

Abstract
Enhanced osteoclast-mediated bone resorption and diminished formation may promote bone loss. Pleckstrin homology (PH) domain and leucine-rich repeat protein phosphatase 1 (Phlpp1) regulates protein kinase C (PKC) and other proteins in the control of bone mass. Germline Phlpp1 deficiency reduces bone volume, but the mechanisms remain unknown. Here, we found that conditional Phlpp1 deletion in murine osteoclasts increases their numbers, but also enhances bone mass. Despite elevating osteoclasts, Phlpp1 deficiency did not increase serum markers of bone resorption, but elevated serum markers of bone formation. These results suggest that Phlpp1 suppresses osteoclast formation and production of paracrine factors controlling osteoblast activity. Phlpp1 deficiency elevated osteoclast numbers and size in ex vivo osteoclastogenesis assays, accompanied by enhanced expression of proto-oncogene C-Fms (C-Fms) and hyper-responsiveness to macrophage colony-stimulating factor (M-CSF) in bone marrow macrophages. Although Phlpp1 deficiency increased TRAP+ cell numbers, it suppressed actin-ring formation and bone resorption in these assays. We observed that Phlpp1 deficiency increases activity of PKCζ, a PKC isoform controlling cell polarity, and that addition of a PKCζ pseudosubstrate restores osteoclastogenesis and bone resorption of Phlpp1-deficient osteoclasts. Moreover, Phlpp1 deficiency increased expression of the bone-coupling factor collagen triple helix repeat-containing 1 (Cthrc1). Conditioned growth medium derived from Phlpp1-deficient osteoclasts enhanced mineralization of ex vivo osteoblast cultures, an effect that was abrogated by Cthrc1 knockdown. In summary, Phlpp1 critically regulates osteoclast numbers, and Phlpp1 deficiency enhances bone mass despite higher osteoclast numbers because it apparently disrupts PKCζ activity, cell polarity, and bone resorption and increases secretion of bone-forming Cthrc1.
Keywords: cell polarity; phosphatase; Akt PKB; cytoskeleton; protein kinase C (PKC); bone resorption; collagen triple helix repeat containing 1 (CTHRC1); colony-stimulating factor 1 receptor (CSF1R), mineralization; macrophage colony-stimulating factor (M-CSF); protein kinase C zeta (PKC ζ)
Introduction
Bone modeling, remodeling, and repair in response to injury all occur through coordinated bone resorption and bone deposition; thus, cellular activities of osteoclasts and osteoblasts must be tightly coupled to maintain optimal bone health. Osteoclasts function in these processes through direct bone resorbing activity, but also via secretion of paracrine factors that stimulate osteoblast-mediated bone production. Because disruptions of osteoclast differentiation and/or activity impact bone resorption and coupling to bone deposition, a better understanding of osteoclast biology will help design strategies to limit bone loss.
Osteoclasts arise from the fusion of monocyte/macrophage progenitors. This process is facilitated by the actions of two cytokines, macrophage colony-stimulating factor (M-CSF)2 (Csf1) and receptor activator of nuclear factor κB (RANKL), which are necessary and sufficient for osteoclastogenesis. Whereas RANKL is required for definitive osteoclast differentiation, M-CSF promotes the proliferation, survival, and differentiation of osteoclast precursors (1). M-CSF exerts its actions through the C-fms receptor encoded by the Csf1r gene. Binding of M-CSF to the C-fms receptor induces receptor autophosphorylation and activation of downstream signaling kinases including Akt and Mek/Erk.
Once formed, osteoclasts become highly polarized cells. They resorb bone through the secretion of H+ ions and matrix-degrading enzymes through a specialized portion of the basal plasma membrane known as the ruffled border (2). To limit bone resorption to a defined area, osteoclasts must tightly adhere to the bone surface via formation of actin ring structures creating resorption lacunae. Disruptions in osteoclast polarity suppress bone resorption (3).
Phlpp1 (Phlpp, Scop, Plekhe1, Ppm3a) was identified in 2005 by searching the NCBI database for pleckstrin homology (PH) domain containing proteins. It is a member of the underexplored type 2C protein phosphatase family that is insensitive to traditional phosphatase inhibitors (4). Although Phlpp1 is broadly expressed, its levels are controlled by numerous cellular mechanisms including transcription and proteolysis (5). Phlpp1 is poised to be a critical regulator of bone mass because it dephosphorylates and inactivates numerous pathways that promote bone cell function. These substrates include Akt, Raf1, Histone 3, Mst1, p70 S6K, and both the typical (e.g. α/β) and atypical (e.g. ι/ζ) PKC isoforms (4–9). Germline knockout mice of this gene are viable, but show enhanced growth factor responsiveness, cellular proliferation, and survival (4). Our group has shown that Phlpp1 deficiency decreases body size, long bone length, and bone volume (10); however, the cell-type specific functions of Phlpp1 could not be elucidated with this model. Here we define the osteoclast-specific functions of Phlpp1 and demonstrate that deletion of Phlpp1 in mature bone resorbing osteoclasts enhances bone volume and mineral density.
Results
Conditional deletion of Phlpp1 in Ctsk-expressing cells increases bone volume and bone mineral density
Germline deletion of Phlpp1 causes reduction in body size, limb length, and bone mass (10, 11). Although changes in chondrocyte proliferation were noted, the effects of Phlpp1 deletion in other skeletal cell types on the gross phenotype were not determined. To examine the cell type-specific effects of Phlpp1, we generated a novel mouse model that allows for its conditional deletion. LoxP sequences were inserted in introns flanking exon 4 with TALEN technology (Fig. 1A). Sequencing demonstrated successful insertion of the loxP sites. To determine the function of Phlpp1 in mature, bone resorbing osteoclasts, we crossed Phlpp1fl/fl mice with mice expressing Cre recombinase under the control of the Ctsk promoter (Ctsk–Cre) (12). To visualize recombination at the Phlpp1 locus, we isolated DNA from bone marrow macrophage-derived osteoclasts of Cre+ control, Phlpp1fl/+:Cre− and Phlpp1 cHetCtsk mice. Successful recombination of the allele was observed in mature Phlpp1 cHetCtsk osteoclasts (Fig. 1B). Reduction in Phlpp1 levels via qPCR and Western blotting were also noted (Fig. 9, E and G).
Figure 1.

Generation of a Phlpp1-floxed allele. A, LoxP sites were added to the intronic sequence surrounding Exon 4 of the Phlpp1 gene via a TALEN-mediated approach. Cre-mediated recombination of the floxed allele generates a premature stop codon. Sequencing confirmed the presence of both LoxP sites and that no mutations were generated within Exon 4. Genotyping primer locations are noted (FP, forward primer; RP, reverse primer). B, genomic DNA was isolated from mature (day 4) osteoclasts derived from Phlpp1fl/+: Cre−, Phlpp1fl/+: Cre+ and Cre+ mice and assessed by PCR.
Figure 9.

Deletion of Phlpp1 in Ctsk Cre-expressing cells promotes bone formation via production of Cthrc1. Female Phlpp1 cKOCtsk mice (n = 3) and Cre+ control littermates (n = 3) were aged to 12 weeks. Femora were decalcified, embedded, and sectioned. Masons trichrome staining was performed (A) and the number of osteoblasts per total area (B) and the number of osteoblasts per bone perimeter (C) was evaluated by histomorphometry. *, p < 0.05. D, female Phlpp1 cKOCtsk mice (n = 3) and Cre+ control littermates (n = 3) were aged to 12 weeks and calcein was injected 4 days and 24 h prior to euthanasia. Femora were plastic embedded, sectioned, and dynamic histomorphometry was performed to determine the mineralizing surface per bone surface (MS/BS). Bone marrow macrophages were collected from 12-week-old female Phlpp1 cKOCtsk and Cre+ control littermates. Cells were cultured in the presence of 60 ng/ml of RANKL and 25 ng/ml of M-CSF for 4 days. Expression of (E) Phlpp1 and (F) Cthrc1 was determined by qPCR. **, p < 0.01. G, Western blotting was performed as indicated. H, schematic depiction of experiment performed in I and J. I, bone marrow macrophages derived from WT or Phlpp1 cKOCtsk female mice were transfected with siRNAs targeting Cthrc1 or control siRNAs. Western blotting was performed to confirm Cthrc1 knockdown. J, WT calvarial osteoblasts were cultured in conditioned medium derived from female Phlpp1 cKOCtsk or WT littermate osteoclasts transfected with a siRNAs targeting Cthrc1 or control siRNAs (PBS). Alizarin Red staining of male and female WT calvaria was performed.
We next assessed the bone phenotype of 12-week-old Phlpp1 cKOCtsk mice. Radiographic analyses showed no overall reductions in hind limb length or gross abnormalities of male or female mice (Fig. 2A). No cancellous bone changes were noted in male mice; however, micro-CT analyses of the distal femur trabeculae revealed a 55% increase the BV/TV of female Phlpp1cKOCtsk mice as compared with their Cre+ control littermates (Fig. 2D). Likewise, bone mineral density was elevated by 29% in female Phlpp1 cKOCtsk mice compared with their Cre+ control littermates (Fig. 2E). This was accompanied by an increase in trabecular number and a corresponding decrease in trabecular spacing (Fig. 2, F–H). Trabecular thickness was modestly increased (Fig. 2I). Reconstructions of female Phlpp1 cKOCtsk and Cre+ control littermates are shown in Fig. 2, B and C.
Figure 2.

Conditional deletion of Phlpp1 using Ctsk–Cre increases distal femur bone volume and bone mineral density. Female Phlpp1 cKOCtsk mice (n = 3) and Cre+ control littermates (n = 3) were aged to 12 weeks. A, representative radiographs of Cre+ control and Phlpp1 cKOCtsk hind limbs were generated. Micro-CT analyses were performed at the distal femur. Shown are (B) reconstructions from Cre+ control and (C) Phlpp1 cKOCtsk distal femurs, (D) bone volume per total volume, (E) bone mineral density, (F) bone volume, (G) trabecular number, (H) trabecular spacing, and (I) trabecular thickness. ***, p < 0.005; **, p < 0.01; *, p < 0.05.
Other groups have documented activity of Ctsk–Cre drivers within mesenchymal lineage cells including perichondrial cells, hypertrophic chondrocytes, within the groove of Ranvier and potentially by cells within the ovaries and testes (12–17). Phlpp1 was expressed within the growth plate of Phlpp1 cKOCtsk mice and was present within osteoblasts and osteocytes of trabecular and cortical bone (Fig. S1). Recombination of the Phlpp1 allele was not observed within the ovaries, testes, or liver. No changes in Phlpp1 expression within the ovaries, testes, and bone shaft, which is primarily populated by osteocytes, were observed (Fig. S1). No changes in cortical bone were noted in male or female mice. Osteoblasts obtained from Phlpp1 cKOCtsk mice mineralized normally (Fig. S2).
Bone histomorphometric analyses were conducted and confirmed that female Phlpp1 cKOCtsk mice have a 2-fold increase in BV/TV as compared with Cre+ control littermate controls (Fig. 3, A–C). TRAP staining demonstrated increased osteoclast number and osteoclasts per bone surface (Fig. 3, A, B, D, and E). No alteration of osteoclasts per osteoclast perimeter was observed, indicating that osteoclast size was not altered in vivo (Fig. 3F).
Figure 3.

Osteoclast-specific deletion of Phlpp1 increases femoral osteoclast number. Female Phlpp1 cKOCtsk mice (n = 3) and Cre+ control littermates (n = 3) were aged to 12 weeks. A and B, TRAP staining was performed; and C, BV/TV; D, the number of osteoclasts; E, osteoclasts per bone perimeter; and F, osteoclasts per osteoclast perimeter within the distal femur were evaluated by histomorphometry. **, p < 0.01; *, p < 0.05.
We also evaluated the effect of Phlpp1 osteoclast-specific deletion on vertebral bone. Connective density (19%) and trabecular number (14%) were increased within the cranial region of L5 vertebral bodies of female mice (Fig. 4, A and B). Trabecular spacing was suppressed by 17% (Fig. 4C). Modest to negligible changes were observed in trabecular thickness, structure model index, and BV/TV (Fig. 4, D–F).
Figure 4.

Osteoclast-specific deletion of Phlpp1 increases vertebral body bone volume. Female Phlpp1 cKOCtsk mice (n = 3) and Cre+ control littermates (n = 3) were aged to 12 weeks and μCT analyses of the L5 cranial region of the vertebrae were performed. Shown are: (A) connective density, (B) trabecular number, (C) trabecular spacing, (D) trabecular thickness, (E) bone volume and (F) SMI. ***, p < 0.005; *, p < 0.05. SMI, structure model index.
Deletion of Phlpp1 enhances osteoclast numbers and M-CSF responsiveness
To evaluate possible mechanisms for enhanced osteoclast number in Phlpp1 cKOCtsk mice, we collected bone marrow macrophages from Phlpp1fl/fl mice. Cells were transduced with AdGFP or AdCre on day 0 and osteoclastogenesis assays were performed. Genomic DNA was collected from multinucleated cells on day 4 and the recombined Phlpp1 allele in the presence of AdCre transduction was validated (Fig. 5A). AdCre-infected cells expressed less Phlpp1 (Fig. 5, B and C) and increased phosphorylation (p) of Phlpp1 targets, including, pSer-660 PKC and pSer-10 histone 3 (Fig. 5C).
Figure 5.

Phlpp1 deficiency increases ex vivo osteoclastogenesis. Bone marrow macrophages were collected from Phlpp1fl/fl 6-week-old female mice and infected with AdGFP or AdCre (m.o.i. = 300). A, genomic DNA was collected and PCR was performed to validate recombination of the Phlpp1-floxed allele. B, expression of Phlpp1 was determined by qPCR. C, Western blotting was performed as indicated. D, bone marrow macrophages were cultured in the presence of 60 ng/ml of RANKL and increasing amounts of M-CSF (0, 5, 15, 25, and 50 ng/ml) for 4 days. TRAP staining was performed and (E) the number of osteoclasts per field and the (F) average osteoclast area was determined. The dotted line represents a logarithmic curve fit. **, p < 0.01
Phlpp1 deficiency increases receptor tyrosine kinase expression, such as epidermal growth factor receptor, by mouse embryonic fibroblasts (18). To test the hypothesis that Phlpp1 suppression in osteoclasts enhances the receptor tyrosine kinase c-fms expression, we performed M-CSF dose-response assays on Phlpp1fl/fl BMMs transduced with AdGFP or AdCre. M-CSF dose-dependently increased TRAP+ osteoclast numbers and area in AdGFP-transduced cells on day 4 of culture (Fig. 5, D–F). Cells transduced with AdCre were more sensitive to M-CSF at all concentrations tested above 0 and showed an ∼2.7-fold increase in osteoclast number and 1.5-fold increase in osteoclast size compared with AdGFP-transduced cells (Fig. 5, D–F).
We next sought to determine whether Phlpp1 deficiency enhanced M-CSF signaling during osteoclastogenesis. BMMs from female WT or Phlpp1−/− mice were cultured for 3 days with RANKL and M-CSF to select for pre-osteoclast cultures. These cells were then cultured in serum-free medium for 1 h and exposed to M-CSF for the indicated times (Fig. 6A). Phosphorylation of Akt2, Mek1/2, and Erk1/2 was higher following exposure to M-CSF in Phlpp1−/− pre-osteoclasts (Fig. 6A). c-fms expression was higher in Phlpp1−/− BMMs (Fig. 6B), as well as in mature osteoclasts from Phlpp1−/− (Fig. 6C).
Figure 6.

Phlpp1 deficiency increases ex vivo osteoclastogenesis. Bone marrow macrophages were collected from female Phlpp1−/− or WT littermate 6-week-old mice. A, cells were cultured in the presence of 60 ng/ml of RANKL and 25 ng/ml of M-CSF for 3 days. Cultures were then serum starved in the absence of RANKL and M-CSF for 1 h. Following serum starvation, 25 ng/ml of M-CSF was added back to cultures for the indicated times (minutes) and Western blotting was performed. B, Western blotting was performed using bone marrow macrophages. C, cells were cultured in the presence of 30 ng/ml of RANKL and 25 ng/ml of M-CSF for 4 days and c-fms immunofluorescence was performed.
Phlpp1 promotes osteoclast polarization, bone resorption, and represses PKCζ activity
Enhanced responsiveness to M-CSF could in part account for the increase in osteoclast number exhibited by Phlpp1 cKOCtsk mice, but this does not explain increased BV/TV and bone mineral density also observed. Phlpp1 dephosphorylates and destabilizes multiple PKC isoforms. Among those, Phlpp1 inactivates the atypical isoform PKCζ to control cell polarity (19). Establishment of cell polarity is crucial in actin ring formation, development of resorption lacunae, and secretion of bone resorbing enzymes and H+ ions required for osteoclast-mediated bone resorption (20, 21). Osteoclasts derived from Phlpp1 cKOCtsk bone marrow macrophages exhibited disrupted actin rings of lesser thickness as visualized by phalloidin binding (Fig. 7A). This was accompanied by a 37% decreased formation of resorption pits when Phlpp1 cKOCtsk osteoclasts were cultured on bovine bone slices (Fig. 7C). Elevated osteoclast numbers in vivo were confirmed by increased circulating serum TRAP levels (Fig. 7E), but serum CTX-1 levels were not changed in 12-week-old Phlpp1 cKOCtsk female mice (Fig. 7F). In contrast, serum P1NP was elevated by 36%, indicating increased bone formation of the Phlpp1 cKOCtsk female mice (Fig. 7G). Osteoclasts generated from 12-week-old female Phlpp1 cKOCtsk mice also had increased phosphorylation of PKCζ at the activation loop (Ser-410) and Ser-560 within the turn motif as compared with Cre+ control osteoclasts (Fig. 7D).
Figure 7.

Osteoclast cell polarity and bone resorption are diminished by Phlpp1 deficiency. Bone marrow macrophages were collected from female 12-week-old Phlpp1 cKOCtsk and Cre+ control littermates. Cells were cultured in the presence of 60 ng/ml of RANKL and 25 ng/ml of M-CSF for 4 days and (A) phalloidin Alexa 488 and (B) DAPI staining was performed. C, cells were seeded onto bovine bone slices and cultured in the presence of 60 ng/ml of RANKL and 25 ng/ml of M-CSF for 10 days. Resorption pits were visualized by toluidine blue staining. D, Western blotting was performed as indicated. Serum was collected from female 12-week-old Phlpp1 cKOCtsk and WT:Cre+ mice and used in (E) TRAP, (F) CTX-1, and (G) P1NP ELISA. **, p < 0.01.
We next determined if the effects of Phlpp1 deficiency on osteoclastogenesis and bone resorption were due to increased PKCζ activity. Bone marrow macrophages were collected from WT or Phlpp1 cKOCtsk mice and cultured with M-CSF and RANKL. On day 3 of differentiation, cells were exposed to a PKCζ pseudosubstrate (1 μm) for 24 h. On day 4, actin ring formation was visualized by phalloidin Alexa 488 binding. The PKCζ pseudosubstrate partially restored actin ring formation in Phlpp1 cKOCtsk osteoclasts (Fig. 8A). The increase in osteoclast number induced by Phlpp1 deficiency was attenuated by the PKCζ pseudosubstrate (Fig. 8, B and D). We then determined if blocking PKCζ activity likewise restored bone resorption of Phlpp1 cKOCtsk osteoclasts. Blocking PKCζ activity also restored the number and size of pits formed by Phlpp1-deficient osteoclasts (Fig. 8, C, E, and F).
Figure 8.

PKCζ is a downstream target of Phlpp1 that controls osteoclastogenesis and bone resorption. Bone marrow macrophages were collected from female Phlpp1 cKOCtsk and Cre+ control littermates. Cells were cultured in the presence of 60 ng/ml of RANKL and 25 ng/ml of M-CSF. On day 3, cells were exposed to a PKCζ pseudosubstrate for 24 h and (A) phalloidin Alexa 488, DAPI, and phase-contrast images were collected. TRAP staining was performed (B) and the number of osteoclasts was determined (D). *, p < 0.05. C, cells were seeded onto bovine bone slices and cultured in the presence of 60 ng/ml of RANKL and 25 ng/ml of M-CSF for 14 days in the presence of the PKCζ pseudosubstrate or vehicle control (PBS). E and F, resorption pits were visualized by toluidine blue staining and the number of pits (E) and percent of each bone slice resorbed (F) was evaluated using ImageJ software. *, p < 0.05.
Phlpp1 suppresses the coupling of bone resorption to bone formation
Decreased osteoclast activity along with increases in bone suggested that Phlpp1 deficiency also regulated coupling to ossification. We therefore assessed osteoblast numbers within Cre+ control and Phlpp1 cKOCtsk 12-week-old mice and found that osteoblast number per total area was increased by 3.2-fold and osteoblast numbers per bone surface were elevated by 2.4-fold in Phlpp1-deficient females (Fig. 9, A–C). Although there was a trend toward increased mineralizing surface per bone surface (Fig. 9D), measures of dynamic histomorphometry (e.g. MAR, BFR/BS) were unchanged (Fig. S2). We surveyed a panel of known osteoclast coupling factors (Bmp6, Cardiotrophin1, Cthrc1, Ephrin B2, Semaphorin D, Sphk1/2, and Wnt10b) and found that expression of Cthrc1 was elevated by 3.5-fold in Phlpp1 cKOCtsk osteoclasts (Fig. 9, E–G). Expression of all other coupling factors was unchanged by Phlpp1 deficiency. To determine whether increased Cthrc1 expression by Phlpp1-deficient osteoclasts was responsible for enhanced bone formation, we performed an ex vivo coupling experiment (Fig. 9H). Phlpp1 cKOCtsk osteoclast-conditioned medium enhanced Alizarin Red staining of female calvarial osteoblasts. Knockdown of Cthrc1 in Phlpp1 cKOCtsk osteoclast cultures attenuated the effect of conditioned medium on Alizarin Red staining (Fig. 9, I and J). No increase in Alizarin Red staining was observed in male calvarial osteoblasts cultured in Phlpp1 cKOCtsk osteoclast-derived conditioned medium. These data demonstrate that enhanced osteoclast Cthrc1 secretion induced by Phlpp1 deficiency promotes ex vivo osteoblast mineralization.
Discussion
We previously demonstrated that germline deletion of the serine/threonine phosphatase Phlpp1 diminished bone volume, but the mechanism for this phenotype was unclear. To define the cell-specific functions of Phlpp1, we generated a Phlpp1-floxed allele. In this report, we show that deletion of Phlpp1 in Ctsk–Cre-expressing cells causes increases in bone volume and mineral density. Despite elevated osteoclast numbers, bone formation of Phlpp1 cKOCtsk mice is also enhanced. We attribute these findings to decreased osteoclast-mediated bone resorption due to disruption of osteoclast polarity with a concomitant increase in coupling to ossification (see Fig. 10 for a model). The enhanced bone mass and coupling to ossification phenotype is specific to female mice. Phlpp1 controls the activity of nongenomic estrogen signaling mediators (e.g. Akt, Erk1/2); thus, future work will be aimed at determining if the sex-dependent phenotype of Phlpp1 cKOCtsk mice occurs via this mechanism.
Figure 10.
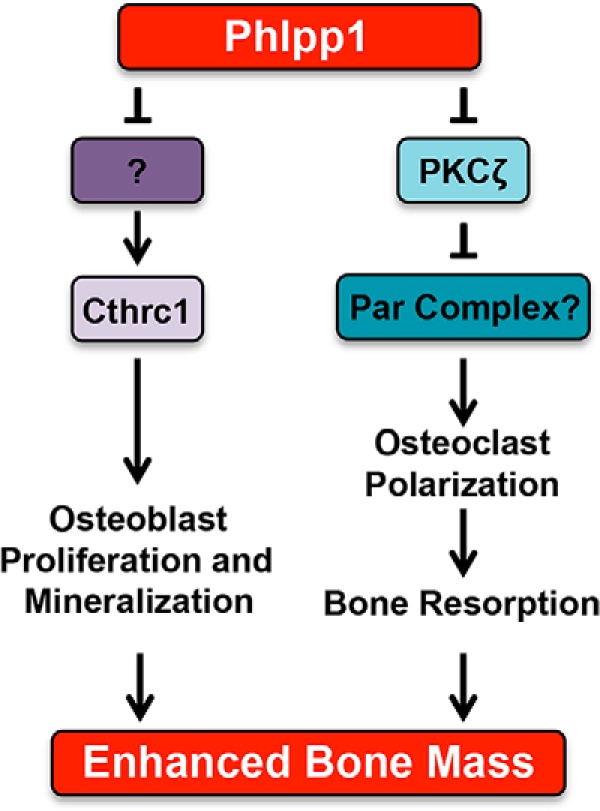
Model depicting how Ctsk-mediated Phlpp1 deletion may affect osteoclastogenesis and bone mass.
The Ctsk–Cre transgene is active in osteoclasts and we have observed recombination of the Phlpp1 allele in osteoclasts using this Cre driver (12). Prior reports demonstrate that Ctsk–Cre drivers can be active within mesenchymal lineage cells including perichondrial cells, hypertrophic chondrocytes, and within the groove of Ranvier (13–15); however, the specific mesenchymal cell populations expressing the Ctsk–Cre driver were not consistent between these studies. We did not see a cortical bone phenotype or change in limb length that was observed with germline deletion of Phlpp1 (10). We therefore attribute our findings to the effects of Phlpp1 deficiency within osteoclasts.
Germline deletion of Phlpp1 results in decreased bone volume. Phlpp1 is widely expressed throughout the body and could impact a number of cell types controlling bone volume (e.g. osteoclast, osteoblasts, brain, and endocrine tissues). To delineate the effects of Phlpp1 within these different cell types we generated the floxed allele described here. The effects of Phlpp1 deletion within other cell types will be explored in future studies.
In this report we show that Phlpp1 represses the receptor tyrosine kinase c-fms. Phlpp1 deficiency was previously shown to decrease histone phosphorylation and acetylation leading to changes in growth factor receptor and RTK expression (18). We attribute increased osteoclastogenesis observed in our ex vivo Phlpp1-deficient cultures to enhanced c-fms expression. Likewise, hyper-responsiveness to M-CSF and enhanced signaling following exposure to M-CSF could be due to increased receptor density. We did observe two Phlpp1 bands in Western blots derived from D3 cells (Fig. 6A), whereas osteoclast precursors or mature D4 cells exhibited a single band (Figs. 5C, 6B, and 7G). There are two Phlpp1 isoforms, so this observation could be explained by a change in Phlpp1 isoform expression. Future work will be aimed at understanding the specific role of each Phlpp1 isoform and how each controls c-fms expression and commitment of progenitor cells to the osteoclast lineage. Increased osteoclast number with concomitant increase in bone is a phenotype that has been observed previously, including in mice with c-Src deficiency (22). Src is a nonreceptor tyrosine kinase, as such it would be interesting to evaluate the effects of Phlpp1 on c-Src expression and/or activity.
Here we show Phlpp1 deficiency leads to enhanced PKCζ activity within osteoclasts concomitant with disruptions in actin ring formation and bone resorption that are restored by the addition of a PKCζ pseudosubstrate. Osteoclast-mediated bone resorption depends on cellular polarization, with the basal cell membrane forming a tight seal against the bone via actin ring formation. Osteoclasts also establish the ruffled cell border along this membrane that promotes the secretion of matrix degrading enzymes and H+ ions. In other cell types, establishment of cell polarity is controlled by PKCζ activity (19). Full PKCζ kinase activity requires phosphorylation of the activation loop and the turn motif at Ser-410 and Ser-560, respectively (23–25). This is antagonized by Phlpp1-mediated dephosphorylation of PKCζ, leading to disruption of the Par complex that facilitates cell polarity (19, 26, 27). Likewise, in macrophages PKCζ regulates actin polymerization during phagocytosis (28). The PKCζ-Par complex has been observed in osteoclasts (20); but determining the requirement of PKCζ activity and Par-complex function has not been evaluated and is a subject for future study.
Increased bone mass and mineral density that occurred along with increased osteoclast numbers also suggested that Phlpp1 deficiency within Ctsk-expressing cells enhanced coupling to bone formation. We surveyed a panel of known ossification coupling factors produced by osteoclasts, including Bmp6, Cardiotrophin1, Cthrc1, Ephrin B2, Semaphorin D, Sphk1/2, and Wnt10b. Of these, only Cthrc1 expression was elevated by Phlpp1-deficient osteoclasts. Cthrc1 is a hormone produced by bone cells and functions within the Wnt/PCP pathway (29–31). Germline deletion of Cthrc1 diminishes bone mass and bone formation; in contrast, forced Cthrc1 expression by osteoblasts increases bone mass (30). Cthrc1 is also produced by osteoclasts, and expression markedly increases during differentiation. Osteoclast-specific deletion of Cthrc1 decreases bone mass, whereas osteoblast-specific deletion does not (31). Our results demonstrate that Phlpp1 suppresses expression of the bone-coupling factor Cthrc1. The mechanism by which Phlpp1 controls Cthrc1 transcript levels will be explored in future studies. Together our data demonstrate that Phlpp1 deficiency enhances bone mass while preserving osteoclast numbers due to loss of cell polarity and bone resorption concomitant with increased bone formation coupling facilitated by Cthrc1.
Experimental procedures
Generation of Phlpp1 conditional knockout mice
Phlpp1-floxed/floxed (Phlpp1fl/fl) mice were generated by insertion of loxP sites surrounding exon 4 using a TALEN-mediated approach (Fig. 1). Phlpp1fl/fl mice were crossed with mice expressing Cre-recombinase under the control of the Ctsk promoter to delete Phlpp1 within Ctsk-expressing cells (12). Mice were genotyped for Cre as previously described (32) or the Phlpp1-floxed allele using the following primers: forward: 5′-CAGTGGATATCTGGATAATC-3′, reverse: 5′-GATGAGTGTTTTCATGAGGA-3′. Conditional knockout animals from these crossings are referred to as Phlpp1 cKOCtsk mice and are on the C57Bl/6 background. Cre+ control littermates from crossings were used as controls as appropriate. Phlpp1−/− animals were genotyped as previously described (11). Animals were housed in an accredited facility under a 12-h light/dark cycle and provided water and food ad libitum. All animal research was conducted according to guidelines provided by the National Institute of Health and the Institute of Laboratory Animal Resources, National Research Council. The Mayo Clinic Institutional Animal Care and Use Committee approved all animal studies.
Radiographs and micro-computed tomography
Radiographs of the right hind limb of 12-week-old mice were collected using a Faxitron X-ray imaging cabinet (Faxitron Bioptics, Tucson, AZ). Femurs from 12-week-old male and female Phlpp1 cKOCtsk mice (n = 3) and their Cre+ control littermates (n = 3) were isolated and fixed in 10% neutral buffered formalin for 48 h. Femurs were then stored in 70% ethanol prior to scanning at 70 kV, 221 ms with a 10.5-μm voxel size using a Scanco Viva40 micro-CT. For cortical bone analyses, a region of interest was defined at 10% of total femur length beginning at the femoral midpoint; defining the outer cortical shell and running a midshaft analysis with 260-threshold air filling correction analyzed samples. For trabecular measurements, a region of interest was defined at 10% of total femur length starting immediately proximal to the growth plate; samples were analyzed using a 220-threshold air filling correction.
Histology and static and dynamic bone histomorphometry
Following micro-CT analyses, femurs from 12-week-old mice were decalcified in 15% EDTA for 14 days. Tissues were paraffin embedded and 5-μm sections were collected and TRAP/Fast Green stained (Sigma, number 387A-1KT). Standardized histomorphometry was performed using Osteomeasure software (33). Calcein injections (10 mg/kg, i.p.) were also administered to mice 4 days and 1 day prior to euthanasia at 12 weeks of age as previously described (32). Femurs from calcein-labeled mice were fixed in 10% neutral buffered formalin for 48 h, transferred to 70% ethanol, and embedded in plastic. Calcein labeling was evaluated using standard histomorphometric techniques as previously described (32).
ELISAs for serum markers of bone formation and resorption
Serum was collected from 12-week-old Phlpp1 cKOCtsk (n = 3) female mice and their Cre+ control littermates (n = 3) and stored at −80 °C. An enzyme-linked immunosorbent assays (ELISA) for bone resorption (CTX-1) was performed in duplicate using 20 μl of serum from each mouse according to the manufacturer's specifications (Ratlaps (CTX-1) number AC-06F1, Immunodiagnostics Systems). Bone formation was assessed using the ELISA for serum P1NP performed in duplicate using 5 μl of serum from each mouse as described by the manufacturer (Ratlaps (P1NP), number AC-33F1, Immunodiagnostics Systems).
Ex vivo osteoclastogenesis, transfection, and bovine bone resorption assays
Bone marrow macrophages were collected from 4- to 6-week-old WT, Phlpp1 cKOCtsk, or Phlpp1fl/fl male or female mice as previously described (34). Cells were cultured in phenol red-free α-MEM overnight in the presence of 35 ng/ml of rM-CSF (number 410-ML, R&D Systems, Minneapolis, MN). Nonadherent cells were collected and cultured with 60 ng/ml of rRANKL (number 315-11, PreproTech, Rocky Hill, NJ) and 35 ng/ml of M-CSF. For dose-response assays using Phlpp1fl/fl cells, cells were exposed to increasing M-CSF concentrations and infected with adenoviral (Ad) GFP or AdCre at an m.o.i. of 300 as previously described (35–37). The PKCζ pseudosubstrate (number 1791, Tocris, Minneapolis, MN) was used at 1 μm and added at day 3 for counting assays. For bone resorption assays, nonadherent cells were seeded onto bovine bone slices (number NC1309388, Fisher Scientific) cultured with 60 ng/ml of RANKL and 35 ng/ml of M-CSF in 96-well plates. Cells were fed every 3 to 4 days with phenol red-free α-MEM supplemented with 35 ng/ml of M-CSF and 60 ng/ml of RANKL. For PKCζ pseudosubstrate experiments, bone marrow macrophages from WT and Phlpp1 cKOCtsk mice were cultured on bovine bone slices in the presence of M-CSF and RANKL. On day 3, the PKCζ pseudosubstrate was added to cultures. On day 14, cells were lysed with 10% domestic bleach and bone slices were stained with 1% toluidine blue. For knockdown experiments, ON-TARGET plus siRNA smart pools targeting Cthrc1 or nontargeting siRNAs were purchased from Dharmacon (Cthrc1 siRNA ON-TARGETplus SMARTpool, number 68588, ON-TARGETplus Nontargeting Pool, number D-001810-10, Lafayette, CO). Osteoclast precursor cells were transfected on day 1 with each siRNA using Lipofectamine RNAiMax at a 1:1 ratio.
Osteoblast cell mineralization assays
Calvarial osteoblasts were collected from P7 male or female WT mice as previously described (38). Osteoblasts were cultured (0.65 × 106 cells/cm2) in conditioned medium derived from Cthrc1 or Control siRNA-transfected female Phlpp1 cKOCtsk or WT littermate osteoclasts that was diluted 1:1 in fresh α-MEM and supplemented with 20% FBS, 50 μg/ml of ascorbate, 10 mm β-glycerolphosphate, 1 × 107 m dexamethasone. Cultures were fed every 3–4 days with respective conditioned medium plus osteogenic supplements. Cells were fixed and stained with Alizarin Red on day 14.
RNA extraction and semi-quantitative PCR
Total RNA was extracted from primary osteoclasts using TRIzol (Invitrogen) and chloroform, and 2 μg was reverse transcribed using the SuperScript III first-strand synthesis system (Invitrogen). The resulting cDNAs were used to assay gene expression via real-time PCR using the following gene-specific primers: Phlpp1 (5′-CTCCAAGGTTGCATCACAGC-3′, 5′-CGCAGGGCATTGCAAGATAC-3′); Cthrc1 (5′-ATCCCAGGTCGGGATGGATT-3′, 5′-CGTGAATGTACACTCCGCAA-3′); and Tubulin (5′-CTGCTCATCAGCAAGATCAGAG-3′, 5′-GCATTATAGGGCTCCACCACAG-3′) (32). Fold-changes in gene expression for each sample were calculated using the 2−ΔΔCq method relative to control after normalization of gene-specific Cq values to tubulin Cq values (32). Each experiment was performed in triplicate and repeated at least three times. Results from a representative experiment are shown.
Western blotting
Cell lysates were collected in a buffered SDS solution (0.1% glycerol, 0.01% SDS, 0.1 m Tris, pH 6.8) on ice. Total protein concentrations were obtained with the Bio-Rad DC assay (Bio-Rad). Proteins (20 μg) were then resolved by SDS-PAGE and transferred to a polyvinylidene difluoride membrane. Western blotting was performed with antibodies (1:2000 dilution) for phospho-Thr-202/Tyr-204-Erk1/2 (Cell Signaling Technology, number 4370), Erk1/2 (Cell Signaling Technology, number 4695), phospho-Ser-217/221 Mek1/2 (Cell Signaling Technology, number 9154), Mek1/2 (Cell Signaling Technology, number 4694), phospho-Ser-474-Akt2 (Cell Signaling Technology, number 8599), Akt (Cell Signaling Technology, number 4691), phosho-Ser-660-PKC (Cell Signaling Technology, number 9371), PKCζ (Cell Signaling Technology, number 9368), c-fms (Cell Signaling Technology, number 3152), phospho-Ser-10 Histone 3 (Cell Signaling Technology, number 3377), phospho-Ser-410 PKCζ (Invitrogen, number MA5-15060), phospho-Ser-560 PKCζ (Invitrogen, number PA5-38419), Cthrc1 (Invitrogen, number PA5-38051), Phlpp1 (Millipore, number 07-1341), Histone 3 (Millipore, number 05-928), PKCα and PKCβ (BD Biosciences), actin (Sigma, A2228), and tubulin (Developmental Studies Hybridoma Bank, E7) and corresponding secondary antibodies conjugated to horseradish peroxidase (Cell Signaling Technology). Antibody binding was detected with the Supersignal West Femto Chemiluminescent Substrate (Pierce Biotechnology, Rockford, IL). Each experiment was repeated at least three times, and data from a representative experiment are shown.
TRAP and immunofluorescence staining
Cells were fixed in 10% neutral buffered formalin for 10 min and then washed 3 times with phosphate-buffered saline (PBS). Fixed cells were TRAP stained using the Acid Phosphatase, Leukocyte (TRAP) Kit (number 387A-1KT, Sigma). For c-fms immunofluorescence, cells were permeabilized with ice-cold methanol for 10 min at −20 °C, blocked in PBS, 5% normal goat serum, 0.3% Triton X-100, and then washed 3 times with PBS. Cells were then incubated with an anti-c-fms antibody (diluted 1:50 in PBS, 1% BSA, 0.3% Triton X-100) overnight at 4 °C. Cells were then washed 3 times with PBS, and incubated with an Alexa 488-coupled goat anti-rabbit antibody (number 150077, Abcam, Cambridge, MA) diluted 1:200 in PBS, 1% BSA, 0.3% Triton X-100. For phalloidin staining, cells were incubated in 0.33 μm phallodin Alexa 488 (number 8878, Cell Signaling Technology) for 15 min. Cells were washed once with PBS and staining was visualized using wide-field fluorescence. Each experiment was repeated at least three times, and data from a representative experiment are shown.
Imaging and quantification
For osteoclastogenesis experiments, three images were collected using a ×10 objective per cover glass. Three cover glasses were used per experiment. The number and area of each image were quantified using ImageJ software. The average osteoclast area and average number of osteoclasts per field were determined. A logarithmic curve fit was applied to describe osteoclast number and area data resulting from increasing M-CSF concentrations. Each experiment was repeated independently three times.
Statistics
Data obtained are the mean ± S.D. p values were determined with the Student's t test when only one experimental comparison was made. For assessment of significance with greater than two conditions, a one-way analysis of variance was performed. p < 0.05 was considered statistically significant. Statistical analyses were performed using GraphPad Prism 7 software.
Author contributions
A. M. M., M. J. O., J. J. W., and E. W. B. conceptualization; A. M. M., D. L. B., D. H. H. M., M. A. M., and E. W. B. data curation; A. M. M., D. L. B., and E. W. B. investigation; A. M. M. and E. W. B. methodology; A. M. M., D. L. B., D. H. H. M., M. A. M., M. J. O., J. J. W., and E. W. B. writing-review and editing; D. H. H. M., M. A. M., and E. W. B. formal analysis; M. J. O., J. J. W., and E. W. B. supervision; M. J. O., J. J. W., and E. W. B. funding acquisition; J. J. W. resources; J. J. W. project administration; E. W. B. writing-original draft.
Supplementary Material
Acknowledgments
We thank Xiaodong Li and Elizabeth Zars for technical assistance and the Mayo Clinic Bone Histomorphometry Core.
This work supported by National Institutes of Health Research Grants AR065397, AR072634, AR068103, AR065402, and AR067129, the Mayo Clinic Center for Biomedical Discovery, and the Mayo Clinic Foundation. The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
This article contains Figs. S1 and S2.
- M-CSF
- macrophage colony-stimulating factor
- RANKL
- receptor activator of nuclear factor κB
- PH
- pleckstrin homology
- BV/TV
- bone volume per total volume
- AdGFP
- adenoviral GFP
- AdCre
- adenoviral Cre recombinase
- α-MEM
- α-minimal essential medium
- Cq
- quantitation cycle
- TRAP
- tartrate-resistant acid phosphatase
- DAPI
- 4′,6-diamidino-2-phenylindole
- PKC
- protein kinase C
- BMM
- bone marrow macrophages
- qPCR
- quantitative PCR
- PHLPP1
- PH domain and leucine-rich repeat protein phosphatase 1
- CTHRC1
- collagen triple helix repeat-containing 1.
References
- 1. Ross F. P. (2006) M-CSF, c-Fms, and signaling in osteoclasts and their precursors. Ann. N.Y. Acad. Sci. 1068, 110–116 10.1196/annals.1346.014 [DOI] [PubMed] [Google Scholar]
- 2. Coxon F. P., and Taylor A. (2008) Vesicular trafficking in osteoclasts. Semin. Cell Dev. Biol. 19, 424–433 [DOI] [PubMed] [Google Scholar]
- 3. Baron R. (1989) Polarity and membrane transport in osteoclasts. Connect. Tissue Res. 20, 109–120 10.3109/03008208909023879 [DOI] [PubMed] [Google Scholar]
- 4. Grzechnik A. T., and Newton A. C. (2016) PHLPPing through history: a decade in the life of PHLPP phosphatases. Biochem. Soc. Trans. 44, 1675–1682 10.1042/BST20160170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Gao T., Furnari F., and Newton A. C. (2005) PHLPP: a phosphatase that directly dephosphorylates Akt, promotes apoptosis, and suppresses tumor growth. Mol. Cell 18, 13–24 10.1016/j.molcel.2005.03.008 [DOI] [PubMed] [Google Scholar]
- 6. Newton A. C., and Trotman L. C. (2014) Turning off AKT: PHLPP as a drug target. Annu. Rev. Pharmacol. Toxicol. 54, 537–558 10.1146/annurev-pharmtox-011112-140338 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Warfel N. A., and Newton A. C. (2012) Pleckstrin homology domain leucine-rich repeat protein phosphatase (PHLPP): a new player in cell signaling. J. Biol. Chem. 287, 3610–3616 10.1074/jbc.R111.318675 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Brognard J., Sierecki E., Gao T., and Newton A. C. (2007) PHLPP and a second isoform, PHLPP2, differentially attenuate the amplitude of Akt signaling by regulating distinct Akt isoforms. Mol. Cell 25, 917–931 10.1016/j.molcel.2007.02.017 [DOI] [PubMed] [Google Scholar]
- 9. Gao T., Brognard J., and Newton A. C. (2008) The phosphatase PHLPP controls the cellular levels of protein kinase C. J. Biol. Chem. 283, 6300–6311 10.1074/jbc.M707319200 [DOI] [PubMed] [Google Scholar]
- 10. Bradley E. W., Carpio L. R., Newton A. C., and Westendorf J. J. (2015) Deletion of the PH-domain and leucine rich repeat protein phosphatase 1 (Phlpp1) increases fibroblast growth factor (Fgf) 18 expression and promotes chondrocyte proliferation. J. Biol. Chem. 290, 16272–16280 10.1074/jbc.M114.612937 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Masubuchi S., Gao T., O'Neill A., Eckel-Mahan K., Newton A. C., and Sassone-Corsi P. (2010) Protein phosphatase PHLPP1 controls the light-induced resetting of the circadian clock. Proc. Natl. Acad. Sci. U.S.A. 107, 1642–1647 10.1073/pnas.0910292107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Chiu W. S., McManus J. F., Notini A. J., Cassady A. I., Zajac J. D., and Davey R. A. (2004) Transgenic mice that express Cre recombinase in osteoclasts. Genesis 39, 178–185 10.1002/gene.20041 [DOI] [PubMed] [Google Scholar]
- 13. Ruiz P., Martin-Millan M., Gonzalez-Martin M. C., Almeida M., González-Macias J., and Ros M. A. (2016) CathepsinKCre mediated deletion of betacatenin results in dramatic loss of bone mass by targeting both osteoclasts and osteoblastic cells. Sci. Rep. 6, 36201 10.1038/srep36201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Yang W., Wang J., Moore D. C., Liang H., Dooner M., Wu Q., Terek R., Chen Q., Ehrlich M. G., Quesenberry P. J., and Neel B. G. (2013) Ptpn11 deletion in a novel progenitor causes metachondromatosis by inducing hedgehog signalling. Nature 499, 491–495 10.1038/nature12396 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Debnath S., Yallowitz A. R., McCormick J., Lalani S., Zhang T., Xu R., Li N., Liu Y., Yang Y. S., Eiseman M., Shim J. H., Hameed M., Healey J. H., Bostrom M. P., Landau D. A., and Greenblatt M. B. (2018) Discovery of a periosteal stem cell mediating intramembranous bone formation. Nature 562, 133–139 10.1038/s41586-018-0554-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Winkeler C. L., Kladney R. D., Maggi L. B. Jr., and Weber J. D. (2012) Cathepsin K-Cre causes unexpected germline deletion of genes in mice. PloS One 7, e42005 10.1371/journal.pone.0042005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Nakamura T., Imai Y., Matsumoto T., Sato S., Takeuchi K., Igarashi K., Harada Y., Azuma Y., Krust A., Yamamoto Y., Nishina H., Takeda S., Takayanagi H., Metzger D., Kanno J., Takaoka K., Martin T. J., Chambon P., and Kato S. (2007) Estrogen prevents bone loss via estrogen receptor α and induction of Fas ligand in osteoclasts. Cell 130, 811–823 10.1016/j.cell.2007.07.025 [DOI] [PubMed] [Google Scholar]
- 18. Reyes G., Niederst M., Cohen-Katsenelson K., Stender J. D., Kunkel M. T., Chen M., Brognard J., Sierecki E., Gao T., Nowak D. G., Trotman L. C., Glass C. K., and Newton A. C. (2014) Pleckstrin homology domain leucine-rich repeat protein phosphatases set the amplitude of receptor tyrosine kinase output. Proc. Natl. Acad. Sci. U.S.A. 111, E3957–3965 10.1073/pnas.1404221111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Xiong X., Li X., Wen Y. A., and Gao T. (2016) Pleckstrin homology (PH) domain leucine-rich repeat protein phosphatase controls cell polarity by negatively regulating the activity of atypical protein kinase C. J. Biol. Chem. 291, 25167–25178 10.1074/jbc.M116.740639 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Ito Y., Teitelbaum S. L., Zou W., Zheng Y., Johnson J. F., Chappel J., Ross F. P., and Zhao H. (2010) Cdc42 regulates bone modeling and remodeling in mice by modulating RANKL/M-CSF signaling and osteoclast polarization. J. Clin. Investig. 120, 1981–1993 10.1172/JCI39650 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Jurdic P., Saltel F., Chabadel A., and Destaing O. (2006) Podosome and sealing zone: specificity of the osteoclast model. Eur. J. Cell Biol. 85, 195–202 10.1016/j.ejcb.2005.09.008 [DOI] [PubMed] [Google Scholar]
- 22. Boyce B. F., Yoneda T., Lowe C., Soriano P., and Mundy G. R. (1992) Requirement of pp60 c-src expression for osteoclasts to form ruffled borders and resorb bone in mice. J. Clin. Investig. 90, 1622–1627 10.1172/JCI116032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Li X., and Gao T. (2014) mTORC2 phosphorylates protein kinase Cζ to regulate its stability and activity. EMBO Rep. 15, 191–198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Newton A. C. (2003) Regulation of the ABC kinases by phosphorylation: protein kinase C as a paradigm. Biochem. J. 370, 361–371 10.1042/bj20021626 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Suzuki A., Yamanaka T., Hirose T., Manabe N., Mizuno K., Shimizu M., Akimoto K., Izumi Y., Ohnishi T., and Ohno S. (2001) Atypical protein kinase C is involved in the evolutionarily conserved par protein complex and plays a critical role in establishing epithelia-specific junctional structures. J. Cell Biol. 152, 1183–1196 10.1083/jcb.152.6.1183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Durgan J., Kaji N., Jin D., and Hall A. (2011) Par6B and atypical PKC regulate mitotic spindle orientation during epithelial morphogenesis. J. Biol. Chem. 286, 12461–12474 10.1074/jbc.M110.174235 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Linch M., Sanz-Garcia M., Rosse C., Riou P., Peel N., Madsen C. D., Sahai E., Downward J., Khwaja A., Dillon C., Roffey J., Cameron A. J., and Parker P. J. (2014) Regulation of polarized morphogenesis by protein kinase Cι in oncogenic epithelial spheroids. Carcinogenesis 35, 396–406 10.1093/carcin/bgt313 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Allen L. A., and Allgood J. A. (2002) Atypical protein kinase C-zeta is essential for delayed phagocytosis of Helicobacter pylori. Curr. Biol. 12, 1762–1766 10.1016/S0960-9822(02)01216-2 [DOI] [PubMed] [Google Scholar]
- 29. Yamamoto S., Nishimura O., Misaki K., Nishita M., Minami Y., Yonemura S., Tarui H., and Sasaki H. (2008) Cthrc1 selectively activates the planar cell polarity pathway of Wnt signaling by stabilizing the Wnt-receptor complex. Dev. Cell 15, 23–36 10.1016/j.devcel.2008.05.007 [DOI] [PubMed] [Google Scholar]
- 30. Kimura H., Kwan K. M., Zhang Z., Deng J. M., Darnay B. G., Behringer R. R., Nakamura T., de Crombrugghe B., and Akiyama H. (2008) Cthrc1 is a positive regulator of osteoblastic bone formation. PloS One 3, e3174 10.1371/journal.pone.0003174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Takeshita S., Fumoto T., Matsuoka K., Park K. A., Aburatani H., Kato S., Ito M., and Ikeda K. (2013) Osteoclast-secreted CTHRC1 in the coupling of bone resorption to formation. J. Clin. Invest. 123, 3914–3924 10.1172/JCI69493 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Razidlo D. F., Whitney T. J., Casper M. E., McGee-Lawrence M. E., Stensgard B. A., Li X., Secreto F. J., Knutson S. K., Hiebert S. W., and Westendorf J. J. (2000) Histone deacetylase 3 depletion in osteo/chondroprogenitor cells decreases bone density and increases marrow fat. PloS One 5, e11492 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Dempster D. W., Compston J. E., Drezner M. K., Glorieux F. H., Kanis J. A., Malluche H., Meunier P. J., Ott S. M., Recker R. R., and Parfitt A. M. (2013) Standardized nomenclature, symbols, and units for bone histomorphometry: a 2012 update of the report of the ASBMR Histomorphometry Nomenclature Committee. J. Bone Miner. Res. 28, 2–17 10.1002/jbmr.1805 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Bradley E. W., and Oursler M. J. (2008) Osteoclast culture and resorption assays. Methods Mol. Biol. 455, 19–35 10.1007/978-1-59745-104-8_2 [DOI] [PubMed] [Google Scholar]
- 35. Bradley E. W., Ruan M. M., and Oursler M. J. (2008) Novel pro-survival functions of the Kruppel-like transcription factor Egr2 in promotion of macrophage colony-stimulating factor-mediated osteoclast survival downstream of the MEK/ERK pathway. J. Biol. Chem. 283, 8055–8064 10.1074/jbc.M709500200 [DOI] [PubMed] [Google Scholar]
- 36. Bradley E. W., Ruan M. M., and Oursler M. J. (2008) PAK1 is a novel MEK-independent raf target controlling expression of the IAP survivin in M-CSF-mediated osteoclast survival. J. Cell. Physiol. 217, 752–758 10.1002/jcp.21550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Bradley E. W., Ruan M. M., Vrable A., and Oursler M. J. (2008) Pathway crosstalk between Ras/Raf and PI3K in promotion of M-CSF-induced MEK/ERK-mediated osteoclast survival. J. Cell. Biochem. 104, 1439–1451 10.1002/jcb.21719 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. McGee-Lawrence M. E., Carpio L. R., Bradley E. W., Dudakovic A., Lian J. B., van Wijnen A. J., Kakar S., Hsu W., and Westendorf J. J. (2014) Runx2 is required for early stages of endochondral bone formation but delays final stages of bone repair in Axin2-deficient mice. Bone 66, 277–286 10.1016/j.bone.2014.06.022 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.