

Abstract
Human guanylate kinase (hGMPK) is the only known enzyme responsible for cellular GDP production, making it essential for cellular viability and proliferation. Moreover, hGMPK has been assigned a critical role in metabolic activation of antiviral and antineoplastic nucleoside-analog prodrugs. Given that hGMPK is indispensable for producing the nucleotide building blocks of DNA, RNA, and cGMP and that cancer cells possess elevated GTP levels, it is surprising that a detailed structural and functional characterization of hGMPK is lacking. Here, we present the first high-resolution structure of hGMPK in the apo form, determined with NMR spectroscopy. The structure revealed that hGMPK consists of three distinct regions designated as the LID, GMP-binding (GMP-BD), and CORE domains and is in an open configuration that is nucleotide binding–competent. We also demonstrate that nonsynonymous single-nucleotide variants (nsSNVs) of the hGMPK CORE domain distant from the nucleotide-binding site of this domain modulate enzymatic activity without significantly affecting hGMPK's structure. Finally, we show that knocking down the hGMPK gene in lung adenocarcinoma cell lines decreases cellular viability, proliferation, and clonogenic potential while not altering the proliferation of immortalized, noncancerous human peripheral airway cells. Taken together, our results provide an important step toward establishing hGMPK as a potential biomolecular target, from both an orthosteric (ligand-binding sites) and allosteric (location of CORE domain-located nsSNVs) standpoint.
Keywords: nuclear magnetic resonance (NMR), protein structure, enzyme kinetics, enzyme mutation, lung cancer, guanylate kinase, hGMPK, non-synonymous single nucleotide variants (nsSNV), solution structure
Introduction
Metabolism in cancer cells is altered, resulting in tumor proliferation (1). One outcome of this altered metabolism is elevated levels of the nucleotides GTP and dGTP (2) due to the up-regulation of nucleotide biosynthesis (3). GTP and dGTP are essential precursors of RNA and DNA, respectively, GTP is a vital source of energy for protein biosynthesis and a critical signaling molecule via GTP-binding proteins. An indispensable enzyme involved in GTP and dGTP metabolism is human guanylate kinase (hGMPK,7 gene name GUK1, ATP:GMP phosphotransferase, EC 2.7.4.8). Positioned at the junction of the salvage and the de novo purine nucleotide biosynthesis pathways (Fig. 1A), hGMPK catalyzes the reversible phosphorylation of GMP to GDP by utilizing ATP as a phosphoryl group donor (4–8) (Fig. 1B). Thus, all GDP/GTP production must go through hGMPK, yet chemotherapeutic strategies that focus on depleting GDP/GTP from cancer cells have targeted two de novo purine nucleotide biosynthesis pathway enzymes upstream of hGMPK, IMP dehydrogenase (IMPDH) (9) and guanosine monophosphate synthase (GMPS) (10) (Fig. 1A). Interestingly, the depletion of GDP/GTP levels resulting from the inhibition of IMPDH can be replenished by supplying cells with either guanosine or guanine (11), because the salvage pathway is still operational through the action of hGMPK and nucleoside diphosphate kinases. This observation shows that to fully inhibit GDP/GTP production, an enzyme positioned downstream from the intersection of the salvage and de novo pathways must be targeted. Thus, inhibition of hGMPK could be an effective therapeutic target.
Figure 1.

Summary of hGMPK's role in the cell. A, hGMPK is positioned at the junction of the de novo nucleotide synthesis and salvage pathways. The enzymes IMPDH (9) and GMPS (10) are biomolecular targets for inhibition and are part of the de novo nucleotide synthesis pathway. B, schematic of the hGMPK reaction cycle following a random sequential mechanism (4, 8).
Currently, hGMPK's physiological function is exploited for the activation of numerous antiviral and antineoplastic nucleoside prodrugs (12, 13), most notably 6-thioguanine and its closely related analogue 6-mercaptopurine (14), 9-β-d-arabinofuranosylguanine (15), ganciclovir (16, 17), and acyclovir (18). Despite the importance of hGMPK for metabolic prodrug activation and the documented inefficiency in processing these prodrugs (18–23), the structure and functional dynamics of hGMPK remain virtually unexplored. This scarcity of structural and functional details pertaining to hGMPK is due to difficulties in expressing functional recombinant protein (24) and in producing high-quality crystals for X-ray diffraction (25, 26).
The goal of the current work was to provide a better understanding of the structural basis for hGMPK functionality. Although structural details are essential for understanding how the catalytic activity of hGMPK is achieved, only structures from nonhuman organisms have been available for making deductions concerning the relationship between hGMPK structure and function (26–33). Recently, a small-angle X-ray scattering (SAXS) study on hGMPK presented the first reported structural data on hGMPK demonstrating that ligand binding leads to a compaction of hGMPK by about 2 Å (34). To further investigate the relationship between hGMPK structure and function with more detail, we solved the first high-resolution structure of hGMPK with solution NMR spectroscopy, which is also the first structure with atomic-level resolution of a nucleotide-free mammalian GMPK. A comparison of our hGMPK NMR structure with the yeast and mouse GMPK X-ray structures revealed a conserved structural fold as well as a large-scale interdomain motion associated with nucleotide binding. In addition to solving the hGMPK structure, we explored the impact of active-site distant, nonsynonymous single-nucleotide variants (nsSNVs) on hGMPK's structure and function. We found that CORE domain nsSNVs of hGMPK alter catalytic activity without significantly perturbing the average structure of the apo-enzyme. Finally, we present the first study demonstrating that intracellular inhibition of hGMPK may represent a novel approach to chemotherapy.
Results and discussion
Solution structure of hGMPK
We solved the solution structure of hGMPK using our previously published chemical shift assignment and our measured 15N-resolved 1H,1H NOESY (60-ms mixing time), 13Caliphatic-resolved 1H,1H NOESY (80 ms mixing time), and 13Caromatic-resolved 1H,1H NOESY (80-ms mixing time) spectra (35) (Fig. 2; Protein Data Bank (PDB) accession code 6NUI; see “Experimental procedures” for a detailed description of the structure determination). As a supplement to these data, we measured backbone 15N heteronuclear NOEs (15N-hetNOEs) and a set of residual dipolar couplings (RDCs) (Fig. 3 and Table S1). The 15N-hetNOE data enabled us to justify removal of dihedral angle and RDC constraints for flexible residues with 15N-hetNOE values lower than 0.70 from the structure calculation.
Figure 2.
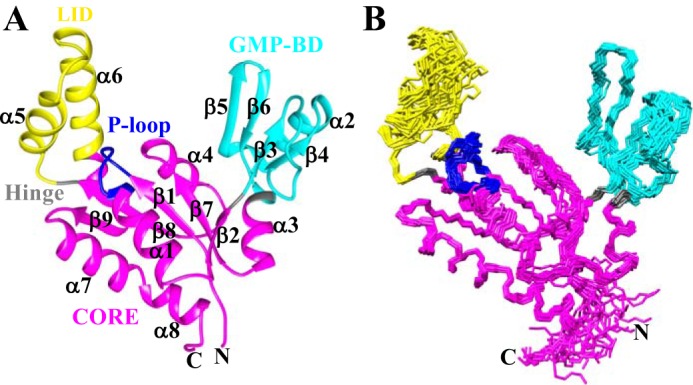
Solution NMR structure of apo-hGMPK. A, ribbon diagram of the lowest-energy structure of hGMPK (PDB code 6NUI). The color scheme is as follows: CORE domain in magenta, GMP-BD in cyan, LID domain in yellow, hinge residues (35, 87, 124, and 157) in gray, and the P-loop (residues 11–18) motif in blue. B, the 20 lowest-energy conformers of hGMPK that were superimposed based on residues 7–193.
Figure 3.

Plot of the steady state heteronuclear [1H]-15N NOE data (black) and the per residue RMSD (red) for hGMPK. Hinge residue positions are indicated by blue arrows. Residues preceding and following the hinge residues have lower than average 15N-hetNOE values (blue circles). In addition to the N and C termini, regions of high residue RMSDs also correspond to lower than average 15N-hetNOE values (gray circles).
Totals of 2430 NOE, 281 backbone dihedral, 89 NHN-RDCs, and 58 hydrogen bond constraints were used as input for the structure calculation and refinement performed with CYANA (36, 37) and AMBER (39). Table 1 summarizes the experimental constraints and the structural statistics for the hGMPK structure. The RMSD (calculated for residues 7–193) of the backbone (heavy) atoms was 0.74 ± 0.16 Å (0.86 ± 0.15 Å). The z-scores of the hGMPK structure were analyzed by PROCHECK (40) and were 0.79/0.12 for the backbone and all atom dihedrals, respectively, signifying a high-quality structure.
Table 1.
Summary of experimental constraints and structural statistics computed for the 20 lowest-energy hGMPK structures
| Parameter | Value |
|---|---|
| NMR-derived constraintsa | |
| Distance constraints | |
| Total NOEs | 2430 |
| Intraresidue | 722 |
| Interresidue | |
| Sequential (|i − j| = 1) | 640 |
| Medium range (|i − j| < 5) | 454 |
| Long range (|i − j| ≥ 5) | 614 |
| Hydrogen bonds | 58 |
| Dihedral constraints | 281 |
| No. of constraints per residue | 14.0 |
| No. of long-range constraints per residue | 3.3 |
| Structure statistics | |
| CYANA target functionb (Å) | 0.66 ± 0.04 |
| Residual distance violationsb | |
| Number ≥ 0.1 Å | 0.7 ± 1.1 |
| Maximum (Å) | 0.1 ± 0.1 |
| Residual dihedral angle violationsb | |
| Number ≥ 2.5° | 0 |
| Maximum (degrees) | 1.5 ± 0.1 |
| Residual dipolar coupling Q-factorb | 0.17 ± 0.01 |
| RMSD from idealized geometryc | |
| Bond lengths (Å) | 0.014 |
| Bond angles (degrees) | 1.9 |
| Average pairwise RMSD to the meanb (Å) | |
| Backbone atomsd | 0.74 ± 0.16 |
| Heavy atomsd | 0.86 ± 0.15 |
| Procheck Ramachandran statistics | |
| Most favored regions (%) | 94.6 |
| Additionally favored regions (%) | 5.3 |
| Generously allowed regions (%) | 0.0 |
| Disallowed regions (%) | 0.1 |
| Procheck quality scorese | |
| Raw score (φ and ψ/all dihedrals) | 0.12/0.02 |
| z-score (φ and ψ/all dihedrals) | 0.79/0.12 |
a As reported by CYANA after using the automated NOE assignment and structure calculation functionalities (36, 37).
b Calculated with CYANA after structural refinement with AMBER.
c Calculated by the Protein Structure Validation Software (PSVS) Suite (38).
d Calculated for residues 7–193.
e Residues 4–11, 17–115, 117–136,138–140, and 142–193 were selected by the PSVS Suite for the calculation.
We proceeded to ascertain the location of structural domains within the hGMPK structure. For this purpose, we utilized the program HingeProt (41), which identified 4 hinge residues within the hGMPK structure: Ser-35, Ala-87, Pro-124, and Glu-157. Based on this result, we defined three distinct structural domains: domain 1, spanning residues 1–34, 88–123, and 158–197; domain 2, spanning residues 36–86; and domain 3, spanning residues 125–156 (Fig. 2). In support of the HingeProt results, the measured 15N-hetNOEs preceding and following the hinge residues show lower than average values that signify more flexibility in the picosecond to nanosecond time scale (Fig. 3). These hinge residues are similar to what was previously inferred from a comparison of the GMP + ADP–bound mouse GMPK (mGMPK; 1LVG) X-ray structure with the apo- and GMP-bound yeast GMPK (yGMPK; 1EX6 and 1EX7, respectively) X-ray structures: hinge 1, residues 32–36; hinge 2, residues 90–96; hinge 3, residues 124–125; and hinge 4, residues 157–164 (26). Interestingly, mutating Ser-35 to proline has been shown to significantly reduce catalytic activity by preventing closure of the GMP-binding domain (GMP-BD) (42), which also supports the identification of Ser-35 as a hinge residue. The three domains described here are very similar to the domains identified from nonhuman GMPKs. Namely, domains 1, 2, and 3 have been defined previously as the central CORE domain, the GMP-BD, and the LID domain (this domain's name refers to how it closes like a lid onto the CORE domain during catalysis), respectively, nomenclature that we will utilize for our hGMPK structure.
The sequence arrangement of the regular secondary structure elements is β1-α1-β2-β3-β4-α2-β5-β6-α3-β7-α4-β8-α5-α6-β9-α7-α8 (Fig. 2A; see Fig. S1 for a schematic plot of the short-range NOE connections). Within the CORE domain, a four-stranded twisted parallel β-sheet consisting of β9-β8-β1-β7 is situated in between the helices α1, α7, and α8 on one side and α4 and a portion of α6 from the LID domain on the other side. The canonical P-loop motif (11GXXXXGK(T/S)18), involved in ATP binding, bridges β1 and α1, whereas β2 connects the CORE domain to the GMP-BD and is oriented slightly perpendicular to the central parallel β-sheet. A dense network of hydrophobic contacts is present in the region between α1-α7-α8 and β9-β8-β1-β7-β2 involving residues Val-7 and Leu-9 from β1; Lys-17, Leu-21, Leu-24, and Leu-25 from α1; Phe-32 from β2; Ile-97 and Val-99 from β7; Ile-118, Ile-120, and Val-122 from β8; Val-167 and Ile-169 from β9; Ala-177, Leu-181, and Ala-184 from α7; Leu-185 in between α7 and α8; and Glu-188, Ile-189, and Ala-192 from α8 (Fig. S2). The GMP-BD consists of a twisted antiparallel sheet involving strands β3-β5-β6 with β4 oriented parallel to β3 and α2 bridging β4 and β5. A long loop spanning residues 40–52 connects β3 and β4. Joining the GMP-BD back to the CORE is α3, which possesses the hinge residue Ala-87. Finally, the LID domain consists of two α-helices, α5 and α6, that are connected by a flexible loop. The higher RMSDs for residues within this LID domain loop are supported by the corresponding 15N-hetNOE values, especially for residues 137–142 (Fig. 3 and Table S1), although it should be noted that dihedral angle and RDC constraints were removed from the structure calculation for these residues due to low 15N-hetNOEs values.
The electrostatic charge distribution of hGMPK indicates that nucleotide binding is mediated by electrostatic interactions (Fig. 4). A highly positive patch positioned between the LID domain and the P-loop from the CORE domain includes residues that have been shown to coordinate ATP in the X-ray structure of mGMPK (Figs. S3 and S4). As for GMP binding, both a positive (GMP-BD) and negative patch (GMP-BD and CORE) encompass residues that interact with the phosphate group and the guanine base, respectively. In fact, the orientation of the side chains involved in coordinating both GMP and ATP are very similar between the human and mouse structures (Fig. S3), supporting the importance of these electrostatic patches for hGMPK function.
Figure 4.
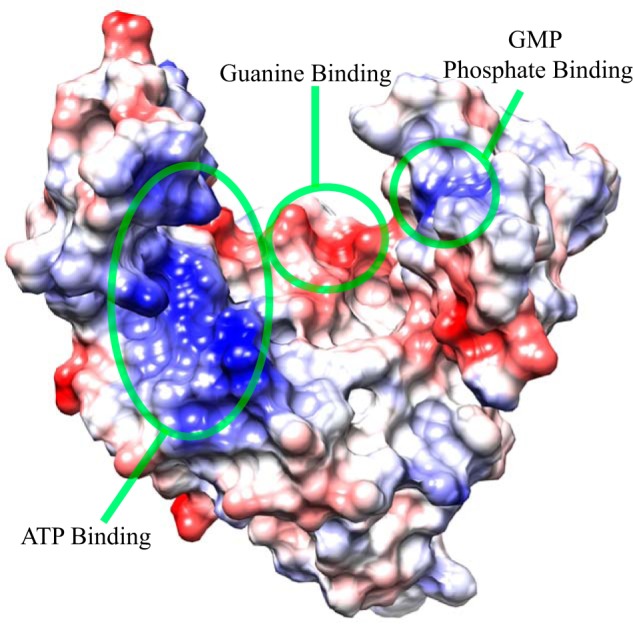
Electrostatic charge distribution of the hGMPK surface. Residues identified as interacting with nucleotides from both sequence homology (Fig. S4) and inspection of the GMP + ADP–bound mouse GMPK X-ray structure (Fig. S3) are located in hGMPK electropositive and electronegative regions. The lowest-energy structure of hGMPK is depicted, where positive and negative charges are colored blue and red, respectively.
Comparison of the hGMPK structure with yeast and mouse GMPK
Next, we then compared the domain orientations from our hGMPK NMR structure with the yGMPK and mGMPK X-ray structures (Fig. 5). For the structural comparison, we utilized multiple sequence alignment to select the same residues from each species (Fig. S4 and Table S2). It should be noted that hGMPK and mGMPK share 88% sequence identity, equating to a total of 24 of 197 different residues; however, the orientations of the different side chains remain relatively similar (Fig. S5). The main structural differences related to these residue substitutions, aside from the N and C termini, are primarily located around the hinge regions, namely G30S, V86E, and Q89R, where domain movement has been shown to be associated with function (see below). An interesting exception is L25F within the center of α1, with the leucine side chain pointing away from the interior of hGMPK, whereas the phenylalanine side chain of mGMPK points inward (Fig. S5).
Figure 5.
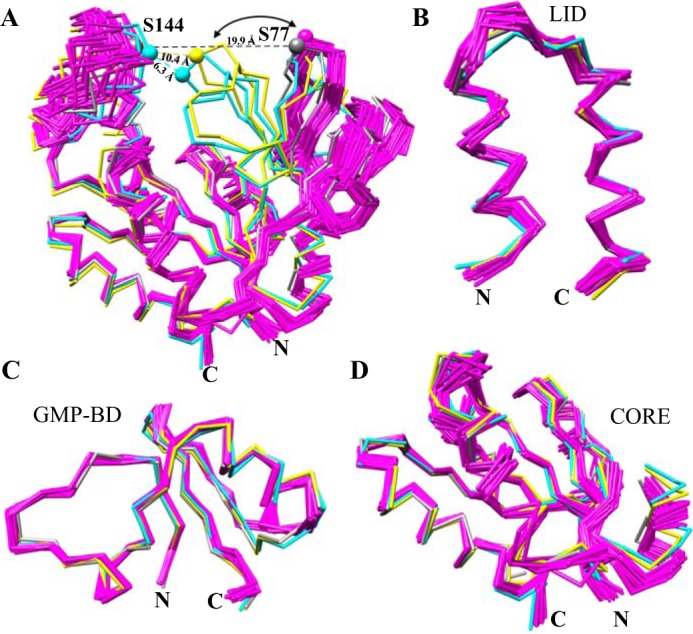
Structural comparison of hGMPK with nucleotide-free and bound GMPK X-ray structures indicates a large-scale domain motion associated with nucleotide binding. A, superposition of the 20-member ensemble of hGMPK (magenta) and three X-ray structures of GMP + ADP mouse GMPK (mGMPK; cyan; PDB code 1LVG) and apo-(gray; PDB code 1EX6) and GMP-bound (yellow; PDB code 1EX7) yeast yGMPK based on residues 6–188 (hGMPK numbering) with a backbone RMSD of 6.2 ± 1.0 Å. Distances between the Cα atoms of Ser-77 (from GMP-BD) and Ser-144 (from the LID domain) in hGMPK and mGMPK, which correspond to Ser-74 and Ser-142 in yGMPK, respectively, are 22.9 ± 1.1 Å for apo-hGMPK, 6.3 Å for mGMPK, 19.9 Å for apo-yGMPK, and 10.4 Å for GMP-bound yGMPK. B, overlay of the LID domain with a backbone RMSD of 1.09 ± 0.18 Å. C, superposition of the GMP-BD with a backbone RMSD of 1.19 ± 0.19 Å. D, superposition of the CORE domain with a backbone RMSD of 1.45 ± 0.18 Å. Highly flexible N- and C-terminal regions are not shown here. Table S2 indicates the residues used from each GMPK for structural superposition.
Immediately apparent from the hGMPK, yGMPK, and mGMPK structural bundle is the orientation of the GMP-BD (Fig. 5A). With ligands bound, the GMP-BD is positioned closer to the LID domain, as evident by the distance between Ser-77 and Ser-144 in the GMP-bound yGMPK structure (10.4 Å) and mGMPK (6.3 Å), in preparation for catalysis. In the absence of ligands, the GMP-BD is positioned 19.9 and 22.9 ± 1.1 Å away from the LID domain for apo-yGMPK and hGMPK, respectively. A complete lack of NOEs connecting the GMP-BD and the LID domain (Table S3) supports the open conformation present in the hGMPK structure. The open state depicted by our structure is also reinforced by the recently published hGMPK SAXS study (34), where the SAXS profile of apo-hGMPK yielded a radius of gyration (Rg) of 21.1 Å, whereas the SAXS profile of hGMPK bound to the bisubstrate inhibitor Ap5G resulted in a more compact Rg of 18.1 Å. Finally, we superimposed the three GMPK domains using the lowest energy NMR structure as a reference (Fig. 5, B–D). The relatively similar backbone conformations for each domain across the three species, whether ligand-free or bound, points to a rigid-body motion bringing the GMP-BD and the LID domains together for catalysis.
As is the case for many other enzymes (43), our structure indicates that large-scale domain motions are required for hGMPK functionality. It has been shown that preventing closure of the two nucleotide-binding domains in yGMPK, whether through mechanical stress (44) or mutation of either Ser-35 (a hinge residue identified in this study) or Ser-37 to a proline, considerably reduces catalytic efficiency (42). This relationship between domain motions and function, specifically the GMP-BD and LID domains, is a feature that has been known for GMPKs (26, 27, 42, 45) and other nucleoside monophosphate kinases from various organisms (46–49).
nsSNVs of hGMPK indicate a regulatory role for the CORE domain
Whereas the positioning of the GMP-BD and LID domains is clearly important for functionality, we were curious whether the CORE domain possesses a regulatory role in hGMPK catalysis. We focused on a selection of nsSNVs of hGMPK, as reported in the Catalogue of Somatic Mutations in Cancer (COSMIC) database (50), particularly nsSNVs not directly involved in nucleotide binding within the CORE domain. Because changing one residue within a protein can alter functionality and cellular viability (51, 52), we reasoned that functional site–distant nsSNVs were physiologically viable mutants of hGMPK and could report on a potential regulatory role for the CORE. We overexpressed the following nsSNVs in Escherichia coli: S2L, G3A, L25P, V91M, R96H, R116Q, S121F, and S186Y (Fig. 6A, Fig. S6, and Table S4). According to multiple nsSNV predictors curated by the consensus classifier PredictSNP (53), contradictory results for the same mutation arose, where some of the hGMPK nsSNVs are reported to be both tolerated and damaging to protein function (Table S5). It should be noted that these nsSNV predictors utilize sequence conservation, structure, and machine learning to forecast the impact of nsSNVs on protein functionality (54); however, there still exists a significant gap between experiment and computational prediction (55, 56).
Figure 6.

CORE domain nsSNVs modulate the functionality of hGMPK. A, location of the CORE domain, nsSNVs, and active site residues depicted in the lowest-energy conformer of apo-hGMPK. B, SV-AUC of wt-hGMPK without and with Ap5G. Relative distribution of hGMPK species in solution (c(s)) versus sedimentation coefficient (s20,w). Black curve, 20 μm hGMPK; orange curve, 20 μm hGMPK and 5 μm Ap5G; purple curve, 20 μm hGMPK and 40 μm Ap5G. C, DSF of wt-hGMPK and hGMPK nsSNVs. First derivative versus temperature DSF plots for the hGMPK series. D, steady-state kinetics plots of wt-hGMPK and hGMPK nsSNVs. The kinetic data were fit to the Michaelis–Menten kinetics model. E, plot of the fitted Michaelis–Menten parameters Km versus kcat. The high correlation between these parameters suggests kcat–Km compensation. F, plot of the specificity constant (kcat/Km) versus s20,w. The correlation between the two parameters suggests that compaction of the kinase leads to a reduction in enzymatic efficiency. The color-coding for all panels is as follows: wt (black), S2L (blue), G3A (cyan), V91M (brown), R96H (magenta), R116Q (green), S121F (yellow), and S186Y (red).
To assess the monomeric/oligomeric state and compactness of the hGMPK variants, we first performed sedimentation velocity analytical ultracentrifugation (SV-AUC). We hypothesized that the SV-AUC–measured Svedberg coefficient (S) would be able to detect subtle changes in the open-to-closed hGMPK transition. We first fit our NMR structure to the previously published apo-hGMPK and Ap5G-hGMPK SAXS data and found that our NMR structure fit better to the apo-hGMPK SAXS profile with a reduced χ2 of 1.7 compared with a reduced χ2 of 3.1 for the Ap5G–hGMPK complex SAXS profile (34). With our hypothesis supported by the better fit to the apo-hGMPK SAXS data, we carried out an SV-AUC–detected titration of wt-hGMPK with Ap5G. The S value for apo-hGMPK was 2.04 ± 0.03, which is remarkably close to the theoretical S value of 2.07 ± 0.02 calculated for our solution structure of hGMPK using the program HydroPro (57). Upon the addition of Ap5G, an appreciable shift to larger S values (at 2:1 molar ratio of Ap5G to hGMPK, the S value is 2.40 ± 0.03) was observed as well as a narrowing of the S distribution (Fig. 6B). HydroPro predicts a 0.05 increase in the S value entirely due to an increase in molecular weight (Mr = 932 Da for the free acid) upon Ap5G binding, which suggests that SV-AUC can detect subtle shifts in conformational equilibria for hGMPK. Because an increase in the S value designates a more compact structure for a system with the same Mr, Ap5G-bound wt-hGMPK is more closed than the apo form, in line with the SAXS data (34).
For the SV-AUC measurements of the hGMPK nsSNVs, almost all of the hGMPK mutants behaved as monomers in solution and shifted the conformational equilibrium to a slightly more closed average macroscopic structure when compared with the WT (ranging from 2.12 to 2.23 S; Table 2). The only exception was mutant L25P, which led to a significant amount of aggregation (Fig. S7) and was forecast to have deleterious effects on hGMPK functionality (Table S5), probably due to the helix-disrupting proline substitution in the CORE domain (Fig. 6A). For the rest of the nsSNVs, the SV-AUC results indicate a change in the macroscopic structure toward a more compact state.
Table 2.
Sedimentation coefficients, thermal stabilities, and kinetic parameters for WT apo-hGMPK and nsSNVs of hGMPK
All measurements on WT and the mutants were performed in triplicate, and errors were taken from the S.D. of the three measurements. The kinetic data were collected from a coupled enzymatic assay where the ATP concentration was held constant at 2 mm, and the GMP concentration was varied from 10 to 200 μm (Fig. 6D). The kinetic parameters were extracted from a fit of the data to the Michaelis–Menten equation, and the error in the fitted parameters was determined from 1000 Monte Carlo simulations.
| hGMPK | s20,w | Tm | kcat | Km | kcat/Km |
|---|---|---|---|---|---|
| S | °C | s−1 | μm | s−1 μm−1 | |
| WT | 2.04 ± 0.03 | 47.4 ± 0.4 | 58.5 ± 0.3 | 20.7 ± 0.3 | 2.8 ± 0.1 |
| S2L | 2.22 ± 0.03 | 45.5 ± 0.5 | 77.7 ± 1.1 | 31.5 ± 0.7 | 2.5 ± 0.1 |
| G3A | 2.15 ± 0.03 | 46.0 ± 0.7 | 184.4 ± 2.8 | 78.3 ± 2.1 | 2.4 ± 0.1 |
| V91M | 2.14 ± 0.03 | 44.4 ± 0.8 | 132.2 ± 2.2 | 54.7 ± 1.9 | 2.4 ± 0.1 |
| R96H | 2.18 ± 0.03 | 42.4 ± 0.7 | 100.6 ± 2.4 | 29.6 ± 1.6 | 3.4 ± 0.2 |
| R116Q | 2.23 ± 0.03 | 45.6 ± 0.4 | 127.0 ± 5.9 | 54.3 ± 4.1 | 2.3 ± 0.2 |
| S121F | 2.22 ± 0.03 | 43.8 ± 0.2 | 101.4 ± 5.7 | 48.6 ± 3.8 | 2.1 ± 0.2 |
| S186Y | 2.14 ± 0.03 | 44.6 ± 0.2 | 96.5 ± 0.7 | 36.8 ± 0.7 | 2.6 ± 0.1 |
To assess the effect of each nsSNV on hGMPK's catalytic properties, we evaluated their thermostability with differential scanning fluorimetry (DSF). DSF demonstrated that the WT is the most thermostable species, with a Tm of 47.4 °C, whereas the hGMPK nsSNVs show only a slight decrease in thermostability (Table 2 and Fig. 6C). Next, we characterized their catalytic activity with a coupled-enzyme kinetic assay (4, 34) (Table 2 and Fig. 6D). Unexpectedly, the hGMPK nsSNVs possess more catalytic activity, as shown by an increase in kcat, than the WT. When plotting the two Michaelis–Menten parameters, it is apparent that kcat–Km compensation (Fig. 6E; ρ = 0.97, p < 0.001) is occurring, a phenomenon that, to our knowledge, has only been observed for the enzymes adenylate kinase (58) and DNA repair demethylase AlkB (59). This tight regulation of the specificity constant, kcat/Km, could be a built-in evolutionary feature of enzymes to accommodate nsSNVs and maintain an optimal level of performance. Finally, there is a correlation between the specificity constant (kcat/Km) and the Svedberg coefficient (Fig. 6F; ρ = −0.80, p < 0.001, excluding R96H), suggesting an inverse relationship between hGMPK compactness and enzymatic efficiency. Only R96H is the outlier, which has a slightly higher kcat/Km than the WT (3.4 versus 2.8 μm−1 s−1), while being the least thermostable hGMPK variant. It is intriguing to note that the nsSNV predictors imply that S2L and R96H are detrimental to protein function, yet they are functional kinases (Table 2), highlighting the difficulties in predicting functional outcomes for active/binding site–distant nsSNVs.
To explore whether the enhanced kinetic activity is a result of perturbations to the average hGMPK structure on the atomic level, we measured 2D 1H,15N HSQC NMR spectra for the WT and five of the hGMPK nsSNVs (S2L, G3A, R96H, R116Q, and S186Y; Fig. S8). An analysis of our NMR structure reveals that the side chains of Arg-96, Arg-116, and Ser-186 are solvent-exposed, and S2L and G3A are part of the flexible N terminus (Fig. 6A). Fig. 7, A–D, shows plots of combined chemical shift differences Δδdiff resulting from the mutations S2L (blue) (A), G3A (cyan) (B), R96H (magenta) (C), and S186Y (red) (D), as calculated with Equation 1.
| (Eq. 1) |
Figure 7.

hGMPK nsSNVs result in chemical shift differences localized near the site of mutation. The NMR data suggest that the nSNVs do not lead to significant structural changes when compared with wt-hGMPK. Plots of combined chemical shift differences Δδdiff resulting from the mutations S2L (blue) (A), G3A (cyan) (B), R96H (magenta) (C), and S186Y (red) (D), as calculated with Equation 1 (60, 61). The threshold for considering a chemical shift significantly different was set at 1 S.D. value (0.026 ppm) of all Δδdiff across the entire spectral series (including R116Q; see Fig. S9), excluding Δδdiff for G3A (1.62 ppm). Depicted in the plots are the locations of Δδdiff that are considered significantly different from the wt-hGMPK chemical shift mapped on the lowest energy conformer from the NMR structure.
The spectra show minimal variations in chemical shifts (R116Q does not possess any significant chemical shift differences from wt-hGMPK; Fig. S9), and those shifts that do display a sizable change are primarily located around the site of the mutation, signifying a shared average structure across the nsSNV series (Fig. 7). This conclusion is supported by the similar melting temperatures measured by DSF for the hGMPK variants (Fig. 6C and Table 2). Interestingly, G3A is the only exception. This mutation within the flexible N terminus perturbs residues near the Ala-87 hinge residue and the GMP-BD residues Glu-72 and His-73 (Fig. 7B). Taken together with the SV-AUC data, the 2D 1H,15N HSQC NMR spectrum for G3A may be detecting a slight compaction of hGMPK due to a shift in hGMPK's conformational landscape, although the structural changes are probably subtle in nature given that the chemical shift differences for a majority of the residues are within 2 S.D. values from the mean. These observations do suggest that this set of nsSNVs can modulate catalytic activity without causing substantial structural changes, indicating a previously unknown regulatory role for residues within the CORE domain.
The NMR data indicate that the solution structure of wt-hGMPK alone cannot rationalize the observed catalytic enhancement caused by the nsSNVs. Because the enhancement only spans a range of 1.2–3.2-fold more activity than wt-hGMPK, we acknowledge that a strictly structural explanation for the observed variations in function would be challenging at best. That said, single-residue mutations within the AMP-binding domain of adenylate kinase can lead to subtle enhancements in the measured kcat, which was attributed to a modulation of the conformational equilibria between folded and unfolded conformations for the AMP-binding domain (62). Although we cannot ascribe the variations in hGMPK activity caused by the CORE domain mutants to also be associated with a shift in an unfolding/folding equilibrium, the results do suggest that allosteric networking links the CORE domain to the nucleotide-binding domains. This conclusion is supported by work demonstrating that allegedly nonallosteric proteins possess allosteric sites, well-described by the ensemble view of allostery (63, 64). It is still quite difficult to forecast the effect of an active site–distant, nonconserved nsSNV on the processes of molecular recognition, enzymatic catalysis, and signal transduction (65). This is especially evident in the experimental LacI/GalR transcription regulation system, where multiple substitutions at nonconserved positions resulted in “rheostatic” behavior that defies predictability (52, 65). Thus, a continued focus on the atomic level features defining the mechanisms of action for nsSNVs, especially those that may act as proxies for allosteric processes, are necessary to decipher and predict the outcome of nsSNVs at nonconserved positions, especially when they are present in diseases originating from identified gene alterations.
Exploring the functional role of hGMPK for lung adenocarcinoma cellular viability
Given our high-resolution NMR structure and our results indicating a regulatory role for the CORE domain, we were interested in examining whether hGMPK could be a biomolecular target. Due to the elevation of GTP in cancer cells (2), we posited that levels of hGMPK might be higher in cancer samples than in adjacent normal tissue. To investigate this possibility, we first surveyed a number of publicly available data sets and found that the genomic locus encoding hGMPK, GUK1, is gained or amplified in more than half of lung cancers analyzed (Fig. 8A), suggesting an enhanced role for hGMPK in the proliferation of cancer cells. Next, we then determined whether expression of hGMPK is required for survival and growth of human lung adenocarcinoma cell lines using RNAi techniques. Remarkably, we observed that loss of hGMPK decreases cell proliferation and clonogenic potential for all of the lung adenocarcinoma cells tested but not in immortalized human peripheral airway (HPL1D) cells (Fig. 8, B–D), suggesting that hGMPK may be a potential target for novel target cell type–selective cancer therapeutics. In the case of A549 cells, cleavage of the active poly(ADP-ribose) polymerase (PARP) following loss of hGMPK indicates that the mechanism of cell death is apoptosis (Fig. 8E) (66, 67).
Figure 8.

hGMPK is a novel tumor cell vulnerability target. A, lung cancer tumors possess multiple copies of the hGMPK (GUK1) gene, as indicated by data representing a gain (3–5 copies) and an amplification (>5 copies) of the gene. These findings suggest that cancer cells increase GTP production via enhancement of hGMPK activity in the cell through more copies of the gene being available for expression. Copy number (Genomic Identification of Significant Targets in Cancer (GISTIC) alteration status (68)) data for hGMPK (GUK1) was downloaded for 412 lung adenocarcinoma samples (230 from the Cancer Genome Atlas (TCGA) and 182 from the Broad Institute) from cBioPortal (69). Data were graphed according to whether the hGMPK (GUK1) locus was lost, diploid, gained, or amplified. B, loss of hGMPK activity by gene silencing decreases relative cell numbers in the lung adenocarcinoma cell lines A549, HOP62, H1650, and H322 but not in immortalized HPL1D cells. The positive control is si_Kif11, which silences the translation of kinesin family member 11 (Kif11). Kif11 is a molecular motor protein that is essential for mitotic processes (70). si_NT is the nontargeting control siRNA, and si_hGMPK is the hGMPK-targeting siRNA. C, Western blotting at 72 h post-transfection shows that hGMPK expression is eliminated or significantly reduced when compared with the controls. rhGMPK, recombinantly produced hGMPK. GAPDH is shown as a control for consistent antibody detection. D, loss of hGMPK alters the clonogenic potential (an assay for observing the survival and proliferation of cells) of A549 and HOP62 lung adenocarcinoma cells but not of HPL1D cells. D, the mechanism of cell death caused by the loss of hGMPK is apoptosis in A549 lung adenocarcinoma cells, as suggested by cleavage of PARP, whose cleavage is a marker for the activation of caspases (66, 67). Error bars, S.D.
These results denote a differential effect on normal and cancerous cell lines when depleting hGMPK by siRNA, signifying a potential therapeutic window for targeting hGMPK. This differential effect is most likely due to the higher requirement of GTP in rapidly dividing cancer cells and the absence of the only enzyme that converts GMP to GDP. In fact, silencing hGMPK translation coupled with turnover of the enzyme already present in the treated cancer cell lines appears to lead directly to cell death by apoptosis. To date, only a few competitive inhibitors have been described for hGMPK (30, 71). Current efforts toward depleting cells of GTP and GDP have focused primarily on IMPDH, an enzyme that acts a few steps upstream of hGMPK in the de novo synthesis of nucleotides (Fig. 1A). This focus on IMPDH continues, although inhibiting IMPDH can affect ATP and pyrimidine levels, leading to even more pleiotropic outcomes (72). Our results suggest that targeting the production of GDP/GTP at a more proximal step, such as at hGMPK-catalyzed phosphorylation, will lower the number of cellular processes affected while still specifically targeting cancer cell growth. Future work in our laboratory will focus on the cellular processes and signaling pathways that are affected by inhibition of hGMPK and will attempt to develop small-molecule inhibitors of hGMPK using our NMR structure.
Conclusions
In the current work, we solved the structure of hGMPK with atomic resolution for the first time by solution NMR spectroscopy, a structure that we anticipate will provide an important starting point for the development of therapeutically relevant hGMPK inhibitors and for improving the efficiency of prodrug phosphorylation. Furthermore, we offer support for the existence of allosteric networking linking nonactive site CORE domain residues to the nucleotide-binding domains via sites of nsSNV mutations. In addition, our results on depleting hGMPK in multiple lung adenocarcinoma cell lines targeted by siRNA strongly suggest that loss of hGMPK is detrimental to lung cancer cell lines due to adverse effects on cellular viability, proliferation, and clonogenic potential. Most importantly, hGMPK inhibition was well-tolerated in the nontransformed lung epithelial cell line (HPL1D), signifying a potential therapeutic window for targeting hGMPK. Taken together, our results provide an important step forward in establishing hGMPK as a potential biomolecular target.
Experimental procedures
Site-directed mutagenesis of hGMPK
The E. coli expression plasmid, pET-14bSUMOΔThr, containing the WT hGMPK (34) insert with N-terminal His6-SUMO (small ubiquitin-related modifier) tag, was used as a template for introducing 10 amino acid substitutions, which include S2L, G3A, L25P, V91M, R96H, R116Q, S121F, and S186Y. These nsSNVs were generated using the Q5 Site-Directed Mutagenesis kit (New England Biolabs) (73). Twenty mutagenic oligonucleotides containing the desired point mutations were designed for this purpose and were synthesized by Integrated DNA Technologies (Table S4). All oligonucleotides were 18–34 bases in length, Tm ≤ 76 °C, with GC contents in the range of 50–78%. Each 25-μl reaction mixture contained 15 ng of hGMPK pET-14bSUMOΔThr template (∼5.5 kb), Q5 Hot Start High-Fidelity 1× Master Mix, 0.5 μm forward and reverse primers. The cycling parameters used were initial denaturation at 98 °C for 30 s, followed by 25 cycles of denaturation at 98 °C for 10 s, annealing of primers at 57–61 °C for 25 s, and extension at 72 °C for 2.5 min. The final extension was at 72 °C for 2 min. To check for sufficient amplification, 10 μl of the PCR product was loaded on 1% agarose gel. Once verified, the amplified product, 1 μl of the PCR product, 1× KLD (kinase, ligase, and DpnI) Enzyme Mix, and 1× KLD Reaction Buffer were mixed in a 10-μl final volume and incubated at 25 °C for 5 min. The KLD enzyme mix ensures room temperature circularization of the PCR product and removal of the template DNA. For transformation of competent E. coli XL1-Blue cells, 5 μl of the KLD mix was used. The mutated plasmids were purified by Qiagen Plasmid Mini kit according to the manufacturer's protocol. The desired mutations in the hGMPK inserts were sequence-verified by the DNA Core Facility (University of Louisville, Louisville, KY).
Expression and purification of hGMPK
E. coli C41(DE3) cells were transformed with the WT and mutant hGMPK in the pET-14bSUMOΔThr vector (34). For the overexpression of 15N,13C-labeled wt-hGMPK in M9 minimal medium, the protocol mentioned previously (35) was used, followed by chromatographic purification that includes affinity and gel filtration chromatography, as stated in the supplemental information of Ref. 34. One extra purification step of ion-exchange chromatography was added using a Mono Q 10/100 GL column (GE Healthcare) according to the manufacturer's protocol. The starting buffer was 20 mm Tris, pH 8.0, containing 2 mm DTT, and the elution buffer was 20 mm Tris, pH 8.0, containing 2 mm DTT, and 1 m NaCl. The gradient applied was 50% of the target elution buffer in 80 min (160 ml) with the flow rate of 2 ml/min using the ÅKTAprime Plus system (GE Healthcare). For the expression of unlabeled WT and nsSNVs of hGMPK, cells were grown in Terrific Broth at 37 °C until A600 0.65, and overexpression was induced with 0.5 mm isopropyl β-d-1-thiogalactopyranoside (Gold Biotechnology), incubating for an additional 4 h. Cells were harvested by centrifugation at 5000 × g for 15 min at 4 °C. The purification procedure was the same as described above. Purity of the samples was tested by SDS-PAGE (Fig. S6).
NADH-dependent spectroscopic assay
The activity of wt-hGMPK and its nsSNVs was determined by the NADH-dependent enzyme-coupled assay using a JASCO V-550 UV-visible spectrophotometer (4). ADP and GDP produced by the hGMPK-catalyzed reaction were coupled to two additional reactions catalyzed by the helper enzymes pyruvate kinase and lactate dehydrogenase, respectively. The absorbance change of the reaction mixture due to oxidation of NADH (ϵ = 6.22 mm−1 cm−1) to NAD+ was monitored at 340 nm. The NADH conversion to NAD+ in the third step of the assay is directly proportional to the phosphorylation of GMP to GDP in the first step. The assay measurements were performed in triplicate with 10 nm hGMPK at 25 °C in a buffer containing 100 mm Tris, pH 7.5, 100 mm KCl, and 10 mm MgCl2. For each hGMPK variant, the ATP concentration was held constant at 2 mm, and the GMP concentration was varied from 10 to 200 μm. Specific activities and turnover rates were calculated. The kinetic data were fit to the Michaelis–Menten equation for determining kcat and Km, and the error in the fitted parameters was determined from 1000 Monte Carlo simulations using Mathematica.
NMR spectroscopy
The NMR measurements (15N-resolved 1H,1H NOESY, τmix = 60 ms; 13Caliphatic-resolved 1H,1H NOESY, τmix = 80 ms; 13Caromatic-resolved 1H,1H NOESY, τmix = 80 ms) providing the distance restraints for the structure determination of hGMPK were previously measured and described (35). The buffer used for all measurements was 20 mm phosphate, 100 mm sodium chloride, 1 mm tris(2-carboxyethyl)phosphine, 3 mm sodium azide, 10% D2O, pH 7.2 (referred to as NMR buffer).
Standard pulse sequences were used to record the steady-state [1H]-15N NOE data on a sample of 500 μm 15N,13C-labeled hGMPK in NMR buffer at 25 °C and on a Bruker Avance Neo 600-MHz instrument equipped with a nitrogen-cooled Prodigy TCI cryoprobe. The steady-state [1H]-15N NOE spectra were recorded in an interleaved manner with a 10-s 1H saturation time and a 10-s recovery time for the reference experiment (74). Both measurements were preceded by an additional 1-s recovery time. The spectral parameters were 512 and 128 complex points in the direct (t2) and indirect (t1) dimensions, respectively, and 66 scans per t1 increment. The t1,max and t2,max were 62.0 and 62.4 ms, respectively. Steady-state [1H]-15N NOE values were calculated from the ratio of peak heights in a pair of NMR spectra acquired with and without proton saturation. The signal/noise ratio in each spectrum was used to estimate the experimental uncertainty.
For the RDC sample, Pf1 phage (ASLA Ltd., Riga, Latvia) was suspended in NMR buffer and pelleted three times at 140,000 g for 1 h. To the Pf1 phage pellet, a solution of 400 μm 15N-labeled hGMPK was added, resulting in a final phage concentration of 12 mg/ml (75). Backbone amide RDC data for the anisotropic sample, as well as the isotropic reference experiment for measuring backbone amide scalar couplings, were measured using 2D-IPAP-15N,1H HSQC experiments (76) at 25 °C and on a Bruker Avance Neo 600-MHz instrument equipped with a nitrogen-cooled Prodigy TCI cryoprobe. Both the anisotropic and isotropic spectra were recorded with 1024 and 50 complex points in the direct (t2) and indirect (t1) dimensions, respectively, with 352 and 160 scans per t1 increment for the aligned and unaligned samples, respectively. The t1,max and t2,max were 69.6 and 125 ms, respectively. Measured RDCs and scalar couplings were derived from the difference in coupling between aligned and isotropic samples. To provide a lower limit for the accuracy of the measurements, an estimate of the error in the RDCs was calculated by dividing the average peak line width by the signal/noise ratio and the measurement error propagated throughout the calculation of the coupling constants, yielding an average error of 0.8 Hz for the extracted RDCs (77). This error represents the lower limit for the uncertainty in the RDC calculation.
2D 1H,15N HSQC measurements of the hGMPK nsSNV series (S2L, G3A, R96H, R116Q, and S186Y) were carried out at 800 MHz and 25 °C on a Varian Inova spectrometer equipped with a 5-mm inverse triple resonance pulsed-field gradient probe. The concentration of the hGMPK nsSNVs was 100 μm in 90%/10% H2O/D2O in NMR buffer. The spectra were recorded with 1024 and 100 complex points in the direct (t2) and indirect (t1) dimensions, respectively, with 256 scans per t1 increment. The t1,max and t2,max were 34.3 and 98.5 ms, respectively.
Frequency discrimination in the indirectly detected dimension was achieved with the States-TPPI scheme (78). All spectra were processed using NMRPipe (79) and analyzed with CARA (80).
Structure calculation and analysis
The chemical shift assignment was determined manually as described previously (35). Backbone dihedral angle restraints from 1H, 15N, 13Cα, 13Cβ, 13C′ chemical shifts were calculated by TALOS-N (81) and reported previously (35). It should be noted that 15N-hetNOE values <0.7 are associated with backbone amide vectors that experience rapid picosecond to nanosecond fluctuations and are generally more disordered. Residues with 15N-hetNOE values >0.7 primarily sense the molecule's overall tumbling and are more ordered. Therefore, we removed dihedral and RDC constraints for those residues with 15N-hetNOE values <0.7 from the structure calculation. Automatic NOE assignment and initial structure calculation were performed with CYANA (version 3.97) (36, 37), generating the 20 lowest energy conformers of hGMPK from 100 structures using 2430 NOE distance constraints and 281 dihedral constraints.
From this initial structure, we identified hydrogen bonds with an automatic algorithm implemented in CYANA, which does not make any assumptions about the secondary structure or the protein topology. This routine identified all potential backbone acceptor–donor pairs that are less than 3.5 Å apart in at least 50% of the structures. Hydrogen bond distance constraints were then generated for those hydrogen atoms that are involved in exactly one potential acceptor–donor pair and were present in all 20 structures. As additional support for the generated hydrogen bond constraints, we utilized a 2D 1H,15N HSQC spectrum of hGMPK in 100% D2O (following 1 day of immersion in 100% D2O), where the presence of cross-peaks indicates protection from deuterium for that backbone amide (Fig. S10). Two upper and two lower distance limits are imposed for each hydrogen bond to restrict the HN–O distance to the range 1.8–2.0 Å and the N–O distance to 2.7–3.0 Å. Finally, we performed the structure calculation again, this time including 58 hydrogen bond pairs and 89 RDCs as constraints in addition to the list of assigned NOEs and dihedral constraints. We calculated 2500 structures and selected 100 with the lowest target functions for further refinement.
NMR-restrained molecular dynamics (RMD) simulations were performed by the AMBER 16 package using the AMBER ff14SB force field (39). Distance, dihedral angle, and RDC restraints were introduced by the respective makeDIST_RST, makeANG_RST, and makeDIP_RST modules of AmberTools 17. Refinement and RMD were performed under generalized Born (GB) model for implicit solvation (igb = 1) conditions with a distance cut-off of 12 Å throughout. NMR distance and angle constraints were implemented with a force constant of 300 kcal/mol/Å2, and the RDC constraints were weighted by a factor of 0.05. Calculation of the initial RDC alignment tensor was first determined by setting freezemol = .true. Initial refinement was carried out for the 100 best structures derived from the CYANA calculations. The 20 best structures (based on lowest target function) were subjected to the same alignment/refinement procedure followed by 1 ns of RMD at 300 K, followed by subsequent minimization and analysis. These 20 structures constituted the final structure.
SAXS data analysis
The previously published hGMPK SAXS data (34) were analyzed with the ATSAS software package (82). The theoretical scattering from the individual structures was calculated using the program CRYSOL.
Analytical ultracentrifugation
Sedimentation velocity experiments were carried out in a Beckman Coulter ProteomeLab XL-A analytical ultracentrifuge (Beckman Coulter Inc., Brea, CA) at 20 °C and 50,000 rpm in standard two-sector cells. Buffer density was determined on a Mettler/Paar Calculating Density Meter DMA 55A at 20 °C, and viscosity was measured using an Anton Parr AMVn Automated Microviscometer at 20 °C. Data were analyzed with the program Sedfit (http://www.analyticalultracentrifugation.com)8 using the continuous c(s) distribution model. The partial specific volume of hGMPK was calculated from the amino acid composition (0.734 ml/g) using the Protparam tool in ExPASy (http://web.expasy.org/protparam). Frictional ratios were calculated by Sedfit assuming no bound water. Experimental sedimentation coefficients were corrected to s20,w using the corrections based on the measured density and viscosity. Analytical runs were performed with at least three independent samples for each condition studied. Sedimentation coefficients were calculated from atomic coordinates using the program winHYDROPRO10 (http://leonardo.inf.um.es/macromol/programs/hydropro/hydropro.htm)8 (57) with the residue/shell model.
DSF
DSF experiments were carried out using Applied Biosystems StepOne Plus real-time PCR system (83, 84). Melting curves were determined in 96-well plates using a temperature ramp from 20 °C to 99 °C at a constant increase of 0.2 °C/min. The assay mixture contained wt-hGMPK or nsSNVs (5 μm), SYPRO Orange dye (5× final concentration; Thermo Fisher Scientific), and PBS, pH 7.4, to a final volume of 20 μl. Samples were loaded in triplicate onto the 96-well plate, sealed, and centrifuged at 1350 rpm for 2 min. Each plate was run in duplicate. The Tm was determined from the first derivative of the melt curve (fluorescence versus temperature).
Antibodies used in this study
Antibodies used were as follows: GAPDH (catalog no. 2118), PARP (catalog no. 9532) (Cell Signaling Technologies Inc., Danvers, MA), human guanylate kinase (C-terminal peptide) (catalog no. ab198816, Abcam, Cambridge, MA).
Cell culture, siRNA transfection, and protein analysis
Human lung adenocarcinoma cell lines A549, H3255, H1650, and HOP62 and HPL1D were purchased from American Type Culture Collection (ATCC, Manassas, VA) and cultured in RPMI medium supplemented with 10% fetal bovine serum (Invitrogen) and 1% antibiotic/antimycotic solution (Sigma). All cell lines were routinely subcultured every 3–5 days. siRNA transfections were performed using Dharmafect1 (catalog no. T-2001-03, Thermo Fisher Scientific) as per the manufacturer's protocol. 72 h post-transfection, cells were harvested in cell extraction buffer (CEB) lysis buffer (catalog no. FNN0011, Invitrogen, Life Technologies). Protein was quantitated by using Pierce's BCA Protein Assay Reagent Kit (catalog no. 23227) as per the manufacturer's protocol.
siRNA sequences used in this study
siRNAs used in this study were ordered from Thermo Fisher Scientific and Dharmacon: nontargeting siRNA (si_NT), UAAGGCUAUGAAGAGAUACAA; Kif11 siRNA (si_Kif11), ON-TARGET plus SMART pool (catalog no. L-003317-00); hGMPK siRNA (si_hGMPK), ON-TARGET plus SMART pool (catalog no. L-006734–00).
Western blot analysis
CEB (catalog no. FNN0011, Invitrogen) was used to lyse the cells, supplemented with Complete Mini Protease Inhibitor tablets (Roche Applied Science). Protein quantification was performed, and an equal amount of protein (40 μg) from each sample was added to SDS loading buffer, boiled, and resolved on a 4–12% SDS-polyacrylamide gel and transferred onto a membrane. Blots were probed with different antibodies, and immunoreactive proteins were visualized using the SuperSignal West Femto Maximum Sensitivity Substrate (catalog no. PI34095) from Fisher, according to the manufacturer's instructions. The membrane was stripped by using Western blotting stripping reagent (Bio-Rad) and reprobed with GAPDH to normalize the variation in loading of samples.
Cell viability and clonogenic assay
A549, H3255, H1650, HOP62, and HPL1D cells were cultured in 60-mm culture plates. After 24 h of transfection either with nontargeting siRNA (si_NT), positive control Kif11 siRNA (si_Kif11), or with siRNA targeting hGMPK (si_hGMPK), cells were trypsinized and counted, and 1000 cells were reseeded per well in 96-well plates. Cell viability was analyzed for 4 successive days using alamarBlue (catalog no. DAL1100, Invitrogen Detection Technologies, Eugene, OR). Also following transfection, 1000 cells were reseeded per well in 6-well plates in triplicate for each transfection. Cells were allowed to grow on 6-well plates for 10 days; they were supplemented with fresh media after every 2–3 days. After 10 days, colonies formed were washed once with PBS, fixed with 70% methanol, and stained with 0.25% Coomassie Brilliant Blue stain R (catalog no. B 7920, Sigma-Aldrich) and photographed.
Author contributions
N. K., P. P. S., D. B., P. T.-M., M. G. C., L. D., W. L. D., J. O. T., L. J. B., M. K., D. L., and T. M. S. formal analysis; N. K., P. P. S., D. B., P. T.-M., M. G. C., L. D., W. L. D., J. O. T., L. J. B., D. L., and T. M. S. investigation; N. K., L. J. B., and T. M. S. writing-original draft; N. K., P. P. S., D. B., P. T.-M., M. G. C., L. D., W. L. D., J. O. T., L. J. B., M. K., D. L., and T. M. S. writing-review and editing; D. B., L. J. B., M. K., D. L., and T. M. S. conceptualization; J. O. T., D. L., and T. M. S. methodology; D. L. and T. M. S. supervision; D. L. and T. M. S. project administration; T. M. S. resources; T. M. S. data curation; T. M. S. funding acquisition.
Supplementary Material
Acknowledgments
We thank Christian Griesinger (MPI for Biophysical Chemistry) and J. Bradford Chaires (University of Louisville) for valuable discussions related to the project. We thank Simone Techert and Rohit Jain (MPI for Biophysical Chemistry) for supplying unpublished SAXS data on hGMPK.
This research was supported by a Deutscher Akademischer Austauschdienst (DAAD) scholarship (to N. K.), by the Max Planck Society, and by funds provided by the James Graham Brown Cancer Center. The authors declare that they have no conflicts of interest with the contents of this article.
The atomic coordinates and structure factors (code 6NUI) have been deposited in the Protein Data Bank (http://wwpdb.org/).
Resonance assignments have been deposited in the BMRB database under accession number 27151.
This article contains Tables S1–S4 and Figs. S1–S10.
Please note that the JBC is not responsible for the long-term archiving and maintenance of this site or any other third party hosted site.
- hGMPK or GUK1
- human guanylate kinase
- GMP-BD
- GMP-binding domain
- GMPS
- guanosine monophosphate synthase
- HPL1D
- immortalized human peripheral airway cells
- IMPDH
- inosine-5′-monophosphate dehydrogenase
- mGMPK
- mouse guanylate kinase
- nsSNVs
- non-synonymous single nucleotide variants
- PARP
- poly(ADP ribose) polymerase
- yGMPK
- yeast guanylate kinase
- GAPDH
- glyceraldehyde 3-phosphate dehydrogenase
- SAXS
- small-angle X-ray scattering
- 15N-hetNOE
- 15N heteronuclear NOE
- RDC
- residual dipolar coupling
- RMSD
- root mean square deviation
- SV-AUC
- sedimentation velocity analytical ultracentrifugation
- wt-hGMPK
- WT hGMPK
- DSF
- differential scanning fluorimetry
- 2D
- two-dimensional
- HSQC
- heteronuclear single quantum coherence
- SUMO
- small ubiquitin-like modifier
- RMD
- NMR-restrained molecular dynamics
- GB
- generalized Born
- CEB
- cell extraction buffer.
References
- 1. Cantor J. R., and Sabatini D. M. (2012) Cancer cell metabolism: one hallmark, many faces. Cancer Discov. 2, 881–898 10.1158/2159-8290.CD-12-0345 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Traut T. W. (1994) Physiological concentrations of purines and pyrimidines. Mol. Cell. Biochem. 140, 1–22 10.1007/BF00928361 [DOI] [PubMed] [Google Scholar]
- 3. Shuvalov O., Petukhov A., Daks A., Fedorova O., Vasileva E., and Barlev N. A. (2017) One-carbon metabolism and nucleotide biosynthesis as attractive targets for anticancer therapy. Oncotarget 8, 23955–23977 10.18632/oncotarget.15053 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Agarwal K. C., Miech R. P., and Parks R. E. Jr. (1978) Guanylate kinases from human erythrocytes, hog brain, and rat liver. Methods Enzymol. 51, 483–490 10.1016/S0076-6879(78)51066-5 [DOI] [PubMed] [Google Scholar]
- 5. Hall S. W., and Kühn H. (1986) Purification and properties of guanylate kinase from bovine retinas and rod outer segments. Eur. J. Biochem. 161, 551–556 10.1111/j.1432-1033.1986.tb10477.x [DOI] [PubMed] [Google Scholar]
- 6. Konrad M. (1992) Cloning and expression of the essential gene for guanylate kinase from yeast. J. Biol. Chem. 267, 25652–25655 [PubMed] [Google Scholar]
- 7. Gaidarov I. O., Suslov O. N., and Abdulaev N. G. (1993) Enzymes of the cyclic GMP metabolism in bovine retina. I. Cloning and expression of the gene for guanylate kinase. FEBS Lett. 335, 81–84 10.1016/0014-5793(93)80444-Y [DOI] [PubMed] [Google Scholar]
- 8. Li Y., Zhang Y., and Yan H. (1996) Kinetic and thermodynamic characterizations of yeast guanylate kinase. J. Biol. Chem. 271, 28038–28044 10.1074/jbc.271.45.28038 [DOI] [PubMed] [Google Scholar]
- 9. Hedstrom L. (2009) IMP dehydrogenase: structure, mechanism, and inhibition. Chem. Rev. 109, 2903–2928 10.1021/cr900021w [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Bianchi-Smiraglia A., Wawrzyniak J. A., Bagati A., Marvin E. K., Ackroyd J., Moparthy S., Bshara W., Fink E. E., Foley C. E., Morozevich G. E., Berman A. E., Shewach D. S., and Nikiforov M. A. (2015) Pharmacological targeting of guanosine monophosphate synthase suppresses melanoma cell invasion and tumorigenicity. Cell Death Differ. 22, 1858–1864 10.1038/cdd.2015.47 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Li G., Segu V. B. G., Rabaglia M. E., Luo R.-H., Kowluru A., and Metz S. A. (1998) Prolonged depletion of guanosine triphosphate induces death of insulin-secreting cells by apoptosis. Endocrinology 139, 3752–3762 10.1210/endo.139.9.6207 [DOI] [PubMed] [Google Scholar]
- 12. Deville-Bonne D., El Amri C., Meyer P., Chen Y., Agrofoglio L. A., and Janin J. (2010) Human and viral nucleoside/nucleotide kinases involved in antiviral drug activation: structural and catalytic properties. Antiviral Res. 86, 101–120 10.1016/j.antiviral.2010.02.001 [DOI] [PubMed] [Google Scholar]
- 13. Karran P., and Attard N. (2008) Thiopurines in current medical practice: molecular mechanisms and contributions to therapy-related cancer. Nat. Rev. Cancer. 8, 24–36 10.1038/nrc2292 [DOI] [PubMed] [Google Scholar]
- 14. Miller R. L., Adamczyk D. L., and Spector T. (1977) Reassessment of interactions of guanylate kinase and 6-thioguanosine 5′-phosphate. Biochem. Pharmacol. 26, 1573–1576 10.1016/0006-2952(77)90071-5 [DOI] [PubMed] [Google Scholar]
- 15. Johansson M., Amiri M., and Karlsson A. (2005) Phosphorylation of 9-β-d-arabinofuranosylguanine monophosphate by Drosophila melanogaster guanylate kinase. Biochem. Pharmacol. 70, 987–992 10.1016/j.bcp.2005.06.023 [DOI] [PubMed] [Google Scholar]
- 16. Ashton W. T., Karkas J. D., Field A. K., and Tolman R. L. (1982) Activation by thymidine kinase and potent antiherpetic activity of 2′-nor-2′-deoxyguanosine (2′NDG). Biochem. Biophys. Res. Commun. 108, 1716–1721 10.1016/S0006-291X(82)80109-5 [DOI] [PubMed] [Google Scholar]
- 17. Field A. K., Davies M. E., DeWitt C., Perry H. C., Liou R., Germershausen J., Karkas J. D., Ashton W. T., Johnston D. B., and Tolman R. L. (1983) 9-([2-Hydroxy-1-(hydroxymethyl)ethoxy]methyl)guanine: a selective inhibitor of herpes group virus replication. Proc. Natl. Acad. Sci. U.S.A. 80, 4139–4143 10.1073/pnas.80.13.4139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Miller W. H., and Miller R. L. (1980) Phosphorylation of acyclovir (acycloguanosine) monophosphate by GMP kinase. J. Biol. Chem. 255, 7204–7207 [PubMed] [Google Scholar]
- 19. Gentry B. G., Gentry S. N., Jackson T. L., Zemlicka J., and Drach J. C. (2011) Phosphorylation of antiviral and endogenous nucleotides to di- and triphosphates by guanosine monophosphate kinase. Biochem. Pharmacol. 81, 43–49 10.1016/j.bcp.2010.09.005 [DOI] [PubMed] [Google Scholar]
- 20. Miller W. H., Daluge S. M., Garvey E. P., Hopkins S., Reardon J. E., Boyd F. L., and Miller R. L. (1992) Phosphorylation of carbovir enantiomers by cellular enzymes determines the stereoselectivity of antiviral activity. J. Biol. Chem. 267, 21220–21224 [PubMed] [Google Scholar]
- 21. Boehme R. E. (1984) Phosphorylation of the antiviral precursor 9-(1,3-dihydroxy-2-propoxymethyl)guanine monophosphate by guanylate kinase isozymes. J. Biol. Chem. 259, 12346–12349 [PubMed] [Google Scholar]
- 22. Auvynet C., Topalis D., Caillat C., Munier-Lehmann H., Seclaman E., Balzarini J., Agrofoglio L. A., Kaminski P. A., Meyer P., Deville-Bonne D., and El Amri C. (2009) Phosphorylation of dGMP analogs by vaccinia virus TMP kinase and human GMP kinase. Biochem. Biophys. Res. Commun. 388, 6–11 10.1016/j.bbrc.2009.07.089 [DOI] [PubMed] [Google Scholar]
- 23. Ardiani A., Goyke A., and Black M. E. (2009) Mutations at serine 37 in mouse guanylate kinase confer resistance to 6-thioguanine. Protein Eng. Des. Sel. 22, 225–232 10.1093/protein/gzn078 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Brady W. A., Kokoris M. S., Fitzgibbon M., and Black M. E. (1996) Cloning, characterization, and modeling of mouse and human guanylate kinases. J. Biol. Chem. 271, 16734–16740 10.1074/jbc.271.28.16734 [DOI] [PubMed] [Google Scholar]
- 25. Prinz H., Lavie A., Scheidig A. J., Spangenberg O., and Konrad M. (1999) Binding of nucleotides to guanylate kinase, p21ras, and nucleoside-diphosphate kinase studied by nano-electrospray mass spectrometry. J. Biol. Chem. 274, 35337–35342 10.1074/jbc.274.50.35337 [DOI] [PubMed] [Google Scholar]
- 26. Sekulic N., Shuvalova L., Spangenberg O., Konrad M., and Lavie A. (2002) Structural characterization of the closed conformation of mouse guanylate kinase. J. Biol. Chem. 277, 30236–30243 10.1074/jbc.M204668200 [DOI] [PubMed] [Google Scholar]
- 27. Blaszczyk J., Li Y., Yan H., and Ji X. (2001) Crystal structure of unligated guanylate kinase from yeast reveals GMP-induced conformational changes. J. Mol. Biol. 307, 247–257 10.1006/jmbi.2000.4427 [DOI] [PubMed] [Google Scholar]
- 28. Stehle T., and Schulz G. E. (1992) Refined structure of the complex between guanylate kinase and its substrate GMP at 2.0 Å resolution. J. Mol. Biol. 224, 1127–1141 10.1016/0022-2836(92)90474-X [DOI] [PubMed] [Google Scholar]
- 29. Hible G., Renault L., Schaeffer F., Christova P., Zoe Radulescu A., Evrin C., Gilles A.-M., and Cherfils J. (2005) Calorimetric and crystallographic analysis of the oligomeric structure of Escherichia coli GMP kinase. J. Mol. Biol. 352, 1044–1059 10.1016/j.jmb.2005.07.042 [DOI] [PubMed] [Google Scholar]
- 30. Hible G., Daalova P., Gilles A.-M., and Cherfils J. (2006) Crystal structures of GMP kinase in complex with ganciclovir monophosphate and Ap5G. Biochimie 88, 1157–1164 10.1016/j.biochi.2006.04.002 [DOI] [PubMed] [Google Scholar]
- 31. Hible G., Christova P., Renault L., Seclaman E., Thompson A., Girard E., Munier-Lehmann H., and Cherfils J. (2006) Unique GMP-binding site in Mycobacterium tuberculosis guanosine monophosphate kinase. Proteins 62, 489–500 10.1002/prot.20662 [DOI] [PubMed] [Google Scholar]
- 32. Vedadi M., Lew J., Artz J., Amani M., Zhao Y., Dong A., Wasney G. A., Gao M., Hills T., Brokx S., Qiu W., Sharma S., Diassiti A., Alam Z., Melone M., et al. (2007) Genome-scale protein expression and structural biology of Plasmodium falciparum and related Apicomplexan organisms. Mol. Biochem. Parasitol. 151, 100–110 10.1016/j.molbiopara.2006.10.011 [DOI] [PubMed] [Google Scholar]
- 33. El Omari K., Dhaliwal B., Lockyer M., Charles I., Hawkins A. R., and Stammers D. K. (2006) Structure of Staphylococcus aureus guanylate monophosphate kinase. Acta Crystallogr. Sect. F Struct. Biol. Cryst. Commun. 62, 949–953 10.1107/S174430910603613X [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Jain R., Khan N., Menzel A., Rajkovic I., Konrad M., and Techert S. (2016) Insights into open/closed conformations of the catalytically active human guanylate kinase as investigated by small-angle X-ray scattering. Eur. Biophys. J. 45, 81–89 10.1007/s00249-015-1079-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Khan N., Ban D., Trigo-Mourino P., Carneiro M. G., Konrad M., Lee D., and Sabo T. M. (2018) 1H, 13C and 15N resonance assignment of human guanylate kinase. Biomol. NMR Assign. 12, 11–14 10.1007/s12104-017-9771-6 [DOI] [PubMed] [Google Scholar]
- 36. Güntert P. (2004) Automated NMR structure calculation with CYANA. Methods Mol. Biol. 278, 353–378 [DOI] [PubMed] [Google Scholar]
- 37. Güntert P., and Buchner L. (2015) Combined automated NOE assignment and structure calculation with CYANA. J. Biomol. NMR 62, 453–471 10.1007/s10858-015-9924-9 [DOI] [PubMed] [Google Scholar]
- 38. Bhattacharya A., Tejero R., and Montelione G. T. (2007) Evaluating protein structures determined by structural genomics consortia. Proteins 66, 778–795 10.1002/prot.21165 [DOI] [PubMed] [Google Scholar]
- 39. Case D. A., Cerutti D. S., Cheatham T. E. 3rd, Darden T. A., Duke R. E., Giese T. J., Gohlke H., Goetz A. W., Greene D., Homeyer N., Izadi S., Kovalenko A., Lee T. S., LeGrand S., Li P., et al. (2018) AMBER 2018, University of California, San Francisco [Google Scholar]
- 40. Laskowski R. A., MacArthur M. W., Moss D. S., and Thornton J. M. (1993) PROCHECK: a program to check the stereochemical quality of protein structures. J. Appl. Crystallogr. 26, 283–291 10.1107/S0021889892009944 [DOI] [Google Scholar]
- 41. Emekli U., Schneidman-Duhovny D., Wolfson H. J., Nussinov R., and Haliloglu T. (2008) HingeProt: automated prediction of hinges in protein structures. Proteins 70, 1219–1227 10.1002/prot.21613 [DOI] [PubMed] [Google Scholar]
- 42. Johnston C. A., Whitney D. S., Volkman B. F., Doe C. Q., and Prehoda K. E. (2011) Conversion of the enzyme guanylate kinase into a mitotic-spindle orienting protein by a single mutation that inhibits GMP-induced closing. Proc. Natl. Acad. Sci. U.S.A. 108, E973–E978 10.1073/pnas.1104365108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Kovermann M., Rogne P., and Wolf-Watz M. (2016) Protein dynamics and function from solution state NMR spectroscopy. Q. Rev. Biophys. 49, e6 10.1017/S0033583516000019 [DOI] [PubMed] [Google Scholar]
- 44. Choi B., and Zocchi G. (2007) Guanylate kinase, induced fit, and the allosteric spring probe. Biophys. J. 92, 1651–1658 10.1529/biophysj.106.092866 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Whitney D. S., Volkman B. F., and Prehoda K. E. (2016) Evolution of a protein interaction domain family by tuning conformational flexibility. J. Am. Chem. Soc. 138, 15150–15156 10.1021/jacs.6b05954 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Eisenmesser E. Z., Millet O., Labeikovsky W., Korzhnev D. M., Wolf-Watz M., Bosco D. A., Skalicky J. J., Kay L. E., and Kern D. (2005) Intrinsic dynamics of an enzyme underlies catalysis. Nature 438, 117–121 10.1038/nature04105 [DOI] [PubMed] [Google Scholar]
- 47. Hazra S., Szewczak A., Ort S., Konrad M., and Lavie A. (2011) Post-translational phosphorylation of serine 74 of human deoxycytidine kinase favors the enzyme adopting the open conformation making it competent for nucleoside binding and release. Biochemistry 50, 2870–2880 10.1021/bi2001032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Segura-Peña D., Lichter J., Trani M., Konrad M., Lavie A., and Lutz S. (2007) Quaternary structure change as a mechanism for the regulation of thymidine kinase 1-like enzymes. Structure 15, 1555–1566 10.1016/j.str.2007.09.025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Segura-Peña D., Sekulic N., Ort S., Konrad M., and Lavie A. (2004) Substrate-induced conformational changes in human UMP/CMP kinase. J. Biol. Chem. 279, 33882–33889 10.1074/jbc.M401989200 [DOI] [PubMed] [Google Scholar]
- 50. Forbes S. A., Bindal N., Bamford S., Cole C., Kok C. Y., Beare D., Jia M., Shepherd R., Leung K., Menzies A., Teague J. W., Campbell P. J., Stratton M. R., and Futreal P. A. (2011) COSMIC: mining complete cancer genomes in the Catalogue of Somatic Mutations in Cancer. Nucleic Acids Res. 39, D945–D950 10.1093/nar/gkq929 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Michielssens S., Peters J. H., Ban D., Pratihar S., Seeliger D., Sharma M., Giller K., Sabo T. M., Becker S., Lee D., Griesinger C., and de Groot B. L. (2014) A designed conformational shift to control protein binding specificity. Angew. Chem. Int. Ed. Engl. 53, 10367–10371 10.1002/anie.201403102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Meinhardt S., Manley M. W. Jr., Parente D. J., and Swint-Kruse L. (2013) Rheostats and toggle switches for modulating protein function. PLoS One 8, e83502 10.1371/journal.pone.0083502 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Bendl J., Stourac J., Salanda O., Pavelka A., Wieben E. D., Zendulka J., Brezovsky J., and Damborsky J. (2014) PredictSNP: robust and accurate consensus classifier for prediction of disease-related mutations. PLOS Comput. Biol. 10, e1003440 10.1371/journal.pcbi.1003440 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Niroula A., and Vihinen M. (2016) Variation interpretation predictors: principles, types, performance, and choice. Hum. Mutat. 37, 579–597 10.1002/humu.22987 [DOI] [PubMed] [Google Scholar]
- 55. Miosge L. A., Field M. A., Sontani Y., Cho V., Johnson S., Palkova A., Balakishnan B., Liang R., Zhang Y., Lyon S., Beutler B., Whittle B., Bertram E. M., Enders A., Goodnow C. C., and Andrews T. D. (2015) Comparison of predicted and actual consequences of missense mutations. Proc. Natl. Acad. Sci. U.S.A. 112, E5189–E5198 10.1073/pnas.1511585112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Gallion J., Koire A., Katsonis P., Schoenegge A. M., Bouvier M., and Lichtarge O. (2017) Predicting phenotype from genotype: improving accuracy through more robust experimental and computational modeling. Hum. Mutat. 38, 569–580 10.1002/humu.23193 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Ortega A., Amorós D., and García de la Torre J. (2011) Prediction of hydrodynamic and other solution properties of rigid proteins from atomic- and residue-level models. Biophys. J. 101, 892–898 10.1016/j.bpj.2011.06.046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Ådén J., Verma A., Schug A., and Wolf-Watz M. (2012) Modulation of a pre-existing conformational equilibrium tunes adenylate kinase activity. J. Am. Chem. Soc. 134, 16562–16570 10.1021/ja3032482 [DOI] [PubMed] [Google Scholar]
- 59. Yu B., and Hunt J. F. (2009) Enzymological and structural studies of the mechanism of promiscuous substrate recognition by the oxidative DNA repair enzyme AlkB. Proc. Natl. Acad. Sci. U.S.A. 106, 14315–14320 10.1073/pnas.0812938106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Schumann F. H., Riepl H., Maurer T., Gronwald W., Neidig K.-P., and Kalbitzer H. R. (2007) Combined chemical shift changes and amino acid specific chemical shift mapping of protein-protein interactions. J. Biomol. NMR 39, 275–289 10.1007/s10858-007-9197-z [DOI] [PubMed] [Google Scholar]
- 61. Williamson M. P. (2013) Using chemical shift perturbation to characterise ligand binding. Prog. Nucl. Magn. Reson. Spectrosc. 73, 1–16 10.1016/j.pnmrs.2013.02.001 [DOI] [PubMed] [Google Scholar]
- 62. Saavedra H. G., Wrabl J. O., Anderson J. A., Li J., and Hilser V. J. (2018) Dynamic allostery can drive cold adaptation in enzymes. Nature 558, 324–328 10.1038/s41586-018-0183-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Motlagh H. N., Wrabl J. O., Li J., and Hilser V. J. (2014) The ensemble nature of allostery. Nature 508, 331–339 10.1038/nature13001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Liu J., and Nussinov R. (2016) Allostery: An overview of its history, concepts, methods, and applications. PLoS Comput. Biol. 12, e1004966 10.1371/journal.pcbi.1004966 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Miller M., Bromberg Y., and Swint-Kruse L. (2017) Computational predictors fail to identify amino acid substitution effects at rheostat positions. Sci. Rep. 7, 41329 10.1038/srep41329 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Kaufmann S. H., Desnoyers S., Ottaviano Y., Davidson N. E., and Poirier G. G. (1993) Specific proteolytic cleavage of poly(ADP-ribose) polymerase: an early marker of chemotherapy-induced apoptosis. Cancer Res. 53, 3976–3985 [PubMed] [Google Scholar]
- 67. Tewari M., Quan L. T., O'Rourke K., Desnoyers S., Zeng Z., Beidler D. R., Poirier G. G., Salvesen G. S., and Dixit V. M. (1995) Yama/CPP32-β, a mammalian homolog of CED-3, is a CrmA-inhibitable protease that cleaves the death substrate poly(ADP-ribose) polymerase. Cell 81, 801–809 10.1016/0092-8674(95)90541-3 [DOI] [PubMed] [Google Scholar]
- 68. Mermel C. H., Schumacher S. E., Hill B., Meyerson M. L., Beroukhim R., and Getz G. (2011) GISTIC2.0 facilitates sensitive and confident localization of the targets of focal somatic copy-number alteration in human cancers. Genome Biol. 12, R41 10.1186/gb-2011-12-4-r41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Gao J., Aksoy B. A., Dogrusoz U., Dresdner G., Gross B., Sumer S. O., Sun Y., Jacobsen A., Sinha R., Larsson E., Cerami E., Sander C., and Schultz N. (2013) Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci. Signal. 6, pl1 10.1126/scisignal.2004088 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Wojcik E. J., Buckley R. S., Richard J., Liu L., Huckaba T. M., and Kim S. (2013) Kinesin-5: cross-bridging mechanism to targeted clinical therapy. Gene 531, 133–149 10.1016/j.gene.2013.08.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Navé J. F., Eschbach A., and Halazy S. (1992) 9-(Phosphonoalkyl)guanine derivatives as substrates or inhibitors of guanylate kinase. Arch. Biochem. Biophys. 295, 253–257 10.1016/0003-9861(92)90515-X [DOI] [PubMed] [Google Scholar]
- 72. Qiu Y., Fairbanks L. D., Rückermann K., Hawrlowicz C. M., Richards D. F., Kirschbaum B., and Simmonds H. A. (2000) Mycophenolic acid-induced GTP depletion also affects ATP and pyrimidine synthesis in mitogen-stimulated primary human T-lymphocytes. Transplantation 69, 890–897 [DOI] [PubMed] [Google Scholar]
- 73. Zheng L., Baumann U., and Reymond J.-L. (2004) An efficient one-step site-directed and site-saturation mutagenesis protocol. Nucleic Acids Res. 32, e115 10.1093/nar/gnh110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Lakomek N.-A., Ying J., and Bax A. (2012) Measurement of N-15 relaxation rates in perdeuterated proteins by TROSY-based methods. J. Biomol. NMR 53, 209–221 10.1007/s10858-012-9626-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Zweckstetter M., and Bax A. (2001) Characterization of molecular alignment in aqueous suspensions of Pf1 bacteriophage. J. Biomol. NMR 20, 365–377 10.1023/A:1011263920003 [DOI] [PubMed] [Google Scholar]
- 76. Ottiger M., Delaglio F., and Bax A. (1998) Measurement of J and dipolar couplings from simplified two-dimensional NMR spectra. J. Magn. Reson. 131, 373–378 10.1006/jmre.1998.1361 [DOI] [PubMed] [Google Scholar]
- 77. Bax A., Kontaxis G., and Tjandra N. (2001) Dipolar couplings in macromolecular structure determination. Methods Enzymol. 339, 127–174 10.1016/S0076-6879(01)39313-8 [DOI] [PubMed] [Google Scholar]
- 78. Marion D., Ikura M., Tschudin R., and Bax A. (1989) Rapid recording of 2D NMR spectra without phase cycling: application to the study of hydrogen exchange in proteins. J. Magn. Reson. 85, 393–399 10.1016/0022-2364(89)90152-2 [DOI] [Google Scholar]
- 79. Delaglio F., Grzesiek S., Vuister G. W., Zhu G., Pfeifer J., and Bax A. (1995) NMRPipe: a multidimensional spectral processing system based on UNIX pipes. J. Biomol. NMR 6, 277–293 [DOI] [PubMed] [Google Scholar]
- 80. Keller R. L. (2004) Optimizing the Process of Nuclear Magnetic Resonance Analysis and Computer Aided Resonance Assignment, Ph.D. thesis, Institute of Molecular Biology and Biophysics, ETH, Zürich [Google Scholar]
- 81. Shen Y., and Bax A. (2013) Protein backbone and sidechain torsion angles predicted from NMR chemical shifts using artificial neural networks. J. Biomol. NMR 56, 227–241 10.1007/s10858-013-9741-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Franke D., Petoukhov M. V., Konarev P. V., Panjkovich A., Tuukkanen A., Mertens H. D. T., Kikhney A. G., Hajizadeh N. R., Franklin J. M., Jeffries C. M., and Svergun D. I. (2017) ATSAS 2.8: a comprehensive data analysis suite for small-angle scattering from macromolecular solutions. J. Appl. Crystallogr. 50, 1212–1225 10.1107/S1600576717007786 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Huynh K., and Partch C. L. (2015) Analysis of protein stability and ligand interactions by thermal shift assay. Curr. Protoc. Protein Sci. 441, 28.9.1–28.9.14 10.1002/0471140864.ps2809s79 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Ericsson U. B., Hallberg B. M., Detitta G. T., Dekker N., and Nordlund P. (2006) Thermofluor-based high-throughput stability optimization of proteins for structural studies. Anal. Biochem. 357, 289–298 10.1016/j.ab.2006.07.027 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.