

Abstract
The Notch receptor is a key mediator of developmental programs and cell-fate decisions. Imbalanced Notch signaling leads to developmental disorders and cancer. To fully characterize the Notch signaling pathway and exploit it in novel therapeutic interventions, a comprehensive view on the regulation and requirements of Notch signaling is needed. Notch is regulated at different levels, ranging from ligand binding, stability to endocytosis. Using an array of different techniques, including reporter gene assays, immunocytochemistry, and ChIP-qPCR we show here, to the best of our knowledge for the first time, regulation of Notch signaling at the level of the nuclear pore. We found that the nuclear pore protein Nup214 (nucleoporin 214) and its interaction partner Nup88 negatively regulate Notch signaling in vitro and in vivo in zebrafish. In mammalian cells, loss of Nup88/214 inhibited nuclear export of recombination signal-binding protein for immunoglobulin κJ region (RBP-J), the DNA-binding component of the Notch pathway. This inhibition increased binding of RBP-J to its cognate promoter regions, resulting in increased downstream Notch signaling. Interestingly, we also found that NUP214 fusion proteins, causative for certain cases of T-cell acute lymphatic leukemia, potentially contribute to tumorigenesis via a Notch-dependent mechanism. In summary, the nuclear pore components Nup88/214 suppress Notch signaling in vitro, and in zebrafish, nuclear RBP-J levels are rate-limiting factors for Notch signaling in mammalian cells, and regulation of nucleocytoplasmic transport of RBP-J may contribute to fine-tuning Notch activity in cells.
Keywords: Notch pathway, nuclear pore, nuclear transport, leukemia, cell compartmentalization, cancer, cell signaling, gene regulation, nucleoporin 214 (Nup214), nucleoporin 88 (Nup88), recombination signal–binding protein for immunoglobulin kappa J region (RBP-J), T-cell acute lymphatic leukemia (T-ALL)
Introduction
Notch is required for cell–cell communication and regulates cell fate (1–3). Notch undergoes three proteolytical cleavage events, first by Furin (4), then, after ligand-binding, by ADAM/TACE (5); and finally by γ-secretase (6). γ-Secretase releases the Notch intracellular domain (NICD),2 which is imported into the nucleus. There, it interacts with the DNA-binding protein RBP-J, also called CSL, and Mastermind proteins to control gene expression (7, 8). To fine-tune and control Notch signaling, a tight network of Notch activity control instances has evolved. Notch signal modulation can include acetylation (9), glycosylation (10), methylation (11), or ubiquitination (12–16) of the receptor. NICD activity can be regulated via phosphorylation by several kinases (17–20). Apart from post-translational modifications, the cleavage of Notch may also be regulated (21, 22).
Nucleocytoplasmic transport is mediated by exportins and importins that shuttle proteins through the nuclear pore (23, 24). Nuclear pore protein (Nup, or NUP in case of humans; we use Nup throughout the text for mammalian Nup and nup for zebrafish Nup) 214 is a peripheral FG-repeat Nup and is located on the cytoplasmic face of the nuclear pore. Together with its binding partner Nup88, Nup214 was shown to be involved in nuclear import and export (25–28). Interestingly, Nup214 fusion proteins were found in ∼10% of T-cell acute lymphatic leukemia (T-ALL) cases, a fatal leukemia that is strongly associated with aberrant Notch signaling (29, 30).
We here identified Nup88 and Nup214 as novel negative regulators of Notch signaling in vitro and in vivo. Nup88/214 negatively regulates the Notch co-factor RBP-J by decreasing its nuclear levels, thus reducing NICD-dependent transcription. T-ALL–associated Nup214 fusion proteins phenocopied a loss of Nup214 and up-regulated Notch, potentially contributing to tumorigenesis.
Results and discussion
Knockdown of Nup88/214 increases Notch signaling in vitro and in vivo
Here we describe a novel role for the nuclear pore components Nup88 and Nup214 in Notch signaling. Using a constitutively active Notch (NotchΔE) tagged with EGFP (31), we screened siRNAs for modulators of Notch trafficking. Surprisingly, siRNAs against Nup214 did not change trafficking of NotchΔE/NICD-EGPF but increased Notch signaling.
To further get insights into the mechanism, we transfected human cells with siRNAs against Nup214 and a luciferase-based Notch reporter with 12× CSL-binding sites (31). Stability of Nup214 depends on its binding partner Nup88 (32), therefore knockdown (KD) of Nup88 was also tested. KD of either Nup strongly induced Notch signaling, whereas the KD of an unrelated Nup, Nup358, had only a small effect (Fig. 1A). The induction of Notch signaling upon KD of Nup88 or Nup214 was blocked by the γ-secretase inhibitor (GSI) DAPT (Fig. 1A). This suggests that the KD affects canonical NICD- and CSL/RBP-J–mediated Notch signaling.
Figure 1.

Nup88 and Nup214 negatively regulate canonical Notch signaling. A–C, PC3 cells were transfected with 12× CSL luciferase Notch reporter and siRNAs as indicated. 24 h later cells were harvested and subjected to luciferase assay (A) or probed with the indicated antibodies for protein expression (B) and immunofluorescence (C). A displays the means ± S.E. of n = 3 independent experiments. Arrows depict nuclear rim staining indicative for nuclear pores. Mab414, anti–FG-repeat antibody. Scale bar, 10 μm. NPC, nuclear pore complex. D, PC3 cells transfected with increasing amounts of Nup214 siRNA were subjected to qPCR with primers specific for Nup214 and HES1. Displayed are the means of two independent experiments and S.E. E, PC3 cells transfected with 12× CSL luciferase Notch reporter were treated for 24 h with 10 μm DAPT and additionally 10 nm LMB where indicated and subjected to luciferase assay. Displayed are the means ± S.E. of n = 3 independent experiments. The asterisk indicates p < 0.05, Student's t test. F and G, C2C12 cells were transfected with 12× CSL luciferase Notch reporter and siRNAs as indicated. 24 h later, the cells were harvested and subjected to luciferase assay (F) or probed with indicated antibodies for protein expression (G). The graph displays the means ± S.E. from three independent experiments. The asterisk indicates p < 0.05, Students t test. For full-size blots of B and G, see supporting Fig. S6. MW, molecular mass.
All siRNAs specifically down-regulated their targets (Fig. 1B, additional siRNA against Nup214 was tested in Fig. S3, C and D). The KD of Nup88 influenced protein levels of Nup214 and vice versa, as published before (28, 33). Fig. 1C demonstrates that KD of Nup88 or Nup214 did not lead to general nuclear pore collapse, as indicated by staining with an anti–FG-repeat antibody (Mab414).
Because both KDs of Nup88 and Nup214 had the same effect, because of the mutual dependence, we focused on Nup214. We transfected increasing amounts of Nup214 siRNA and measured mRNA expression of Nup214 and a canonical Notch target, HES1 (34). Fig. 1D shows a dose-dependent KD of Nup214 mRNA and a concomitant HES1 up-regulation.
Nup214 plays a role in CRM1-mediated nuclear export (27, 28, 33), but a connection to Notch was not reported before. We therefore tested whether CRM1-mediated nuclear export is involved and inhibited this transport pathway by the selective CRM1 inhibitor leptomycin B (LMB) (35). Incubation of PC3 cells with LMB resulted in a similar, GSI-sensitive up-regulation of Notch signaling as the KD of Nup214 (Fig. 1E). The effects are not cell type–specific, because KD of Nup214 induced Notch signaling also in the mouse myoblast cell line C2C12 (Fig. 1, F and G). Together these results suggest that the CRM1–Nup214 axis is involved in regulating Notch signaling.
The CSL-luciferase assay in PC3 cells measured endogenous Notch signaling activity, but not its physiological consequences. We therefore analyzed the differentiation of C2C12 cells with or without KD of Nup214. Upon serum withdrawal, these cells differentiate into myotubes, and this differentiation is prevented by Notch signaling (36). KD of Nup214 inhibited proliferation of C2C12 cells (1.7-fold proliferation after 2 days in siControl-treated cells, −0.9-fold in siNup214 treated cells, n = 3 experiments), confirming earlier findings in HeLa cells (28). In addition KD of Nup214 delayed or reduced differentiation of C2C12 cells (Fig. 2A) as measured by the ratio of myosin heavy chain (MyHC) positive cells to total cells (Fig. 2B), the fusion index (not shown) and a reduction on MyHC protein levels (Fig. 2, C and D). Differentiation could be rescued by additional KD of Hes1, a major Notch downstream gene in myogenesis (37, 38), suggesting that the differentiation block by KD of Nup214 was mediated via Notch signaling (Fig. 2, A–D). Taken together, the data suggested that KD of Nup214 prevented differentiation of C2C12 cells by increasing Notch signaling.
Figure 2.
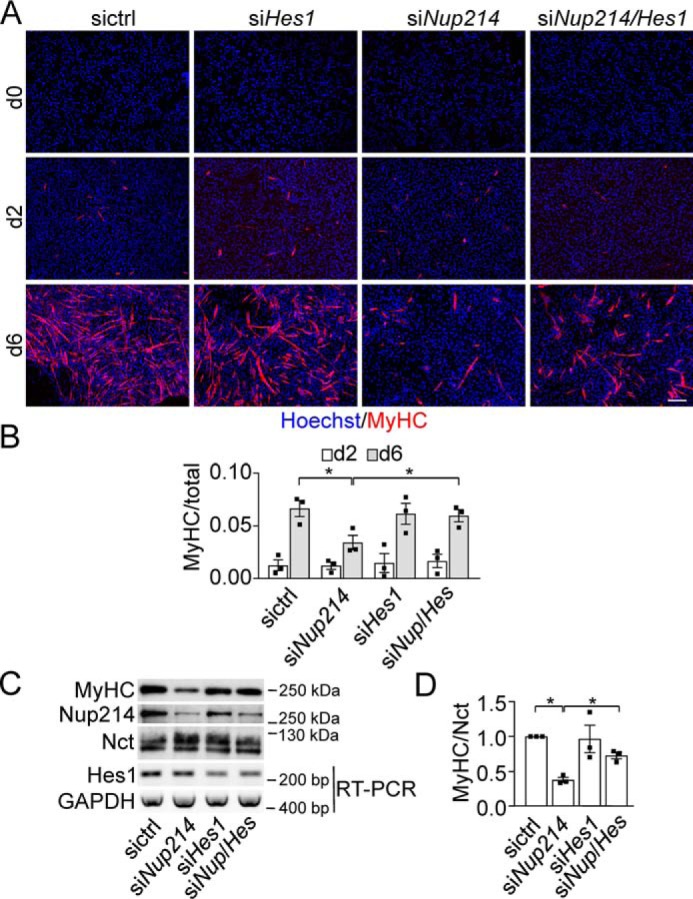
Nup214 KD delays or prevents differentiation of C2C12 cells. A, C2C12 cells transfected with siRNA as indicated were differentiated for 0, 2, and 6 days; fixed; and processed for immunofluorescence with MyHC antibodies. The nuclei were stained with Hoechst 33342. Scale bar, 200 μm. B, quantitation of at least 800 nuclei/condition. Displayed are the mean ratios ± S.E. from three independent experiments of MyHC positive to total cells. C, lysates from C2C12 cells treated as in A were immunoblotted for MyHC, Nup214, and Nicastrin (Nct), and mRNA levels were determined by RT-PCR for Hes1 and GAPDH. D, displays the relative intensities of MyHC to loading control Nct. siControl levels were set to 1. The graph displays means ± S.E. of three independent experiments. Asterisks indicate p < 0.05, Student's t test. For full-size blots of C, see supporting Fig. S6.
To analyze the role of Nup214 in vivo, we used zebrafish. In zebrafish, Notch signaling is involved in differentiation of many tissues, for example in notochord formation, somitogenesis, and hypochord specification (39). RT-PCR for the single zebrafish nup214 orthologue demonstrated that nup214 mRNA is maternally supplied to the zygote and that nup214 is strongly expressed in early stages of development (Fig. 3A). In situ hybridization confirmed the expression in early stages. After 17 h postfertilization and even more pronounced after 24 h postfertilization, nup214 displayed an increased tissue-specific expression pattern with strongest expression in the developing brain (Fig. 3B).
Figure 3.

Nup214 is differentially expressed in vivo and negatively regulates Notch-signaling. A–D, expression analysis of Nup214 in zebrafish. A, RT-PCR with nup214-specific primers on zebrafish embryos and larvae from indicated time points. B, in situ hybridization of zebrafish embryos with a nup214-specific antisense probe. Scale bar, 150 μm. C, RT-PCR with nup214-specific primers on tissues from adult zebrafish tissues. D, in situ hybridization with a nup214 probe in adult zebrafish. Arrows point to localized mRNA in later stages of oogenesis. Scale bar, 100 μm. E and F, Nup214 inactivation by morpholinos activates Notch. E, scheme of nup214 pre-mRNA. Black bars indicate the binding site of the splice MO targeting exon 4/intron 4 splice site and the 5′-UTR MO (ATG MO). Arrows indicate the binding sites for the primers used to demonstrate the efficacy of knockdown, displayed in the agarose gel below. 215 bp, correctly spliced mRNA; 601 bp, mRNA with intron 4 inclusion. −RT, control condition without reverse transcriptase. Below the gel are the number of injected fish and the percentage of fish with the phenotype shown in F. F, whole mount in situ hybridization of col2a1a of 22-h-old zebrafish embryos injected with control or nup214 MOs (0.4 pm). For number of injected fish and percentage of phenotype, see E. The boxed areas are enlarged on the right, and the line scans at the indicated positions (blue bars) show intensity profiles (arbitrary units). Arrowhead, hypochord; arrow, floorplate. Scale bar, 200 μm, 50 μm in enlarged area. ctr, control.
Interestingly, in adult zebrafish nup214 expression is restricted to a number of tissues like ovary, brain, kidney, and gills (Fig. 3, C and D). In the ovary, nup214 is expressed in oocytes of early stages, whereas in maturing oocytes nup214 message is localized in a stripe-like subcortical domain (Fig. 3D). Such asymmetrical localization was also observed for other maternally supplied mRNAs like notch (40). Of note, in Arabidopsis, the only other organism for which we found a tissue expressing study, the Nup214 orthologue LNO1 is also highly expressed in reproductive tissue and very low in vegetative tissue (41).
To address the function of nup214 in zebrafish development, we performed a morpholino (MO) based knockdown approach. RT-PCR analysis showed that injection of a MO targeting the splice donor site of exon 4 resulted in a dose-dependent reduction of correctly spliced nup214 mRNA (Fig. 3E).
To directly assess whether nup214 KD causes up-regulation of Notch activity in zebrafish, we analyzed the expression of col2a1a by in situ hybridization. Normally, col2a1a expressing cells are confined to a single cell layer in the trunk hypochord and floorplate of zebrafish embryos. After injection of the nup214 splice MO, col2a1a is expressed now also in the area of the trunk notochord (Fig. 3F, compare line scans of both conditions). As shown before (39), this expansion into the notochord area is a clear indication of Notch activation.
The knockdown effect was validated using a second nup214 MO that targets the 5′ UTR, interfering with translation (ATG MO; Fig. 3E). Injection of the ATG-MO dose-dependently induced the same expansion into the notochord as seen with the splice morpholino (Fig. S1). These data strongly suggested that Nup214 functions as a negative regulator of Notch signaling also in vivo during zebrafish development.
To further substantiate the in vivo data we analyzed an additional target of Notch, her4 (42). Injection of the splice MO against Nup214 induced an up-regulation of her4, suggestive of an increased Notch signaling (Fig. S2).
Together, these data demonstrate that Nup88/214 negatively regulate Notch activity and, as LMB treatment has a similar effect, raise the intriguing possibility that Nup88/214 is involved in nuclear export of a Notch activator. KD of Nup88/214 (or LMB treatment) keeps this putative activator in the nucleus, resulting in increased Notch signaling.
Nup214 regulates Notch signaling via trafficking of RBP-J
An essential component of canonical Notch signaling is the DNA-binding factor RBP-J (CSL), which together with NICD forms the co-activator complex that drives Notch-dependent transcription (43). To test whether Nup214 is involved in RBP-J trafficking and whether this affects Notch-signaling, we used RBP-VP16, a RBP-J mutant that drives RBP-J/CSL-promoter genes independently of NICD (44). Transfection of RBP-VP16 in the presence or absence of GSI showed that it indeed induced transcription of a CSL-based luciferase reporter independent of NICD (Fig. 4A). KD of Nup214 further increased luciferase expression more than 3-fold, suggesting that Nup214 is directly or indirectly involved in nuclear trafficking of RBP-J.
Figure 4.

Nup214 exports RBP-J out of the nucleus. A, PC3 cells transfected with 12× CSL luciferase-based reporter and Notch-independent RBP-VP16 were transfected with control siRNA or Nup214 siRNA and treated with DAPT as indicated. Luciferase assays were performed after 72 h. The values from reporter and control-siRNA transfected cells were set to 1, and other conditions were related to that. Shown are the means ± S.E. of at least three independent experiments. B, localization of endogenous RBP-J with or without KD of endogenous Nup214 in C2C12 cells differentiated for 3 days was visualized by antibody staining and fluorescence microscopy. The boxed areas are magnified on the right, and examples of intensity profiles are shown. Note that the cytoplasmic intensity is halved in KD conditions. C, 180 cells from three independent experiments respectively were analyzed, and the ratio of extranuclear/nuclear RBP-J fluorescence was determined. Displayed are the means ± S.E. D, Western blotting of cells used in B and C probed with indicated antibodies. Top row, displayed is the mean RPB-J expression normalized to PARP and related to the siControl values set to 1. Displayed are the means ± S.E., n = 3 independent experiments. For full-size blots, see supporting Fig. S6. E, C2C12 cells transfected with control or Nup214 siRNA were subjected to ChIP-qPCR with RBP-J antibody (clone 1F1, Merck) and primers specific for a control region (enh PIIA) and three dynamic RBP-J binding sites (prom Hes, UTR Hes, and enh Hey). Error bars display the S.E. of three technical replicates. One of n = 2 independent experiments is shown. F and G, PC3 cells transfected with AID-GFP were transfected with control or Nup214 siRNA for 72 h or treated with 10 nm LMB for 24 h, fixed, and analyzed by immunofluorescence microscopy (F) or lysed and processed for immunoblotting with antibodies as indicated (G). For full-size blots, see supporting Fig. S6. Asterisks indicate p < 0.05, Student's t test. Scale bars in B and F, 10 μm. MW, molecular mass; ctr, control.
To visualize this trafficking, C2C12 cells were treated with or without Nup214 siRNA and processed for immunofluorescence detection of endogenous RBP-J (see Fig. S3 for characterization of antibody). Quantification demonstrated that KD of Nup214 significantly shifted the distribution of RBP-J toward the nucleus (Fig. 4, B and C) while not changing total RBP-J levels (Fig. 4D).
More RBP-J in the nucleus does not necessarily mean more DNA binding, and not all RBP-J binding sites are dynamic (45, 46). We therefore performed ChIP-qPCR with an established RBP-J antibody (45) and analyzed known RBP-J binding sites in Notch target genes (45) compared with control regions (Fig. 4E). KD of Nup214 increased RBP-J binding to Notch target genes, but not to control regions. Very similar results were obtained with a different RBP-J antibody (Fig. S4). Taken together, these data suggested that Nup214 negatively regulates Notch signaling by shuttling RBP-J out of the nucleus. KD of Nup214 increases the level of nuclear RBP-J and its binding to Notch target genes, thereby enhancing Notch activity.
LMB inhibits CRM1-dependent nuclear export, a major export route for proteins and RNA (47), and Nup214 is involved in CRM1-dependent nuclear export (28) of certain cargoes. To test whether KD of Nup214 would cause inhibition of all CRM1-dependent export, we determined the localization of activation-induced deaminase (AID)–GFP, a known CRM1 substrate (48), in PC3 cells. Although LMB clearly inhibited nuclear export of AID-GFP, KD of Nup88 or Nup214 did not, indicating that Nup88/214 are involved in the export of only a subset of CRM1-dependent proteins (Fig. 4, F and G).
Nup214 is up-regulated with increasing cell density (Fig. S5). This, together with the nonubiquitous, highly selective expression in zebrafish (Fig. 3, B–D) and Arabidopsis (41), suggests that Nup214 is not an essential core component of every nuclear pore but has specific roles in export of a subset of cargos. What remains to be shown is to what extent Nup214 isoforms are involved in context-specific transport.
Our data confirmed that Nup214 has a specific set of substrates (27, 28, 33). In agreement with this, expression levels of Nup214 were recently shown to be cell type–specific (49). Nup214 therefore belongs to the growing list of cell type/differentiation status–specific Nups (50–52).
T-ALL associated Nup214 fusion proteins increase Notch signaling
In 50% of T-ALL the tumor is caused by aberrant Notch signaling (29). Interestingly, chromosomal translocations can cause or contribute to T-ALL in around 10% of cases. In all of these translocations, oncogenic fusions of proteins to Nup214 were detected (53–55). This raises the intriguing possibility that the Nup214-fusion proteins are loss-of-Nup214-function mutations that increase Notch signaling, contributing to malignancy.
To test this hypothesis, HEK293T cells were transfected with SET-Nup214, DEK-Nup214, and Nup214-ABL. SET and DEK are fused to the N terminus of Nup214, replacing parts of it. In Nup214-Abl, the Abl is fused to the C terminus of Nup214, replacing parts of it (53–55). After transfection of the fusion proteins, their effect on the distribution of endogenous Nup88 and 214 was assessed. HEK293 cells were used because of their suitability for nuclear pore immunostainings. Fig. 5A confirms that SET-Nup214 localized to nuclear, dense, brightly stained aggregates (56) that are in a dynamic equilibrium with the nucleoplasm (57). In contrast DEK-Nup214 localized to smaller, less bright, uniformly distributed aggregates, as shown before (56). Remarkably, SET-Nup214, but not DEK-Nup214 sequestered endogenous Nup88 and 214 to nuclear aggregates, diminishing their amounts at nuclear pores (Fig. 5A; quantification in Fig. 5B).
Figure 5.
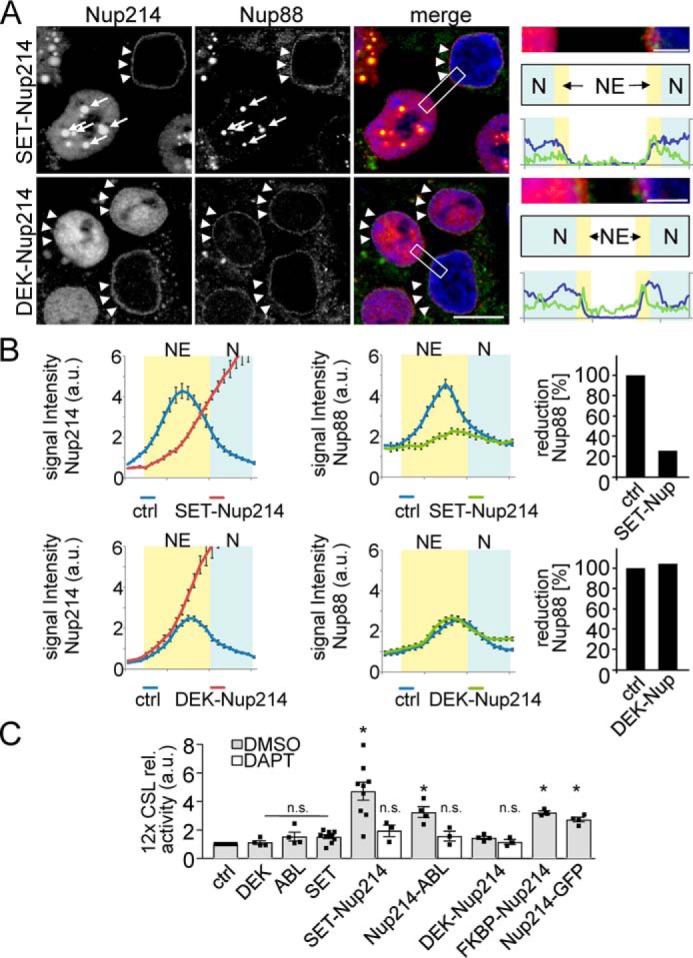
T-ALL causing Nup214 fusion proteins cause Notch up-regulation. A, PC3 cells transfected with SET-Nup214 or DEK-Nup214 were fixed after 24 h and processed for immunofluorescence with antibodies against Nup214 (staining the endogenous Nup214, as well as the overexpressed Nup214 fusion) and Nup88. Arrows depict SET-Nup214 nuclear aggregates; arrowheads indicate intact endogenous Nup88/214 staining at nuclear pores. Note that the SET-Nup214 aggregates in the Nup214 channel are overexposed to visualize also the endogenous Nup214 in the untransfected cell. The boxed areas in the merge images are enlarged on the right panel to demonstrate that SET-Nup214 but not DEK-Nup214 dislocates endogenous Nup88/214. The positions of nucleoplasm (N, blue) and nuclear envelope (NE, yellow) are schematized below, and line scans depicting Nup88 and DAPI staining are shown. Scale bar for main panels, 10 μm; Scale bar for enlargements, 2 μm. B, quantification using line scans of cells from A. 60 cells from n = 3 independent experiments were analyzed as described under “Experimental procedures.” Error bars depict S.E. The percentage of Nup88 reduction refers to the area under the curves at the position of the NE, where control area was set to 100%. C, PC3 cells transfected with plasmids as indicated and 12× CSL luciferase reporter were processed for Notch activity detection. The activity in control (ctrl) cells was set to 1, and the other values were related to that. Displayed are the means ± S.E. of n = 3–9 independent experiments. Asterisks indicate significance (p < 0.05) related to control, Student's t test. n.s., not significant.
In contrast, Nup214-ABL, which contains the N terminus of Nup214, is recruited to nuclear pores and partially replaces endogenous Nup214 as published before (Fig. 6) (58). Nup88 is not changed under these conditions (Fig. 6). This “trapping” of endogenous Nup88/214 by SET-Nup214 away from the nuclear pores and the partial replacement of Nup214 by Nup214-ABL mimics the KD of Nup214 we described above. Therefore, we tested whether the expression of the fusion proteins would up-regulate Notch signaling. Indeed, in a CSL-dependent reporter assay, SET-Nup214 up-regulated Notch almost 4.6-fold, DEK-Nup214 up-regulated Notch 1.4-fold, and Nup214-ABL up-regulated Notch 2.9-fold (Fig. 5C). To analyze whether it is the Nup214 part that is responsible for the effect on Notch signaling, we replaced the fusion partners of Nup214 by GFP or an FK506-binding protein domain. In both cases the expression of the artificial fusion proteins resulted in a clear up-regulation of Notch, indicating that the Nup214-part of the fusion protein is causing the effect on Notch signaling (Fig. 5C).
Figure 6.
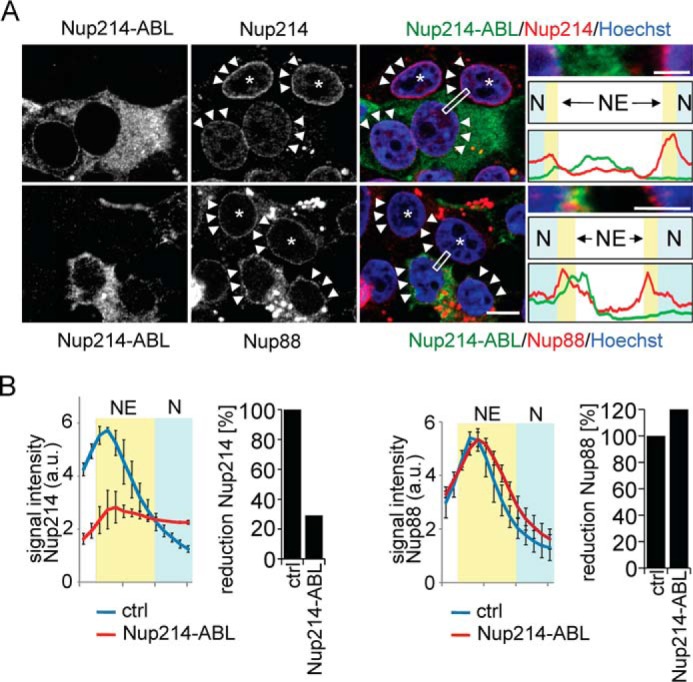
Nup214-ABL displaces endogenous Nup214 but not Nup88. A, HEK293T cells were transfected with Nup214-ABL, fixed after 24 h, and processed for immunofluorescence against myc (the epitope in Nup214 recognized by the Nup214 antibody we use is deleted) and Nup88 or Nup214 as indicated. Nuclear pore localization was determined as depicted in Fig. 5. Note that transfected cells show clear loss of fluorescence signal of Nup214 at the nuclear rim (arrowheads, upper panel) but not of Nup88 (arrowheads, lower panel). Asterisk, untransfected cells. Scale bar for main panels, 10 μm; Scale bar for enlargements, 2 μm. The boxed areas in the merge images are enlarged in the right panel. The positions of nucleoplasm (N, blue) and nuclear envelope (NE, yellow) are schematized, and line scans depicting Nup214-ABL (green), Nup214 (red, upper graph), or Nup88 (red, lower graph) are shown. B, quantification using line scans of cells from A. 60–80 cells from n = 3 independent experiments were analyzed as described under “Experimental procedures.” Error bars depict S.E. The percentage of Nup88/214 reduction refers to the area under the curves at the position of the NE (yellow), where the control area was set to 100%. ctrl refers to untransfected cells.
Up-regulated Notch signaling is very important in T-ALL development (29). Our data suggest that in T-ALL patients with translocations resulting in Nup214 fusion proteins, an up-regulation of Notch could contribute to tumorigenesis. Our overexpression experiments with SET-Nup214 and DEK-Nup214 may exaggerate the patient situation.
Interestingly, the level of up-regulation of Notch coincides with the incidence of the respective mutations in T-ALL. SET-Nup214, with the highest up-regulation of Notch, is found in 6% of T-ALL patients (59), followed by Nup214-ABL, in 5% (54), and finally DEK-Nup214, which only weakly influences Notch and is actually found in AML, not T-ALL (60). It should also be noted that Nup214 translocations are rarely found in patients with direct mutations of Notch (59).
In summary, we showed that the nuclear pore components Nup88/214 negatively regulate Notch signaling in vitro and in vivo in zebrafish. Although the importance of the nuclear transport machinery and its regulation/misregulation in development and disease has been widely appreciated (reviewed in Refs. 52, 61, and 62), its importance in Notch signaling was not yet recognized. Our data suggest that nuclear RBP-J is rate-limiting for Notch signaling and that regulation of nucleocytoplasmic transport is an additional measure for fine-tuning Notch activity, adding another level of complexity to an already complex signaling mechanism.
Inhibiting nuclear export is a promising strategy in cancer therapy (63, 64). However, an unwanted side effect might be an up-regulation of Notch signaling, which, given its role in proliferation and cancer, could be detrimental. It remains to be shown to what extent the up-regulation of Notch by oncogenic Nup214 fusion proteins indeed contributes to tumorigenesis of T-ALL and/or whether additional functions of Nup88/214 like mRNA export (27, 28, 57, 65) are involved.
Experimental procedures
For antibodies, primers, and morpholino sequences, see Tables S1 and S2.
Cell lines
HeLa Kyoto, HEK293T, C2C12 (ATCC CRL-1772), PC3 (ATCC® CRL-1435TM), cells were maintained in Dulbecco's modified Eagle's medium + GlutaMAX (Invitrogen) supplemented with 10% FBS and incubated at 37 °C, 95% relative humidity, and 5% CO2.
Quantitative RT-PCR
Expression levels of Nup214 and HES1 mRNA were quantified using real-time PCR according to standard protocols. cDNAs were reverse-transcribed from 2 μg of total RNA. The PCR (denaturation 95 °C for 2 min followed by 40 cycles of 95 °C for 15 s and 60 °C for 1 min) was performed using Sybr Green PCR Core Reagents (PE Applied Biosystems).
Nup214 fusion plasmids
DEK-Nup214 (66) was kindly provided by Carl Sandén from LUND University and was subcloned in via EagI restriction site into pCMV-Myc. Human DEK cDNA (accession no. 5122743, Invitrogen) was subcloned via EcoRI and XhoI restriction sites into pCMV-Myc. SET-Nup214 according to the sequence published in Ref. 53 was constructed by overlap extension PCR and cloned in pcDNA4/myc-HisA vector (Invitrogen). The SET coding sequence up to the end of exon7 was amplified using Phusion polymerase (Fynnzymes) and primers SET-F and SET-Nup-R from a MGC verified human SET cDNA clone (accession no. 5587291, Thermo Scientific). The Nup214 coding sequence starting from exon 18 was amplified from a human Nup214 full-length construct using primers SET-Nup-F and Nup214-R. Nup214-ABL, Nup214-GFP, and FKBP-Nup214 were constructed via Gibson assembly cloning kit (NEB), and primers were generated with NEBuilder®. For Nup214-ABL (sequence according to Ref. 54), Nup214 was amplified from start codon to exon29 and ABL1 from exon2 to stop codon from an ABL1 cDNA in pCR TOPO2.1 isolated from PC3 lysates. The sequences were assembled together with pcDNA3.1-Myc vector (Invitrogen). For Nup214-GFP the same Nup214 sequence was amplified as for Nup214-ABL. GFP was amplified from pEGFP-C1 vector, and the fragments were assembled together with EcoRI and XhoI digested pcDNA3.1 vector. For FKBP-Nup214 the same Nup214 sequence was amplified as for SET-Nup214 or DEK-Nup214, FKBP was amplified from pC4-Fv2E vector (Clontech), and the fragments were assembled together with StuI- and XhoI-digested pCMV-Myc vector. All vectors contain a cytomegalovirus promotor. All primer sequences are listed in the supporting information.
Reporter assays
Notch activity was analyzed using a 12× CSL-luciferase plasmid as described before (31). Briefly, for KD experiments PC3 cells plated in a 24-well format were transfected for 2 days with 10 nm siRNA (siCtrl), siNUP214, siNUP88 (SMARTpool, Thermo Fisher), siNUP358 (ON-TARGET plus, Dharmacon) using Dharmafect followed by plasmid transfection using Lipofectamine 2000 (Invitrogen), 300 ng of pGL4.20–12× CSL-luciferase reporter plasmid, and 6 ng of pGL4.74-[hRluc/TK] Renilla luciferase internal control plasmid. Firefly and Renilla luciferase activities were measured in cell lysates prepared 24 h after transfection with the Promega Dual-Luciferase kit via manufacturer's instructions, using a Mithras LB 940.
For NUP214 fusion overexpression experiments, PC3 cells were plated in 6-well format and transfected for 24 h with equimolar Plasmid DNA and 500 ng of pGL4.20–12× CSL-luciferase reporter plasmid using Polyethylenglycol (PEI). Firefly luciferase activity was measured in cell lysates prepared 24 h after transfection with the Promega Dual-Luciferase kit. Firefly values were normalized against the protein content analyzed with Pierce BCA protein assay kit via the manufacturer's instructions, using a Mithras LB 940.
C2C12 transfection and differentiation
Control siRNA or Nup214 or Hes1 siRNA (details in the supporting information) were transfected in C2C12 cells by using Amaxa® Cell Line Nucleofector® kit V and the Nucleofector® II device (Lonza). For differentiation, the cells were plated at a density of 6 × 104 cells/cm2 and starved in DMEM with 1% FCS. For quantification of differentiation, MyHC-positive cells were counted manually with ImageJ and Hoechst-stained nuclei automatically with CellProfiler. The ratios of MyHC-positive/total number of cells, subtracted with the value for d.0 of differentiation, were calculated.
Immunocytochemistry and microscopy of cells
For immunofluorescence cells were grown on coverslips. After incubation the cells were fixed with 4% formalin and processed for immunofluorescence with antibodies as indicated. The nuclei were stained with Hoechst 33342 (Invitrogen H1399). Images were acquired on a Zeiss Axiovert200 using 40× or 63× objective and Axiovision and Zen2012 software. For some settings a confocal-like Apotome slider was used. For the quantification of the nuclear-cytoplasmic ratio of RBP-J, images were analyzed in ZEN2012 software (Zeiss). The bar scan tool was applied across single cells (30 cells per condition per experiment), and the resulting signal intensity profiles were exported to Excel 2010 (Microsoft). The RBP-J signal intensity in the cytoplasm and in the nucleus was averaged, and the ratio was calculated. To avoid ambiguities in allocation of intensities to either nucleus or cytoplasm at the transition nucleus–cytoplasm, RBP-J signals from that area were excluded from the calculations. The transition region was defined to be 0.25 μm in both directions of the line where the Hoechst signal is half-maximal. Images were assembled and processed in Adobe Photoshop. In some cases nonlinear γ-settings of <1 were used to match the image perception by eye, but care was taken that identical settings were applied to conditions from the same experiment.
Zebrafish maintenance and staging
WT zebrafish (strain AB Tübingen) were maintained under standard conditions for zebrafish husbandry (67). Embryos were staged according to morphological criteria (68).
Morpholino antisense oligonucleotides
Nup214 MOs (Gene Tools, LLC, Philomath, OR) were designed to target either the translational start (nup214 ATG) or the splice donor site of exon 4 (nup214 splice). As a negative control a standard MO that targets a human β-globin intron mutation was used. MOs were diluted in water to a working concentration of 1 mm with 0.05% phenol red (Sigma) as a tracer. Injections into the yolk of 1- or 2-cell embryos were performed using a manual micromanipulator (type M1, Saur, Germany). For each MO different volumes were injected, calculated by using a graticule (Pyser-SGI, Edenbridge, UK). To analyze knockdown efficiency of the splice MO, total RNA was isolated from 24-h-old zebrafish embryos using the RNeasy mini kit (Qiagen), reverse-transcribed with the iScriptTM cDNA synthesis kit (Bio-Rad), and analyzed by RT-PCR.
In situ hybridization
For generation of the nup214 probe, a cDNA fragment comprising exons 1–6 served as a template. The col2a1a probe (69) was generously provided by John H. Postlethwait (University of Oregon). The cDNA fragment that was used to synthesize the antisense RNA probe for her4.1 was amplified with the following primers: AGAAAGCCCATGGTGGAGAAG (forward), GCTTTCCATTGTTGCTTGATTGTA (reverse) and gave rise to an 785-bp amplicon. Whole-mount in situ hybridizations were carried out as previously described (70). In 1situ hybridization on longitudinal sections of adult zebrafish was performed essentially as described (71). Images were captured using a SteREO Discovery.V8 microscope (Zeiss) equipped with an Achromat-S objectives and an AxioCam MRC camera using the AxioVision software. Images were processed with Adobe Photoshop. Intensity profiles were obtained using Zen software (Zeiss).
ChIP-qPCR
ChIP was performed on 3 days serum-starved (DMEM + 1% FBS) C2C12 cells. The cells from three 10-cm dishes were fixed with 1% formaldehyde in 1× PBS at room temperature for 10 min, quenched with 0.125 m glycin for 5 min, washed two times with ice-cold 1× PBS, and harvested in ice-cold PBS containing Protease inhibitor mixture. To release the nuclei, the cells were pelleted at 500 relative centrifugal force for 10 min at 4 °C and resuspended in 5 ml of hypotonic buffer (20 mm Tris, pH 7.4, 2 mm MgCl2, 5% glycerol) containing protease inhibitor cocktail and 0.6% Nonidet P-40. The suspension was incubated on ice for 4 min with occasional mild vortexing. The nuclei were pelleted at 1000 rcf for 5 min at 4 °C and resuspended in ChIP lysis buffer (50 mm HEPES-KOH, pH 7.5, 140 mm NaCl, 1 mm EDTA, pH 8, 1% Triton X-100, 0.1% sodium deoxycholate, 0.1% SDS) for 10 min. The lysate was sonicated to shear the DNA into 300–800-bp fragments. The protein–DNA complexes were detected using 30–35 μg of DNA and 5 μg of Ab against RBP-J (clone 1F1; Merck Chemicals; MABE982 or Abcam; ab25949) or isotype control (rat IgG) in 1 ml of chip lysis buffer and incubated for 1 h at 4 °C under rotating conditions. The complexes were precipitated by an overnight incubation at 4 °C on a rotator with 30 μl of magnetic Dynabeads protein G (Thermo Fischer). The protein–DNA complexes were washed two times for 5 min with low-salt wash buffer (0.1% SDS, 1% Triton X-100, 2 mm EDTA, 20 mm Tris-HCl, pH 8.0, 150 mm NaCl) followed by high salt (0.1% SDS, 1% Triton X-100, 2 mm EDTA, 20 mm Tris-HCl, pH 8.0, 500 mm NaCl) and LiCl (0.25 m LiCl, 1% Nonidet P-40, 1% sodium deoxycholate, 1 mm EDTA, 10 mm Tris-HCl, pH 8.0) wash buffer always by incubating at 4 °C on a rotation wheel. The protein–DNA complexes were eluted and reverse cross-linked, and the DNA was purified via phenol chloroform extraction. The specific DNA enrichment was quantified by real-time PCR.
Statistics
Arithmetic means of the numerical data were compared using the Student's t test. A difference in means was considered statistically significant as indicated in the figures. Error bars depict the S.E., and numbers of independent replicates are indicated in the figure legends.
Author contributions
B. K., C. Englert, and C. K. conceptualization; B. K., C. V., A. K., B. P., C. Engelmann, L. B., D. K., C. Englert, and C. K. investigation; B. K., C. V., A. K., B. P., C. Engelmann, L. B., D. K., B.J., R. H. K., F. O., C. Englert, and C. K. methodology; B. K., C. Englert, and C. K. writing-original draft; B. K., L. B., B.J., R. H. K., F. O., C. Englert, and C. K. writing-review and editing; A. K. and B. P. visualization; L. B. and C. K. data curation; L. B. and C. K. formal analysis; D. K., B.J., R. H. K., F. O., and C. Englert resources; D. K., B.J., F. O., C. Englert, and C. K. supervision; C. Englert and C. K. project administration.
Supplementary Material
Acknowledgments
We are grateful to Christina Ebert and Eric Rivera-Milla for technical assistance, to John Postlethwait for hybridization probes, and to Carl Sandén (Lund University) for DEK-NUP. We acknowledge the support from the FLI imaging facility and Torsten Kroll from the functional genomics facility.
This work was supported by Grant KA1751/4-1 from the Deutsche Forschungsgemeinschaft (to C. K. and C. E.) and a Pakt für Forschung und Innovation grant from the Leibniz Gemeinschaft (to C. K.). Further support came from Deutsche Forschungsgemeinschaft Collaborative Research Centers SFB 1074/A3 and 860/B8 (to F. O. and R. H. K.), respectively and by the Bundesministerium für Bildung und Forschung Research Nucleus SyStAR (to F. O.). The authors declare that they have no conflicts of interest with the contents of this article.
This article contains Tables S1 and S2 and Figs. S1–S6.
- NICD
- Notch intracellular domain
- T-ALL
- T-cell acute lymphatic leukemia
- KD
- knockdown
- LMB
- leptomycin B
- MO
- morpholino
- GSI
- γ-secretase inhibitor
- MyHC
- myosin heavy chain
- AID
- activation-induced deaminase
- qPCR
- quantitive PCR
- DAPT
- N-[N-(3,5-difluorophenacetyl-L-alanyl)]-(S)-phenylglycine t-butyl ester.
References
- 1. Artavanis-Tsakonas S., Matsuno K., and Fortini M. (1995) Notch signaling. Science 268, 225–232 10.1126/science.7716513 [DOI] [PubMed] [Google Scholar]
- 2. Bray S. J. (2006) Notch signalling: a simple pathway becomes complex. Nat. Rev. Mol. Cell Biol. 7, 678–689 10.1038/nrm2009 [DOI] [PubMed] [Google Scholar]
- 3. Fiúza U.-M., and Arias A. M. (2007) Cell and molecular biology of Notch. J. Endocrinol. 194, 459–474 10.1677/JOE-07-0242 [DOI] [PubMed] [Google Scholar]
- 4. Logeat F., Bessia C., Brou C., LeBail O., Jarriault S., Seidah N. G., and Israël A. (1998) The Notch1 receptor is cleaved constitutively by a furin-like convertase. Proc. Natl. Acad. Sci. 95, 8108–8112 10.1073/pnas.95.14.8108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Brou C., Logeat F., Gupta N., Bessia C., LeBail O., Doedens J. R., Cumano A., Roux P., Black R. A., and Israël A. (2000) A novel proteolytic cleavage involved in Notch signaling: the role of the disintegrin-metalloprotease TACE. Mol. Cell 5, 207–216 10.1016/S1097-2765(00)80417-7 [DOI] [PubMed] [Google Scholar]
- 6. De Strooper B., Annaert W., Cupers P., Saftig P., Craessaerts K., Mumm J. S., Schroeter E. H., Schrijvers V., Wolfe M. S., Ray W. J., Goate A., and Kopan R. (1999) A presenilin-1-dependent γ-secretase-like protease mediates release of Notch intracellular domain. Nature 398, 518–522 10.1038/19083 [DOI] [PubMed] [Google Scholar]
- 7. Borggrefe T., and Oswald F. (2009) The Notch signaling pathway: transcriptional regulation at Notch target genes. Cell. Mol. Life Sci. 66, 1631–1646 10.1007/s00018-009-8668-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Kopan R., and Ilagan M. X. (2009) The canonical Notch signaling pathway: unfolding the activation mechanism. Cell 137, 216–233 10.1016/j.cell.2009.03.045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Kim M.-Y., Ann E.-J., Kim J.-Y., Mo J.-S., Park J.-H., Kim S.-Y., Seo M.-S., and Park H.-S. (2007) Tip60 histone acetyltransferase acts as a negative regulator of Notch1 signaling by means of acetylation. Mol. Cell Biol. 27, 6506–6519 10.1128/MCB.01515-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Acar M., Jafar-Nejad H., Takeuchi H., Rajan A., Ibrani D., Rana N. A., Pan H., Haltiwanger R. S., and Bellen H. J. (2008) Rumi is a CAP10 domain glycosyltransferase that modifies Notch and is required for Notch signaling. Cell 132, 247–258 10.1016/j.cell.2007.12.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Hein K., Mittler G., Cizelsky W., Kühl M., Ferrante F., Liefke R., Berger I. M., Just S., Sträng J. E., Kestler H. A., Oswald F., and Borggrefe T. (2015) Site-specific methylation of Notch1 controls the amplitude and duration of the Notch1 response. Sci. Signal. 8, ra30 10.1126/scisignal.2005892 [DOI] [PubMed] [Google Scholar]
- 12. Le Bras S., Loyer N., and Le Borgne R. (2011) The multiple facets of ubiquitination in the regulation of Notch signaling pathway. Traffic 12, 149–161 10.1111/j.1600-0854.2010.01126.x [DOI] [PubMed] [Google Scholar]
- 13. Wu G., Lyapina S., Das I., Li J., Gurney M., Pauley A., Chui I., Deshaies R. J., and Kitajewski J. (2001) SEL-10 is an inhibitor of Notch signaling that targets Notch for ubiquitin-mediated protein degradation. Mol. Cell Biol. 21, 7403–7415 10.1128/MCB.21.21.7403-7415.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Oberg C, Li J., Pauley A., Wolf E., Gurney M., and Lendahl U. (2001) The Notch intracellular domain is ubiquitinated and negatively regulated by the mammalian Sel-10 homolog. J. Biol. Chem. 276, 35847–35853 10.1074/jbc.M103992200 [DOI] [PubMed] [Google Scholar]
- 15. Gupta-Rossi N., Le Bail O., Gonen H., Brou C., Logeat F., Six E., Ciechanover A., and Israël A. (2001) Functional interaction between SEL-10, an F-box protein, and the nuclear form of activated Notch1 receptor. J. Biol. Chem. 276, 34371–34378 10.1074/jbc.M101343200 [DOI] [PubMed] [Google Scholar]
- 16. McGill M. A., and McGlade C. J. (2003) Mammalian numb proteins promote Notch1 receptor ubiquitination and degradation of the Notch1 intracellular domain. J. Biol. Chem. 278, 23196–23203 10.1074/jbc.M302827200 [DOI] [PubMed] [Google Scholar]
- 17. Ishitani T., Hirao T., Suzuki M., Isoda M., Ishitani S., Harigaya K., Kitagawa M., Matsumoto K., and Itoh M. (2010) Nemo-like kinase suppresses Notch signalling by interfering with formation of the Notch active transcriptional complex. Nat. Cell Biol. 12, 278–285 10.1038/ncb2028 [DOI] [PubMed] [Google Scholar]
- 18. Fryer C. J., White J. B., and Jones K. A. (2004) Mastermind recruits CycC:CDK8 to phosphorylate the Notch ICD and coordinate activation with turnover. Mol. Cell 16, 509–520 10.1016/j.molcel.2004.10.014 [DOI] [PubMed] [Google Scholar]
- 19. Song J., Park S., Kim M., and Shin I. (2008) Down-regulation of Notch-dependent transcription by Akt in vitro. FEBS Lett. 582, 1693–1699 10.1016/j.febslet.2008.04.024 [DOI] [PubMed] [Google Scholar]
- 20. Ranganathan P., Vasquez-Del Carpio R., Kaplan F. M., Wang H., Gupta A., VanWye J. D., and Capobianco A. J. (2011) Hierarchical phosphorylation within the ankyrin repeat domain defines a phosphoregulatory loop that regulates Notch transcriptional activity. J. Biol. Chem. 286, 28844–28857 10.1074/jbc.M111.243600 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Fassa A., Parisiadou L., Robakis N. K., and Efthimiopoulos S. (2007) Novel processing of Notch 1 within its intracellular domain by a cysteine protease. Neurodegener. Dis. 4, 148–155 10.1159/000101839 [DOI] [PubMed] [Google Scholar]
- 22. Tagami S., Okochi M., Yanagida K., Ikuta A., Fukumori A., Matsumoto N., Ishizuka-Katsura Y., Nakayama T., Itoh N., Jiang J., Nishitomi K., Kamino K., Morihara T., Hashimoto R., Tanaka T., et al. (2008) Regulation of Notch signaling by dynamic changes in the precision of S3 cleavage of Notch-1. Mol. Cell Biol. 28, 165–176 10.1128/MCB.00863-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Pumroy R. A., and Cingolani G. (2015) Diversification of importin-α isoforms in cellular trafficking and disease states. Biochem. J. 466, 13–28 10.1042/BJ20141186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Cautain B., Hill R., de Pedro N., and Link W. (2015) Components and regulation of nuclear transport processes. FEBS J. 282, 445–462 10.1111/febs.13163 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. van Deursen J., Boer J., Kasper L., and Grosveld G. (1996) G2 arrest and impaired nucleocytoplasmic transport in mouse embryos lacking the proto-oncogene CAN/Nup214. EMBO J. 15, 5574–5583 10.1002/j.1460-2075.1996.tb00942.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Trotman L. C., Mosberger N., Fornerod M., Stidwill R. P., and Greber U. F. (2001) Import of adenovirus DNA involves the nuclear pore complex receptor CAN/Nup214 and histone H1. Nat. Cell Biol. 3, 1092–1100 10.1038/ncb1201-1092 [DOI] [PubMed] [Google Scholar]
- 27. Bernad R., Engelsma D., Sanderson H., Pickersgill H., and Fornerod M. (2006) Nup214-Nup88 nucleoporin subcomplex is required for CRM1-mediated 60 S preribosomal nuclear export. J. Biol. Chem. 281, 19378–19386 10.1074/jbc.M512585200 [DOI] [PubMed] [Google Scholar]
- 28. Hutten S., and Kehlenbach R. H. (2006) Nup214 is required for CRM1-dependent nuclear protein export in vivo. Mol. Cell. Biol. 26, 6772–6785 10.1128/MCB.00342-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Koch U., and Radtke F. (2011) Notch in T-ALL: new players in a complex disease. Trends Immunol. 32, 434–442 10.1016/j.it.2011.06.005 [DOI] [PubMed] [Google Scholar]
- 30. Belver L., and Ferrando A. (2016) The genetics and mechanisms of T cell acute lymphoblastic leukaemia. Nat. Rev. Cancer 16, 494–507 10.1038/nrc.2016.63 [DOI] [PubMed] [Google Scholar]
- 31. Huenniger K., Krämer A., Soom M., Chang I., Köhler M., Depping R., Kehlenbach R. H., and Kaether C. (2010) Notch1 signaling is mediated by importins α 3, 4, and 7. Cell Mol. Life Sci. 67, 3187–3196 10.1007/s00018-010-0378-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Fornerod M., van Deursen J., van Baal S., Reynolds A., Davis D., Murti K. G., Fransen J., and Grosveld G. (1997) The human homologue of yeast CRM1 is in a dynamic subcomplex with CAN/Nup214 and a novel nuclear pore component Nup88. EMBO J. 16, 807–816 10.1093/emboj/16.4.807 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Bernad R., van der Velde H., Fornerod M., and Pickersgill H. (2004) Nup358/RanBP2 attaches to the nuclear pore complex via association with Nup88 and Nup214/CAN and plays a supporting role in CRM1-mediated nuclear protein export. Mol. Cell. Biol. 24, 2373–2384 10.1128/MCB.24.6.2373-2384.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Sasai Y., Kageyama R., Tagawa Y., Shigemoto R., and Nakanishi S. (1992) Two mammalian helix–loop–helix factors structurally related to Drosophila hairy and enhancer of split. Genes Dev. 6, 2620–2634 10.1101/gad.6.12b.2620 [DOI] [PubMed] [Google Scholar]
- 35. Kudo N., Wolff B., Sekimoto T., Schreiner E. P., Yoneda Y., Yanagida M., Horinouchi S., and Yoshida M. (1998) Leptomycin B inhibition of signal-mediated nuclear export by direct binding to CRM1. Exp. Cell Res. 242, 540–547 10.1006/excr.1998.4136 [DOI] [PubMed] [Google Scholar]
- 36. Kopan R., Nye J. S., and Weintraub H. (1994) The intracellular domain of mouse Notch: a constitutively activated repressor of myogenesis directed at the basic helix–loop–helix region of MyoD. Development 120, 2385–2396 [DOI] [PubMed] [Google Scholar]
- 37. Kuroda K., Tani S., Tamura K., Minoguchi S., Kurooka H., and Honjo T. (1999) Delta-induced Notch signaling mediated by RBP-J inhibits MyoD expression and myogenesis. J. Biol. Chem. 274, 7238–7244 10.1074/jbc.274.11.7238 [DOI] [PubMed] [Google Scholar]
- 38. Lahmann I., Bröhl D., Zyrianova T., Isomura A., Czajkowski M. T., Kapoor V., Griger J., Ruffault P. L., Mademtzoglou D., Zammit P. S., Wunderlich T., Spuler S., Kühn R., Preibisch S., Wolf J., et al. (2019) Oscillations of MyoD and Hes1 proteins regulate the maintenance of activated muscle stem cells. Genes Dev. 33, 524–535 10.1101/gad.322818.118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Latimer A. J., and Appel B. (2006) Notch signaling regulates midline cell specification and proliferation in zebrafish. Dev. Biol. 298, 392–402 10.1016/j.ydbio.2006.05.039 [DOI] [PubMed] [Google Scholar]
- 40. Bally-Cuif L., Schatz W. J., and Ho R. K. (1998) Characterization of the zebrafish Orb/CPEB-related RNA binding protein and localization of maternal components in the zebrafish oocyte. Mech. Dev. 77, 31–47 10.1016/S0925-4773(98)00109-9 [DOI] [PubMed] [Google Scholar]
- 41. Braud C., Zheng W., and Xiao W. (2012) LONO1 encoding a nucleoporin is required for embryogenesis and seed viability in Arabidopsis. Plant Physiol. 160, 823–836 10.1104/pp.112.202192 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Takke C., Dornseifer P., v Weizsäcker E., and Campos-Ortega J. A. (1999) her4, a zebrafish homologue of the Drosophila neurogenic gene E(spl), is a target of NOTCH signalling. Development 126, 1811–1821 [DOI] [PubMed] [Google Scholar]
- 43. Tamura K., Taniguchi Y., Minoguchi S., Sakai T., Tun T., Furukawa T., and Honjo T. (1995) Physical interaction between a novel domain of the receptor Notch and the transcription factor RBP-Jκ/Su(H). Curr. Biol. 5, 1416–1423 10.1016/S0960-9822(95)00279-X [DOI] [PubMed] [Google Scholar]
- 44. Waltzer L., Bourillot P. Y., Sergeant A., and Manet E. (1995) RBP-Jκ repression activity is mediated by a co-repressor and antagonized by the Epstein–Barr virus transcription factor EBNA2. Nucleic Acids Res. 23, 4939–4945 10.1093/nar/23.24.4939 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Castel D., Mourikis P., Bartels S. J., Brinkman A. B., Tajbakhsh S., and Stunnenberg H. G. (2013) Dynamic binding of RBPJ is determined by Notch signaling status. Genes Dev. 27, 1059–1071 10.1101/gad.211912.112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Wang H., Zang C., Taing L., Arnett K. L., Wong Y. J., Pear W. S., Blacklow S. C., Liu X. S., and Aster J. C. (2014) NOTCH1-RBPJ complexes drive target gene expression through dynamic interactions with superenhancers. Proc. Natl. Acad. Sci. U.S.A. 111, 705–710 10.1073/pnas.1315023111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Hutten S., and Kehlenbach R. H. (2007) CRM1-mediated nuclear export: to the pore and beyond. Trends Cell Biol 17, 193–201 10.1016/j.tcb.2007.02.003 [DOI] [PubMed] [Google Scholar]
- 48. McBride K. M., Barreto V., Ramiro A. R., Stavropoulos P., and Nussenzweig M. C. (2004) Somatic hypermutation is limited by CRM1-dependent nuclear export of activation-induced deaminase. J. Exp. Med. 199, 1235–1244 10.1084/jem.20040373 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Ori A., Banterle N., Iskar M., Andrés-Pons A., Escher C., Khanh Bui H., Sparks L., Solis-Mezarino V., Rinner O., Bork P., Lemke E. A., and Beck M. (2013) Cell type-specific nuclear pores: a case in point for context-dependent stoichiometry of molecular machines. Mol. Syst. Biol. 9, 648 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. D'Angelo M. A., Gomez-Cavazos J. S., Mei A., Lackner D. H., and Hetzer M. W. (2012) A change in nuclear pore complex composition regulates cell differentiation. Dev. Cell 22, 446–458 10.1016/j.devcel.2011.11.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Raices M., and D'Angelo M. A. (2012) Nuclear pore complex composition: a new regulator of tissue-specific and developmental functions. Nat. Rev. Mol. Cell Biol. 13, 687–699 10.1038/nrm3461 [DOI] [PubMed] [Google Scholar]
- 52. Gomez-Cavazos J. S., and Hetzer M. W. (2012) Outfits for different occasions: tissue-specific roles of nuclear envelope proteins. Curr. Opin. Cell Biol. 24, 775–783 10.1016/j.ceb.2012.08.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Van Vlierberghe P., van Grotel M., Tchinda J., Lee C., Beverloo H. B., van der Spek P. J., Stubbs A., Cools J., Nagata K., Fornerod M., Buijs-Gladdines J., Horstmann M., van Wering E. R., Soulier J., Pieters R., et al. (2008) The recurrent SET-NUP214 fusion as a new HOXA activation mechanism in pediatric T-cell acute lymphoblastic leukemia. Blood 111, 4668–4680 10.1182/blood-2007-09-111872 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Graux C., Cools J., Melotte C., Quentmeier H., Ferrando A., Levine R., Vermeesch J. R., Stul M., Dutta B., Boeckx N., Bosly A., Heimann P., Uyttebroeck A., Mentens N., Somers R., et al. (2004) Fusion of NUP214 to ABL1 on amplified episomes in T-cell acute lymphoblastic leukemia. Nat. Genet. 36, 1084–1089 10.1038/ng1425 [DOI] [PubMed] [Google Scholar]
- 55. Gorello P., La Starza R., Di Giacomo D., Messina M., Puzzolo M. C., Crescenzi B., Santoro A., Chiaretti S., and Mecucci C. (2010) SQSTM1-NUP214: a new gene fusion in adult T-cell acute lymphoblastic leukemia. Haematologica 95, 2161–2163 10.3324/haematol.2010.029769 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Fornerod M., Boer J., van Baal S., Jaeglé M., von Lindern M., Murti K. G., Davis D., Bonten J., Buijs A., and Grosveld G. (1995) Relocation of the carboxyterminal part of CAN from the nuclear envelope to the nucleus as a result of leukemia-specific chromosome rearrangements. Oncogene 10, 1739–1748 [PubMed] [Google Scholar]
- 57. Port S. A., Mendes A., Valkova C., Spillner C., Fahrenkrog B., Kaether C., and Kehlenbach R. H. (2016) The oncogenic fusion proteins SET-Nup214 and sequestosome-1 (SQSTM1)-Nup214 form dynamic nuclear bodies and differentially affect nuclear protein and poly(A)+ RNA export. J. Biol. Chem. 291, 23068–23083 10.1074/jbc.M116.735340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. De Keersmaecker K., Rocnik J. L., Bernad R., Lee B. H., Leeman D., Gielen O., Verachtert H., Folens C., Munck S., Marynen P., Fornerod M., Gilliland D. G., and Cools J. (2008) Kinase activation and transformation by NUP214-ABL1 is dependent on the context of the nuclear pore. Mol. Cell 31, 134–142 10.1016/j.molcel.2008.05.005 [DOI] [PubMed] [Google Scholar]
- 59. Ben Abdelali R., Roggy A., Leguay T., Cieslak A., Renneville A., Touzart A., Banos A., Randriamalala E., Caillot D., Lioure B., Devidas A., Mossafa H., Preudhomme C., Ifrah N., Dombret H., et al. (2014) SET-NUP214 is a recurrent gammadelta lineage-specific fusion transcript associated with corticosteroid/chemotherapy resistance in adult T-ALL. Blood 123, 1860–1863 10.1182/blood-2013-08-521518 [DOI] [PubMed] [Google Scholar]
- 60. von Lindern M., Fornerod M., van Baal S., Jaegle M., de Wit T., Buijs A., and Grosveld G. (1992) The translocation (6;9), associated with a specific subtype of acute myeloid leukemia, results in the fusion of two genes, dek and can, and the expression of a chimeric, leukemia-specific dek-can mRNA. Mol. Cell. Biol. 12, 1687–1697 10.1128/MCB.12.4.1687 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Poon I. K., and Jans D. A. (2005) Regulation of nuclear transport: central role in development and transformation? Traffic 6, 173–186 10.1111/j.1600-0854.2005.00268.x [DOI] [PubMed] [Google Scholar]
- 62. Köhler A., and Hurt E. (2010) Gene regulation by nucleoporins and links to cancer. Mol. Cell 38, 6–15 10.1016/j.molcel.2010.01.040 [DOI] [PubMed] [Google Scholar]
- 63. Gravina G. L., Senapedis W., McCauley D., Baloglu E., Shacham S., and Festuccia C. (2014) Nucleo-cytoplasmic transport as a therapeutic target of cancer. J. Hematol. Oncol. 7, 85 10.1186/s13045-014-0085-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Sun Q., Chen X., Zhou Q., Burstein E., Yang S., and Jia D. (2016) Inhibiting cancer cell hallmark features through nuclear export inhibition. Signal Transduct. Target Ther. 1, 16010 10.1038/sigtrans.2016.10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Saito S., Cigdem S., Okuwaki M., and Nagata K. (2016) Leukemia-associated Nup214 fusion proteins disturb the XPO1-mediated nuclear–cytoplasmic transport pathway and thereby the NF-κB signaling pathway. Mol. Cell. Biol. 36, 1820–1835 10.1128/MCB.00158-16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Sandén C., Ageberg M., Petersson J., Lennartsson A., and Gullberg U. (2013) Forced expression of the DEK-NUP214 fusion protein promotes proliferation dependent on upregulation of mTOR. BMC Cancer 13, 440 10.1186/1471-2407-13-440 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Westerfield M. (1995) The Zebrafish Book: A Guide for the Laboratory Use of Zebrafish (Danio rerio), University of Oregon Press, Eugene, OR [Google Scholar]
- 68. Kimmel C. B., Ballard W. W., Kimmel S. R., Ullmann B., and Schilling T. F. (1995) Stages of embryonic development of the zebrafish. Dev. Dyn. 203, 253–310 10.1002/aja.1002030302 [DOI] [PubMed] [Google Scholar]
- 69. Yan Y. L., Hatta K., Riggleman B., and Postlethwait J. H. (1995) Expression of a type II collagen gene in the zebrafish embryonic axis. Dev. Dyn. 203, 363–376 10.1002/aja.1002030308 [DOI] [PubMed] [Google Scholar]
- 70. Perner B., Englert C., and Bollig F. (2007) The Wilms tumor genes wt1a and wt1b control different steps during formation of the zebrafish pronephros. Dev. Biol. 309, 87–96 10.1016/j.ydbio.2007.06.022 [DOI] [PubMed] [Google Scholar]
- 71. Leimeister C., Bach A., and Gessler M. (1998) Developmental expression patterns of mouse sFRP genes encoding members of the secreted frizzled related protein family. Mech. Dev. 75, 29–42 10.1016/S0925-4773(98)00072-0 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.