direct role of Cx43 in ERK regulation. In addition, we found altered phosphorylation of Akt substrates in Cx43CK1 mice and cells, including a dramatic difference in the level of Akt phosphorylation on the protein NDRG1. Myocardial protection from IRI via the reperfusion injury salvage kinase (RISK) pathway has been shown to involve both ERK and Akt (19). Indeed, we found that hearts from old Cx43CK1 mice were resistant to IRI. These findings indicate that Cx43-mediated effects on cell behavior are not merely downstream effects of kinase activation but, rather, may exert control over the kinases themselves.
Results
Cx43CK1 mice are protected from IRI despite decreased Cx43 expression
Myocardium from aged WT mice show both diminished Cx43 expression (29) and increased susceptibility to myocardial infarction compared with young mice (30, 31). Similarly, hearts from knock-in mice containing Cx43 with mutations at CK1 phosphorylation sites (Cx43CK1) contain less total Cx43 protein than those from WT syngeneic mice and show increased susceptibility to arrhythmia (27). We were interested in how the aging and Cx43CK1 phenotypes would interact in terms of both protein expression and susceptibility to myocardial infarction. We compared the extent of myocardial infarction in response to IRI in hearts from young (3–6 months old) and old (>12 months old; average, 16 months) mice expressing Cx43CK1 compared with syngeneic WT mice. We isolated the hearts on a Langendorff apparatus and subjected them to 30 min of no-flow normothermic global ischemia, followed by 2 h of reperfusion. Hearts were treated with triphenyl tetrazolium chloride (TTC) to stain live tissue red; dead tissue appears white. Images of stained heart slices were analyzed by thresholding on the TTC signal and measuring the area of living tissue (positively stained with the red TTC dye in Fig. 1A). We found that the protocol caused partial ischemic damage but no difference in the area of living tissue between young WT and Cx43CK1 animals (Fig. 1B, 45.9% versus 44.4% TTC-positive). However, hearts from old WT animals were much more prone to ischemic damage, with a significant decrease in living tissue (Fig. 1B; reduced to 17.9%, p = 0.009 for young versus old). Remarkably, older Cx43CK1 animals were relatively protected, showing levels similar to the young animals (40.5% TTC-positive).
Figure 1.
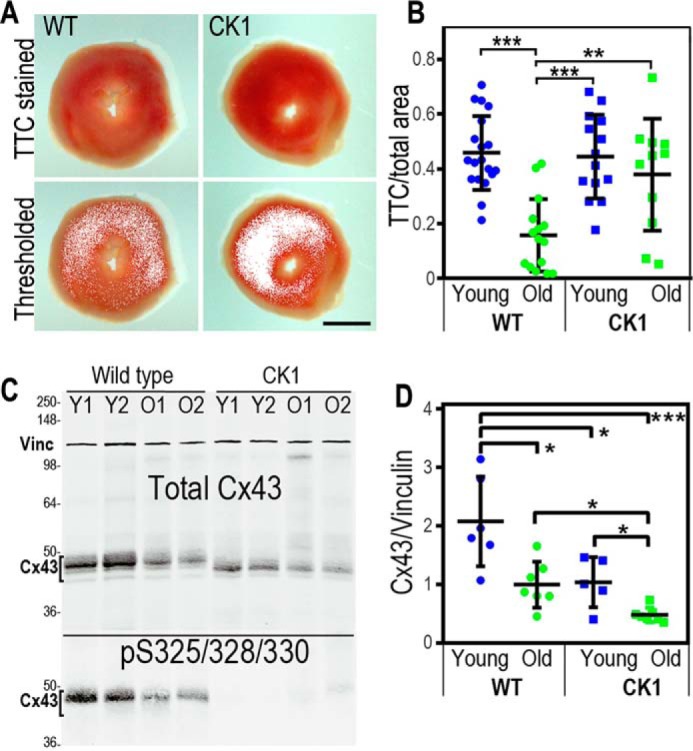
Aged Cx43CK1 mice are protected from IRI despite diminished Cx43 expression. A and B, representative image of hearts from young and old WT or Cx43CK1 (CK1) mice that were exposed to IRI, sliced and stained with TTC to identify live tissue (A, red). Images were thresholded to identify and measure live tissue and are quantified in B as a percentage of live tissue to total area. At least four slices from each heart and at least six hearts from each age and genotype were analyzed. C, immunoblots showing levels of total Cx43 and vinculin (Vinc) in lysates from hearts of WT and Cx43CK1 mutant mice (CK1). Lysates from two different young (Y1 and Y2) and old (O1 and O2) animals are shown. Bottom panel, immunoblot probed for Cx43 phosphorylation on Ser-325/328/330 using a phospho-specific antibody. D, quantification of Cx43 levels normalized to vinculin. At least 4 hearts of each type were analyzed and are presented as mean ± S.E. Significance was determined by two-tailed unpaired t tests. *, p < 0.05; **, p < 0.01; ***, p < 0.001. Scale bar = 1 mm.
To determine whether Cx43 expression levels were playing a role in this effect, we examined how levels changed in response to both genotype and age. Immunoblotting was performed to measure Cx43 expression in heart tissue (Fig. 1C) from mice 3–6 months old (young) and over 18 months in age (old). We found that these two phenotypes were additive; in both young and old mice, Cx43 levels in Cx43CK1 mice were about half that of WT mice (p = 0.022 and 0.012, respectively), and both genotypes showed a similar decrease in Cx43 expression with age (WT: 2.07 ± 0.38 versus 1.00 ± 0.19 a.u., p = 0.016; Cx43CK1: 1.05 ± 0.19 versus 0.42 ± 0.06 a.u., p = 0.042) (Fig. 1D). Because we observed the same relative decrease in protein levels in response to age, this difference in protein levels appears to be at least partially independent of Ser-325/328/330 phosphorylation. Indeed, relative levels of Ser-325/328/330 phosphorylation were not significantly affected by age (1.15 ± 0.20 versus 1.47 ± 0.56 a.u., p = 0.20) (Fig. 1A).
To determine whether these differences were due to effects on transcription, we performed quantitative PCR for Cx43. Equal amounts of total RNA were analyzed. We found that WT animals had similar Cx43 mRNA levels regardless of age (young, 1.01 ± 0.08 a.u.; old, 0.92 ± 0.15 a.u.). We did find that young Cx43CK1 animals trended toward having less mRNA for Cx43 compared with young WT animals (0.87 ± 0.08 a.u., p = 0.11) and with Cx43CK1 old animals (1.01 ± 0.06 a.u., p = 0.08). However, it seems unlikely that this difference would be sufficient to explain the nearly 50% difference in protein levels in young animals, and in the case of young versus old Cx43CK1 animals, mRNA level differences are actually in the opposite direction compared with protein levels. Thus, the effects observed appear to be posttranscriptional. Phosphorylation of Cx43 is critical to both gap junction function and protein stability; thus, we hypothesized that kinase interactions might be altered in response to age and genotype.
Cx43 phosphorylation by ERK is increased in Cx43CK1 myocardium
Consistent with data reported previously (26, 27), we found that lack of Ser-325/328/330 in Cx43CK1 mice did not modify the distribution of cardiac Cx43 by immunohistochemistry (data not shown). In all genotypes and ages, Cx43 was seen predominantly at the intercalated disc, arguing that these mutations did not dramatically alter the ability of Cx43 to traffic and assemble into gap junctions. This supports the idea that altered signaling may be driving the protection from IRI we observed. Indeed, Cx43 is directly phosphorylated by some of the most critical kinases involved in both cardiac damage and protection from IRI, including ERK, Akt, and PKC (19–21). Immunoblotting of Cx43 from young and old myocardium using phosphoantibodies to these sites showed that both genotype and age altered phosphorylation on Cx43 by ERK. Using phospho-specific antibodies to the known ERK sites pSer-262 and pSer-279/282, we found that Cx43CK1 mice had increased phosphorylation on these sites (Fig. 2, A and C). Ser-262 phosphorylation was increased almost 2.5-fold in both young and old animals (0.80 ± 0.16 versus 2.16 ± 0.22, p = 0.002 for young and 0.65 ± 0.12 versus 2.07 ± 0.10, p < 0.0001 for old), with an even more dramatic increase in phosphorylation on Ser-279/282 driven by genotype (1.13 ± 0.77 versus 7.23 ± 0.72, p = 0.0006 for young animals and 4.47 ± 3.52 versus 16.64 ± 3.16, p = 0.0004 for old animals) (Fig. 2, A and C). Thus, by eliminating phosphorylation of Cx43 by CK1, we increased phosphorylation by ERK on sites 50 residues upstream, showing that these events are co-regulated in some manner.
Figure 2.
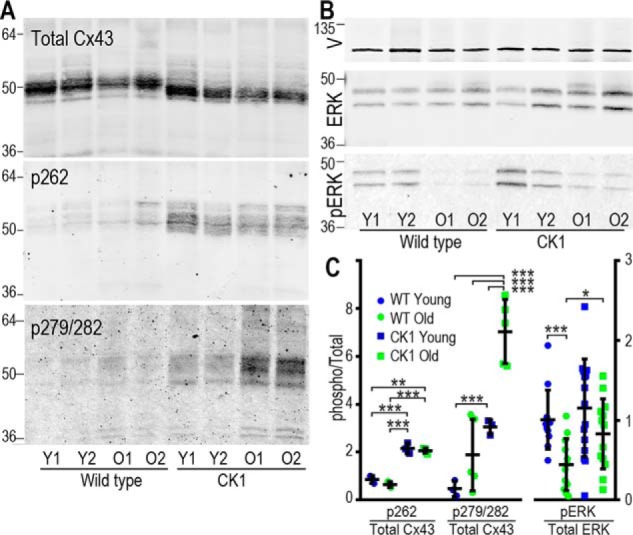
Phosphorylation of Cx43 and ERK are altered in Cx43CK1 mouse hearts. A, immunoblots showing levels of total Cx43 (Cx43NT1) and Cx43 phosphorylated on Ser-262 and Ser-279/282 in lysates from hearts of WT and Cx43CK1 (CK1) mutant mice. Lysate volumes in these blots were adjusted to show equal levels of Cx43 to highlight changes in phosphorylation. 64-, 50-, and 36-kDa molecular mass markers are indicated. Y, young; O, old. B, immunoblots showing levels of vinculin (V), ERK, and pERK in mouse heart lysates. 36- and 64-kDa molecular mass markers are indicated. C, quantification of phospho-forms normalized to total levels of Cx43 or ERK. At least four hearts of each type were analyzed and are presented as mean ± S.E. Significance was determined by two-tailed unpaired t tests. *, p < 0.05; **, p < 0.01; ***, p < 0.001.
Cx43 phosphorylation status affects ERK activation
Given that Ser-262 and Ser-279/282 are ERK substrates, we also examined the expression and activation of ERK in these animals and saw a dramatic decrease in ERK phosphorylation (pERK) with age in WT animals (1.01 ± 0.19 versus 0.45 ± 0.16 in young versus old, p = 0.0007), whereas old Cx43CK1 animals maintained higher levels of ERK phosphorylation with age (1.15 ± 0.31 versus 0.83 ± 0.22 in young versus old, not significantly different) (Fig. 2, B and C). To further examine this interaction, we utilized Cx43-deficient Madin-Darby canine kidney (MDCK) cells stably expressing either WT or mutant Cx43 with all CK1 sites mutated to alanine (i.e. CK1) or each site mutated separately (S325A, S328A, and S330A) and were able to recapitulate the effect on ERK activation (Fig. 3A). CK1 mutant–expressing cells showed 60% higher levels (p = 0.009) of ERK phosphorylation compared with WT Cx43–expressing cells (Fig. 3A). Furthermore, we were able to isolate this effect to residue Ser-330. We found that cells expressing the single S330A mutation showed a significant 42% ± 12% increase in phosphorylated ERK compared with WT cells (p = 0.005), whereas mutations at Ser-325 or Ser-328 appeared to have little effect on ERK activation. We hypothesized that this may be due to direct binding of ERK to Cx43 in a manner dependent on dephosphorylated Ser-330. To determine whether the region around the CK1 sites could directly impact ERK binding to Cx43, we performed experiments utilizing a GST-Cx43CT fusion protein where residues 320–340 were deleted (CTΔ330) and examined its ability to bind to and pull down ERK from a cell lysate. We found that the GST-CTΔ330 construct could pull down less than a quarter of the amount of ERK compared with GST-CT, a construct containing the full C terminus of Cx43 (22.8% ± 5.6%, p = 0.0001) (Fig. 3B). We also found that we could much more efficiently co-immunoprecipitate ERK from cells expressing the S330A mutant compared with WT cells using two different Cx43 antibodies (Cx43IF1 generated in mouse and RbAb generated in rabbit) (Fig. 3C). These data argue that CK1 phosphorylation events play a negative role in regulating the ERK:Cx43 interaction, consistent with its role in gap junction assembly and stability. Furthermore, in combination with the results in tissue, we show that the phosphorylation status of Cx43 could regulate whole cellular ERK phosphorylation and activation in vivo.
Figure 3.

Phosphorylation of ERK is elevated in cells expressing Cx43 mutants, likely through a mechanism involving direct interaction. A, immunoblots showing levels of vinculin, ERK, pERK and Cx43 in cell lysates from MDCK cells stably expressing Cx43 mutants where Ser-325, Ser-328, Ser-330, or all three (CK1) were converted to alanine. Quantification of phospho-ERK/total ERK levels are shown in the graph. B, immunoblot showing result of a GST pulldown assay where cell lysates were incubated with either GST alone or GST fusion proteins containing GST-CT or a deletion mutant where amino acids 320–340 were deleted (CTΔ330). The GST blot shows signal using a mouse anti-GST antibody, whereas the ERK blot shows signal from a rabbit anti-ERK antibody that was pulled down from the cell lysate. C, immunoprecipitation (IP) was performed on lysates from cells expressing either WT or S330A mutant Cx43 using a rabbit anti-Cx43 antibody (RbAb) or a mouse anti-Cx43 antibody (Cx43IF1). Immunoblots (IB) show levels of Cx43 or ERK that were immunoprecipitated. 36- and 64-kDa molecular mass markers are indicated. **, p < 0.01.
Cx43 phosphorylation status affects Akt substrate specificity in myocardium and cells
ERK and Akt have been shown to be critical kinases involved in cardiomyocyte responses to IRI, acting through a pathway referred to as the RISK pathway (19). Thus, we also examined Akt expression and phosphorylation in the mouse myocardium and cells but saw no consistent effect because of genotype or age (data not shown). However, when we probed immunoblots using an antibody that recognizes phospho-Akt substrates (PAS antibody, Cell Signaling Technology, catalog no. 9614; binds to peptides corresponding to Akt substrate motifs), we found that there was a dominant substrate in the 50- to 60-kDa range that was highly phosphorylated in both myocardium and cell lines (Fig. 4; note that other clear bands were present near the dye front and at ∼40, 70, and 110 kDa). Similar to what we saw for phospho-ERK, we found diminished phosphorylation on this 50- to 60-kDa protein in old WT animals compared with young WT animals (0.83 ± 0.24 versus 1.70 ± 0.62 a.u., p = 0.04) but saw that old Cx43CK1 animals maintained levels of PAS signal similar to young animals (Fig. 4A, 1.32 ± 0.28 a.u.; p = 0.04, comparing old WT and old Cx43CK1 mice). Similarly, when we examined cells expressing Cx43 with single or all three CK1 sites mutated to alanine, we found that CK1 and S330A also had elevated levels of PAS (WT versus CK1, 5.16× ± 1.35, p = 0.002, WT versus S330A, 5.71× ± 0.91, p = 0.009) (Fig. 4B). These data indicate that altering the phosphorylation of Cx43 led to changes in signaling through ERK and Akt; we hypothesize that these changes may contribute to the cardioprotection observed in the old Cx43CK1 animals.
Figure 4.
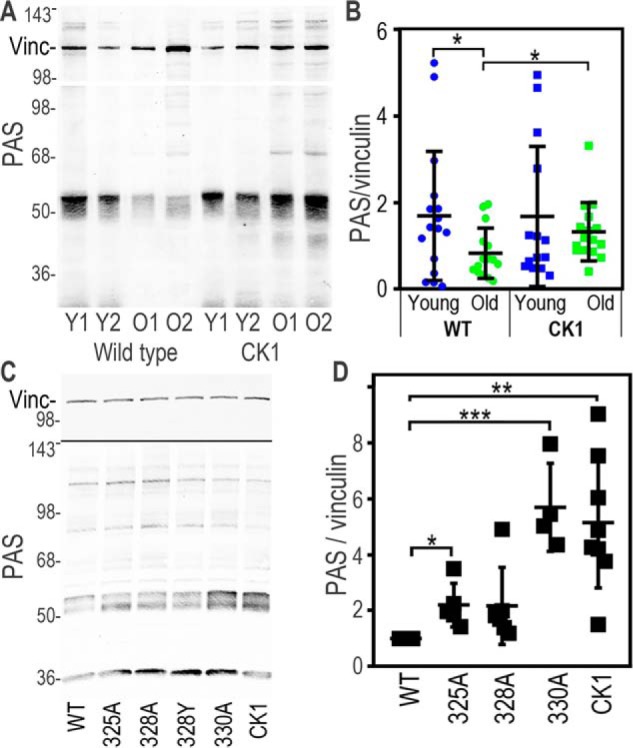
Cx43 mutants show elevated levels of a 50- to 60-kDa Akt substrate in tissue and in cells. A, immunoblots of lysates from mouse hearts, showing levels of vinculin (Vinc) and signal from an antibody that recognizes proteins phosphorylated by Akt (PAS). Y, young; O, old. B, quantification of PAS levels normalized to vinculin. C, immunoblots showing levels of PAS and vinculin in cell lysates from MDCK cells stably expressing Cx43 mutants where Ser-325, Ser-328, Ser-330, or all three (CK1) have been converted to alanine. Note that the blot was first probed for PAS and then reprobed for vinculin. D, quantification of PAS levels normalized to vinculin. *, p < 0.05; **, p < 0.01; ***, p < 0.001.
Cx43 interacts with and alters the expression of the Akt substrate NDRG1
Knowing the approximate molecular weight of our Cx43 regulated PAS substrate, we examined the literature and Cell Signaling Technology PhosphoSite Plus to try to identify the protein recognized by the PAS antibody. We tried several potential candidates (e.g. GSK3β), but NDRG1 was our best match, as it was of the correct molecular size and has been shown to alter its migration on SDS-PAGE with changes in phosphorylation, similar to the banding pattern we saw with the PAS antibody (Fig. 4C). NDRG1 has been shown to be increased and promote cell survival in response to hypoxia, which is functionally consistent with our findings (32, 33). Based on our PAS data, we anticipated a change in phosphorylation in response to the expression of Cx43 mutants, but instead we found that total NDRG1 protein levels closely mimicked our results with the PAS antibody (Fig. 5A). In these experiments, we included a phosphomimetic Cx43 mutant where Ser-325, Ser-328, and Ser-330 were converted to glutamic acid (CK1-E). As with the PAS antibody, we found that both CK1-A– and S330A mutant–expressing cell lines had elevated NDRG1 levels compared with the WT (WT: 1.05 ± 0.14; CK1-A: 2.28 ± 0.52, p = 0.0005; S330A: 3.92 ± 0.79 a.u., p < 0.0001), whereas CK1-E cell lines responded in the opposite direction, showing both diminished NDRG1 expression (CK1-E: 0.75 ± 0.14, p = 0.04) and decreased PAS signal (WT: 0.99 ± 0.19; CK1-E: 0.45 ± 0.08, p = 0.0002).
Figure 5.
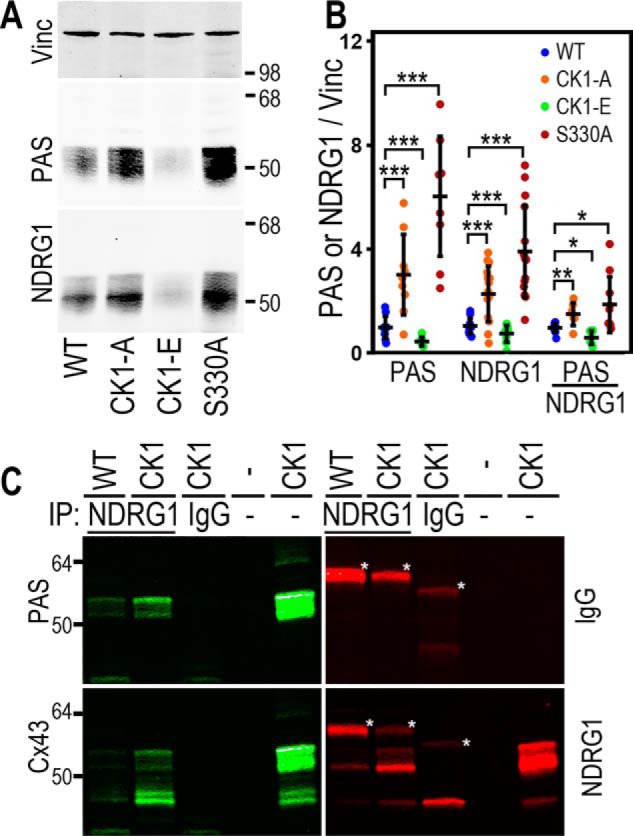
Cx43 mutants alter the stability and phosphorylation of NDRG1. A, immunoblots show levels of PAS and corresponding NDRG1 signal from cells expressing WT Cx43 or mutants with Ser-325/328/330 or Ser-330 alone (S330A) converted to alanine to eliminate phosphorylation (CK1-A) or glutamic acid to mimic phosphorylation (CK1-E). Vinc, vinculin. B, quantification of PAS and NDRG1 signal normalized to vinculin and PAS normalized to NDRG1. C, co-immunoprecipitations (IP) were performed on lysates from cells expressing either WT or CK1 mutant Cx43 using a mouse anti-NDRG1 antibody (NDRG1, first and second lanes). An anti-mouse IgG antibody was used for CK1 lysates only as a negative control (IgG, third lane). The fourth lane is blank, and the fifth lane shows the CK1 lysate used in immunoprecipitation. The immunoblot was sequentially probed with Alexa Fluor 680 anti-mouse secondary antibody to identify signal due to the immunoprecipitating antibodies (IgG in red, bands noted with an asterisk) and then anti-PAS antibody and Alexa Fluor 790 anti-rabbit secondary antibody (PAS in green). Next the blots were probed with rabbit anti-Cx43 and Alexa Fluor 790 anti-rabbit secondary antibody (Cx43) and mouse anti-NDRG1 and Alexa Fluor 680 anti-mouse secondary antibody (NDRG1). *, p < 0.05; **, p < 0.01; ***, p < 0.001.
To determine whether NDRG1 and Cx43 interact with each other, we performed co-immunoprecipitations using the NDRG1 antibody from cells expressing either WT Cx43 or CK1-A. Fig. 5C shows an example of a single immunoblot probed with three different antibodies; the top left panel was probed with the PAS antibody (made in rabbit), and the first and second lanes show that the NDRG1 antibody immunoprecipitated more PAS from CK1-A lysates than from WT lysates. Mouse IgG control antibody immunoprecipitation (Fig. 5C, third lane) showed no bands, whereas the fifth lane shows the PAS signal from the CK1-A lysate alone (the fourth lane is empty). Subsequent probing of the blot with a Cx43 antibody (Fig. 5C, bottom left panel) shows that the NDRG1 antibody also co-precipitated Cx43. Fig. 5C, top right panel, shows the signal (marked with an asterisk) from the immunoprecipitating antibody recognized by the mouse secondary antibody. Fig. 5C, bottom right panel, shows that more NDRG1 was immunoprecipitated from the CK1-A lysate. Taken together, it appears that a direct interaction between Cx43 and NDRG1 can somehow stabilize NDRG1 protein levels.
Like Cx43, NDRG1 migrates as multiple isoforms via SDS-PAGE, in part because of differences in phosphorylation (34, 35). Although all NDRG1 isoforms are increased or decreased in the Cx43 mutants, the effects are most evident on the high-migrating isoform that is strongly recognized by the PAS antibody (Fig. 6, A and B). We have shown previously that Cx43 phosphorylation and localization can be stabilized via proteasome inhibition, as seen in Fig. 6C, where MG132 leads to an increase in the slow-migrating phospho-isoform in WT cells. This effect is not due to direct inhibition of Cx43 degradation but, rather, is a result of Akt stabilization and subsequent Cx43 phosphorylation (17). To determine whether Akt may play a role in stabilizing NDRG1, we treated cells with the proteasome inhibitor MG132 and the Akt inhibitor MK2206. Indeed, we found that in WT cells, the PAS/NDRG1 signal nearly doubled after 2 h of MG132 treatment (0.93 ± 0.11 versus 1.81 ± 0.39 a.u., p = 0.012), whereas Akt inhibition dramatically diminished the signal (down to 0.16 ± 0.05 a.u., p < 0.0001, at 2 h but apparent as early as 30 min after treatment). Although we saw a similar shift in migratory isoforms, the effect was less dramatic in CK1-A cell lysates, with a 55% increase with MG132 (control: 1.25 ± 0.39 versus 1.94 ± 0.9 a.u.) and just less than a 50% decrease with MK2206 (0.68 ± 0.40 a.u.), neither of which reached significance. We hypothesize that this is due to the enhanced interaction with the CK1-A mutant leading to increased interaction with Akt and/or protecting it from proteasomal degradation. CK1-E cells also responded to the drugs, with proteasome inhibition increasing PAS/NDRG1 levels to WT control levels (0.93 ± 0.34 a.u.), whereas the already low levels were essentially abolished with Akt inhibition (down to 0.08 ± 0.02, p = 0.0007). Similar to what we saw with Cx43, in contrast to phosphorylation levels, total NDRG1 levels were not dramatically affected by these drug treatments. This may be due to increased stabilization of Akt in response to MG132 (17).
Figure 6.
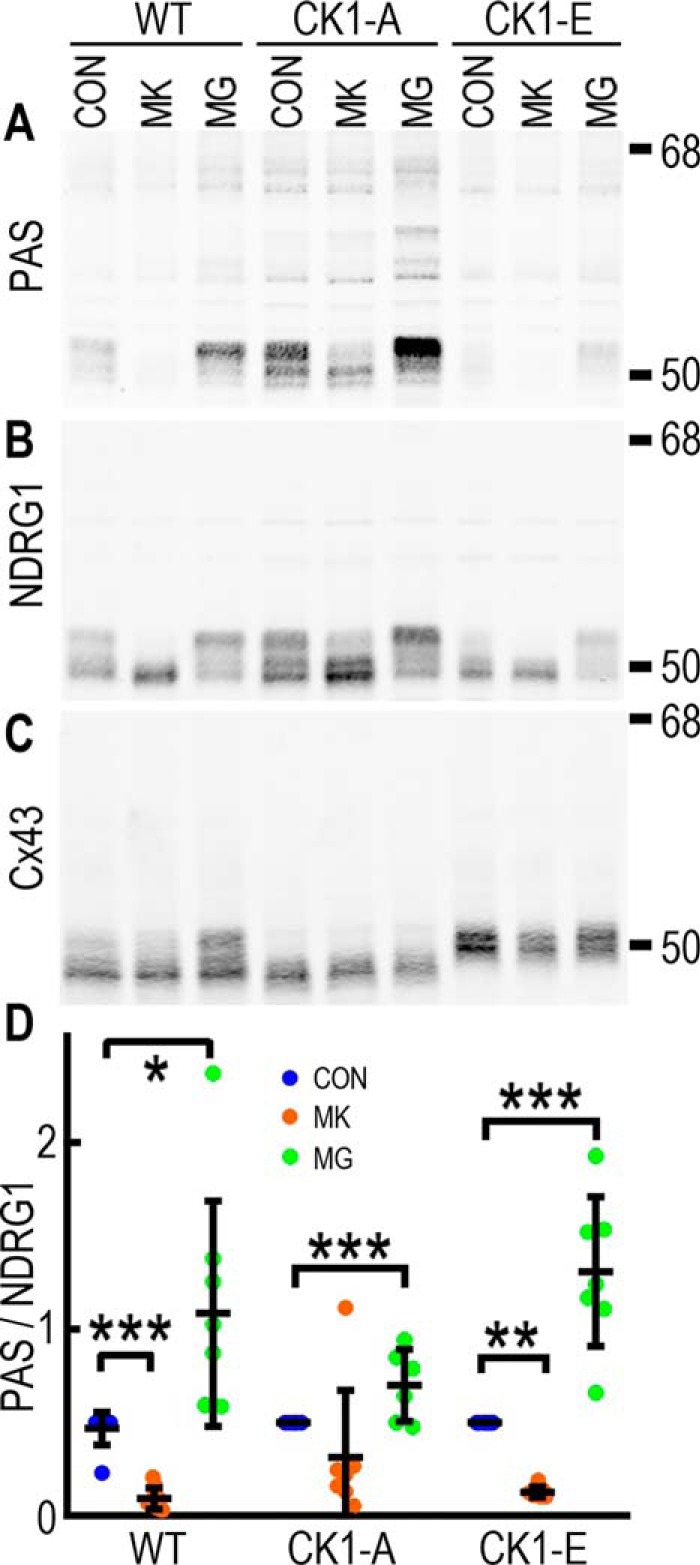
Cx43 interaction with NDRG1 regulates the stability of Akt-phosphorylated NDRG1. A–C, immunoblots showing changes in PAS (A), NDRG1 (B), and total Cx43 (C) in response to 2-h treatment of cells with the Akt inhibitor MK2206 (MK) and the proteasome inhibitor MG132 (MG). CON, control. D, quantification of PAS levels relative to vinculin. *, p < 0.05; **, p < 0.01; ***, p < 0.001.
Previous reports of NDRG1 regulation have focused on phosphorylation by GSK3β and (serum- and glucocorticoid-stimulated kinase) SGK; our data indicate that Akt phosphorylation of NDRG1 occurs in cells and is regulated via interaction with Cx43. We hypothesize that this may be a novel signaling node regulated by gap junctions and are continuing these studies to elucidate all components of this pathway and the functional consequences of these interactions.
Discussion
Gap junctions play a role in cell survival and are involved in cellular responses to injury, including response of the myocardium to IRI. Although intercellular communication certainly plays a role in these effects, Cx43 also interacts with a variety of kinases that are important in cell survival and signaling, including ERK and Akt. Rather than just being a downstream effector of these pathways, the data presented here show that Cx43, specifically Cx43 phosphorylation, can play a role in regulating these pathways. We have shown that, in cardiac tissue, mutation of the CK1 phosphorylation sites reduced Cx43 expression by over 50% and increased Cx43 phosphorylation on Ser-279/282 and Ser-262, sites phosphorylated by ERK, regardless of age. In young mice, hearts were resistant to damage via IRI and expressed high levels of phospho-ERK. Aged WT mouse hearts exhibited much higher levels of myocardial damage in conjunction with a more than 50% decrease in phospho-ERK. Old CK1 mice, however, were similar to young animals both in terms of reduced tissue damage and ability to preserve phospho-ERK. Using the PAS antibody, we found that old CK1 animals also maintained high levels of a 50- to 60-kDa protein phosphorylated by Akt. These effects were recapitulated in cell lines where we observed that ERK could interact directly with Cx43 in a manner dependent on Ser-330 being nonphosphorylated. Further, we identified the Akt substrate to be a novel Cx43-interacting protein, NDRG1, whose stability and phosphorylation by Akt was dramatically affected by Cx43 phosphorylation; elimination of CK1 sites led to more than double the expression level and Akt phosphorylation of NDRG1, whereas a phosphomimetic diminished expression. Co-immunoprecipitation showed that NDRG1 preferentially interacts with nonphosphorylated Cx43, arguing that this interaction could protect NDRG1 from degradation.
To put these results in context, our laboratory and others have shown that acute coordination of these kinases affects gap junction stability and degradation, but here we show that the converse is also true; Cx43 interactions with kinases can affect their ability to signal. Specifically, we found that mutation of the CK1 phosphorylation sites led to ERK activation and increased Cx43 phosphorylation on Ser-279/282 and Ser-262. Phosphorylation on these sites is typically associated with gap junction degradation, although Cx43CK1 hearts clearly maintain a reduced level of gap junctions at the intercalated disc. Previous studies have shown that Cx43CK1 mice are prone to induced cardiac arrhythmia (27). Because phosphorylation on Ser-279/282 regulates channel gating, our finding of these additional changes in Cx43 phosphorylation indicates that they cooperate with the CK1 mutation to contribute to the arrhythmia phenotype. Phosphorylation on Ser-262 in cardiomyocytes, however, has been shown to correlate with protection from IRI, particularly in response to fibroblast growth factor (24). However, ERK activation correlates better with IRI protection than phosphorylation on Cx43 at Ser-262 and Ser-279/282, both of which were generally higher in CK1 mice regardless of age. Obviously, the aging process is complex, with many factors affected, including altered growth signaling, mitochondrial dysfunction, matrix remodeling, impaired calcium homeostasis, and other processes (36), so making a firm conclusion about their relative importance for IRI protection is not yet possible. For example, coordinated signaling through ERK and Akt has been shown to provide protection from IRI, referred to as the RISK pathway (19). Indeed, our results show that ERK phosphorylation itself is elevated in IRI-resistant Cx43CK1 animals. Protection was only apparent in old animals, where IRI induced more extensive injury compared with young animals. This was, at least in part, likely due to the fact that young animals have high levels of pERK that, in our studies and another (37), dropped dramatically with age. However, the Cx43CK1 mutant mostly preserved pERK levels with age. The mechanism behind this appears to involve a direct interaction between ERK and Cx43 in a manner dependent on residues 320–340 (Fig. 3B) and a strong preference for Cx43 when it is not phosphorylated specifically on Ser-330 (Fig. 3C). Interestingly, it was recently reported that Cx43 expression in osteoblasts could alter ERK activity (38); in this case, Cx43 knockdown diminished ERK phosphorylation, whereas overexpression increased ERK phosphorylation. This is certainly distinct from our data, as we saw an ERK effect in animals expressing the lowest total levels of Cx43 (old Cx43CK1 mice, Fig. 2). We propose that it is not expression but the phosphorylation status of Cx43 that can promote or deter ERK binding, thus altering its ability to interact with other regulators, such as phosphatases.
We have shown previously that Akt can promote gap junction stability and directly phosphorylate Cx43 (17). However, somewhat counterintuitively, this interaction often precedes phosphorylation on Cx43 by ERK and gap junction turnover. This process of regulated turnover appears to be highly coordinated, as at least three phosphorylation events occur sequentially over about 30 min (18, 39). Similar coordination appears to be necessary for ERK and Akt to provide protection from IRI, where promotion or inhibition of cell damage is linked to both the amplitude and duration of ERK and Akt signaling (40). We examined Akt expression and phosphorylation in WT and Cx43CK1 hearts but saw no change in regards to genotype or age (data not shown); however, when we utilized a phospho-epitope antibody that recognized phospho-Akt substrates, we discovered that there was a dominant phosphoprotein that appeared to be co-regulated with ERK. Thus, old Cx43CK1animals appeared to show some level of coordination between ERK activity and Akt substrate specificity, consistent with activation of the RISK pathway (40). As discussed above, Ser-262 phosphorylation, likely by ERK1/2, has been shown to promote cardiomyocyte survival, and we have shown previously that Akt phosphorylation of Cx43 occurs in response to ischemia in mouse myocardium. PKCϵ has also been shown to promote cell survival during IRI in a manner dependent on Cx43 (20, 23, 41). Thus, Cx43 resides at an intersection between these cardiomyocyte survival modulation pathways, leading to both changes in gap junction communication and, as we show here, changes in the signaling pathways themselves.
The novel Cx43-interacting protein we found here, NDRG1, may provide a link to cell survival during hypoxia. NDRG1 has primarily been studied in the context of tumorigenesis, where it appears to be a metastasis suppressor. NDRG1 is regulated by HIF-1α, which is itself regulated by Akt, and is often associated with protection of cells from hypoxic injury (42–44). Like Cx43, NDRG1 interacts with a variety of signaling pathways that regulate cell stasis and survival (45, 46) and migration (44, 47). For example, NDRG1 has been reported to inhibit NF-κB signaling (48), leading to downstream effects on epithelial mesenchymal transition (49, 50) and angiogenesis (48). It has been shown to affect MAPK signaling through regulation of EGF receptor expression and internalization (34, 51) and to stabilize adherens junctions through regulation of E-cadherin (34, 52) and β-catenin (50, 53) localization and degradation. The role of phosphorylation in regulating NDRG1 is not yet clear, although it is known to be phosphorylated by SGK, followed by GSK (54). In one study, phosphorylation on NDRG1 by SGK1 was essential for it to inhibit the NF-κB pathway (55). Phosphorylation of NDRG1 by Akt has not been well studied, but given that Akt and GSK can behave antagonistically (phosphorylation of GSK by Akt is inhibitory (56)), we hypothesize that the ability of Cx43 to specifically modulate Akt phosphorylation on NDRG1 could have important regulatory consequences.
We discovered that, in MDCK cells, CK1 phosphorylation on Cx43 altered both NDRG1 expression and phosphorylation, where elimination of CK1 phosphorylation led to high levels of NDRG1, and, conversely, a Cx43 phosphomimetic displayed reduced NDRG1 levels. Under normal circumstances, essentially all Cx43 at the intercalated disc in the myocardium would be phosphorylated by CK1; however, ischemia leads to loss of CK1 phosphorylation (13), which could then allow accumulation of NDRG1. Because old Cx43CK1 animals maintained higher levels of basal PAS signal compared with the WT, they may be predisposed to protection via NDRG1. Although the Cx43-mediated effects on NDRG1 are clear in cells, we tried three different NDRG1 antibodies on heart tissue that gave little to no signal via immunoblot, possibly for technical reasons. However, an antibody to NDRG1 phosphorylated on Thr-346 did show similar reactivity levels to PAS on heart tissue (Fig. S1). One possibility is that in the heart, we might actually be seeing the closely related family member NDRG2, which shares homologous phosphorylation sites and is more prevalent in heart tissue (54). In cells, we could detect a direct interaction between NDRG1 and Cx43 that was most prevalent when the CK1 sites were not phosphorylated. We propose that this interaction leads to stabilization of the NDRG1 protein, thus the change in protein levels in the CK1 mutants. We hypothesize that interaction with Akt is also involved, as inhibition of proteasomal degradation led to increased NDRG1 phosphorylation via Akt, whereas Akt inhibition diminished this signal, similar to what we see for Cx43 (17).
In this study, we focused on shorter time points (≤2 h) to understand the more acute interactions that might be occurring, in part because of the known short half-life of Cx43 (∼2 h). Typically, any individual Cx43 molecule remains unphosphorylated on CK1 sites only until it reaches the gap junction. This would argue that, under homeostatic conditions, interaction between NDRG1 and Cx43 is relatively short-lived; however, in the CK1-A mutants, the effects of Akt and proteasome inhibition were minimal (Fig. 6; changes in PAS signal did not show significant differences), arguing that the interaction with Cx43 stabilized the Akt phosphoform and isolated it from the effects of these drugs. Thus, it appears that Cx43 could be involved in dynamic regulation of NDRG1 and Akt interaction, particularly under conditions such as hypoxia, where Cx43 phosphorylation is spatiotemporally regulated (12).
Although there is a wealth of data showing that Cx43 and gap junctions can alter cell behavior, the mechanistic underpinnings of these effects remain obscure. Many studies, especially our own, have focused on how signaling affects gap junctions; in these studies we also found the converse to be true, gap junctions act as a signaling scaffold that can promote or inhibit kinase activity and potentially signal cross-talk. Further elucidation of the functional and mechanistic consequences of these processes will yield new insights into how Cx43 exerts effects on cell behaviors such as survival.
Experimental procedures
Reagents and antibodies
All general chemicals, unless otherwise noted, were purchased from Thermo Fisher Scientific. Mouse monoclonal antibodies to total Cx43 (Cx43NT1) and to the C-terminal region of Cx43 (Cx43CT1) and anti-GST were developed by the Fred Hutchinson Cancer Research Center Hybridoma Development Facility (Seattle, WA). The Cx43 phospho-specific rabbit antibodies p325/328/330, p279/282, and pS373 were created as described previously (13, 17, 57). The Cx43 phospho-specific antibody to pSer-262 was purchased from Santa Cruz Biotechnology. Mouse anti-vinculin and rabbit anti-Cx43 were from Millipore Sigma and made in-house (RbAb). Mouse anti-NDRG1 was purchased from Abcam. Mouse anti-p42/44 ERK, rabbit antibodies to phospho-p42/44ERK, PAS, and NDRG1 phospho-specific antibody to Thr-346 were from Cell Signaling Technology.
Mouse strains and tissue processing
All mouse studies were conducted with Institutional Animal Care and Use Committee approval (Fred Hutchinson Cancer Research Center). Cx43CK1 mutant mice were generated as described previously (26) and inbred into a C57Bl6/6J strain background. Young animals were between 2–6 months old, and old animals were 18–24 months old. For IRI studies, animals were euthanized with isoflurane followed by cervical dislocation and rapid excision of the heart. Tissue was either flash-frozen for immunoblotting and quantitative PCR or fixed in 10% normal buffered formalin and embedded in paraffin for analysis as described previously (58).
Quantitative PCR
Total RNA from three hearts of each age and genotype RNA were isolated using Qiagen RNeasy® Mini Kit (Qiagen) and quantified using the RiboGreen® RNA Quantitation Kit (Thermo Fisher Scientific). cDNA was synthesized from total RNA using the High Capacity cDNA Reverse Transcription Kit (Applied Biosystems). Quantitative PCR was performed on an Mx3005P® Multiplex QPCR System (Stratagene) with samples loaded in triplicate using ∼80 ng of cDNA. Quantitative PCR was run in a 10-μl reaction using SYBR® Green PCR Master Mix (Applied Biosystems) (5 μl of 2× Master Mix, 400 nm each primer) with PCR cycling conditions of 95 °C for 10 min and 40 cycles of 95 °C for 15 s and 60 °C for 1 min. After each assay, a dissociation curve was run to confirm the specificity of all PCR amplicons. Cx43 primers (59; forward, 5′-TCCAAGGAGTTCCACCACTT-3′; reverse, 3′-TGGAGTAGGCTTGGACCTTG-5′) were from Integrated DNA Technologies. Pooled cDNA was used for a standard curve as 1:2 serial dilutions. Resulting Ct (cycle threshold) values were converted to nanograms, normalized to total RNA, and expressed as the average of triplicate samples ± 1 S.D.
Plasmids, cell culture, and recombinant protein
MDCK II cells (ATCC) that were screened for lack of Cx43 expression (60) were cultured in DME medium (Mediatech) supplemented with 10% FBS and antibiotics in a humidified 5% CO2 environment. WT and mutant Cx43 were cloned into the pIREShygro vector (Clontech) as described previously (61) and transfected into MDCK cells via Nucleofector (Amaxa). At least two stable clones for each construct were analyzed. GST fusion constructs containing GST alone, the C terminus of WT Cx43 (GST-CT), or a deletion mutant lacking residues 320–340 (Δ330) were used to express and isolate recombinant protein from Escherichia coli for GST pulldown assays as described previously (62).
Immunoblotting and immunoprecipitation
Flash-frozen hearts were homogenized in Laemmli sample buffer containing 1× Complete protease inhibitor mixture, 1× phosphatase inhibitor mixture, and 2 mm PMSF (all from Millipore Sigma) using a glass pestle tissue grinder. Cell lysates were directly lysed in Laemmli sample buffer containing the same inhibitors. After sonication, samples were separated by SDS-PAGE, blotted to nitrocellulose, checked for load with Ponceau S, blocked in milk, and probed with antibodies as noted. Antibodies were detected with appropriate secondary antibodies conjugated to Alexa Fluor 680 or Alexa Fluor 790 (Thermo Fisher Scientific) and directly quantified using the Li-Cor Biosciences Odyssey IR imaging system and associated software. Normalized densitometry values were calculated as a ratio using either vinculin as a loading control or comparing phospho-specific antibody signals to corresponding total protein levels. Vinculin was chosen as a loading control because it showed high correspondence with Ponceau S staining in heart tissue (data not shown). At least six hearts for each age and genotype were included in these analyses, and p values were determined by two-sided Student's t tests.
Langendorff-perfused mouse heart and ischemia reperfusion injury
Perfusion of mouse hearts was performed as described previously (63). Briefly, animals were injected with 200 units/kg heparin, followed by injection with Avertin (0.83 mg/g body weight) and cervical dislocation. Hearts were rapidly excised, and the aortae were cannulated with a 20-gauge cannula. Hearts were perfused in the Langendorff system (ADInstruments) at constant pressure (80 ± 3 mm Hg) with Krebs-Henseleit solution (0.5 mm EDTA, 5.3 mm KCl, 1.2 mm MgSO4, 118 mm NaCl, 25 mm NaHCO3, 2 mm CaCl2, 10 mm glucose, and 0.5 mm pyruvate bubbled with 5% CO2/95% O2). After a 30-min stabilization period, hearts were subjected to 30 min of no-flow normothermic global ischemia, followed by 2 h of reperfusion. For vital staining, hearts were immediately removed from the Langendorff apparatus, weighed, and frozen at −20 °C. The frozen heart was then cut using a heart slicer (Zivic Instruments, Pittsburgh, PA) into seven or eight transverse slices of ∼0.8-mm thickness and stained by incubation in 10% TTC for 20 min at 37 °C. The TTC solution was then replaced with 10% formaldehyde–PBS solution for fixation. The infarcted tissue remained discolored (pale), whereas normal tissue was stained red. The infarct area was traced, and the total area was calculated using ImageJ software (64). Infarct size was expressed as a percentage of total ventricular area.
Data analysis
Data are represented as mean ± S.E. from at least three independent experiments. Statistical analyses were performed using t tests as indicated.
Author contributions
J. L. S., L. M.-R., and P. D. L. conceptualization; J. L. S. and L. M.-R. data curation; J. L. S., L. M.-R., and P. D. L. formal analysis; J. L. S., L. M.-R., and P. D. L. investigation; J. L. S. and P. D. L. methodology; J. L. S. and P. D. L. writing-original draft; J. L. S. and P. D. L. writing-review and editing; P. D. L. supervision; P. D. L. funding acquisition.
Supplementary Material
Acknowledgments
We thank Mark Velasco and Joshua Dawson for technical help with some of the experiments.
This work was funded by NIGMS, National Institutes of Health Grant GM055632 (P.D.L.) and National Institutes of Health Grants P30 DK017047 and P30 CA015704. P. D. L. and J. L. S. receive royalties from sales of the Cx43IF1 and Cx43NT1 antibodies. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
- ERK
- extracellular signal–related kinase
- PKC
- protein kinase C
- IRI
- ischemia reperfusion injury
- RISK
- reperfusion injury salvage kinase
- TTC
- triphenyl tetrazolium chloride
- a.u.
- arbitrary units
- MDCK
- Madin-Darby canine kidney
- PAS
- phospho-Akt substrate
- GSK
- glycogen synthase kinase
- cDNA
- complementary DNA
- RbAb
- rabbit antibody
- SGK
- serum-and glucocorticoid-induced kinase.
References
- 1. Loewenstein W. R. (1981) Junctional intercellular communication: the cell-to-cell membrane channel. Physiol. Rev. 61, 829–913 10.1152/physrev.1981.61.4.829 [DOI] [PubMed] [Google Scholar]
- 2. Willecke K., Eiberger J., Degen J., Eckardt D., Romualdi A., Güldenagel M., Deutsch U., and Söhl G. (2002) Structural and functional diversity of connexin genes in the mouse and human genome. Biol. Chem. 383, 725–737 [DOI] [PubMed] [Google Scholar]
- 3. Saez J. C., Berthoud V. M., Branes M. C., Martinez A. D., and Beyer E. C. (2003) Plasma membrane channels formed by connexins: their regulation and functions. Physiol. Rev. 83, 1359–1400 10.1152/physrev.00007.2003 [DOI] [PubMed] [Google Scholar]
- 4. Laird D. W. (2006) Life cycle of connexins in health and disease. Biochem. J. 394, 527–543 10.1042/BJ20051922 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Crow D. S., Beyer E. C., Paul D. L., Kobe S. S., and Lau A. F. (1990) Phosphorylation of connexin43 gap junction protein in uninfected and Rous sarcoma virus-transformed mammalian fibroblasts. Mol. Cell. Biol. 10, 1754–1763 10.1128/MCB.10.4.1754 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Laird D. W., Puranam K. L., and Revel J. P. (1991) Turnover and phosphorylation dynamics of connexin43 gap junction protein in cultured cardiac myocytes. Biochem. J. 273, 67–72 10.1042/bj2730067 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Berthoud V. M., Ledbetter M. L., Hertzberg E. L., and Sáez J. C. (1992) Connexin43 in MDCK cells: regulation by a tumor-promoting phorbol ester and calcium. Eur. J. Cell Biol. 57, 40–50 [PubMed] [Google Scholar]
- 8. Musil L. S., Beyer E. C., and Goodenough D. A. (1990) Expression of the gap junction protein connexin43 in embryonic chick lens: molecular cloning, ultrastructural localization, and post-translational phosphorylation. J. Membr. Biol. 116, 163–175 10.1007/BF01868674 [DOI] [PubMed] [Google Scholar]
- 9. Brissette J. L., Kumar N. M., Gilula N. B., and Dotto G. P. (1991) The tumor promoter 12-O-tetradecanoylphorbol-13-acetate and the ras oncogene modulate expression and phosphorylation of gap junction proteins. Mol. Cell. Biol. 11, 5364–5371 10.1128/MCB.11.10.5364 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Kadle R., Zhang J. T., and Nicholson B. J. (1991) Tissue-specific distribution of differentially phosphorylated forms of Cx43. Mol. Cell. Biol. 11, 363–369 10.1128/MCB.11.1.363 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Solan J. L., and Lampe P. D. (2009) In Connexins: A Guide (Harris A., and Locke D., eds.) pp. 263–286, Humana Press, New York [Google Scholar]
- 12. Solan J. L., and Lampe P. D. (2018) Spatio-temporal regulation of connexin43 phosphorylation and gap junction dynamics. Biochim. Biophys. Acta Biomembr. 1860, 83–90 10.1016/j.bbamem.2017.04.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Lampe P. D., Cooper C. D., King T. J., and Burt J. M. (2006) Analysis of Connexin43 phosphorylated at S325, S328 and S330 in normoxic and ischemic heart. J. Cell Sci. 119, 3435–3442 10.1242/jcs.03089 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Cooper C. D., and Lampe P. D. (2002) Casein kinase 1 regulates connexin43 gap junction assembly. J. Biol. Chem. 277, 44962–44968 10.1074/jbc.M209427200 [DOI] [PubMed] [Google Scholar]
- 15. Park D. J., Wallick C. J., Martyn K. D., Lau A. F., Jin C., and Warn-Cramer B. J. (2007) Akt phosphorylates Connexin43 on Ser373, a “mode-1” binding site for 14-3-3. Cell Commun. Adhes. 14, 211–226 10.1080/15419060701755958 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Warn-Cramer B. J., Lampe P. D., Kurata W. E., Kanemitsu M. Y., Loo L. W., Eckhart W., and Lau A. F. (1996) Characterization of the MAP kinase phosphorylation sites on the connexin43 gap junction protein. J. Biol. Chem. 271, 3779–3786 10.1074/jbc.271.7.3779 [DOI] [PubMed] [Google Scholar]
- 17. Dunn C. A., and Lampe P. D. (2014) Injury-triggered Akt phosphorylation of Cx43: a ZO-1-driven molecular switch that regulates gap junction size. J. Cell Sci. 127, 455–464 10.1242/jcs.142497 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Solan J. L., and Lampe P. D. (2016) Kinase programs spatiotemporally regulate gap junction assembly and disassembly: effects on wound repair. Semin. Cell Dev. Biol. 50, 40–48 10.1016/j.semcdb.2015.12.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Hausenloy D. J., and Yellon D. M. (2004) New directions for protecting the heart against ischaemia-reperfusion injury: targeting the reperfusion injury salvage kinase (RISK)-pathway. Cardiovasc. Res. 61, 448–460 10.1016/j.cardiores.2003.09.024 [DOI] [PubMed] [Google Scholar]
- 20. Hund T. J., Lerner D. L., Yamada K. A., Schuessler R. B., and Saffitz J. E. (2007) Protein kinase Cϵ mediates salutary effects on electrical coupling induced by ischemic preconditioning. Heart Rhythm 4, 1183–1193 10.1016/j.hrthm.2007.05.030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Gray M. O., Karliner J. S., and Mochly-Rosen D. (1997) A selective ϵ-protein kinase C antagonist inhibits protection of cardiac myocytes from hypoxia-induced cell death. J. Biol. Chem. 272, 30945–30951 10.1074/jbc.272.49.30945 [DOI] [PubMed] [Google Scholar]
- 22. Ek-Vitorin J. F., King T. J., Heyman N. S., Lampe P. D., and Burt J. M. (2006) Selectivity of Connexin 43 channels is regulated through protein kinase C-dependent phosphorylation. Circ. Res. 98, 1498–1505 10.1161/01.RES.0000227572.45891.2c [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Doble B. W., Ping P., and Kardami E. (2000) The epsilon subtype of protein kinase C is required for cardiomyocyte connexin-43 phosphorylation. Circ. Res. 86, 293–301 10.1161/01.RES.86.3.293 [DOI] [PubMed] [Google Scholar]
- 24. Srisakuldee W., Jeyaraman M. M., Nickel B. E., Tanguy S., Jiang Z. S., and Kardami E. (2009) Phosphorylation of connexin-43 at serine 262 promotes a cardiac injury-resistant state. Cardiovasc Res. 83, 672–681 10.1093/cvr/cvp142 [DOI] [PubMed] [Google Scholar]
- 25. Miura T., Ohnuma Y., Kuno A., Tanno M., Ichikawa Y., Nakamura Y., Yano T., Miki T., Sakamoto J., and Shimamoto K. (2004) Protective role of gap junctions in preconditioning against myocardial infarction. Am. J. Physiol. Heart Circ. Physiol. 286, H214–221 10.1152/ajpheart.00441.2003 [DOI] [PubMed] [Google Scholar]
- 26. Huang G. Y., Xie L. J., Linask K. L., Zhang C., Zhao X. Q., Yang Y., Zhou G. M., Wu Y. J., Marquez-Rosado L., McElhinney D. B., Goldmuntz E., Liu C., Lampe P. D., Chatterjee B., and Lo C. W. (2011) Evaluating the role of connexin43 in congenital heart disease: screening for mutations in patients with outflow tract anomalies and the analysis of knock-in mouse models. J. Cardiovasc. Dis. Res. 2, 206–212 10.4103/0975-3583.89804 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Remo B. F., Qu J., Volpicelli F. M., Giovannone S., Shin D., Lader J., Liu F. Y., Zhang J., Lent D. S., Morley G. E., and Fishman G. I. (2011) Phosphatase-resistant gap junctions inhibit pathological remodeling and prevent arrhythmias. Circ. Res. 108, 1459–1466 10.1161/CIRCRESAHA.111.244046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Slavi N., Toychiev A. H., Kosmidis S., Ackert J., Bloomfield S. A., Wulff H., Viswanathan S., Lampe P. D., and Srinivas M. (2018) Suppression of connexin 43 phosphorylation promotes astrocyte survival and vascular regeneration in proliferative retinopathy. Proc. Natl. Acad. Sci. U.S.A. 115, E5934–E5943 10.1073/pnas.1803907115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Boengler K., Konietzka I., Buechert A., Heinen Y., Garcia-Dorado D., Heusch G., and Schulz R. (2007) Loss of ischemic preconditioning's cardioprotection in aged mouse hearts is associated with reduced gap junctional and mitochondrial levels of connexin 43. Am. J. Physiol. Heart Circ. Physiol. 292, H1764–H1769 10.1152/ajpheart.01071.2006 [DOI] [PubMed] [Google Scholar]
- 30. Azhar G., Gao W., Liu L., and Wei J. Y. (1999) Ischemia-reperfusion in the adult mouse heart influence of age. Exp. Gerontol. 34, 699–714 10.1016/S0531-5565(99)00031-5 [DOI] [PubMed] [Google Scholar]
- 31. Quan N., Sun W., Wang L., Chen X., Bogan J. S., Zhou X., Cates C., Liu Q., Zheng Y., and Li J. (2017) Sestrin2 prevents age-related intolerance to ischemia and reperfusion injury by modulating substrate metabolism. FASEB J. 31, 4153–4167 10.1096/fj.201700063R [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Sibold S., Roh V., Keogh A., Studer P., Tiffon C., Angst E., Vorburger S. A., Weimann R., Candinas D., and Stroka D. (2007) Hypoxia increases cytoplasmic expression of NDRG1, but is insufficient for its membrane localization in human hepatocellular carcinoma. FEBS Lett. 581, 989–994 10.1016/j.febslet.2007.01.080 [DOI] [PubMed] [Google Scholar]
- 33. Cangul H. (2004) Hypoxia upregulates the expression of the NDRG1 gene leading to its overexpression in various human cancers. BMC Genet. 5, 27 10.1186/1471-2156-5-27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Menezes S. V., Kovacevic Z., and Richardson D. R. (2019) The metastasis suppressor NDRG1 down-regulates the epidermal growth factor receptor via a lysosomal mechanism by up-regulating mitogen-inducible gene 6. J. Biol. Chem. 294, 4045–4064 10.1074/jbc.RA118.006279 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Park K. C., Menezes S. V., Kalinowski D. S., Sahni S., Jansson P. J., Kovacevic Z., and Richardson D. R. (2018) Identification of differential phosphorylation and sub-cellular localization of the metastasis suppressor, NDRG1. Biochim. Biophys. Acta Mol. Basis Dis. 1864, 2644–2663 10.1016/j.bbadis.2018.04.011 [DOI] [PubMed] [Google Scholar]
- 36. Chiao Y. A., and Rabinovitch P. S. (2015) The aging heart. Cold Spring Harb. Perspect. Med. 5, a025148 10.1101/cshperspect.a025148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Przyklenk K., Maynard M., Darling C. E., and Whittaker P. (2008) Aging mouse hearts are refractory to infarct size reduction with post-conditioning. J. Am. Coll. Cardiol. 51, 1393–1398 10.1016/j.jacc.2007.11.070 [DOI] [PubMed] [Google Scholar]
- 38. Gupta A., Leser J. M., Gould N. R., Buo A. M., Moorer M. C., and Stains J. P. (2019) Connexin43 regulates osteoprotegerin expression via ERK1/2-dependent recruitment of Sp1. Biochem. Biophys. Res. Commun. 509, 728–733 10.1016/j.bbrc.2018.12.173 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Solan J. L., and Lampe P. D. (2014) Specific Cx43 phosphorylation events regulate gap junction turnover in vivo. FEBS Lett. 588, 1423–1429 10.1016/j.febslet.2014.01.049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Hausenloy D. J., Mocanu M. M., and Yellon D. M. (2004) Cross-talk between the survival kinases during early reperfusion: its contribution to ischemic preconditioning. Cardiovasc. Res. 63, 305–312 10.1016/j.cardiores.2004.04.011 [DOI] [PubMed] [Google Scholar]
- 41. Jain S. K., Schuessler R. B., and Saffitz J. E. (2003) Mechanisms of delayed electrical uncoupling induced by ischemic preconditioning. Circ. Res. 92, 1138–1144 10.1161/01.RES.0000074883.66422.C5 [DOI] [PubMed] [Google Scholar]
- 42. Zhang Z., Yao L., Yang J., Wang Z., and Du G. (2018) PI3K/Akt and HIF1 signaling pathway in hypoxia ischemia (review). Mol. Med. Rep. 18, 3547–3554 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Chen B., Nelson D. M., and Sadovsky Y. (2006) N-myc down-regulated gene 1 modulates the response of term human trophoblasts to hypoxic injury. J. Biol. Chem. 281, 2764–2772 10.1074/jbc.M507330200 [DOI] [PubMed] [Google Scholar]
- 44. Kitowska A., and Pawełczyk T. (2010) N-myc downstream regulated 1 gene and its place in the cellular machinery. Acta Biochim. Pol. 57, 15–21 [PubMed] [Google Scholar]
- 45. Wu F., Rom W. N., Koshiji M., Mo Y., Hosomi Y., and Tchou-Wong K. M. (2015) Role of GLI1 and NDRG1 in increased resistance to apoptosis induction. J. Environ. Pathol. Toxicol. Oncol. 34, 213–225 10.1615/JEnvironPatholToxicolOncol.2015013472 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Kurdistani S. K., Arizti P., Reimer C. L., Sugrue M. M., Aaronson S. A., and Lee S. W. (1998) Inhibition of tumor cell growth by RTP/rit42 and its responsiveness to p53 and DNA damage. Cancer Res. 58, 4439–4444 [PubMed] [Google Scholar]
- 47. Sun J., Zhang D., Bae D. H., Sahni S., Jansson P., Zheng Y., Zhao Q., Yue F., Zheng M., Kovacevic Z., and Richardson D. R. (2013) Metastasis suppressor, NDRG1, mediates its activity through signaling pathways and molecular motors. Carcinogenesis 34, 1943–1954 10.1093/carcin/bgt163 [DOI] [PubMed] [Google Scholar]
- 48. Hosoi F., Izumi H., Kawahara A., Murakami Y., Kinoshita H., Kage M., Nishio K., Kohno K., Kuwano M., and Ono M. (2009) N-myc downstream regulated gene 1/Cap43 suppresses tumor growth and angiogenesis of pancreatic cancer through attenuation of inhibitor of κB kinase β expression. Cancer Res. 69, 4983–4991 10.1158/0008-5472.CAN-08-4882 [DOI] [PubMed] [Google Scholar]
- 49. Menezes S. V., Fouani L., Huang M. L. H., Geleta B., Maleki S., Richardson A., Richardson D. R., and Kovacevic Z. (2018) The metastasis suppressor, NDRG1, attenuates oncogenic TGF-β and NF-κB signaling to enhance membrane E-cadherin expression in pancreatic cancer cells. Carcinogenesis 10.1093/carcin/bgy178 [DOI] [PubMed] [Google Scholar]
- 50. Chen Z., Zhang D., Yue F., Zheng M., Kovacevic Z., and Richardson D. R. (2012) The iron chelators Dp44mT and DFO inhibit TGF-β-induced epithelial-mesenchymal transition via up-regulation of N-Myc downstream-regulated gene 1 (NDRG1). J. Biol. Chem. 287, 17016–17028 10.1074/jbc.M112.350470 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Kovacevic Z., Menezes S. V., Sahni S., Kalinowski D. S., Bae D. H., Lane D. J., and Richardson D. R. (2016) The metastasis suppressor, N-MYC downstream-regulated gene-1 (NDRG1), down-regulates the ErbB family of receptors to inhibit downstream oncogenic signaling pathways. J. Biol. Chem. 291, 1029–1052 10.1074/jbc.M115.689653 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Kachhap S. K., Faith D., Qian D. Z., Shabbeer S., Galloway N. L., Pili R., Denmeade S. R., DeMarzo A. M., and Carducci M. A. (2007) The N-Myc down-regulated gene1 (NDRG1) is a Rab4a effector involved in vesicular recycling of E-cadherin. PLoS ONE 2, e844 10.1371/journal.pone.0000844 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Jin R., Liu W., Menezes S., Yue F., Zheng M., Kovacevic Z., and Richardson D. R. (2014) The metastasis suppressor NDRG1 modulates the phosphorylation and nuclear translocation of β-catenin through mechanisms involving FRAT1 and PAK4. J. Cell Sci. 127, 3116–3130 10.1242/jcs.147835 [DOI] [PubMed] [Google Scholar]
- 54. Murray J. T., Campbell D. G., Morrice N., Auld G. C., Shpiro N., Marquez R., Peggie M., Bain J., Bloomberg G. B., Grahammer F., Lang F., Wulff P., Kuhl D., and Cohen P. (2004) Exploitation of KESTREL to identify NDRG family members as physiological substrates for SGK1 and GSK3. Biochem. J. 384, 477–488 10.1042/BJ20041057 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Murakami Y., Hosoi F., Izumi H., Maruyama Y., Ureshino H., Watari K., Kohno K., Kuwano M., and Ono M. (2010) Identification of sites subjected to serine/threonine phosphorylation by SGK1 affecting N-myc downstream-regulated gene 1 (NDRG1)/Cap43-dependent suppression of angiogenic CXC chemokine expression in human pancreatic cancer cells. Biochem. Biophys. Res. Commun. 396, 376–381 10.1016/j.bbrc.2010.04.100 [DOI] [PubMed] [Google Scholar]
- 56. Manning B. D., and Toker A. (2017) AKT/PKB signaling: navigating the network. Cell 169, 381–405 10.1016/j.cell.2017.04.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Solan J. L., and Lampe P. D. (2008) Connexin 43 in LA-25 cells with active v-src is phosphorylated on Y247, Y265, S262, S279/282, and S368 via multiple signaling pathways. Cell Commun. Adhes. 15, 75–84 10.1080/15419060802014016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. King T. J., and Lampe P. D. (2004) Mice deficient for the gap junction protein Connexin32 exhibit increased radiation-induced tumorigenesis associated with elevated mitogen-activated protein kinase (p44/Erk1, p42/Erk2) activation. Carcinogenesis 25, 669–680 10.1093/carcin/bgh071 [DOI] [PubMed] [Google Scholar]
- 59. Montalbano A. P., Hawgood S., and Mendelson C. R. (2013) Mice deficient in surfactant protein A (SP-A) and SP-D or in TLR2 manifest delayed parturition and decreased expression of inflammatory and contractile genes. Endocrinology 154, 483–498 10.1210/en.2012-1797 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Jordan K., Solan J. L., Dominguez M., Sia M., Hand A., Lampe P. D., and Laird D. W. (1999) Trafficking, assembly and function of a connexin43-green fluorescent protein chimera in live mammalian cells. Mol. Biol. Cell 10, 2033–2050 10.1091/mbc.10.6.2033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Solan J. L., Marquez-Rosado L., Sorgen P. L., Thornton P. J., Gafken P. R., and Lampe P. D. (2007) Phosphorylation of Cx43 at S365 is a gatekeeper event that changes the structure of Cx43 and prevents downregulation by PKC. J. Cell Biol. 179, 1301–1309 10.1083/jcb.200707060 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Singh D., Solan J. L., Taffet S. M., Javier R., and Lampe P. D. (2005) Connexin 43 interacts with zona occludens-1 and -2 proteins in a cell cycle stage-specific manner. J. Biol. Chem. 280, 30416–30421 10.1074/jbc.M506799200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Fan G. C., Ren X., Qian J., Yuan Q., Nicolaou P., Wang Y., Jones W. K., Chu G., and Kranias E. G. (2005) Novel cardioprotective role of a small heat-shock protein, Hsp20, against ischemia/reperfusion injury. Circulation 111, 1792–1799 10.1161/01.CIR.0000160851.41872.C6 [DOI] [PubMed] [Google Scholar]
- 64. Schneider C. A., Rasband W. S., and Eliceiri K. W. (2012) NIH Image to ImageJ: 25 years of image analysis. Nat. Methods 9, 671–675 10.1038/nmeth.2089 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.