

Abstract
The cardiac mechanosensitive BK (Slo1) channels are gated by Ca2+, voltage, and membrane stretch. The neuropeptide GsMTx4 is a selective inhibitor of mechanosensitive (MS) channels. It has been reported to suppress stretch-induced cardiac fibrillation in the heart, but the mechanism underlying the specificity and even the targeting channel(s) in the heart remain elusive. Here, we report that GsMTx4 inhibits a stretch-activated BK channel (SAKcaC) in the heart through a modulation specific to mechano-gating. We show that membrane stretching increases while GsMTx4 decreases the open probability (Po) of SAKcaC. These effects were mostly abolished by the deletion of the STREX axis-regulated (STREX) exon located between RCK1 and RCK2 domains in BK channels. Single-channel kinetics analysis revealed that membrane stretch activates SAKcaC by prolonging the open-time duration (τO) and shortening the closed-time constant (τC). In contrast, GsMTx4 reversed the effects of membrane stretch, suggesting that GsMTx4 inhibits SAKcaC activity by interfering with mechano-gating of the channel. Moreover, GsMTx4 exerted stronger efficacy on SAKcaC under membrane-hyperpolarized/resting conditions. Molecular dynamics simulation study revealed that GsMTx4 appeared to have the ability to penetrate deeply within the bilayer, thus generating strong membrane deformation under the hyperpolarizing/resting conditions. Immunostaining results indicate that BK variants containing STREX are also expressed in mouse ventricular cardiomyocytes. Our results provide common mechanisms of peptide actions on MS channels and may give clues to therapeutic suppression of cardiac arrhythmias caused by excitatory currents through MS channels under hyper-mechanical stress in the heart.
Keywords: ion channel, peptides, inhibition mechanism, gating, kinetics, molecular dynamics, structural model, patch clamp, biophysics, heart, arrhythmias, BK channel, lipid–peptide interaction, mechanosensitive channel, single channel kinetics
Introduction
Mechanosensitive (MS)4 channels are membrane proteins that play important roles in multiple sensory processes, including hearing, touching, proprioception, pain, and numerous cellular functions, including gene expression, vesicular transport, and fluid homeostasis (1–5). In the heart, mechanical stress could increase excitability and initiate arrhythmias and failure. Thus, specific inhibition of cardiac MS channels could provide a novel therapy for cardiac arrhythmogenesis (6, 7). Recently, the neuro-active peptide GsMTx4, extracted from the venom of the tarantula Grammostola spatulata (8), was found to specifically inhibit MS channels (9–12). It has become an important pharmacological tool to investigate the functional roles of the MS channel in excitatory systems for normal physiology and pathogenesis (3, 5, 13–16). Nevertheless, the mechanism of inhibition is unclear.
GsMTx4 is a small amphipathic molecule with a conserved inhibitory cysteine-knot (ICK) backbone similar to other channel-sensitive peptides from the spider venom (17, 18). GsMTx4 is known to influence MS channel gating (the processes of opening and closing) by partitioning into the lipid membrane as do other ICK peptides, such as the voltage-sensor toxin (VsTx) (8, 19–22). However, the inhibitory action of GsMTx4 is not stereospecific (19), a property that differs from other ICK peptides (23), implying a different mechanism from others.
BK channels are expressed in nearly all excitable cells, and their activation depends on both voltage and intracellular Ca2+. All types of BK channels are tetramers. Each α subunit contains three functional components: a voltage-sensor domain (VSD, S1–S4); a pore-forming domain (PD: S5–S6); and a large cytosolic tail domain that comprises the two tandem regulators of K+ conductance (RCK) domains (RCK1 and RCK2), referred to as gating-rings (24–27). Some BK (Slo1) channels are mechanosensitive (28–30), including the stretch-activated BK (SAKca) channel that we previously identified in chick heart (31–33). Like other BK channels (24–26), SAKcaC can be activated by both membrane depolarization and intracellular Ca2+. However, SAKcaC demonstrates stronger Ca2+- and mechano-sensitivities than other BK channels (31–33). Molecular cloning and subsequent mutagenesis studies have shown that the pore-forming α-subunit of SAKcaC contains an additional exon located between the RCK1 and RCK2 domains in the conserved BK C terminus (referred to as the stress-axis regulated exon or STREX) (Fig. 1A), which is critical for both the high Ca2+ and mechano-sensitivities of this channel (31–33).
Figure 1.

GsMTx4 inhibits SAKcaC from extracellular side of the cell membrane. A, schematic of SAKCaC. The transmembrane domain (S0–S6) contains a VSD (S1–S4) and a PD (P5–P6). The cytoplasmic domain contains two RCK (RCK1 and RCK2) domains and an extra exon STREX (pink) located between the RCK1 and RCK2 domains. B, sample traces illustrating the mechanosensitivity of SAKcaC. Left, before suction; middle, during suction (−40 mm Hg); right, after suction releasing. C, total histogram events of channel open (O) and closed (C) states corresponding to B were fitted to Gaussian functions. D, statistical comparison of Po for control (before suction), suction (−40 mm Hg), and release (without suction) conditions on the cell membrane (n ≥6). Data show that Po was significantly increased with −40 mm Hg suction (p < 0.01) and was reversed by release. E, typical single channel current traces showing the inhibitory effect of GsMTx4 on SAKcaC at the time points indicated; 100 nm GsMTx4 was back-filled in the pipette. F, total histogram events of single channel open (O) and closed (C) from E were fitted by Gaussian functions. G, time courses of normalized open probability (Po/Po (control)) for control (without GsMTx4) and during GsMTx4 diffusion to the patch membranes. The GsMTx4 concentrations used were 0.1 μm (Vm = −80 mV) and 0.5 μm (Vm +50 mV) as indicated (n = 4–8). Symbols: C beside traces indicates the channel closed levels; O1 and O2 represent the levels of two channels opening. Time points in E, F, and G were measured from the onset of backfilling (see “Experimental procedures”). SAKcaCs were recorded from chick ventricular myocytes. MPs in B and E were held at −80 mV. [Ca2+]i applied in the bath was 1 mm. **, p < 0.01.
The atomic structure of SAKcaC is not known. However, the structure of the related BK channel (34, 35) shows that Ca2+ binding at the two binding sites per subunit stabilizes an expanded conformation of the gating ring, such that the conformational changes are propagated from the gating ring to the channel gates through the S6-RCK1 linkers. This is proposed to open the pore, a mechanism consistent with the spring-based BK gating-ring mode previously proposed by Niu et al. (24). For SAKcaC, membrane tension created by membrane stretching is supposed to be transferred to expand the gating ring (RCK1–STREX–RCK2) and subsequently impact the channel gates by pulling them open in a similar way as the gating-ring mechanism used by Ca2+ in regular BK channels (33). In this study, we investigated the stretch-dependent gating mechanism of SAKcaC and its modulation by GsMTx4 using patch-clamp recordings of the WT channel and its mechano-reducing, STREX-deleted (STREX-del) mutation (31–33). We observed that GsMTx4 failed to inhibit the mechano-reducing mutation as well as the nonmechanosensitive mSlo1 channel. These results are consistent with a necessary role for the channel region encoded by the STREX-exon and lend support to the linker-gating ring hypothesis in which membrane force is transferred to the gates, possibly by an unknown membrane protein (32, 33). We also found that GsMTx4 exerts distinct effects on WT SAKcaC under the condition of membrane depolarization when compared with membrane hyperpolarized/resting states. With the combination of molecular dynamics (MD) simulation technology, we proposed two inhibitory modes for GsMTx4, a shallow- and a deep-inhibitory mode, which differentially impact mechano-gating of the SAKcaC. This paper presents our experimental observations and MD simulation outcomes, and we then present two proposed inhibitory modes as a working hypothesis for the mechano-gating of SAKcaC modulated by both membrane stretch and GsMTx4.
Results
GsMTx4 inhibits stretch-activated BK channel from the extracellular side of cell membrane
Compared with other BK channels, SAKcaC is unique in that its gene contains an extra exon (STREX) encoding a 59-amino acid stretch-sensitive domain of the channel, located between the RCK1 and RCK2 domains (Fig. 1A). Because the cell membrane generates forces automatically upon the formulation of inside-out patch-clamp configurations (36), we first tested the GsMTx4 effect on the SAKcaC under the condition without additional pressure applied on the cell membrane.
Similar to our observations with other BK channels, in inside-out patch-clamp recordings from cultured ventricular myocytes isolated from the chick heart, we observed ongoing spontaneous activity of SAKcaC with a high concentration of Ca2+ in the intracellular side (26), and the channel activity did not inactivate or rundown during the recordings. Furthermore, upon the background force automatically formed, we added −40 mm Hg membrane force and observed that the open probability (Po) of SAKcaC was significantly increased; removal of the force reversed this increase in Po (Fig. 1, B–D), confirming the mechanosensitivity of this channel. The single channel conductance of SAKcaC was ∼270 pS (Fig. S1), consistent with the “big” conductance character of BK channels as characterized previously (31–33).
We subsequently tested the effect of GsMTx4 on the ventricular myocyte SAKcaC by the back-fill method (See “Experimental procedures”), in which GsMTx4 gradually disperses to the outer surface of the ion channel protein (37, 38). Fig. 1E shows typical single channel currents at different time points following the onset of 100 nm GsMTx4 backfilled in the pipette with 1 mm Ca2+ in the bath (facing the intracellular side of the cell membrane). The data indicated that 100 nm GsMTx4 decreased Po of SAKcaC in a time-dependent way (Fig. 1, E and F; time courses of inhibition are summarized in Fig. 1G). The observed gradual decrease in Po for SAKcaC reflects the diffusion of GsMTx4 to the surface of the patch membrane (37, 38). A higher concentration of GsMTx4 (500 nm) completely inhibited SAKcaC at the hyperpolarized voltage (−80 mV). However, the magnitude of channel inhibition by the peptide toxin was reduced by membrane depolarization, such as at +50 mV (Fig. 1G), 500 nm GsMTx4 produced incomplete inhibition, whereas a 5-fold lower concentration produced nearly complete inhibition at −80 mV (Fig. 1G; also see Fig. 9). This indicates that the peptide action is altered by voltages across the cell membrane (also see Fig. 9). Notably, the ion selectivity (K+/Na+) and single channel conductance were not altered by GsMTx4 (Fig. S1, A and B), consistent with the idea that GsMTx4 acts as a gating modifier on MS channels.
Figure 9.

Membrane depolarization reduced the inhibitory effect of GsMTx4 on SAKcaCs. A, typical single channel current traces showing the effect of 100 nm GsMTx4 on SAKcaC at a depolarized voltage (+30 mV). Traces were obtained at the time points as indicated following the onset of back-filling. The cartoons on the left show that GsMTx4 was applied from the pipette with tension (Pm) automatically formed by membrane deformation upon the excised inside-out patch-clamp configuration. B, total amplitude histogram events of channels open (O) and closed (C) states corresponding to A were fitted by Gaussian functions. Po values were 93.1 (upper panel) and 72.4 (lower panel), respectively. C, time courses of normalized Po (Po/Po (control)) during GsMTx4 diffusion to the patch membranes upon backfilling in the pipette. Vm was held at +30 mV or −80 mV as indicated. D, statistical comparison of the inhibition effects (%) for GsMTx4 between depolarized (+30 mV) versus hyperpolarized (−80 mV) potentials. E, macroscopic currents recorded for BK (mSlo1) channels in the absence (black) or presence (blue) of GsMTx4 back-filled in the pipette. F, time courses for normalized BK currents during GsMTx4 diffusion to the patch membranes. The extracellular GsMTx4 concentrations used were 100 nm for SAKcaC (A–D) and 10 μm for mSlo1 (E and F). [Ca2+]i was 1 mm for SAKcaC (A–D) and 300 μm for mSlo1 (E and F). The time points were measured from the onset of back-filling from the extracellular side of the cell membrane. n ≥6. ***, p < 0.001. SAKcaCs currents (A–D) were recorded from CHO-expressing system, and mSlo1 (E and F) was injected in Xenopus oocytes.
We also compared GsMTx4 effects on SAKcaC under two different [Ca2+]i concentrations. With 0.8 μm Ca2+ in the intracellular side of the cell membrane, 50 nm GsMTx4 inhibited SAKcaC by ∼69.8% of the Po (Po was decreased from 61.9 ± 10.1 to 18.7 ± 6.8%). For comparison, the same concentration of GsMTx4 inhibited SAKcaC Po up to ∼51.1% with 1 mm Ca2+ in the intracellular side of the cell membrane (Po was decreased from 85.1.9 ± 6.8 to 42.7 ± 8.5%, Fig. S2). These results suggested that the effects of GsMTx4 on SAKcaC were not significantly impacted by the intracellular Ca2+ concentration.
Deletion of the STREX-exon mostly abolishes the mechanosensitivity and GsMTx4 inhibition on SAKcaC
To investigate whether GsMTx4 acts by influencing channel mechano-gating, rather than interacting with the VSD as the voltage-sensor toxins do (e.g. VsTx1 and VsTx3) (23, 39), we tested the impact of the toxin on STREX-del mutant channel (Fig. 2). Fig. 2A shows the linear map of STREX-del mutation, of which the STREX-exon located between RCK1 and RCK2 in the C terminus of SAKcaC is removed. However, deletion of this domain did not change the unitary single channel conductance nor the ion (K+/Na+) selectivity (Fig. S3) of the channel. Up to −40 mm Hg of negative pressure applied on the cell membrane had no significant effect on channel activation at the voltages we tested (Fig. 2, B and C), confirming that the region encoded by STREX-exon acts as a mechanosensitive domain for SAKcaC. More interestingly, in the absence of the mechanosensitive domain, application of even a saturated concentration of GsMTx4 (7.5 μm) from the extracellular side of the channel did not inhibit channel activity at any of the tested membrane potentials (Fig. 2, E–J). This result suggests that the “STREX” domain in the C terminus is critical for the inhibitory action of the peptide toxin on SAKcaC.
Figure 2.

STREX-deleted mutation (STREX-del) abolishes both the mechanical and GsMTx4 sensitivities of SAKcaC. A, linear map of STREX-del channel, in which the STREX-exon is deleted from wildtype (WT) SAKcaC. B, sample traces illustrating the loss of nearly all mechanosensitivity of the STRETX-del channel. Left, before suction; right, under suction (−40 mm Hg). MP was held at −80 mV. C and D, total histogram events of channel open (O) and closed (C) states corresponding to B were fitted to Gaussian functions. Each number in the y axis is times 1000. Po values are 1.9 (C) and 1.8 (D), respectively. E, sample traces showing the effect of GsMTx4 on the STREX-del channel at the time points indicated following backfilling. 7.5 μm GsMTx4 was applied in the pipette. Time was measured from the onset of backfilling. MP was held at +50 mV. F–H, total amplitude histogram events of channel open (O) and closed (C) states corresponding to E were fitted to Gaussian functions, showing the time-dependent effect of GsMTx4. Po values are 0.90, 0.92, and 0.93, respectively. I, time courses for the changes in the normalized Po during diffusion of GsMTx4 (7.5 μm) to the patch membrane following backfilling at −30 and +30 mV, respectively (n = 4–6). J, Po–V relationships for STREX-del channels with 0 and 50 nm and 7.5 μm GsMTx4 back-filled in the pipette. The solid lines are fittings to the standard Boltzmann function: Po = Po (max)/{1 + exp(−(Vm − V½)/K)}, where V½ represents the voltage required for half of the maximum channel opening, and K represents the slope factor. Data show that even a saturation concentration of GsMTx4 had no effect on STREX-del mutation channel in the range of voltages examined. STREX-del mutation currents were recorded from the CHO-expressing system.
To examine an alternative mechanism in which the STREX-exon influences peptide interaction with voltage-sensing, we prepared a chimeric construct, mSlo1–STREX, in which the STREX-exon was inserted between RCK1 and RCK2 domains in mSlo1 (Fig. 3A). We then compared the effects of voltage-sensor toxin-3 (VsTx3) on mSlo1 and mSlo1–STREX. Macro-current recordings show that 100 and 300 nm VsTx3 inhibited both mSlo1 and mSlo1–STREX to a similar extent (Fig. 3, B–D). For example, at V½ where channels were half-maximally opened, 100 nm VsTx3 inhibited mSlo1 by 51.8 ± 8.8% at +50 mV (n = 5), and 53.1 ± 6.7% for mSlo1 + STREX (n = 4) at +20 mV (V½ was 54.1 ± 6.3 mV for mSlo1, n = 5, and 19.9 ± 3.5 mV for mSlo1 + STREX, n = 7). In addition, at the full-opening states for both channels, e.g. at + 120 mV, 100 nm VsTx3 inhibited mSlo1 by 29.7 ± 6.2% and mSlo1 + STREX by 28.5 ± 4.1% (Fig. 3F), respectively. Furthermore, 100 nm VsTx3 rightward-shifted G-V curves 12.5 ± 5.7 mV for mSlo1 (V½ was shifted from 54.1 ± 6.3 to 66.5 ± 7.0 mV, n = 5) and 16.1 ± 4.6 mV for mSlo1–STREX (V½ was rightward-shifted from 19.9 ± 3.5 to 35.9 ± 6.4 mV, n = 4–7) (Fig. 3G). Despite the fact that the STREX domain influenced V½ for channel activation, a peptide toxin that functions by inhibiting voltage-dependent gating would have a similar efficacy in the presence or absence of the STREX domain in the channel. Thus, we concluded that the STREX insert between RCK1 and RCK2 domains in the BK channel does not affect the action of voltage-sensor toxin. The relatively weak blocking effects of VsTx3 on both mSlo1 and mSlo1–STREX channels are possible because the voltage-sensor toxin was reported to have high channel specificity (36).
Figure 3.

STREX insert does not alter the effects of voltage-sensor toxin (VsTx3) on BK channels. A, linear map for mSlo1 + STREX (mSlo1–STREX) chimeric channel, in which the STREX-exon was inserted between the RCK1 and RCK2 domains in the BK (mSlo1) C terminus. B and C, sample current traces for mSlo1 (A) and mSlo1–STREX chimera (C) channels, showing the effects of 100 nm VsTx3. The inset at top left shows the voltage protocol used for current recordings. For clarity, the currents shown are at the voltages with a 20-mV increase from −140 to +160 mV. For easy comparison, the red-colored traces on the left set highlight the currents activated at +120 mV, and cyan-colored traces at right show those in the presence of 100 nm VsTx3 applied from the extracellular side of cell membrane. D and E, normalized G-V curves for mSlo1 (D) and mSlo1–STREX (E) channels at the concentrations of VsTx3 as indicated. The solid lines are fits to the Boltzmann equation (see “Experimental procedures”). The V½ obtained for mSlo1 is 54.1 ± 7.3 mV for control, 61.1 ± 9.2 mV for 25 nm, 66.5 ± 7.0 mV for 100 nm, and 64.9 ± 6.6 mV for 300 nm VsTx3 applied from the extracellular side. The V½ obtained for mSlo1–STREX chimera are 19.9 ± 3.5 mV for control, 33.5 ± 2.1 mV for 25 nm, 35.9 ± 7.2 mV for 100 nm, and 37.3 ± 6.8 mV for 300 nm VsTx3. F, summarized blocked currents (blocked I) for the effects of 100 nm VsTx3 on mSlo1 and mSlo1–STREX as indicated. The blocking effects were compared at the voltages near V½ (at +50 mV for mSlo1 and at +20 mV for chimera) or at 120 mV, where both channels were fully opened. Blocked I = (1 −IVsTx3/IControl) × 100%. G, comparisons of the effects of 100 nm VsTx3 on the voltage-dependent activation between mSlo1 and mSlo1–STREX. ΔV½ = V½(VsTx3) − V½(Control). n.s., not significant. n = 4–7. mSlo1 and mSlo1–STREX currents were recorded from Xenopus oocytes with outside-out patch configuration. VsTx3 was applied from bath (the extracellular side of the cell membrane).
GsMTx4 does not inhibit stretch-activated BK channel from intracellular side of the cell membrane
To further explore the mechanism of GsMTx4 action, we tested whether GsMTx4 inhibits SAKcaC by directly interacting with the STREX domain of SAKcaC, which is located on the intracellular side of the membrane. GsMTx4 is known to inhibit MS channels by partitioning into the lipid bilayer (19, 21, 40) as do other ICK peptides (e.g. HaTxI and VsTx). Because there are several hydrophobic residues in the STREX-exon, it would seem possible for this domain to be partially attached on or bound to the inner leaflet of the membrane bilayer. If so, GsMTx4 might inhibit SAKcaC more potently from the intracellular side of the channel protein because GsMTx4 would have better access to the STREX domain when applied directly from the intracellular side of the cell membrane. Therefore, we tested the GsMTx4 effect on SAKcaC by direct perfusion from the bath, which accessed the intracellular side of the ion channel in the excised inside-out patch configuration. Fig. 4 shows that 100 nm GsMTx4 in the pipette (the extracellular side of the cell membrane) strongly inhibited SAKcaC Po at −80 mV, but even a 5-fold higher concentration of 500 nm GsMTx4 applied from the intracellular side did not show a significant effect on SAKcaC under the same conditions.
Figure 4.

GsMTx4 does not show significant effect on SAKcaC when applied from the intracellular side of the channel. A, typical single channel current traces showing strong inhibition by 100 nm GsMTx4 on SAKcaCs applied from the extracellular side of the cell membrane. Traces were obtained at the time points as indicated following the onset of back-filling. The cartoon on the left shows the back-filled GsMTx4 (GT) in the pipette with the tension (Pm) automatically formed by the excised inside-out patch configuration. B, total amplitude histogram events of channels open (O1 and O2) and closed (C) states corresponding to A were fitted to Gaussian functions. Po values were 0.48 (upper panel) and 0.05 (lower panel), respectively. C, typical single channel current traces showing the effect of GsMTx4 from the intracellular side of the cell membrane on SAKcaCs. Traces were obtained at the time points as indicated following the onset of back-filling. The cartoon on the left represents that GT (GsMTx4) was directly applied from the bath under the tension (Pm) automatically formed by membrane deformation. GsMTx4 concentration applied from bath was 500 nm. D, total amplitude histogram events of channels open (O1, O2, and O3) and closed (C) states corresponding to C were fitted to Gaussian functions. Po values are 0.42 (upper panel) and 0.40 (lower panel), respectively. E, time courses of normalized Po (Po/Po(control)) during GsMTx4 diffusion to the patch membrane upon the onset of backfilling in the pipette (olive squares) or perfusion from bath (green squares). The time points were measured from the onset of back-filling from the extracellular side of the cell membrane (olive squares) or application from bath (green squares). GsMTx4 concentrations used were 100 nm (olive squares) for backfilling or 500 nm (green circles) applied from the bath (n = 6–8). GsMTx4 concentrations used were 100 nm in A and B and 500 nm in C and D, respectively. MPs were held at −80 mV in all experiments. SAKcaCs were recorded from the CHO-expressing system.
Perhaps surprisingly, there was also no significant effect of GsMTx4 on SAKcaC with higher concentrations applied to the extracellular side of the cell membrane using the cell-attached configuration (data not shown). This may be because the resting tension near the channel protein in the on-cell configuration is minimal (36), consistent with the specific effect of GsMTx4 on MS channels when mechanical stretch (and channel activation by this mechanism) is present.
Extracellular GsMTx4 inhibits SAKcaC gating in a dose-dependent manner
To investigate peptide-induced changes in the SAKcaC gating properties, we analyzed the kinetics of opening and closing in single channel recordings (41–43). With 1 mm Ca2+ in a bath that was used to limit BK states (43), SAKcaC showed a high level of activity and high frequencies of opening and closing transitions, known as “flickering” (Fig. 5A, upper panel). Following the application of 50 or 100 nm GsMTx4 extracellularly, the channel opened less frequently and showed dose-dependent decreases in channel flickerings (Fig. 5A, middle and lower panels). The kinetic characteristics from the representative experiments are shown in Fig. 5B. Curve-fitting results (see “Experimental procedures”) indicated that the distribution of the open-time constant consisted of only one component (τO), and the closed (or inhibited)-time distribution consisted of two components referred to as the fast (τC1) and slow (τC2) components. GsMTx4 significantly abbreviated the open-time duration (τO) (Figs. 5B, left, and 4C) and prolonged both the fast (τC1) and the slow (τC2) closed-time components (Figs. 5B, right, and 4D) in a dose-dependent manner, accompanied by a significant increase in the relative weight of AC1, which reflects the percentage of the closed events for τC1 (Fig. 5E). However, channels exhibited a decreased Po because τC1 was significantly prolonged. In summary, the net effect of GsMTx4 was to abbreviate open-time duration and prolong closed-time constants, resulting in shorter and less frequent opening.
Figure 5.

GsMTx4 inhibits SAKcaC activity through abbreviating the open-time and prolonging the closed-time constants in a dose-dependent manner. A, typical single channel current traces showing the detailed gating properties of SAKcaC modulated by GsMTx4. Red bars above indicate the most frequent opening durations for control with 0 nm (upper panel), 50 nm (middle panel), and 100 nm (lower panel) GT (GsMTx4) applied from the extracellular side of the cell membrane. B, histograms of SAKcaC open- (left) or closed (right)-times constants corresponding to A are presented. Dashed lines in the right panels represent the distributions of the closed-time components (τC1 and τC2) determined by the likelihood ratio test (see “Experimental procedures”). The vertical dashed lines indicate the peaks of one open- (τO, left) or two closed (τC1 and τC2, right)-time components. The open-time constants (τO) obtained were 8.1 ms for control (upper panel, left), 5.2 ms for 50 nm GsMTx4 (middle panel, left), and 1.8 ms for 100 nm GsMTx4 (lower panel, left). The two closed-time constants (τC1 and τC2) were 2.5 and 13.8 ms for control (upper panel, right), 5.3 and 19.2 ms for 50 GT (middle panel, right), and 33.2 and 140.2 ms for 100 nm GT (lower panel, right), respectively. C, statistical comparison of open-time constants (τO) at the different conditions as indicated. D, statistical comparison for the two closed-time constants, τC1 (left) and τC2 (right), at different conditions as indicated. E, summarized relative weight area (AC1 (%)) showing the percentage of the fast closed-time component (τC1): AC1 (%) = (AC1/(AC1+ AC2)) × 100%. Data for GsMTx4 effects were obtained 25 min later following the onset of back-filling when drugs were completely diffused to the cell membrane. MPs were held at −50 mV. Data points at each condition represent at least three determinations (n >3); *, p < 0.01; **, p < 0.001; n.s, not significant. SAKcaCs were recorded from isolated chick ventricular myocytes.
GsMTx4 inhibits SAKcaC Po increased by membrane tension
We next considered how GsMTx4 action specifically targets SAKcaC mechano-gating. Because it is not possible to estimate the resting tension automatically formed from the excised inside-out patch configuration, we studied the GsMTx4 effect on channel activation induced by additional membrane stretch (caused by suction of the membrane patch). As shown in Fig. 6, A–C, −40 mm Hg applied on the cell membrane increased SAKcaC Po dramatically, consistent with our previous data on the channel's mechanosensitivity (31–33). Interestingly, application of a low concentration of GsMTx4 (50 nm) from the extracellular side (with continued membrane stretch of −40 mm Hg) fully reversed the stretch-induced channel activation and backed to the resting level. As also shown in Fig. 2, the STREX-del channel failed to respond to the additional membrane stretch and failed to show inhibition by the same concentration of GsMTx4 (Fig. 6, A, lower panel, and C). Moreover, −40 mm Hg of membrane stretch caused a marked leftward shift of the Po–V curve in the hyperpolarized direction by about 40 mV, whereas subsequent application of 50 nm GsMTx4 from the extracellular side of the cell membrane (in the presence of continuous membrane stretching of −40 mm Hg) shifted the curve in the opposite direction by ∼40 mV, as if GsMTx4 antagonized the gating induced by membrane stretch (Fig. 6D). Finally, GsMTx4 (50 nm) applied in the absence of additional suction induced a rightward shift of the channel's Po–V curve, indicating that even the resting levels of membrane stretch-induced channel activity are antagonized by the toxin.
Figure 6.

GsMTx4 inhibits SAKcaC activation induced by membrane stretch without changing voltage dependence. A, typical current traces showing the effects of membrane stretch (−40 mm Hg, middle panel) and 50 nm GT (with continuous membrane stretching at −40 mm Hg on the patch membrane, right) for SAKca (upper panel) and STREX-del mutant (lower panel) channels. MPs were held at −90 mV for SAKcaC and −40 mV for STREX-del, where Po values for both channels are similar, to minimize the effects of membrane potentials on channel activation. Data show that SAKcaC is activated by membrane stretch (−40 mm Hg) and inhibited by GT, whereas GT has no effect on the STREX-del mutant channel. B, total amplitude histogram events corresponding to A were fitted by Gaussian functions for SAKcaC (upper panel) and STREX-del mutation (lower panel) channels. The horizontal bar in A (above) represents the continuous negative pressure (−40 mm Hg) applied on the patch membrane. C, statistical comparison of normalized Po (Po/Po(control)) at the conditions indicated for SAKcaC wildtype (WT, left) or STREX-del mutation (right) channels. N >6; D, Po–V relationships for SAKcaC under the conditions of control (□, n = 8), suction (○, −40 mm Hg n = 8), or GT + suction (●, 50 nm GT with a membrane stretch of −40 mm Hg, n = 6). For comparisons, the Po–V relationship under the condition of 50 nm GT alone (without membrane stretch) is also shown (♦, 50 nm GT). The solid lines are fittings to the standard Boltzmann function: Po = Po (max)/{1 + exp(−(Vm − V½)/K)}, where V½ represents the voltage required for half of the maximum channel opening, and K represents the slope factor. The V½ and K−1 obtained were: −88.6 ± 0.9 mV and 22.7 ± 0.9 for control (n = 8), −122.8 ± 2.8 mV and 21.2 ± 1.0 for suction (−40 mm Hg), −81.4 ± 1.7 mV and 24.5 ± 1.5 for GT (with continuous suction), and 39.8 ± 1.9 mV and 22.3 ± 1.7 for GT only (without suction). **, p < 0.001. SAKcaCs was recorded from native chick ventricular myocytes. STREX-del channels were recorded from CHO-expressing system.
Next, we examined whether GsMTx4 itself could affect SAKcaC activity through the modulation of membrane tension. Fig. 7A shows SAKcaC activation induced by negative pressure (−40 mm Hg) applied to the cell membrane. Although Po was decreased to 6.9% with 50 nm GsMTx4 applied from the extracellular side, negative pressure (−40 mm Hg) still increased Po to 22.9 for this particular patch recording (Fig. 7B). However, there was no significant difference among the inhibited channel activations by GsMTx4 under the different membrane tensions (Fig. 7, C and D). We thus concluded that GsMTx4 itself does not control channel activation as does membrane stretch.
Figure 7.

GsMTx4 does not control channel activation through changes in membrane tension. A, left, typical single channel current traces showing the effect of membrane stretch (−40 mm Hg) on SAKcaC current. Right, total amplitude histogram events of channels open (O) and closed (C) states corresponding to left were fitted by Gaussian functions. Po values were 32.5 (upper panel) and 83.3 (lower panel), respectively. B, same as in A (with −40 mm Hg) but with 50 nm GsMTx4 back-filled in the pipette. Po values were 6.9 (upper panel) and 22.9 (lower panel), respectively. C, Po –pressure (−mm Hg) relationships for SAKcaC in the absence (○) or presence of 50 nm GsMTx4 (●) applied from the extracellular side. The inset represents y axis in log scale. D, statistical comparison of the inhibited (%) by GsMTx4 on SAKcaC under different membrane pressures as indicated. Inhibited (%) = ((Po(control) − Po GsMTx4))/Po(control)) × 100 (%) under different pressure conditions. Note: no significant difference was observed on the inhibited currents. SAKcaC was expressed in CHO cells. Single channel currents were obtained at −80 mV with 1 mm [Ca2+]i in the intracellular side. n = 4–8.
GsMTx-4 inhibits SAKcaC through the specific gating modulated by membrane stretch
To explore the biophysical mechanism of how GsMTx4 modulates the mechano-gating, we performed detailed analysis of single channel kinetics, which directly reflects the gating characteristics of the channel. Fig. 8 shows SAKcaC channel kinetics induced by membrane stretch alone (−40 mm Hg) or membrane stretch in the presence of GsMTx4 (50 nm). The results show that membrane stretch alone (−40 mm Hg) significantly prolongs SAKcaC open-time duration (τO) at all membrane potentials to a similar fold increase, and this was subsequently reduced approximately to the control levels following addition of an extracellular 50 nm GsMTx4 (with continued stretch in the pipette) (Fig. 8, A–C, left, and D). Furthermore, membrane stretch (−40 mm Hg) significantly shortened the closed-time constants (τC1 and τC2) (Fig. 8E) but only at hyperpolarization or resting potentials (e.g. at −80 or −50 mV, Fig. 8, E and F); there was no measurable effect of stretch on the closed-time constants at more depolarized voltages (e.g. at −20 mV). Again, the channel gating induced by membrane stretch was reversed by GsMTx4 at hyperpolarized/resting potential (Figs. 8, A–C, right, 6, E and F). Finally, membrane tension did not have a significant effect on the relative area of AC2 (e.g. at −20 mV), which reflects the percentage of the closed events for τC2, but tension dramatically decreased AC2 at the membrane potentials close to resting or hyperpolarization levels (for example, AC2 was decreased more than 2.0-fold by −40 mm Hg at −80 mV from 44.5 ± 7.1 to 20.3 ± 3.3%). Again, these effects were almost reversed by further application of 50 nm GsMTx4. Together, these results from single channel analysis suggest that GsMTx4 targets the mechano-gating of SAKcaC.
Figure 8.

Changes in kinetic parameters of mechano-gating by suction are consistently antagonized by GsMTx4. A–C, histograms of SAKcaC open (left) or closed (right) times for control (A), suction (B, −40 mm Hg), and 50 nm GsMTx4 (with continuous suction of −40 mm Hg) are presented. The membrane potentials were held at −20 mV (upper panel), −50 mV (middle panel), and −80 mV (lower panel), respectively. Dashed lines at right in each panel represent the distributions of the closed-time components (τC1 and τC2) determined by the likelihood ratio test (see “Experimental procedures”). D, statistical comparison of the open-time constants (τO) among control, suction, and GsMTx4 with continuous suction at the voltages as indicated. E and F, statistical comparison of the closed-time constants τC1 (E) and τC2 (F) for control, suction, and GsMTx4 with continuous suction on the cell membrane. G, summarized relative weight area (AC1(%)) showing the percentage of the first closed component τC1. AC1 (%) = (AC1/(AC1 + AC2)) × 100%. The histograms in C were obtained from the single channel traces 25 min later following the back-filling when GsMTx4 was completely diffused to the patch membrane. Data points in D–G at each membrane potential represent at least three determinations. *, p < 0.05; **, p < 0.01. SAKcaCs were recorded from isolated chick ventricular myocytes.
In summary, the net effects of membrane stretch on SAKcaC at resting/hyperpolarized voltages (such as −50 and −80 mV) resulted in a prolongation in channel opening (τO) and a decrease in the closed-time constants with a concomitant decrease in the slow-closed events (indicated by AC2 in Fig. 8G), all of which led to an increased Po. However, 50 nm GsMTx4 (under the continuous suction of −40 mm Hg in the pipette) reversed all these effects induced by membrane tension, an effect that could be explained by GsMTx4 inhibition of SAKcaC activation by mechanic stretching. Nevertheless, at more depolarized voltages (e.g. at −20 mV), both membrane stretch (−40 mm Hg) and GsMTx4 (50 nm) did not significantly affect the factors for the closed-time components (e.g. τC1, τC2, and AC2), resulting in a less inhibitory effect of GsMTx4 on SAKcaC (see “Discussion”). These results also suggested that SAKcaC channel gating is modulated by both membranes stretch and the peptide toxin by a mechanism that depends on membranes potential (see “Discussion”).
Taken together, these results demonstrated that membrane stretch activated SAKcaC as a positive-gating modifier, whereas the spider peptide GsMTx4 antagonized the effect of membrane stretch and thus acted as a negative-gating modifier. We concluded that GsMTx4 inhibited SAKcaC gating through the specific modification of the channel mechano-gate.
GsMTx4 shows more efficiency on SAKcaC under hyperpolarized/resting conditions
In addition to being sensitive to membrane stretch, SAKcaC is also regulated by membrane voltages as the normal BK (Slo1) channels (25, 26, 31). We compared the effects of GsMTx4 at a voltage range from hyperpolarization (e.g. at −80 mV) to depolarization potential (e.g. at +30 mV), where the channel reaches maximal levels of opening with 1 mm Ca2+ in the intracellular side (see Fig. 6D). Although 100 nm GsMTx4 inhibited SAKcaC open probability approximately ∼74.5% at −80 mV, the same concentration of GsMTx4 decreased Po by only ∼20% at +30 mV (Fig. 9, A–D). To exclude the possibility that GsMTx4 exerts its effect by interaction with the VSD as is the case for the voltage-sensor toxins (44, 45), we further tested for an effect of GsMTx4 on mSlo1, the regular BK channel that contains VSDs just like SAKcaC, but lacking the STREX-exon in the C terminus. Under similar conditions, concentrations as high as 10 μm GsMTx4 did not show a significant effect on mSlo1 (Fig. 9, E and F), confirming again that GsMTx4 effect was not achieved by interacting with the VSDs.
Lipid–peptide interaction is influenced by membrane voltages
With a belt of positively charged residues (+5e) around the periphery and a hydrophobic protrusion (18, 19), GsMTx4 is proposed to interact with carbonyl oxygen atoms of both leaflets of the lipid bilayer membrane, similar to the action of HaTx1 and VsTx. Using MD simulations under free production runs, Nishizawa and Nishizawa (40) have suggested two peptide–lipid interaction modes: a shallow binding mode in which the positively charged residues of GsMTx4 interact with the outer leaflet lipid, and the deep-binding mode in which the electrostatic interactions bring the positively charged groups of GsMTx4 into the vicinity of the inner leaflet of the cell membrane. To examine the impact of membrane voltage on the inhibitory effect of the toxin, we performed MD simulations to mimic toxin's actions at different voltages. Fig. 10 shows the representative trajectories and the corresponding snapshots under depolarized (A–C) and hyperpolarized (D–F) states. GsMTx4 com (the center of GsMTx4) was initially put at the location of ∼0.5 nm above the center of the DPPC bilayer (Fig. 10, inset). Following a voltage-restricted production run (see “Experimental procedures”), GsMTx4 was driven outward (under depolarized state) or inward (under hyperpolarized/resting states), respectively, by the electrostatic force. At 60–70 ns of the system runs, the GsMTx4 com positions corresponded to two different modes of interactions, namely the shallow interaction mode (under depolarization condition, Fig. 10, A–C) and the deep interaction mode (under the hyperpolarized/resting states, Fig. 10, D–F), respectively. Under the depolarized state (shallow interaction mode), GsMTx4 com moved upward (Fig. 10, A–C), and the positively charged residues (e.g. Lys-8, Lys-15, Lys-18, Lys-20, Lys-22, Lys-25, and Lys-28) become stabilized near the headgroups of the outer leaflet DPPC by interaction with the carbonyl oxygen atoms (Fig. S4). This mode is consistent with the shallow binding mode suggested by Nishizawa and Nishizawa (40). By contrast, under the hyperpolarized condition (deep-binding mode), GsMTx4 com moves downward (Fig. 10, D–F), and the positively charged residues become split into two functional regions, one interacting with the outer leaflet (Lys-18, Lys-20, and Lys-22) and the other interacting with the inner leaflet (e.g. Lys-15, Lys-8, Lys-28, and Lys-25). This later mode is consistent with the deep-binding mode (40). In the deep-binding mode, GsMTx4-induced membrane deformation was uneven around the peptide; it inserted deeply and interacted with the inner monolayer where the mechano-sensor domain (STREX-exon) of SAKcaC is close (or attached) to (see “Discussion”).
Figure 10.

MD simulations revealed two lipid–peptide interaction modes corresponding to depolarized versus hyperpolarized/resting conditions. A, trajectories of the GsMTx4 com (center) under depolarized state. Each gray curve corresponds to an independent simulation, showing the distance between GsMTx4 com and the inner monolayer. The red curve represents the averaged position of GsMTx4 com to the inner monolayer from three independent runs. B and C, shallow peptide–lipid interaction mode under depolarized conditions. Snapshots were taken from representative trajectories at the indicated time points of the runs in A. D, same as in A, but under the hyperpolarized state. The red curve represents the averaged position of GsMTx4 com from three independent runs. E and F, deep peptide–lipid interaction mode under hyperpolarized state. Snapshots were taken from representative trajectories at the indicated time points of the runs in D. The initial position of GsMTx4 is the same as that under depolarized condition (as in B). The inset at top illustrates the method of the distance measurement between GsMTx4 com and inner monolayer measured in A and D. Ocher spheres represent phosphorus atoms, and the red represents carbonyl oxygen atoms; GsMTx4 is represented in surface structure. Basic residues (Arg and Lys) are blue, and acidic residues (Asp and Glu) are red. Trp residues are orange; Phe residues are yellow, and other hydrophobic residues (Ala, Cys, Ile, Leu, Met, Pro, and Val) are green. For clarity, water molecules and chloride ions are not shown.
BK variants containing STREX insert are expressed in mouse cardiac myocyte membrane
To investigate whether SAKcaC is also expressed in mouse ventricular myocytes, we used an anti-STREX Ab, which is specific against STREX-exon in the BK C terminus, to detect STREX-containing BK variants. Notably, immunostaining of STREX indicates a relatively high STREX signal in the heart, in both atrium and ventricular myocytes (Fig. 11, A–F). The overlay of the STREX signals with the plasma membrane marker (WGA), as indicated by white arrows in Fig. 11, I and J, further confirmed the co-localization of STREX with WGA on the plasma membrane of mouse ventricular myocytes (Fig. 11L).
Figure 11.
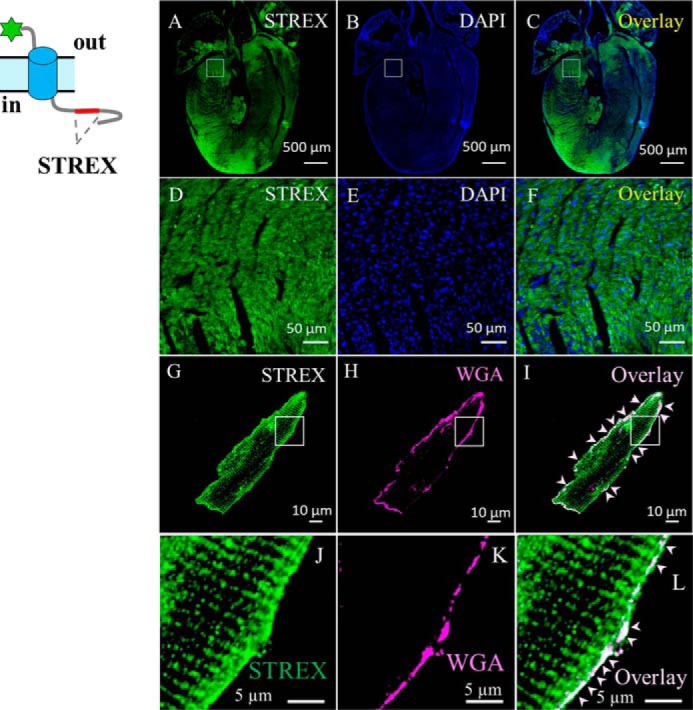
BK variants containing STREX-exon are located at the plasma membrane of mouse cardiac myocytes. A and B, confocal images of mouse heart section labeled with anti-STREX (green, A), which is specific against STREX between RCK1 and RCK2 domains in the BK channel, or DAPI (blue, B); C, overlay of A and B. D–F, zoom-out of the squared regions in A–C, respectively. G and H, ventricular myocytes loaded with plasma membrane marker (WGA), fixed, permeabilized, and labeled with anti-STREX. I, overlay of G and H, showing location of BK variants containing STREX-exons in BK channels. J–L, amplification of the squared regions as indicated in G–I. Cartoon at left shows BK variant with the tag at the N terminus. For clarity, white arrows in I and L highlight the surface expression of BK variants containing STREX-exon in the BK C terminus.
Recently, different BK variants, e.g. BK–STREX, BK–DEC, and BK–STREX–DEC, were identified in ventricular cardiomyocytes (46–49). Among them, the BK variant containing both STREX and DEC (BK–STREX–DEC) was found to be mechanosensitive. To test the possible contribution of DEC-containing BK variants to the native SAKcaC (BK–STREX) currents we obtained, we used a poly-cloned antibody against DEC at the end of the BK α subunit, generated by the same antigen peptide used by Singh et al. (46). Although BK variants containing STREX inserts (e.g. BK–STREX and/or BK–STREX–DEC) are clearly observed in ventricular myocyte membranes (Fig. 11), as shown in Fig. S5, we did not observe signals of overlaid DEC with WGA in ventricular myocytes (Fig. S5), suggesting that BK variants containing DEC, e.g. BK–DEC and BK–STREX–DEC, are not located on the surface of ventricle myocytes (Fig. S5). Thus, we concluded BK–STREX–DEC did not contribute to native cell currents that we recorded from the ventricular myocytes.
Discussion
GsMTx4 inhibits mechanosensitive channels (MSCs) by interacting with the lipid bilayer rather than physically occupying the channel pore (19). MD simulation with free production runs suggested two binding modes of GsMTx4 interaction with the lipid bilayer, which may influence the magnitude of GsMTx4 effects on the MSC gating (22, 40). In this study, we presented data to address the specific inhibitory mechanism that GsMTx4 acts directly on the mechano-gating of a stretch-activated BK channel (SAKcaC). Validation of the expression of these channels in mouse ventricular myocyte membranes (Fig. S5) provides justification for understanding. We also explored the peptide–lipid interaction modes, under depolarized versus hyperpolarized/resting states, to interpret the observations that GsMTx4 inhibited SAKcaC stronger at hyperpolarized/resting states compared with that at the depolarized condition. Each mode appears in a voltage-dependent way parallel to the two inhibitory modes.
Mechano-sensing domain, STREX in SAKcaC, is the target for GsMTx4 inhibition
The mechanosensitive channel inhibitor, GsMTx4, belongs to the same peptide family as voltage sensor toxins, and they have structural similarity with hydrophobic patches in the surface and a belt of positively charged residues around the periphery (18). It is known that the voltage-sensor toxins inhibit voltage-dependent ion channels by partitioning into the bilayer to interact with the voltage sensor paddle (VSP) buried in the lipids (39, 44, 45). SAKcaC possesses a similar VSD to those of regular BK (Slo1) and voltage-dependent K+ channels. Thus, the question first to consider was whether GsMTx4 inhibits SAKcaC by directly targeting its VSD domains.
Generally, the mechanism of BK channels regulated by membrane potentials is explained by having the four voltage sensors move in (or through) the fluid membrane interior, as for voltage-gated Kv channels (24, 27, 35). Judging from the previous reports that GsMTx4 inhibits MS channels by partitioning into the cell membrane to modulate channel gating (19, 40), a reasonable hypothesis is that GsMTx4 inhibits SAKcaC by acting at the VSDs, as in the case of VsTx. This hypothesis is also attractive because the positively charged GsMTx4 (+5e) could repel electrostatically the positively charged VSDs upon its portioning into the cell membrane, thus facilitating the VSDs moving from the exterior (activated state) to the interior (inactivated state), an action similar to that achieved by membrane hyperpolarization. However, the observation that even at saturating concentrations GsMTx4 did not inhibit mSlo1 (Fig. 9, E and F) nor the STREX-del mutant (Figs. 2 and 6) clearly excluded the possibility that GsMTx4 achieves its effect through interaction with the channel's VSDs, because both mSlo1 and STREX-del mutant possess VSDs similar to those of regular Kv channels. Rather, our data support an alternative hypothesis that the STREX-exon located between RCK1 and RCK2 domains in the SAKcaC C terminus is the target (direct or indirect) for the inhibition by GsMTx4. Key data in support of this explanation is that deletion of STREX, one of the mechano-sensing domains (or sites) in SAKcaC, completely abolishes the effect of the peptide at all voltage ranges tested (from −100 to +70 mV). Nevertheless, we did not observe a significant inhibitory effect of GsMTx4 on mSlo1–STREX, a chimeric construct containing the STREX insert between the RCK1 and RCK2 domains in the mSlo1 channel, suggesting that the different structures between mSlo1 and SAKcaC may also account for GsMTx4 effects.
Recently, stretch activation of the BK–STREX–DEC variant was also noticed in cardiac myocytes (46, 47). We thus considered whether this channel variant could have contributed to the results obtained from native myocytes. The SAKcaC clone used in our experiments does not contain the DEC insert in the end of C terminus, and the characteristics of the channel were consistent with our results from experiments done in native myocytes (31, 33). We also failed to detect a signal against the DEC region at the surface of myocytes isolated from newborn mice (Fig. S5). This is consistent with the view that STREX and DEC are located in different places. DEC-containing splice variants are reported to be specifically localized to the mitochondria (46). Thus, there is no evidence that BK variants containing the DEC exon played a role in our experiments.
GsMTx4 inhibits SAKcaC through the specific modulation of the mechano-gating
GsMTx4 has been widely used to detect MS channel functions in many excitable systems because of its specific effects on MS channels. Nevertheless, the mechanism of how this peptide targets the mechano-gates of MS channels is unknown. In this study we show that the Po of SAKcaC was significantly increased by membrane stretch (Figs. 1B, and 6, A and B), which caused a leftward shift of the Po–V curves (Fig. 6C), suggesting that the channel gating was positively modulated by membrane stretch. This “shift” was subsequently reversed by further application of GsMTx4 (Figs. 6 and 8), indicating that GsMTx4 acts as a negative gating modifier to antagonize the opening of channel mechano-gates activated by membrane tension.
GsMTx4 is reported to induce membrane (bilayer) stretch by membrane deformation like an immersible wedge; hence, the deformation of lipid membrane is different from that generated by ordinary membrane stretch, e.g. negative pressure. Thus, GsMTx4 could not induce changes in channel kinetics to control channel activation as membrane stretch does. It only acts to antagonize the effect of membrane stretch. In addition, the extent (%) of the inhibition by GsMTx4 was also not altered by changes in membrane tensions (Fig. 7), suggesting again that GsMTx4 itself could not induce membrane stretch to modulate channel gating. To clarify our understanding of the gating relationships between membrane stretch and GsMTx4, we performed detailed kinetic analysis of the single channel gating. The observations that the prolonged channel open-time (τO) and shortened closed-time constants (τC1 and τC2) by membrane stretch are consequently reversed by further application of GsMTx4 (Fig. 8) confirm that GsMTx4 inhibits SAKcaC through the specific modification of the channel mechano-transduction gating. Furthermore, this is consistent with the elimination of GsMTx4 effects at all voltages on the STREX-del mutant channel. Based on the above observations, we thus hypothesize that stretching of the cell membrane generates a force on channel gate through the expansion of the gating-ring complex (RCK1–STREX–RCK2 in SAKcaC), which opens the channel gates (Fig. 12, A and B), a mechanism analogous to what Ca2+ does on BK channel mode (24, 27, 35). However, the peptide GsMTx4 likewise generates compression forces by specifically targeting the mechanical tension on the channel gates, through the same pathway (gating-ring complex), to push the SAKcaC gate to close (additional details below). There is still the possibility that the toxin binds directly to the STREX domain and thus disrupts the communication between sensing the stretch stimulus and opening the channel. Nevertheless, this seems unlikely, because GsMTx4 shows no inhibitory action when applied to the intracellular side of the cell membrane (Fig. 4), which gives the toxin access directly to the STREX domain.
Figure 12.

Proposed SAKcaC gating modes modulated by membrane stretch and peptide GsMTx4 primarily based on the spring model proposed for BK and SAKcaCs (24, 33). A and B, membrane force (Fm) first pulls SAKcaC gate opening through STREX and MP (33). C, deep inhibition mode: under hyperpolarized/resting conditions, GsMTx4 was driven down (inward) along the electrochemical gradients upon partitioning into the lipid bilayer. It was placed at a deep position to interact with both inner and outer monolayers and induce strong membrane deformation as observed in the MD simulation. Thus, GsMTx4 has the ability to push STREX back strongly through MP, to close the channel gate firmly. D, membrane depolarization drives VSP upward, which generates pulling force to further open channel gate (from B). E, shallow inhibition mode: under the depolarization condition, GsMTx4 was driven back (move outward) by membrane potential. Alternatively, the outward electrostatic forces may also prevent the absorption of GsMTx4 from the extracellular side into the lipid bilayer. In either case, GsMTx4 interacts with the outer monolayer only, and induces weak membrane deformation as observed in the MD simulation. The SAKcaC gating mode is drawn based on BK gating mode (24, 27, 33, 35), in which VSP and STREX-exon are both connected to the channel gate.
Two inhibition modes gated by membrane stretch and GsMTx4 inhibition
We have presented data to show that GsMTx4 inhibits SAKcaC by specifically targeting the mechano-gate without interaction with VSDs. Our hypothesis for the different inhibitory effects of GsMTx4 on SAKcaC between membrane hyperpolarization and depolarization (Fig. 9) is that this may arise from the different lipid–peptide interaction modes.
Recently, with MD simulation technology, Nishizawa and Nishizawa (40) showed two binding modes of GsMTx4 with the lipid bilayer followed by a free 30-ns production run: the shallow binding mode where all the positively charged residues of GsMTx4 interact with the outer leaflet lipid, and the deep-binding mode where the electrostatic interaction brings the positively charged groups into the inner leaflet lipids, accompanied by membrane deformation (40). To explore the possible different peptide–lipid interaction modes, we also performed MD simulation to mimic the action of GsMTx4 inside the lipid bilayer under hyperpolarizing/resting versus depolarizing conditions.
Under the depolarized state, GsMTx4 that partitions into the cell membrane was driven back along the electric field toward the outer leaflet, resulting in binding shallowly with the outer monolayer. This induces weaker membrane deformation, which we call the shallow lipid–peptide interaction mode (Figs. 10, A–C, and 12). In contrast, under membrane hyperpolarization/resting states, the positively charged GsMTx4 moves down the electric field and binds deeply with the inner monolayer of lipid bilayer (Fig. 10). This resulted in deep interaction of GsMTx4 with the inner monolayer, where the mechano-sensing domain (STREX-exon) of SAKcaC is closed (or attached); we call this the deep interaction mode (Fig. 10, D–F). Based on our observations and BK-gating mechanisms established previously (24, 27, 33, 35), we thus predict two inhibition modes, referred as “deep inhibition” (Fig. 12C) and “shallow inhibition” (Fig. 12E), to further interpret our observations.
In terms of the two modes in Fig. 12, membrane tension first turns the passive spring of the gating ring (RCK1–STREX–RCK2 in SAKcaC) into a force-generating machine with a characteristic force that uses the free energy to expand the gating ring (four RCK1–STREX–RCK2 in one α subunit) and thus opens the channel gates, as predicted previously in BK channel (24, 35). Under membrane hyperpolarizing/resting conditions (Fig. 12B), where negative charges accumulated in the inner monolayer (the intracellular side of cell membrane) and the positive charges outside the outer monolayer (the extracellular side of cell membrane), the positively charged (+5e) peptide is driven down deeply along the electrochemical gradients to bind with both leaflets of the lipid bilayer based on our observations from MD simulation outcome (Fig. 10, D–F). This generates a large closing force and strongly pushes the unknown membrane protein (MP) back (as we predicted previously (33)), thus reversing the effects induced by membrane stretching (Fig. 12, B and C). In this mode, GsMTx4 is further stabilized deeply inside the lipid bilayer by interaction simultaneously with the carbonyl oxygen atoms in both leaflets of the lipid bilayer as observed in MD simulation. It is placed deeply and binds tightly with the MP/inner monolayer to reverse the membrane force generated by membrane stretch that expands the four-gating rings (in one SAKcaC α subunit) to open the channel gate.
Under the depolarization condition, the outwardly moved VSD further opens the S6 gate of SAKcaC (Fig. 12, B–D) as observed in BK channel. Our hypothesis is that the less inhibition of GsMTx4 on SAKcaC is reached by the release of lipid–peptide interaction, from the deep interaction mode (Fig. 12C) to the shallow interaction one (Fig. 12E). As we observed in the MD simulation outcome, GsMTx4 is driven back along the electrochemical gradients, thus the closing forces generated by GsMTx4 on the inner monolayer of cell membrane is certainly released, resulting in less inhibitory effect of GsMTx4 on SAKcaC unless a higher concentration of GsMTx4 is used to neutralize the negative charges accumulated outside of the cell membrane. Alternatively, membrane depolarization prevents the positively charged extracellular peptide from partitioning into and moving down against the electrochemical gradients, thus generating less closing force on the inner leaflet of the cell membrane (Fig. 12E). Six lysines in GsMTx4 have been proposed to be important in affecting lipid–peptide binding (21, 22, 40). In our MD simulation outcome, lysines (e.g. Lys-8, Lys-15, Lys-25, and Lys-28) are interacting with carbonyl oxygen atoms to cause membrane deformation (Fig. S4); thus, it is not difficult for one to imagine that mutation of these positively charged lysines in GsMTx4 could reduce the interaction force between lipid–peptide and thus decrease membrane deformation and release the closing force generated by partitioning of peptide. Therefore, the final results of mutating the lysines would result in decreased efficiency of peptide inhibition on SAKcaC.
In these modes, because SAKcaC structure is not known, it is not certain how the mechano-sensing domain STREX-exon in the intracellular side of the SAKcaC C terminus is communicating with the lipid bilayer. Although the hydrophobic residues in STREX-exon (Fig. S3A) provide the possible interaction of STREX with the inner monolayer, an unknown MP is supposed to be in the modes to serve as the communicator to link the peptide and STREX-exon, as predicted previously (32, 33). Although we observed a weak response of the STREX-del mutation to membrane stretch at higher voltages, this does not affect our conclusion for the specific effect of GsMTx4.
Independent and synergistic gating by voltage-sensor domain and membrane stretch/GsMTx4
In the modes of SAKcaC we presented in Fig. 12, the movement of VSDs and the expansion/compression of cytosolic gating-ring complex (four in one α subunit) are relatively independent of one another. However, as the gating ring and the VSDs in SAKcaC are finally converged on the same gate (S6), the gating by each of the voltage sensors in SAKcaC and membrane stretch/GsMTx4 would be synergistic as observed in regular BK channels, for which there are independent and synergistic effects on the gating ring due to the modulation of the VSD moving and Ca2+ binding (24, 27, 33, 35).
Experimental procedures
Cell culture, mutagenesis, and heterologous expression
Native SAKcaC was recorded from chick ventricular myocytes, which were dissected from 10- to 12-day-old White Leghorn embryos as described previously (31, 33). The chick SAKcaC gene was cloned from a cDNA library made from chick embryonic hearts (31–33). STREX-del mutation was verified by sequencing as published previously (32, 33).
Cloned SAKcaC or STREX-del cDNAs were transiently transfected in Chinese hamster ovary (CHO-K1) cells using the LipofectamineTM 2000 transfection reagent (Thermo Fisher Scientific) according to the manufacturer's protocol. pEGFP (Clontech) was co-expressed with the channel cDNA at a ratio of 5:1 (weight/weight) to identify successfully transfected cells as used previously (31–33).
Mouse Slo1 (mSlo1) cDNA clone was a gift from Dr. Christopher Lingle (Washington University in Saint Louis). cRNA was transcribed in vitro with MessageMachine kit SP6 (Ambion) and injected in Xenopus oocytes as described previously (50, 51). No endogenous BK channel activation was detected in untransfected CHO-K1 or uninjected Xenopus oocytes.
Electrophysiology
Single channel currents were recorded from embryonal chick ventricular myocytes from (Figs. 1, 5, 6, and 8) or from heterologously expressed CHO cells (Figs. 2, 4, 7, and 9, A–D). Single channel currents were recorded under a standard excised inside-out patch configuration with an EPC9 patch-clamp amplifier (HEKA Elektronik, Lambrecht, Germany) or A&M amplifier (model 2400). Data were filtered at 10 kHz. The standard pipette/extracellular solution was as follows (in mm): 130 K-gluconate, 15 KCl, 1 EGTA, 10 HEPES, and 5 glucose with pH 7.4 adjusted with NaOH. The bath/intracellular solutions were the same, except [Ca2+]i concentrations were buffered as described previously (31, 32).
Macroscopic currents for BK (mSlo1) and mSlo1–STREX chimera were recorded from Xenopus laevis oocytes under standard excised inside-out (Fig. 9, C and D) or outside-out (Fig. 3) configurations (25, 52) with an A-M 2400 patch-clamp amplifier (A-M Systems, Inc.). pClamp (Molecular Devices) was used to drive stimulus protocols and digitize currents (26). Whether for outside-out or inside-out patches, the standard extracellular solution was as follows (in mm): 140 KMES (methanesulfonate), 20 KOH, 10 HEPES, 2 MgCl2, pH 7.0. The composition of internal solution contained the following (in mm): 140 KMES, 20 KOH, 10 HEPES, 5 EGTA, pH 7.0. The [Ca2+]i solutions were buffered as described previously (25, 26). To obtain the conductance–voltage (G-V) curves, currents were elicited by voltage pulses from −140 to −160 mV (20 ms) with 10-mV increments, whereas the voltages before and after the pulses were held at −140 mV to close the channels.
The extracellular GsMTx4 effect (facing the external side of ion channels) was tested by application in the pipette solution using the standard back-fill method established previously (37, 38). In brief, the tips of the electrodes were first filled with the normal pipette solution and then backfilled with the same solution containing a concentration of GsMTx4. The intracellular GsMTx4 effect (facing the internal side of channels) was examined by directly perfusion of GsMTx4 in the bath solution. No significant difference was observed for SAKcaC recorded from chick ventricular myocytes and CHO cells. The intracellular GsMTx4 effect (facing the internal side of channels) was examined by direct perfusion of GsMTx4 in the bath solution. Membrane stretch was reached by negative pressure applied to the patch pipette using a pneumatic transducer tester (DPM-IB, BioTek Instruments Inc., Winooski, VT) (31, 33). All experiments were performed at room temperature (22–25 °C).
Single channel analysis
Single channel conductance was determined by the slope of current–voltage (I-V) curves from −80 to +80 mV, where I-V data could be fitted to a linear line. Amplitude histograms were measured in inside-out patches with 1–4 channels. Histograms were fitted with a Gaussian function using the software pCLAMPFIT®. The Po (%) was determined as described previously (51, 53). The Po was calculated using the area under each peak (aj) at each current level (j) in the histogram along with the number of channels (N) as shown in Equation 1,
| (Eq. 1) |
The channel number (N) in each patch was defined as the largest open channel numbers that can be observed at +80 mV under 1 mm [Ca2+]i. To assess the effect of GsMTx4 on the kinetic properties of SAKcaC gating, patches with only one single channel were used. The opening and closing transitions of channels were detected by the half-amplitude threshold method after Gaussian filtering at 1 kHz. Open or closed channel events with durations of <0.6 × 10−3 ms were eliminated from the data set, of which this amount of “flickering” increases with depolarization but was not changed either by membrane stretch or by peptide inhibition. These events were log binned into open- or closed-time histograms for better understanding the mechanism of gating states (10 bins/decade (41, 42)). The open and closed (or inhibited) times were fitted to exponential functions using the Marquardt-Levenberg algorithm (Igor Pro, WaveMetrics). To minimize the number of single channel states, the saturating [Ca2+]i concentration of 1 mm was used (43).
Immunohistochemistry
Newborn Kunming mice were used to detect the expression of SAKcaC (BK–STREX variant) in the hearts. All animal protocols were approved by the Animal Experiment Administration Committee in Xuzhou Medical University.
Heart slides were prepared and stained as described previously (54). In brief, mouse hearts were fixed in 4% paraformaldehyde, equilibrated in 10% sucrose, and embedded in OCT. Then cryosections of 6–10 μm thick were prepared and incubated overnight with individual antibodies at 4 °C, followed by 1-h secondary antibody incubations at 37 °C. The slides were mounted with or without the unclear counterstain (DAPI, Beyotime).
Simulation of GsMTx4 interaction with lipid bilayer
The peptide–lipid bilayer system was built by CHARMM-GUI (55, 56). The coordinates for GsMTx4 (PDB code 1TYK) were obtained from RCSB Protein Data Bank (18). The simulation programs, conditions, and water models were basically the same as those described previously by Nishizawa et al. (22, 40). Specifically, the system was composed of 64/64 DPPC bilayers (64 molecules in the upper/lower leaflets), one peptide, and 3895 H2O along with Cl ions that were used to neutralize the positive charges of the peptides. The XYZ axes of the simulation box (Å) was 65 × 65 × 110, and the z-axis was defined as the bilayer normal. GsMTx4 com was embedded into DPPC in the bilayer center (the plane that is parallel to the membrane and contains the com of the membrane). MD simulation was performed with an 80-ns production run followed by an energy minimization and an equilibration run. The temperature was set at 323.15 K with Nose-Hoover coupling. The pressure was controlled by the Parrinello-Rahman at 1 atm with the independent (semi-isotropic) coupling in the xy and z directions. For the simulations containing a bilayer, the bond lengths of z-axis were constrained with LINCS. For the simulations of GsMTx4 interaction with lipid bilayer under the hyperpolarized/depolarized conditions, the systems were settled in the electric fields along the z axis as used by Su and Guo (57). A time step of 2 fs was used. Trajectories were saved every 5 ps for analysis. The simulation was carried out at the National Supercomputer Center, LvLiang, China. All molecular images were made with Visual Molecular Dynamics.
Data analysis
Data acquisition and analysis were carried out using pClamp9 (Molecular Devices), PULSE+PULSEFIT TAC 4.0 (HEKA Elektronik), ANA.EXE and Origin 7.5 software. Data in all figures are presented as means ± S.E. Statistical significance was evaluated by a two-sample t test and p ≤ 0.05 was considered as significant.
Antibodies and chemicals
A SREX-specific antibody (anti-STREX) was raised in rabbits targeting STREX-exon in BK variants containing the STREX insert. Primary antibody for BK was raised in guinea pigs against the C-terminal end of the BK channel encoded by the Kcnma1 gene (catalog no. AGP-014, Alomone Labs). The surface membrane was labeled by WGA (Thermo Fisher Scientific). The peptides GsMTx4 and VsTx3 were purchased from Alomone Labs. The aqueous stock solutions were prepared at 5 or 10 mm and kept at −20 °C. An appropriate amount of the stock aliquot was diluted to the concentration used on the day of the experiment. The working solutions had concentrations of 50, 100, and 500 nm and 7.5 and 10 μm for GsMTx4 and 20, 100, and 300 nm for VsTx3. Other chemicals were purchased from Sigma.
Author contributions
H. L., J. X., Z.-S. S., G.-M. W., M. T., X.-R. D., Y.-T. L., J.-J. W., F.-F. Z., Z. Q., Z. Z., and Q.-Y. T. data curation; H. L., J. X., Z.-S. S., G.-M. W., M. T., X.-R. D., Y.-T. L., J.-J. W., F.-F. Z., Z. Q., Z. Z., and Q.-Y. T. formal analysis; H. L., J. X., Z.-S. S., G.-M. W., M. T., X.-R. D., J.-J. W., F.-F. Z., Z. Z., M. S., and Q.-Y. T. investigation; Z. Z., M. S., and Q.-Y. T. methodology; M. S. and Q.-Y. T. software; M. T., Z. Z., M. S. and Q.-Y. T. funding acquisition; M. S., and Q.-Y. T. project administration; M. S. and Q.-Y. T. resources; M. S. and Q.-Y. T. supervision; M. S. and Q.-Y. T. validation; M. S. and Q.-Y. T. writing-review and editing; M. S. and Q.-Y. T. conceptualization; Q.-Y. T. writing-original draft.
Supplementary Material
Acknowledgments
We are grateful to Dr. Linda Boland (University of Richmond, Richmond, VA) and Dr. Diomedes E. Logothetis (Northeastern University, Boston, MA) for valuable suggestions and comments on this work, and Dr. Clive M. Baumgarten (Virginia Commonwealth University, Richmond, VA) for critical reading and comments on our earlier work.
This work was supported by NSFC grants 81450064 and 31671212 (to Q.-Y. T.), NSFC grants 81471314 and 81671090 (to Z. Z.), Xuzhou Science and Technology Program KC16H0230 (to Q.-Y. T.), JSPS KAKENHI JP15H05936 (to M. S.), Key Project in Sichuan province department of education 16ZA0196 (to M. X. T.), and Jiangsu Provincial Special Program of Medical Science BL2014029 (to W. R). The authors declare that they have no conflicts of interest with the contents of this article.
This article contains Figs. S1–S5.
- MS
- mechanosensitive
- MSC
- mechanosensitive channel
- SAC
- stretch-activated ion channel
- SAKcaC
- stretch-activated BK channel
- VSD
- voltage-sensor domain
- Vm
- membrane potential
- RCK
- regulators of K+ conductance
- ICK
- inhibitory cysteine-knot
- MD
- molecular dynamics
- DAPI
- 4,6-diamidino-2-phenylindole
- VsTx
- voltage-sensor toxin
- MP
- membrane protein
- Com
- center of mass
- WGA
- wheat germ agglutinin
- PD
- pore-forming domain
- GT
- GsMTx4
- VSP
- voltage sensor paddle.
References
- 1. Eyckmans J., Boudou T., Yu X., and Chen C. S. (2011) A hitchhiker's guide to mechanobiology. Dev. Cell 21, 35–47 10.1016/j.devcel.2011.06.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Tyler W. J. (2012) The mechanobiology of brain function. Nat. Rev. Neurosci. 13, 867–878 10.1038/nrn3383 [DOI] [PubMed] [Google Scholar]
- 3. Gu Y., and Gu C. (2014) Physiological and pathological functions of mechanosensitive ion channels. Mol. Neurobiol. 50, 339–347 10.1007/s12035-014-8654-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Kocer A. (2015) Mechanisms of mechanosensing–mechanosensitive channels, function and re-engineering. Curr. Opin. Chem. Biol. 29, 120–127 10.1016/j.cbpa.2015.10.006 [DOI] [PubMed] [Google Scholar]
- 5. He B. H., Christin M., Mouchbahani-Constance S., Davidova A., and Sharif-Naeini R. (2017) Mechanosensitive ion channels in articular nociceptors drive mechanical allodynia in osteoarthritis. Osteoarthritis Cartilage 25, 2091–2099 10.1016/j.joca.2017.08.012 [DOI] [PubMed] [Google Scholar]
- 6. Bode F., Sachs F., and Franz M. R. (2001) Tarantula peptide inhibits atrial fibrillation. Nature 409, 35–36 10.1038/35051165 [DOI] [PubMed] [Google Scholar]
- 7. Wang J., Ma Y., Sachs F., Li J., and Suchyna T. M. (2016) GsMTx4-D is a cardioprotectant against myocardial infarction during ischemia and reperfusion. J. Mol. Cell Cardiol. 98, 83–94 10.1016/j.yjmcc.2016.07.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Suchyna T. M., Johnson J. H., Hamer K., Leykam J. F., Gage D. A., Clemo H. F., Baumgarten C. M., and Sachs F. (2000) Identification of a peptide toxin from Grammostola spatulata spider venom that blocks cation-selective stretch-activated channels. J. Gen. Physiol. 115, 583–598 10.1085/jgp.115.5.583 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Bae C., Sachs F., and Gottlieb P. A. (2011) The mechanosensitive ion channel Piezo1 is inhibited by the peptide GsMTx4. Biochemistry 50, 6295–6300 10.1021/bi200770q [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Coste B., Mathur J., Schmidt M., Earley T. J., Ranade S., Petrus M. J., Dubin A. E., and Patapoutian A. (2010) Piezo1 and Piezo2 are essential components of distinct mechanically activated cation channels. Science 330, 55–60 10.1126/science.1193270 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Coste B., Xiao B., Santos J. S., Syeda R., Grandl J., Spencer K. S., Kim S. E., Schmidt M., Mathur J., Dubin A. E., Montal M., and Patapoutian A. (2012) Piezo proteins are pore-forming subunits of mechanically activated channels. Nature 483, 176–181 10.1038/nature10812 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Suchyna T. M. (2017) Piezo channels and GsMTx4: Two milestones in our understanding of excitatory mechanosensitive channels and their role in pathology. Prog. Biophys. Mol. Biol. 130, 244–253 10.1016/j.pbiomolbio.2017.07.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Hara M., Tabata K., Suzuki T., Do M. K., Mizunoya W., Nakamura M., Nishimura S., Tabata S., Ikeuchi Y., Sunagawa K., Anderson J. E., Allen R. E., and Tatsumi R. (2012) Calcium influx through a possible coupling of cation channels impacts skeletal muscle satellite cell activation in response to mechanical stretch. Am. J. Physiol. Cell Physiol. 302, C1741–C1750 10.1152/ajpcell.00068.2012 [DOI] [PubMed] [Google Scholar]
- 14. Bagriantsev S. N., Gracheva E. O., and Gallagher P. G. (2014) Piezo proteins: regulators of mechanosensation and other cellular processes. J. Biol. Chem. 289, 31673–31681 10.1074/jbc.R114.612697 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Blumenthal N. R., Hermanson O., Heimrich B., and Shastri V. P. (2014) Stochastic nanoroughness modulates neuron-astrocyte interactions and function via mechanosensing cation channels. Proc. Natl. Acad. Sci. U.S.A. 111, 16124–16129 10.1073/pnas.1412740111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Lee W., Leddy H. A., Chen Y., Lee S. H., Zelenski N. A., McNulty A. L., Wu J., Beicker K. N., Coles J., Zauscher S., Grandl J., Sachs F., Guilak F., and Liedtke W. B. (2014) Synergy between Piezo1 and Piezo2 channels confers high-strain mechanosensitivity to articular cartilage. Proc. Natl. Acad. Sci. U.S.A. 111, E5114–E5122 10.1073/pnas.1414298111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Norton R. S., and Pallaghy P. K. (1998) The cystine knot structure of ion channel toxins and related polypeptides. Toxicon 36, 1573–1583 10.1016/S0041-0101(98)00149-4 [DOI] [PubMed] [Google Scholar]
- 18. Oswald R. E., Suchyna T. M., McFeeters R., Gottlieb P., and Sachs F. (2002) Solution structure of peptide toxins that block mechanosensitive ion channels. J. Biol. Chem. 277, 34443–34450 10.1074/jbc.M202715200 [DOI] [PubMed] [Google Scholar]
- 19. Suchyna T. M., Tape S. E., Koeppe R. E. 2nd, Andersen O. S., Sachs F., and Gottlieb P. A. (2004) Bilayer-dependent inhibition of mechanosensitive channels by neuroactive peptide enantiomers. Nature 430, 235–240 10.1038/nature02743 [DOI] [PubMed] [Google Scholar]
- 20. Peng A. W., Gnanasambandam R., Sachs F., and Ricci A. J. (2016) Adaptation independent modulation of auditory hair cell mechanotransduction channel open probability implicates a role for the lipid bilayer. J. Neurosci. 36, 2945–2956 10.1523/JNEUROSCI.3011-15.2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Gnanasambandam R., Ghatak C., Yasmann A., Nishizawa K., Sachs F., Ladokhin A. S., Sukharev S. I., and Suchyna T. M. (2017) GsMTx4: mechanism of inhibiting mechanosensitive ion channels. Biophys. J. 112, 31–45 10.1016/j.bpj.2016.11.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Nishizawa K., Nishizawa M., Gnanasambandam R., Sachs F., Sukharev S. I., and Suchyna T. M. (2015) Effects of Lys to Glu mutations in GsMTx4 on membrane binding, peptide orientation, and self-association propensity, as analyzed by molecular dynamics simulations. Biochim. Biophys. Acta 1848, 2767–2778 10.1016/j.bbamem.2015.09.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Bosmans F., and Swartz K. J. (2010) Targeting voltage sensors in sodium channels with spider toxins. Trends Pharmacol. Sci. 31, 175–182 10.1016/j.tips.2009.12.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Niu X., Qian X., and Magleby K. L. (2004) Linker-gating ring complex as passive spring and Ca2+-dependent machine for a voltage- and Ca2+-activated potassium channel. Neuron 42, 745–756 10.1016/j.neuron.2004.05.001 [DOI] [PubMed] [Google Scholar]
- 25. Tang Q. Y., Zeng X. H., and Lingle C. J. (2009) Closed-channel block of BK potassium channels by bbTBA requires partial activation. J. Gen. Physiol. 134, 409–436 10.1085/jgp.200910251 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Tang Q. Y., Zhang Z., Meng X. Y., Cui M., and Logothetis D. E. (2014) Structural determinants of phosphatidylinositol 4,5-bisphosphate (PIP2) regulation of BK channel activity through the RCK1 Ca2+ coordination site. J. Biol. Chem. 289, 18860–18872 10.1074/jbc.M113.538033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Hite R. K., Tao X., and MacKinnon R. (2017) Structural basis for gating the high-conductance Ca2+-activated K+ channel. Nature 541, 52–57 10.1038/nature20775 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Dopico A. M., Kirber M. T., Singer J. J., and Walsh J. V. Jr. (1994) Membrane stretch directly activates large conductance Ca2+-activated K+ channels in mesenteric artery smooth muscle cells. Am. J. Hypertens. 7, 82–89 10.1093/ajh/7.1.82 [DOI] [PubMed] [Google Scholar]
- 29. Mallouk N., and Allard B. (2000) Stretch-induced activation of Ca2+-activated K+ channels in mouse skeletal muscle fibers. Am. J. Physiol. Cell Physiol. 278, C473–C479 10.1152/ajpcell.2000.278.3.C473 [DOI] [PubMed] [Google Scholar]
- 30. Kawakubo T., Naruse K., Matsubara T., Hotta N., and Sokabe M. (1999) Characterization of a newly found stretch-activated KCa,ATP channel in cultured chick ventricular myocytes. Am. J. Physiol. 276, H1827–H1838 10.1152/ajpheart.1999.276.6.H1827 [DOI] [PubMed] [Google Scholar]
- 31. Tang Q. Y., Qi Z., Naruse K., and Sokabe M. (2003) Characterization of a functionally expressed stretch-activated BKca channel cloned from chick ventricular myocytes. J. Membr. Biol. 196, 185–200 10.1007/s00232-003-0637-8 [DOI] [PubMed] [Google Scholar]
- 32. Naruse K., Tang Q. Y., and Sokabe M. (2009) Stress-axis regulated exon (STREX) in the C terminus of BK(Ca) channels is responsible for the stretch sensitivity. Biochem. Biophys. Res. Commun. 385, 634–639 10.1016/j.bbrc.2009.05.105 [DOI] [PubMed] [Google Scholar]
- 33. Zhao H., and Sokabe M. (2008) Tuning the mechanosensitivity of a BK channel by changing the linker length. Cell Res. 18, 871–878 10.1038/cr.2008.88 [DOI] [PubMed] [Google Scholar]
- 34. Yuan P., Leonetti M. D., Hsiung Y., and MacKinnon R. (2011) Open structure of the Ca2+ gating ring in the high-conductance Ca2+-activated K+ channel. Nature 481, 94–97 10.1038/nature10670 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Tao X., Hite R. K., and MacKinnon R. (2017) Cryo-EM structure of the open high-conductance Ca2+-activated K+ channel. Nature 541, 46–51 10.1038/nature20608 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Schmidt D., and MacKinnon R. (2008) Voltage-dependent K+ channel gating and voltage sensor toxin sensitivity depend on the mechanical state of the lipid membrane. Proc. Natl. Acad. Sci. U.S.A. 105, 19276–19281 10.1073/pnas.0810187105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Auerbach A. (1991) Single-channel dose-response studies in single, cell-attached patches. Biophys. J. 60, 660–670 10.1016/S0006-3495(91)82095-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Cuevas J., and Adams D. J. (1994) Local anaesthetic blockade of neuronal nicotinic ACh receptor-channels in rat parasympathetic ganglion cells. Br. J. Pharmacol. 111, 663–672 10.1111/j.1476-5381.1994.tb14789.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Milescu M., Bosmans F., Lee S., Alabi A. A., Kim J. I., and Swartz K. J. (2009) Interactions between lipids and voltage sensor paddles detected with tarantula toxins. Nat. Struct. Mol. Biol. 16, 1080–1085 10.1038/nsmb.1679 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Nishizawa M., and Nishizawa K. (2007) Molecular dynamics simulations of a stretch-activated channel inhibitor GsMTx4 with lipid membranes: two binding modes and effects of lipid structure. Biophys. J. 92, 4233–4243 10.1529/biophysj.106.101071 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Geng Y., and Magleby K. L. (2014) Single-channel kinetics of BK (Slo1) channels. Front. Physiol. 5, 532 10.3389/fphys.2014.00532 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Sigworth F. J., and Sine S. M. (1987) Data transformations for improved display and fitting of single channel dwell time histograms. Biophys. J. 52, 1047–1054 10.1016/S0006-3495(87)83298-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Shelley C., Niu X., Geng Y., and Magleby K. L. (2010) Coupling and cooperativity in voltage activation of a limited-state BK channel gating in saturating Ca2+. J. Gen. Physiol. 135, 461–480 10.1085/jgp.200910331 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Ruta V., and MacKinnon R. (2004) Localization of the voltage-sensor toxin receptor on KvAP. Biochemistry 43, 10071–10079 10.1021/bi049463y [DOI] [PubMed] [Google Scholar]
- 45. Lee S. Y., Lee A., Chen J., and MacKinnon R. (2005) Structure of the KvAP voltage-dependent K+ channel and its dependence on the lipid membrane. Proc. Natl. Acad. Sci. U.S.A. 102, 15441–15446 10.1073/pnas.0507651102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Singh H., Lu R., Bopassa J. C., Meredith A. L., Stefani E., and Toro L. (2013) MitoBK(Ca) is encoded by the Kcnma1 gene, and a splicing sequence defines its mitochondrial location. Proc. Natl. Acad. Sci. U.S.A. 110, 10836–10841 10.1073/pnas.1302028110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Walewska A., Kulawiak B., Szewczyk A., and Koprowski P. (2018) Mechanosensitivity of mitochondrial large-conductance calcium-activated potassium channels. Biochim. Biophys. Acta Bioenerg. 1859, 797–805 10.1016/j.bbabio.2018.05.006 [DOI] [PubMed] [Google Scholar]
- 48. Xu W., Liu Y., Wang S., McDonald T., Van Eyk J. E., Sidor A., and O'Rourke B. (2002) Cytoprotective role of Ca2+-activated K+ channels in the cardiac inner mitochondrial membrane. Science 298, 1029–1033 10.1126/science.1074360 [DOI] [PubMed] [Google Scholar]
- 49. Frankenreiter S., Bednarczyk P., Kniess A., Bork N. I., Straubinger J., Koprowski P., Wrzosek A., Mohr E., Logan A., Murphy M. P., Gawaz M., Krieg T., Szewczyk A., Nikolaev V. O., Ruth P., and Lukowski R. (2017) cGMP-elevating compounds and ischemic conditioning provide cardioprotection against ischemia and reperfusion injury via cardiomyocyte-specific BK channels. Circulation 136, 2337–2355 10.1161/CIRCULATIONAHA.117.028723 [DOI] [PubMed] [Google Scholar]
- 50. Tang Q. Y., Zhang Z., Xia J., Ren D., and Logothetis D. E. (2010) Phosphatidylinositol 4,5-bisphosphate activates Slo3 currents and its hydrolysis underlies the epidermal growth factor-induced current inhibition. J. Biol. Chem. 285, 19259–19266 10.1074/jbc.M109.100156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Tang Q. Y., Zhang F. F., Xu J., Wang R., Chen J., Logothetis D. E., and Zhang Z. (2016) Epilepsy-related slack channel mutants lead to channel over-activity by two different mechanisms. Cell Rep. 14, 129–139 10.1016/j.celrep.2015.12.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Tang Q. Y., Zhang Z., Xia X. M., and Lingle C. J. (2010) Block of mouse Slo1 and Slo3 K+ channels by CTX, IbTX, TEA, 4-AP and quinidine. Channels 4, 22–41 10.4161/chan.4.1.10481 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Zhang Z., Rosenhouse-Dantsker A., Tang Q. Y., Noskov S., and Logothetis D. E. (2010) The RCK2 domain uses a coordination site present in Kir channels to confer sodium sensitivity to Slo2.2 channels. J. Neurosci. 30, 7554–7562 10.1523/JNEUROSCI.0525-10.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Pallante B. A., Giovannone S., Fang-Yu L., Zhang J., Liu N., Kang G., Dun W., Boyden P. A., and Fishman G. I. (2010) Contactin-2 expression in the cardiac Purkinje fiber network. Circ. Arrhythm. Electrophysiol. 3, 186–194 10.1161/CIRCEP.109.928820 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Jo S., Lim J. B., Klauda J. B., and Im W. (2009) CHARMM-GUI membrane builder for mixed bilayers and its application to yeast membranes. Biophys. J. 97, 50–58 10.1016/j.bpj.2009.04.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Brooks B. R., Brooks C. L. 3rd., Mackerell A. D. Jr., Nilsson L., Petrella R. J., Roux B., Won Y., Archontis G., Bartels C., Boresch S., Caflisch A., Caves L., Cui Q., Dinner A. R., Feig M., et al. (2009) CHARMM: the biomolecular simulation program. J. Comput. Chem. 30, 1545–1614 10.1002/jcc.21287 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Su J., and Guo H. (2011) Control of unidirectional transport of single file water molecules through carbon nanotubes in an electric field. ACS Nano 5, 351–359 10.1021/nn1014616 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.