

Abstract
Methyl coenzyme M reductase (MCR) catalyzes the last step in the biological production of methane by methanogenic archaea, as well as the first step in the anaerobic oxidation of methane to methanol by methanotrophic archaea. MCR contains a number of unique post-translational modifications in its α subunit, including thioglycine, 1-N-methylhistidine, S-methylcysteine, 5-C-(S)-methylarginine, and 2-C-(S)-methylglutamine. Recently, genes responsible for the thioglycine and methylarginine modifications have been identified in bioinformatics studies and in vivo complementation of select mutants; however, none of these reactions has been verified in vitro. Herein, we purified and biochemically characterized the radical S-adenosylmethionine (SAM) protein MaMmp10, the product of the methanogenesis marker protein 10 gene in the methane-producing archaea Methanosarcina acetivorans. Using an array of approaches, including kinetic assays, LC-MS–based quantification, and MALDI TOF–TOF MS analyses, we found that MaMmp10 catalyzes the methylation of the equivalent of Arg285 in a peptide substrate surrogate, but only in the presence of cobalamin. We noted that the methyl group derives from SAM, with cobalamin acting as an intermediate carrier, and that MaMmp10 contains a C-terminal cobalamin-binding domain. Given that Mmp10 has not been annotated as a cobalamin-binding protein, these findings suggest that cobalamin-dependent radical SAM proteins are more prevalent than previously thought.
Keywords: S-adenosylmethionine (SAM), archaea, iron–sulfur protein, protein methylation, post-translational modification (PTM), radical, cobalamin, methanogenesis, methyl coenzyme M reductase, methylarginine, radical SAM
Introduction
Methyl coenzyme M reductase (MCR)2 catalyzes the final and rate-limiting step in methanogenesis, which is the conversion of methyl-2-mercaptoethanesulfonate (methyl-coenzyme M, methyl-SCoM) and N-7-mercaptoheptanoylthreonine phosphate (coenzyme B, CoB–SH) to methane and the mixed disulfide, CoBS–SCoM, by methanogenic archaea (Fig. 1) (1–4). The reaction constitutes a key step in the global carbon cycle, given that methanogenic archaea produce about 1 billion tons of methane annually (5, 6). Interestingly, MCR also catalyzes the first step in the pathway for anaerobic methane oxidation by methanotrophic archaea (7).
Figure 1.

Reaction catalyzed by methyl coenzyme M reductase and its nickel hydrocorphinoid cofactor, F430.
MCR is a 300-kDa hexameric protein composed of a dimer of two heterotrimeric subunits (α, β, and γ) and requires the nickel hydrocorphinoid cofactor, F430, which participates intimately in catalysis (Fig. 1) (8). The X-ray crystal structure of MCR from Methanothermobacter thermoautotrophicum, solved to 1.45 Å resolution, and those from other organisms, solved to even higher resolutions (9, 10), revealed the presence of five modified amino acids in its α subunit, McrA, near the active site: 1-N-methylhistidine (MeHis), S-methylcysteine (MeCys), 5-C-(S)-methylarginine (MeArg), 2-C-(S)-methylglutamine (MeGln), and thioglycine (9). Since those initial discoveries, didehydroaspartate, 6-hydroxytryptophan, and 7-hydroxytryptophan have been observed in MCRs of other methanogenic or methanotrophic archaea (11, 12). Of these post-translational modifications, only 1-N-methylhistidine and thioglycine appear to be present in all MCRs analyzed to date.
Recently, genes that encode proteins responsible for two of these modifications have been identified by bioinformatics methods and complementation of select knockouts in genetically tractable organisms. Mass spectrometric characterization of McrA from WT Methanosarcina acetivorans and mutants thereof showed that the thioglycine modification requires the tfuA and/or ycaO gene products (13). Similar studies showed that the 5-C-(S)-methylarginine modification in McrAs from M. acetivorans (14) and Methanococcus maripaludis (15) requires the mmp10 gene, which encodes methanogenesis marker protein 10 (Mmp10).
Metabolic feeding studies indicate that the methyl groups of the MeHis, MeCys, MeArg, and MeGln modifications are all derived from S-adenosylmethionine (SAM) (16). For the MeHis and MeCys modifications, a typical SAM-dependent nucleophilic displacement is assumed, wherein the target nucleophilic atom attacks the activated methyl group of SAM in a polar Sn2 process. Contrarily, the methylation of C5 of Arg or C2 of Gln is less intuitive mechanistically, because the protons on these carbon centers are not sufficiently acidic to generate a carbon nucleophile. Nevertheless, in the absence of additional clues to these reactions, two mechanisms for the formation of MeArg and MeHis were proposed (16); however, they were not particularly satisfying with respect to mechanistic precedent.
The discovery of mmp10's role in MeArg formation is of interest, because Mmp10 is a member of the radical SAM (RS) superfamily of enzymes, which catalyze an amazing array of reactions via substrate-derived radical intermediates (17–19). RS enzymes require a [4Fe-4S]1+ cluster cofactor coordinated by three conserved cysteine residues in the RS domain—typically a full or partial triose-phosphate isomerase barrel—with SAM serving as the fourth ligand to the unique iron site. SAM is then cleaved reductively to methionine and a 5′-deoxyadenosyl 5′-radical (5′-dA•), although one exception to this paradigm has been recently noted (20, 21). Typically, 5′-dA• abstracts a substrate hydrogen atom, which initiates substrate-based catalysis.
One major subgroup of RS enzymes is the methylases, currently categorized into five classes (A, B, C, D, and E), with members of class B perhaps performing the most diverse chemistry (22, 23). Class B RS methylases use methylcobalamin (MeCbl) as the direct donor of the methyl group to the substrate and can methylate both sp2- and sp3-hybridized carbon centers as well as phosphinate phosphorus centers. One of the hallmarks of class B RS methylases is a cobalamin-binding domain located N-terminal to the RS domain. To date, no known deviations from this domain architecture have been reported.
Herein, we show that Mmp10 from M. acetivorans falls under a new subclass of class B RS methylases, which do not contain an N-terminal cobalamin-binding domain, yet still use cobalamin as the methyl donor in generating MeArg in McrA. We show that the M. acetivorans Mmp10 (MaMmp10) readily methylates the equivalent Arg285 of a peptide substrate surrogate, but only in the presence of cobalamin (Fig. 2). As expected, the products of the reaction are 5′-dAH, methionine (Met) S-adenosylhomocysteine (SAH), and the methylated peptide. In the presence of the required low-potential reductant, Ti(III) citrate, MaMmp10 exhibits a kcat of 1.87 min−1, which is comparable with that of other class B methylases that catalyze reactions via a 5′-dA• intermediate (24).
Figure 2.
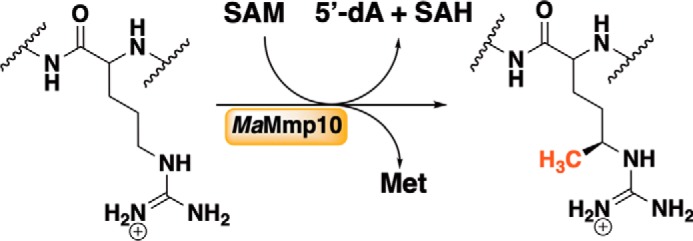
MaMmp10-catalyzed methylation of Arg285 in McrA.
Results
Overexpression, purification, and characterization of as-isolated MaMmp10
The M. acetivorans mmp10 gene was optimized for expression in Escherichia coli and chemically synthesized. The gene was cloned into pET-26b such that the encoded protein would contain a hexahistidine tag separated from its native C-terminal amino acid by a linker of two amino acids. The resulting plasmid, termed pMa4551, was used to transform E. coli harboring pDB1282, a plasmid that houses genes from Azotobacter vinelandii that are important in iron–sulfur (FeS) cluster biosynthesis (25, 26). The transformed bacteria were grown in M9 minimal media before inducing gene expression with isopropyl β-d-thiogalactopyranoside (IPTG) at a final concentration of 200 μm. The protein was purified by immobilized metal affinity chromatography in an anoxic environment due to the known oxygen sensitivity of iron–sulfur (FeS) clusters in RS enzymes. From 16 liters of E. coli cell culture, ∼150 mg of >90% pure protein is obtained (Fig. 3A).
Figure 3.

Characterization of as-purified MaMmp10. A, SDS-PAGE analysis of MaMmp10 purification by immobilized metal-affinity chromatography. Lane 1, molecular mass markers (kDa); lane 2, post-lysis pellet; lane 3, post-lysis supernatant; lane 4, Co-Talon® resin flow-through; lane 5, Co-Talon® start of wash; lane 6, Co-Talon® end of wash; lane 7, eluted MaMmp10. MaMmp10 is denoted by the red box at a theoretical molecular mass of 46.3 kDa. B, UV-visible spectrum of 19.5 μm as-isolated (Ai) MaMmp10 (red trace; A280/A410 = 3.33) and 10.2 μm as-purified (Rc) MaMmp10 (blue trace; A280/A410 = 2.57). C, as-purified MaMmp10 activity with time. 5′-dAH (red squares), SAH (blue squares), and methylated peptide product (black squares) are shown. Reactions were performed in triplicate at 30 °C in a final volume of 200 μl, and they contained the following components: 50 mm HEPES, pH 7.5, 200 mm KCl, 40 μm MaMmp10, 0.5 mm Ti(III) citrate, and 55 μm peptide substrate. Reactions were initiated with 0.5 mm SAM. Error bars indicate the standard deviation of three reactions.
Analysis of as-isolated MaMmp10 by UV-visible spectroscopy shows a spectrum that is characteristic of an FeS cluster-containing protein, exhibiting a broad feature centered at 410 nm (Fig. 3B, red trace). Upon chemical reconstitution of the FeS cluster and subsequent purification of MaMmp10 by size-exclusion chromatography on an ÄKTA LC system housed in an anaerobic chamber, a more prominent feature at 410 nm is observed (Fig. 3B, blue trace). Iron analysis of this reconstituted (Rc) protein reveals it to contain 5.09 ± 0.06 Fe ions per polypeptide after multiplying by a correction factor of 0.9328—obtained by amino acid analysis—for the Bradford method for protein quantification. This form of protein, in which only the FeS cluster has been reconstituted, is termed “as-purified.”
MaMmp10 requires cobalamin for its function
A 13-amino acid peptide (EMLPARRARGPNE) spanning residues 279–291 of the M. acetivorans McrA subunit (site 285 of methylation is shown in bold type) was synthesized by Think Peptides (www.thinkpeptides.com) and was used to assess the ability of as-purified MaMmp10 to methylate Arg285. The concentration of a stock solution of the peptide was determined by amino acid analysis. The calculated mass of the peptide is 1497.7 [M + H]+; however, the unmodified peptide is best detected in its +3 charge state, exhibiting an m/z = 499.9 [M + 3H]3+. In the presence of 0.5 mm SAM, 55 μm peptide substrate, and 0.5 mm Ti(III) citrate, 40 μm MaMmp10 displays very poor activity, generating less than 1 eq of SAH, 5′-dAH, and MeArg-containing peptide (m/z = 504.6 [M + 3H]3+) over a period of 90 min (Fig. 3C). Although 5′-dAH concentrations can sometimes be higher than the theoretical yield because of abortive cleavage of SAM (25, 27, 28), our studies of other RS methylases indicate that SAH and the methylated product are usually strongly coupled (24, 29–32)
The extremely-low activity of MaMmp10 suggested that a necessary component may be missing from the reaction mixtures. Given that cobalamin-dependent RS enzymes are well-known to methylate unactivated sp3-hybridized carbon centers, both MeCbl and hydroxocobalamin (OHCbl) were added to assess their effect on the MaMmp10 reaction, even though MaMmp10 is not annotated as a cobalamin-binding protein. As shown in Fig. 4, the addition of either MeCbl or OHCbl results in a dramatic increase in enzyme activity, as evidenced by the substantial time-dependent increase in SAH, 5′-dAH, and MeArg concentrations. The similar effects of MeCbl and OHCbl on the MaMmp10 reaction suggest that the enzyme readily converts OHCbl to MeCbl, an event that has been observed in other cobalamin-dependent RS enzymes (21, 24, 30).
Figure 4.
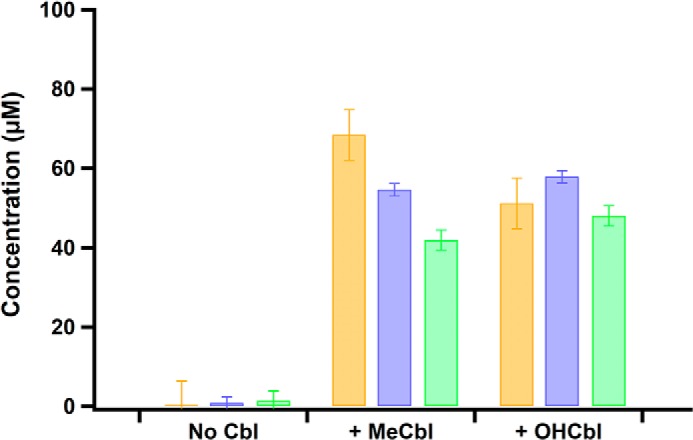
MaMmp10 activity is stimulated by cobalamin. Reaction products of as-purified MaMmp10 are in the presence of MeCbl or OHCbl. SAH (orange bars), 5′-dAH (purple bars), and methylated peptide product (green bars). Reactions were performed in triplicate at 30 °C in a final volume of 200 μl, and they contained the following components: 50 mm HEPES, pH 7.5, 200 mm KCl, 40 μm MaMmp10, 1 mm Ti(III) citrate, and 55 μm peptide substrate. Reactions were initiated with 1 mm SAM and quenched after 30 min. Error bars reflect standard deviation.
Given the effect of cobalamin on the MaMmp10 reaction, as well as the ability of as-purified MaMmp10 to catalyze low amounts of turnover in the absence of added cobalamin, the as-purified MaMmp10 was treated with acid and then analyzed by HPLC with analysis by multiple-reaction monitoring MS (MRM LC-MS) for its cobalamin content. As shown in Fig. S1, m/z values consistent with MeCbl, OHCbl, and adenosylcobalamin (AdoCbl) are observed. Similar to commercially available standards, the cobalamin species bound to MaMmp10 display m/z transitions that are diagnostic for MeCbl (673.0 → 665.0, [M + 2H]2+), OHCbl (664.9 → 635.8, [M + 2H]2+), and AdoCbl (790.6 → 665.6, [M + 2H]2+) and elute with comparable retention times. Quantification based on standards indicates that 1.32 μm AdoCbl, 0.223 μm OHCbl, and 0.193 μm MeCbl are associated with 1560 μm MaMmp10.
MaMmp10 was then reconstituted with its cobalamin cofactor by treating the protein with a 4-fold excess of OHCbl for 2 h before removing unbound molecules by gel-filtration chromatography using a buffer lacking cobalamin. This protein is referred to as cobalamin-reconstituted (Cbl-Rc) MaMmp10. A fraction of the eluted protein was heated with potassium cyanide to convert any bound cobalamin to dicyanocobalamin, which was subsequently quantified by UV-visible spectroscopy (ϵ367 nm = 30,800 m−1 cm−1). After gel filtration, MaMmp10 retains ∼0.8 cobalamins per polypeptide (Fig. 5A). Comparison of UV-visible spectra shows a new, broad feature between 300 and 600 nm that is indicative of cobalamin (Fig. 5B, black trace) (33).
Figure 5.

Characterization of cobalamin-reconstituted MaMmp10 by UV-visible spectroscopy. A, UV-visible spectrum of dicyanocobalamin after incubating MaMmp10 with 4-fold excess OHCbl and gel filtration. 10 μm as-purified (blue trace) or Cbl-Rc (black trace) MaMmp10 was treated with KCN to generate dicyanocobalamin. B, as-purified MaMmp10 (blue trace) and 5.4 μm Cbl-Rc MaMmp10 (black trace). As-purified MaMmp10 was taken from Fig. 3B and graphed for comparison.
MaMmp10 activity was re-assessed by incubating 50 μm Cbl-Rc or as-purified MaMmp10 with 1 mm SAM and 0.28 mm peptide under the reaction conditions described above, but in the absence of added cobalamin. As shown in Fig. S2 (blue squares), the reaction is completed within 50–60 min of incubation time. By contrast, as-purified MaMmp10 catalyzes formation of only ∼20 μm product (less than one turnover) after 120 min (Fig. S2, red squares).
Kinetic analysis of Cbl-Rc MaMmp10
The effect of cobalamin on MaMmp10 activity was substantiated by performing reactions in triplicate using lower enzyme concentrations (10 μm) and saturating concentrations of SAM (1 mm) and peptide substrate (450 μm) to allow the linear phase of the reaction to be captured. Under these conditions, Cbl-Rc MaMmp10 catalyzes the near stoichiometric formation of MeArg, SAH, and 5′-dAH with an apparent kcat value of ∼1.87 min−1 (Fig. 6A). Reactions were also conducted under conditions in which SAM at natural abundance was replaced with S-adenosyl-[methyl-2H3]methionine (d3-SAM) (Fig. 7A). In this instance, the mass of the methylated product increases by 3 mass units, consistent with a C2H3 group incorporated into the peptide with m/z = 505.6 ([M + 3H]3+) rather than 504.6 ([M + 3H]3+) for the peptide containing a CH3 group at natural abundance. This result is consistent with the transfer of an intact methyl moiety from d3-MeCbl and is inconsistent with mechanisms that involve hydrogen atom abstraction from the methyl moiety of SAM, as has been observed in the class A and class C methylases (29, 34, 35). Fig. S3 shows the formation of 5′-dAH (red squares), SAH (blue squares), and MeArg-containing peptide (black squares) in the absence of SAM (Fig. S3A), in the absence of Ti(III) citrate (Fig. S3B), or in the absence of MaMmp10 (Fig. S3C), indicating that each is required for turnover. In addition, turnover is severely diminished when Ti(III) citrate is replaced with dithionite (DDT) as the required low-potential reductant (Fig. 8).
Figure 6.

Activity of Cbl-Rc MaMmp10 and formation of MeCbl intermediate state. A, Cbl-Rc MaMmp10 activity over time (closed markers): 5′-dAH (red squares); SAH (blue squares); and methylated peptide product (black squares). As-purified MaMmp10 activity over time (open markers): 5′-dAH (red squares); SAH (blue squares); methylated peptide product (black squares). B, time-dependent formation of MeCbl and decay of OHCbl. OHCbl m/z transition 664.9 → 635.8 ([M + 2H]2+) (solid traces); MeCbl m/z transition 673.0 → 665.0 ([M + 2H]2+) (dotted traces). Reactions were performed in triplicate at 30 °C in a final volume of 160 μl and contained the following components: 50 mm HEPES, pH 7.5, 200 mm KCl, 10 μm MaMmp10, 1 mm Ti(III) citrate, and 450 μm peptide substrate. Reactions were initiated with 1 mm SAM. Error bars reflect the standard deviation.
Figure 7.

Activity of Cbl-Rc MaMmp10 in the presence of d3-SAM and formation of an MeCbl intermediate state. A, Cbl-Rc MaMmp10 activity with d3-SAM over time (closed markers): 5′-dAH (red squares); SAH (blue squares); d3-methylated peptide product (black squares). Activity of as-purified MaMmp10 (open markers): 5′-dAH (red squares); SAH (blue squares); d3-methylated peptide product (black squares). Data for as-purified MaMmp10 activity was taken from Fig. 6A and graphed for comparison. B, time-dependent formation of d3-MeCbl and decay of OHCbl. OHCbl m/z transition is 664.9 → 635.8 ([M + 2H]2+) (solid traces). d3-MeCbl m/z transition is 674.5 → 665.0 ([M + 2H]2+) (dotted traces). Reactions were performed in triplicate at 30 °C in a final volume of 160 μl and contained the following components: 50 mm HEPES, pH 7.5, 200 mm KCl, 10 μm MaMmp10, 1 mm Ti(III) citrate, and 280 μm peptide substrate. Reactions were initiated with 1 mm d3-SAM. Error bars reflect the standard deviation.
Figure 8.
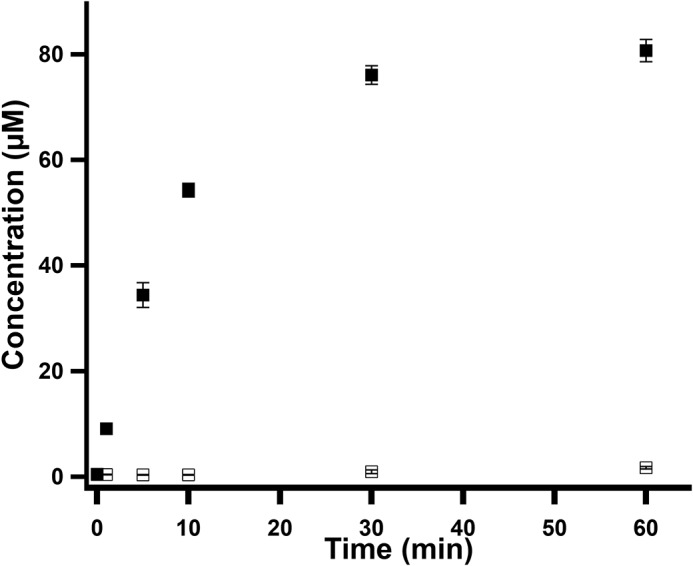
Formation of MeArg-containing peptide in the presence of dithionite or in the presence of Ti(III) citrate. Formation of the MeArg-containing peptide by Cbl-Rc MaMmp10 in the presence of DDT (open squares) or in the presence of Ti(III) citrate (closed squares) is shown. Reactions were performed in triplicate at 30 °C and contained the following components in a final volume of 140 μl: 50 mm HEPES, pH 7.5, 200 mm KCl, 10 μm MaMmp10, 0.5 mm Ti(III) citrate, or 0.5 mm DDT, and 400 μm peptide substrate. Reactions were initiated with 0.5 mm SAM. Error bars reflect the standard deviation.
Analysis of bound OHCbl, MeCbl, and d3-MeCbl under turnover conditions
To assess whether the methyl group from SAM is first transferred to cobalamin before the peptide substrate, the samples from reactions displayed in Figs. 6A and 7A were re-analyzed by LC-MS for the formation of MeCbl or d3-MeCbl, respectively. Given that MaMmp10 was reconstituted with OHCbl in this experiment, the enzyme should release OHCbl into solution upon denaturation with acid. Upon addition of SAM and reductant, OHCbl should be converted into cob(I)alamin, which can receive a methyl group from SAM to afford MeCbl. Therefore, a reduction in OHCbl and an increase in MeCbl should be observed. Fig. 6B shows m/z (+2 charge state) transitions and elution profiles that are characteristic of OHCbl (664.5 → 635, [M + 2H]2+; 3.77 min; solid lines) and MeCbl (673.0 → 665, [M + 2H]2+; 4.91 min; dotted lines), the two major species present during the reaction. At t = 0, cobalamin exists exclusively in its hydroxylated form. After 30 s of incubation time, there is a rapid decrease in the amount of OHCbl and a corresponding increase in the amount of MeCbl. Theoretically, the amount of OHCbl should continue to decrease until all cobalamin exists as MeCbl; however, the lack of complete decay of OHCbl suggests that some of the cofactor does not participate in catalysis.
Samples from Fig. 7A were similarly re-examined to provide evidence that the methyl group is transferred intact from SAM to cobalamin. The samples were monitored for ions with a mass transition of 674.5 → 665.0 ([M + 2H]2+), which would be consistent with a +3.0 mass increase indicative of d3-MeCbl. As detailed above, natural abundance MeCbl exhibits a mass transition of (673.0 → 665, [M + 2H]2+). Fig. 7B shows elution profiles and formation/decay patterns for d3-MeCbl and OHCbl, respectively. These patterns are similar to those observed in the presence of SAM at natural abundance (Fig. 6B). The major cobalamin species exhibit m/z transitions characteristic of d3-MeCbl formation and concomitant OHCbl decay, showing that a methyl group is transferred intact from SAM to cobalamin to yield d3-MeCbl.
Methylcobalamin is an intermediate methyl acceptor in the MaMmp10 reaction
To generate MaMmp10 containing bound MeCbl, the protein was incubated with 5-fold excess SAM and Ti(III) citrate for 30 min and then exchanged into buffer that did not contain SAM or Ti(III) citrate by gel-filtration chromatography. A 40 μm sample of “pre-methylated” MaMmp10 was combined with Ti(III) citrate and the McrA peptide substrate before initiating the reaction with either SAM at natural abundance (NA SAM) or d3-SAM. Fig. 9 shows the results of MaMmp10 turnover using NA SAM (Fig. 9, A and C) or d3-SAM (Fig. 9, B and D) as the co-substrate. Approximately 7.5 μm (∼25%) of bound cobalamin exists as OHCbl (Fig. 9, A and B, crosses), and this percentage does not change significantly throughout the reaction, suggesting that this fraction of MaMmp10 is unreactive or poorly active. Fig. 9A shows that ∼30 μm MaMmp10 is in the pre-methylated state at t = 0 (closed squares) and that the concentration of this form of the enzyme stays relatively consistent throughout the reaction. As-expected, no d3-MeCbl (Fig. 9A, open squares) is observed, given that the experiment was conducted with NA SAM. Accordingly, when the exact same reaction is monitored for product formation, only the MeArg-containing peptide at natural abundance (Fig. 9C, closed circles) is observed during the 10-min incubation time.
Figure 9.

Monitoring cobalamin and product formation under pre-methylation conditions. Observation of cobalamin species and product generated by pre-methylated MaMmp10 using SAM or d3-SAM as co-substrates is shown. A and C, quantification of cobalamin species and product formation using SAM as co-substrate. B and D, quantification of cobalamin species and product formation using d3-SAM as co-substrate. A and B, OHCbl (crosses), MeCbl (closed squares), d3-MeCbl (open squares). C and D, MeArg product (closed circles) and d3-MeArg product (open circles). Reactions were performed in triplicate at 30 °C and contained the following in a final volume of 180 μl: 50 mm HEPES, pH 7.5, 200 mm KCl, 40 μm pre-methylated MaMmp 10, 1 mm Ti(III) citrate, and 400 μm peptide substrate. Reactions were initiated with 1 mm SAM or 1 mm d3-SAM. Error bars denote the standard deviation in the data.
When the pre-methylated MaMmp10 was incubated with d3-SAM (Fig. 9, B and D), rapid formation of 22 μm d3-MeCbl (Fig. 9B, open squares) takes place concomitant with a near-equivalent concentration of NA MeCbl lost over 10 min. Approximately 8 μm cobalamin is not converted into MeCbl, which again may reflect unreactive or poorly-reactive enzyme. When formation of the MeArg-containing peptide is monitored, a burst of the species containing a methyl group at natural abundance is observed, consistent with the form of pre-methylated MaMmp10 used in the reaction (Fig. 9D). The magnitude of the burst is similar in concentration to the starting concentration of pre-methylated MaMmp10 (∼30 μm). This burst is accompanied by an observed lag in d3-MeArg formation, which occurs because NA MeCbl must be consumed before d3-SAM can donate a methyl group to cobalamin to generate d3-MeCbl. d3-MeCbl is then used to generated the d3-MeArg–containing peptide product in all subsequent turnovers. Taken together, Fig. 9 supports the use of cobalamin as an intermediate methyl acceptor in the formation of the MeArg-containing McrA peptide.
Verification of Arg285 as the site of methylation of the peptide substrate analog
To verify that the methyl group is indeed transferred to Arg285 of the peptide substrate, an MaMmp10 reaction was analyzed by high-resolution MS on a Brüker Ultraflextreme MALDI TOF–TOF instrument. Shown in Fig. 10 are the b and y ion series for the unmodified peptide substrate (Fig. 10A) and the methylated peptide product (Fig. 10B) produced by fragmentation of the parent peptide. The y7+ ion for the unmodified peptide displays m/z of 799.42 ([M + H]1+), corresponding to the peptide fragment 285RARGPNE. By contrast, the y7+ ion for the methylated peptide is 14.01 mass units greater, corresponding to the removal of a hydrogen from the substrate (−1) and the addition of a methyl group (+15). Similarly, the b8+ ion for the unmodified peptide displays m/z of 925.50 ([M + H]1+), corresponding to the peptide fragment EMLPARRA (where R indicates Arg285), whereas that of the methylated peptide is 14.02 mass units greater. Importantly, the y6+ ion of the methylated peptide matches that of its theoretical mass based on its sequence, whereas the y7+ ion and those beyond are ∼14.00 mass units greater than they should be based on their sequences. Similarly, the b6+ ion of the methylated peptide matches that of its theoretical mass based on its sequence, whereas the b7+ and b8+ ions and those beyond are ∼14.00 mass units greater than they should be based on their sequences. Taken together, the data show unambiguously that the methyl group is appended to Arg285.
Figure 10.

MALDI TOF–TOF analysis of peptide substrate and methylated peptide product. The 13-amino acid peptide sequence and the b series (orange) and y series (light blue) peptide fragment ions for unmethylated (control) and methylated peptide product are shown. A, unmodified peptide substrate m/z values: y5 = 572.28; y6 = 643.32; y7 = 799.42; y8 = 955.52; y9-NH3 = 1009.53; y10 = 1123.61; b6 = 698.37; b7 = 854.47; b8 = 925.50; b9 = 1081.60; b10 = 1138.63. B, methylated peptide product m/z values: y5 = 572.28; y6 = 643.32; y = 813.43; y8 = 969.53; y9-NH3 = 1023.54; y10 = 1137.62; b6 = 698.37; b7 = 868.48; b8 = 939.52; b9 = 1095.62.
Discussion
MaMmp10 catalyzes the methylation of Arg285 in the α subunit of MCR. Although the absence of MeArg does not drastically influence the activity of MCR, it has been found to increase the stability of the enzyme at elevated temperatures (13). An analysis of the primary structure of MaMmp10 strongly supports its membership in the RS superfamily of enzymes, and in vitro characterization of the isolated enzyme, reported herein, is consistent with this assignment. However, recombinantly expressed and anaerobically purified MaMmp10 methylates a 13-amino acid peptide mimic of McrA poorly. Less than 0.1% of various cobalamin species are bound to MaMmp10, most likely deriving from the E. coli expression host scavenging residual cobalamin from the growth vessels or from the LB starter culture, given that E. coli does not synthesize cobalamin de novo. Because of this poor activity, OHCbl or MeCbl was added to MaMmp10 reactions. When incubated with cobalamin and then purified by size-exclusion chromatography, MaMmp10 retains 0.8 cobalamins per polypeptide. Partial cofactor incorporation could be due to the use of hydroxocobalamin in the reconstitution procedure instead of the native cofactor, Factor III, which contains a 5-hydroxybenzimidazolyl group instead of the typical dimethylbenzimidazole group (Fig. 11) (36, 37). This form of cobalamin is unique to methanogenic archaea (38). Nonetheless, in the presence of cobalamin or with MaMmp10 reconstituted with cobalamin, the enzyme can perform multiple turnovers, exhibiting a kcat value of 1.87 min−1, which is similar to that observed for Fom3 using Ti(III) citrate as a reductant (0.78 min−1) (24). Experiments using d3-SAM support the use of cobalamin as an intermediate methyl carrier in the reaction, accepting an intact methyl group from SAM before transferring it intact to C5 of Arg285.
Figure 11.

Cobalamin and Factor III structures.
Although MaMmp10 is not annotated as a cobalamin-binding protein, the use of this cofactor as a methyl donor in RS reactions is well-precedented (21, 24, 39–44). In-depth bioinformatic analysis of Mmp10s from a variety of organisms does not reveal significant homology to any cluster of characterized RS enzymes and does not suggest any connection with known cobalamin-dependent RS enzymes outside of the homologs that likely perform the same function. In fact, all other cobalamin-dependent RS enzymes have a cobalamin-binding motif N-terminal to the RS motif, whereas MaMmp10's RS motif is at the extreme N terminus of the protein. A sequence similarity network (SSN) of Mmp10s was made through a BLAST analysis of the MaMmp10 amino acid sequence using the enzyme similarity tool on the Enzyme Function Initiative webserver (45). The sequences fell into four major clusters, with several proteins falling outside of these clusters. Although they have been shown in vivo to catalyze the same reaction, MaMmp10 and M. maripaludis Mmp10 (MmMmp10) are located in two different clusters (Fig. 12) (15).
Figure 12.
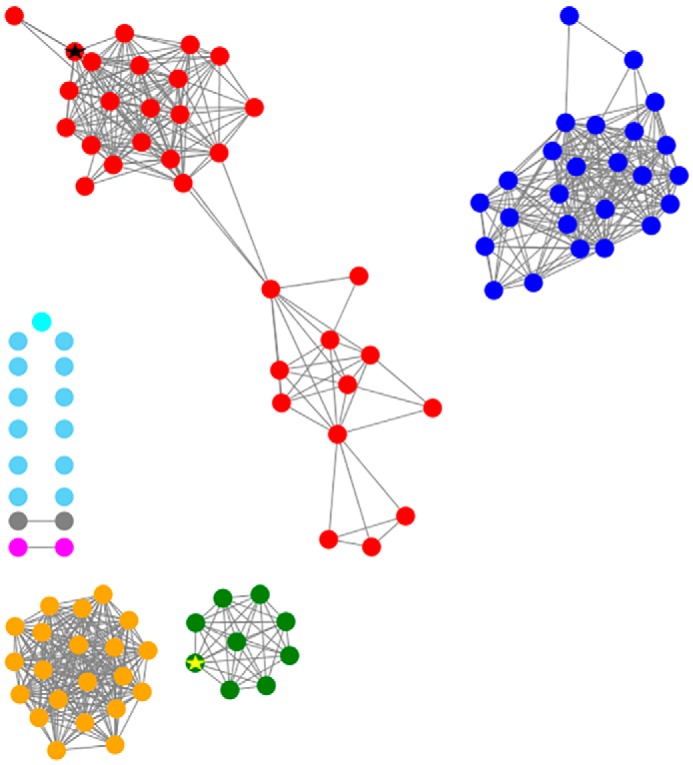
Sequence similarity network for the arginine methylase MaMmp10. Sequences are connected by edges if E <e-150. The results demonstrate that there are four major clusters out of seven, with several proteins not belonging to a specific cluster (light blue). The red cluster represents those sequences that are closely related to MaMmp10 (indicated by a black star), and the green cluster represents the sequences that are related to MmMmp10 (indicated by a yellow star).
A multiple-sequence alignment of MaMmp10 with other known cobalamin-dependent RS enzymes (Fig. S4) does not show significant homology, exhibiting only 15% sequence identity to OxsB, a known cobalamin-dependent RS enzyme that has recently had its X-ray crystal structure determined (46). The alignment shows the known and predicted cobalamin domains of several annotated cobalamin-dependent enzymes, but the domain is clearly not present in MaMmp10. Even the RS motif (CX3CX2C) is not conserved among them (Fig. S4). This analysis suggests that the cobalamin-binding domain in MaMmp10 is located C-terminal to the RS motif, which is a significant finding.
In validating the annotation of MaMmp10 across multiple databases, we discovered one annotation from Integrated Microbial Genomes and Microbiomes (IMG/M) as containing a DUF512 domain (47). DUF512 is a domain of unknown function (DUF) that is typically located C-terminal to an RS domain. However, although the DUF512 domain is annotated in the IMG/M database, MaMmp10 is not present in the proteins that have been grouped into the DUF512-containing family by the InterPro or Pfam databases (48, 49). Additionally, none of the DUF512 proteins have been characterized, so the function of this domain is unknown.
An alignment of MaMmp10 with the proteins curated into the DUF512 domain by InterPro (48) does show a region of homology that is located C-terminal to the RS domain (Fig. S5). The results of the analysis do not confirm that the DUF512 domain is present in Mmp10, but it could represent a possible domain for cobalamin binding, which would expand the subfamily of cobalamin-dependent RS enzymes.
In conclusion, we show that MaMmp10 is a new member of class B RS methylases, using MeCbl to methylate Arg285 of McrA. Initially, the methyl group derives from SAM, with cobalamin acting as an intermediate carrier. Furthermore, we show that MaMmp10 is the first reported cobalamin-dependent RS enzyme that contains a C-terminal cobalamin-binding domain. This new subclass of class B RS methylases is currently made up of proteins only from methanogenic archaea and can be further differentiated into several subclusters (Fig. 12). However, it is likely that they are all involved in the methylation of the arginine residue of MCR. This discovery highlights that Nature's use of cobalamin by RS enzymes is more abundant than previously believed.
Experimental procedures
Chemicals
Enzymes and other reagents for cloning and nucleic acid manipulation were obtained from New England Biolabs (Ipswich, MA). Sodium sulfide (Na2S), SAH, 5′-dAH, lysozyme, β-mercaptoethanol, OHCbl, MeCbl, and AdoCbl were purchased from Sigma. Ferric chloride was obtained from EMD Biosciences (Gibbstown, NJ). Bradford reagent for protein concentration determination as well as the bovine serum albumin (BSA) standard was purchased from Pierce, Thermo Fisher Scientific (Rockford, IL). Dithiothreitol (DTT), l-(+)-arabinose, IPTG, ampicillin, and kanamycin were purchased from Gold Biotechnology (St. Louis, MO). SAM was synthesized from ATP and l-methionine and purified as described previously (50). Ti(III) citrate was prepared as described previously (51). All other materials and reagents have been previously reported (21, 24, 30) or were of the highest available quality.
General methods and instrumentation
DNA sequencing was performed at the Huck Life Sciences Genomics Core Facility at Pennsylvania State University, University Park. Amino acid analysis was performed at the University of California Davis Proteomics Core Facility. UV-visible spectra were recorded on a Cary 50 spectrometer from Varian (Walnut Creek, CA) using the associated WinUV software package. All anaerobic manipulations were conducted in a Coy anaerobic chamber (Grass Lakes, MI) filled with 95% N2 and 5% H2 gases and containing palladium catalysts to maintain the level of oxygen at <1 ppm. HPLC with detection by MS (LC-MS) was conducted using an Agilent Technologies (Santa Clara, CA) 1200 system coupled to an Agilent Technologies 6410 QQQ mass spectrometer. Data collection and analysis were performed using the associated MassHunter software. Size-exclusion chromatography was performed on an ÄKTA system (GE Healthcare) housed in a Coy anaerobic chamber and equipped with a HiPrep 26/60 Sephacryl HR S-200 column (GE Healthcare). SDS-PAGE was performed using a mini-vertical electrophoresis unit from Hoefer (Holliston, MA).
Construction of pMa4551
The gene encoding M. acetivorans Mmp10 was optimized for expression in E. coli and chemically synthesized by Invitrogen GeneArt Gene synthesis (Fig. S6). Mmp10 was designed to contain NdeI and XhoI restriction sites at the 5′ and 3′ termini, respectively. The gene was removed from the GeneArt pMA-T vector using the appropriate restriction enzymes and was ligated into linearized pET26b plasmid using T4 DNA ligase. The resulting plasmid was designated pMa4551. The correct sequence of pMa4551 was verified at the Pennsylvania State Genomics Core Facility.
Overexpression and purification of MaMmp10
pMa4551 was used to transform an E. coli BL-21 (DE3) strain harboring plasmid pDB1282, which contains genes for iron–sulfur (FeS) cluster assembly and trafficking from A. vinelandii (26). Expression of MaMmp10 was conducted using previously described protocols (52), and the resulting protein was purified in a Coy Laboratories anaerobic chamber under a 95% N2 and 5% H2 atmosphere as described previously (52).
A single colony of E. coli BL-21 (DE3) harboring pMa4551 and pDB1282 was used to inoculate 5 ml of LB media starter culture. After 12 h of agitation at 37 °C and 250 rpm, 400 μl of culture was added individually to 4 flasks of 4 liters of M9 minimal media, pH 7.4, supplemented with 50 μg/ml kanamycin and 100 μg/ml ampicillin. At A600 = 0.3, 25 μm FeCl3 was added, and expression of the isc operon was induced with 0.2% arabinose. At A600 = 0.6, 25 μm FeCl3 was added to the media, and the flasks were placed in an ice bath for 40 min. The expression of mmp10 was then induced upon addition of a final concentration of 200 μm isopropyl β-d-1-thiogalactopyranoside. Cells were harvested in a Beckman Coulter Avanti J-26 XP centrifuge at 7000 × g. Approximately 40 g of cells was obtained, which were flash-frozen in liquid nitrogen and stored in a −80 °C ultralow freezer until further use.
Frozen cells were brought into a Coy Laboratories anaerobic chamber and resuspended in “lysis buffer” containing 50 mm HEPES, pH 7.5, 300 mm KCl, 10% w/v glycerol, 10 mm β-mercaptoethanol (BME), and 4 mm imidazole, pH 8.0. After 15 min of stirring, 1 mg/ml lysozyme, 0.1% Triton X-100, 1 protease inhibitor tablet (Sigma), and 0.1 mg/ml DNase were added to the buffer. The solution was allowed to stir for another 20 min or until the cells had completely resuspended. Cell lysis by sonic disruption was preformed using a Fisher Scientific Model 550 sonic dismembrator at 30–40% output. The lysate was subjected to six 45-s bursts and then distributed into Nalgene high-speed centrifuge tubes. The tubes were capped, further sealed with vinyl tape, and brought out of the glovebox. The lysate was clarified by centrifugation in a Beckman Coulter Avanti J-30I centrifuge for 1 h at 4 °C. Clarified lysate was brought back into the anaerobic chamber and loaded onto a 10-ml cobalt–Talon® resin (Clontech) column. The lysate was placed on ice while loading it onto the resin. After loading, the resin was washed with 10 column volumes of cold lysis buffer. The purified MaMmp10 was eluted with buffer containing 50 mm HEPES, pH 7.5, 300 mm KCl, 10% glycerol, and 10 mm BME, and ∼20 ml of protein was collected. The eluate was loaded into Ultra-15 centrifugal units (Merck Millipore Ltd.) with a 10-kDa molecular mass cutoff, and the units were capped, sealed with vinyl tape, and brought out of the anaerobic chamber to concentrate. Upon reducing the protein volume to less than 2.5 ml, the centrifugal units were brought back into the chamber and loaded onto a PD-10 desalting column (GE Biosciences) pre-equilibrated with 15 ml of “storage buffer” containing 50 mm HEPES, pH 7.5, 500 mm KCl, 10% glycerol, and 5 mm DTT. The protein was eluted with storage buffer, aliquoted, and flash-frozen in liquid nitrogen.
Chemical reconstitution of [4Fe-4S] cluster and cobalamin
All steps were performed in an anaerobic chamber unless noted otherwise. Solid DTT was added to a specific volume of storage buffer to a final concentration of 2.5 mm. 250 μm FeCl3 was added to the buffer solution, which was incubated on ice for 5 min before adding MaMmp10 to a final concentration of 50 μm. The protein solution was gently stirred for 20 min on ice. After 20 min, 250 μm Na2S was added incrementally over the span of 2 h. The protein solution was left to incubate on ice overnight in the anaerobic chamber. The following morning, the MaMmp10 was loaded into Ultra-15 centrifugal units, sealed using vinyl tape, and brought out of the chamber to concentrate. The concentrated protein was brought back into the chamber and purified further using an ÄKTA 10 system equipped with a Sephadex S-200 size-exclusion column. The migration of MaMmp10 on the column was monitored using a variable wavelength detector at wavelengths 280 and 410 nm. Fractions corresponding to a peak with significant 410 nm absorbance, excluding aggregated protein (eluting at or near the void-volume of the column), were pooled, concentrated outside of the glovebox using appropriate centrifugal units, brought back into the chamber, and flash-frozen in liquid nitrogen. The resulting protein is labeled as-purified.
As-purified MaMmp10 was incubated with ∼2- or 4-fold excess hydroxocobalamin for 2 h on ice to reconstitute it with its cobalamin cofactor. The protein solution was loaded onto a PD-10 desalting column pre-equilibrated with 15 ml of storage buffer lacking any cobalamin. The band corresponding to MaMmp10 was collected, concentrated, and either immediately assayed or flash-frozen in nitrogen and stored in liquid nitrogen.
Determination of MaMmp10 correction factor and McrA 13-mer peptide concentration
The preparation of samples for amino acid analysis has already been described (52). Lyophilized MaMmp10 and McrA 13-mer peptides were sent to the Molecular Structure Facility at the University of California Davis, where amino acid analysis was conducted on a fee for service basis.
MaMmp10 activity assays
Standard reactions were performed in triplicate and contained 50 mm HEPES, pH 7.5, 200 mm KCl, varying concentrations of MaMmp10, Ti(III) citrate, and peptide substrate. Solutions were heated at 30 °C for 10 min before initiating the reaction by addition of SAM to a final concentration of 0.5 or 1 mm. Reactions were allowed to proceed before being quenched in a solution of 100 mm H2SO4 and 100 μm tryptophan, which was used as an internal standard.
LC-MS–based quantification and analysis
An Agilent Technologies 1200 Series HPLC system coupled to a 6410 QQQ electrospray–ionization mass spectrometer was used to detect and quantify the predicted reaction products, SAH, 5′-dAH, and the MeArg-containing peptide, as well as the internal standard tryptophan. Tryptophan, SAH, and 5′-dAH were detected by MRM in positive mode. Fragmentor voltages of 100, 90, and 130 were set to monitor ion transitions indicative of SAH (384.5 → 136.1 ([M + H]1+) and 384.5 → 134.0 ([M + H]1+)), 5′-dAH (252.1 → 136.0 ([M + H]1+) and 252.1 → 119.0 ([M + H]1+)), and tryptophan (188.0 → 146.1 ([M + H]1+) and 188.0 → 118.0 ([M + H]1+)), respectively. Nonmethylated and methylated peptides were detected and quantified separately using single-ion monitoring in positive mode with tryptophan being used as a standard. The nonmethylated peptide was best detected at a fragmentor voltage of 160 and in the +3 charge state with an m/z of 499.9 ([M + 3H]3+), and the methylated peptide was best detected at an m/z of 504.6 ([M + 3H]3+). Tryptophan was detected at an m/z of 188 ([M + H]1+) at a fragmentor voltage of 130.
Analyte concentrations were calculated using Agilent MassHunter Quantitative Software Version B.7.00. Calibration curves were generated using known concentrations of SAH and 5′-dAH. To quantify the MeArg-containing peptide, a calibration curve of known concentrations of unmethylated peptide was created and used in the detection of m/z = 504.6 ([M + 3H]3+) ions, with the assumption that the methylated and nonmethylated peptides fly similarly in the mass spectrometer.
Determination of the site of methylation by MALDI TOF–TOF MS
To determine the precise site of methylation on the peptide substrate mimic, reaction mixtures were analyzed on a Bruker (Billerica, MA) Ultraflextreme MALDI TOF–TOF instrument equipped with a 355-nm frequency-tripled NdYAG smartbeam-II laser. The mass spectra were acquired using a factory-configured instrument method for the reflector positive-ion detection over the 700–3500 m/z range. Laser power attenuation and pulsed ion extraction times were optimized to achieve the best signal-to-noise ratio. The instrument was calibrated with a BSA tryptic peptide mixture (Protea Biosciences, part no. PS-204-1). Mass spectra were opened in FlexAnalysis (Bruker), smoothed (SavitzkyGolay, 0.2 m/z, 1 cycle), and baseline-subtracted (TopHat), and the mass lists were generated using a Snap peak detection algorithm with the signal–to–noise threshold set at six and using the Averagine SNAP average composition.
The peaks at m/z 1496.8 ([M + H]1+) and 1510.8 ([M + H]1+) were selected for TOF–TOF analysis to establish the position of modification. The resulting MS2 data were processed using the default parameters and interpreted manually. Fragment mass lists were generated using Protein Prospector (http://prospector.ucsf.edu/prospector/cgi-bin/msform.cgi?form=msproduct),3 and the fragment ions were identified based on the best match between the experimental and predicted values.
Bioinformatic analysis of MaMmp10
The MaMmp10 sequence similarity network (SSN) was prepared by a BLAST (53) analysis of the MaMmp10 amino acid sequence (UniProt accession number Q8THG6) on the Enzyme Function Initiative (EFI) website (https://enzymefunction.org) using the enzyme similarity tool (Fig. 10) (45). An e-value of −150 was chosen and resubmitted to the servers. Next, a sequence identity of 70% was chosen for generation of the colored SSN (45). Both SSNs were visualized and manipulated in Cytoscape version 3.5.1 (54).
All of the alignment figures were generated in the following manner. The FASTA sequences were taken from Uniprot (55) and aligned using Clustal Omega (56). The aligned sequences were copied into Microsoft Word® where they were colored manually. The alignment of known cobalamin-dependent proteins (Fig. S4) was generated by including RS enzymes that have been confirmed to use cobalamin. OxsB was used as a reference for the alignment, because it is the only cobalamin-dependent RS enzyme to have its three-dimensional structure determined (46). The alignment of MaMmp10 with representative DUF512-containing proteins (Fig. S5) was constructed by aligning the MaMmp10 and MmMmp10 amino acid sequences against 20 randomly selected DUF512 family of proteins annotated by InterPro, out of a total of 3027 DUF512-containing proteins (48). The highlighting of the PDZ, aldolase-type TIM barrel, and DUF512 domains was approximated based on the predicted ranges from InterPro annotations (48). The percent conservation to decide highlighting was based on all 22 out of 22 sequences containing the particular amino acid for 100% conservation; greater than 19 out of 22 for greater than 85% conservation, and 6 out of 22 for greater than 25% conservation. Hydrophobic, charged, and serine/threonine amino acids were allowed to substitute for each other when conservation less than 100% was being considered.
Author contributions
M. I. R., D. V. M., and S. J. B. conceptualization; M. I. R., D. V. M., T. N. L., and S. J. B. formal analysis; M. I. R. and D. V. M. investigation; M. I. R., D. V. M., and S. J. B. writing-original draft; M. I. R., D. V. M., and S. J. B. writing-review and editing; T. N. L. data curation; T. N. L. software; T. N. L. methodology; S. J. B. supervision; S. J. B. funding acquisition; S. J. B. project administration.
Supplementary Material
Acknowledgment
We thank John Schulze at the Molecular Structure Facility at University of California Davis for amino acid analyses.
This work was supported by NIH GM-122595 (to S. J. B.), the Eberly Family Distinguished Chair in Science (S. J. B.), and The Pennsylvania State University Huck Institute for Life Sciences (T. N. L.). S. J. B is an investigator of the Howard Hughes Medical Institute. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
This article was selected as one of our Editors' Picks.
This article contains Figs. S1–S6 and supporting Refs. 1–3.
Please note that the JBC is not responsible for the long-term archiving and maintenance of this site or any other third party hosted site.
- MCR
- methyl coenzyme M reductase
- 5′-dA•
- 5′-deoxyadenosyl 5′-radical
- 5′-dAH
- 5′-deoxyadenosine
- AdoCbl
- adenosylcobalamin
- DUF
- domain of unknown function
- FeS
- iron–sulfur
- LC-MS
- high-performance liquid chromatography with detection by mass spectrometry
- MeCbl
- methylcobalamin
- Mmp10
- methanogenesis marker protein 10
- MaMmp10
- Mmp10 from M. acetivorans
- MmMm10
- Mmp10 from M. maripaludis
- OHCbl
- hydroxocobalamin
- RS
- radical SAM
- SAH
- S-adenosylhomocysteine
- SAM
- S-adenosylmethionine
- SSN
- sequence similarity network
- IPTG
- isopropyl β-d-thiogalactopyranoside
- NA
- natural abundance
- IMG/M
- Integrated Microbial Genomes and Microbiomes
- MRM
- multiple-reaction monitoring
- Rc
- reconstituted
- BME
- β-mercaptoethanol
- DDT
- dithionite.
References
- 1. Ellefson W. L., and Wolfe R. S. (1981) Component C of the methylreductase system of Methanobacterium. J. Biol. Chem. 256, 4259–4262 [PubMed] [Google Scholar]
- 2. Gunsalus R. P., and Wolfe R. S. (1978) Chromophoric factors F342 and F430 of Methanobacterium thermoautotrophicum. FEMS Microbiol. Lett. 3, 191–193 10.1111/j.1574-6968.1978.tb01916.x [DOI] [Google Scholar]
- 3. Diekert G., Jaenchen R., and Thauer R. K. (1980) Biosynthetic evidence for a nickel tetrapyrrole structure of factor F430 from Methanobacterium thermoautotrophicum. FEBS Lett. 119, 118–120 10.1016/0014-5793(80)81011-8 [DOI] [PubMed] [Google Scholar]
- 4. Ellefson W. L., Whitman W. B., and Wolfe R. S. (1982) Nickel-containing factor F430: chromophore of the methylreductase of Methanobacterium. Proc. Natl. Acad. Sci. U.S.A. 79, 3707–3710 10.1073/pnas.79.12.3707 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Neue H.-U. (1993) Methane emission from rice fields. Bioscience 43, 466–474 10.2307/1311906 [DOI] [Google Scholar]
- 6. Miller S. M., Wofsy S. C., Michalak A. M., Kort E. A., Andrews A. E., Biraud S. C., Dlugokencky E. J., Eluszkiewicz J., Fischer M. L., Janssens-Maenhout G., Miller B. R., Miller J. B., Montzka S. A., Nehrkorn T., and Sweeney C. (2013) Anthropogenic emissions of methane in the United States. Proc. Natl. Acad. Sci. U.S.A. 110, 20018–20022 10.1073/pnas.1314392110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Shima S., and Thauer R. K. (2005) Methyl-coenzyme M reductase and the anaerobic oxidation of methane in methanotrophic Archaea. Curr. Opin. Microbiol. 8, 643–648 10.1016/j.mib.2005.10.002 [DOI] [PubMed] [Google Scholar]
- 8. Wongnate T., Sliwa D., Ginovska B., Smith D., Wolf M. W., Lehnert N., Raugei S., and Ragsdale S. W. (2016) The radical mechanism of biological methane synthesis by methyl-coenzyme M reductase. Science 352, 953–958 10.1126/science.aaf0616 [DOI] [PubMed] [Google Scholar]
- 9. Ermler U., Grabarse W., Shima S., Goubeaud M., and Thauer R. K. (1997) Crystal structure of methyl-coenzyme M reductase: the key enzyme of biological methane formation. Science 278, 1457–1462 10.1126/science.278.5342.1457 [DOI] [PubMed] [Google Scholar]
- 10. Grabarse W., Mahlert F., Shima S., Thauer R. K., and Ermler U. (2000) Comparison of three methyl-coenzyme M reductases from phylogenetically distant organisms: unusual amino acid modification, conservation and adaptation1. J. Mol. Biol. 303, 329–344 10.1006/jmbi.2000.4136 [DOI] [PubMed] [Google Scholar]
- 11. Wagner T., Wegner C.-E., Kahnt J., Ermler U., and Shima S. (2017) Phylogenetic and structural comparisons of the three types of methyl-coenzyme M reductase from Methanococcales. J. Bacteriol. 199, e00197–17 10.1128/JB.00197-17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Wagner T., Kahnt J., Ermler U., and Shima S. (2016) Didehydroaspartate modification in methyl-coenzyme M reductase catalyzing methane formation. Angew. Chem. Int. Ed. Engl. 55, 10630–10633 10.1002/anie.201603882 [DOI] [PubMed] [Google Scholar]
- 13. Nayak D. D., Mahanta N., Mitchell D. A., and Metcalf W. W. (2017) Post-translational thioamidation of methyl-coenzyme M reductase, a key enzyme in methanogenic and methanotrophic Archaea. Elife 6, e29218 10.7554/eLife.29218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Deobald D., Adrian L., Schöne C., Rother M., and Layer G. (2018) Identification of a unique radical SAM methyltransferase required for the sp3-C-methylation of an arginine residue of methyl-coenzyme M reductase. Sci. Rep. 8, 7404 10.1038/s41598-018-25716-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Lyu Z., Chou C., Hao S., Patel R., Duin E. C., and Whitman W. B. (2017) Mmp10 is required for post-translational methylation of arginine at the active site of methyl-coenzyme M reductase. bioRxiv 10.1101/211441 [DOI] [Google Scholar]
- 16. Selmer T., Kahnt J., Goubeaud M., Shima S., Grabarse W., Ermler U., and Thauer R. K. (2000) The biosynthesis of methylated amino acids in the active site region of methyl-coenzyme M reductase. J. Biol. Chem. 275, 3755–3760 10.1074/jbc.275.6.3755 [DOI] [PubMed] [Google Scholar]
- 17. Frey P. A., and Booker S. (1999) Radical intermediates in the reaction of lysine 2, 3-aminomutase. Adv. Free Radic. Chem. 2, 1–43 10.1016/S1874-5237(99)80003-8 [DOI] [Google Scholar]
- 18. Sofia H. J., Chen G., Hetzler B. G., Reyes-Spindola J. F., and Miller N. E. (2001) Radical SAM, a novel protein superfamily linking unresolved steps in familiar biosynthetic pathways with radical mechanisms: functional characterization using new analysis and information visualization methods. Nucleic Acids Res. 29, 1097–1106 10.1093/nar/29.5.1097 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Landgraf B. J., McCarthy E. L., and Booker S. J. (2016) Radical S-adenosylmethionine enzymes in human health and disease. Annu. Rev. Biochem. 85, 485–514 10.1146/annurev-biochem-060713-035504 [DOI] [PubMed] [Google Scholar]
- 20. Pierre S., Guillot A., Benjdia A., Sandström C., Langella P., and Berteau O. (2012) Thiostrepton tryptophan methyltransferase expands the chemistry of radical SAM enzymes. Nat. Chem. Biol. 8, 957–959 10.1038/nchembio.1091 [DOI] [PubMed] [Google Scholar]
- 21. Blaszczyk A. J., Silakov A., Zhang B., Maiocco S. J., Lanz N. D., Kelly W. L., Elliott S. J., Krebs C., and Booker S. J. (2016) Spectroscopic and electrochemical characterization of the iron–sulfur and cobalamin cofactors of TsrM, an unusual radical S-adenosylmethionine methylase. J. Am. Chem. Soc. 138, 3416–3426 10.1021/jacs.5b12592 [DOI] [PubMed] [Google Scholar]
- 22. Zhang Q., van der Donk W. A., and Liu W. (2012) Radical-mediated enzymatic methylation: a tale of two SAMS. Acc. Chem. Res. 45, 555–564 10.1021/ar200202c [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Bauerle M. R., Schwalm E. L., and Booker S. J. (2015) Mechanistic diversity of radical S-adenosylmethionine (SAM)-dependent methylation. J. Biol. Chem. 290, 3995–4002 10.1074/jbc.R114.607044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Wang B., Blaszczyk A. J., Knox H. L., Zhou S., Blaesi E. J., Krebs C., Wang R. X., and Booker S. J. (2018) Stereochemical and mechanistic investigation of the reaction catalyzed by Fom3 from Streptomyces fradiae, a cobalamin-dependent radical S-adenosylmethionine methylase. Biochemistry 57, 4972–4984 10.1021/acs.biochem.8b00693 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Cicchillo R. M., Iwig D. F., Jones A. D., Nesbitt N. M., Baleanu-Gogonea C., Souder M. G., Tu L., and Booker S. J. (2004) Lipoyl synthase requires two equivalents of S-adenosyl-l-methionine to synthesize one equivalent of lipoic acid. Biochemistry 43, 6378–6386 10.1021/bi049528x [DOI] [PubMed] [Google Scholar]
- 26. Zheng L., Cash V. L., Flint D. H., and Dean D. R. (1998) Assembly of iron–sulfur clusters identification of an iscSUA-hscBA-fdx gene cluster from Azotobacter vinelandii. J. Biol. Chem. 273, 13264–13272 10.1074/jbc.273.21.13264 [DOI] [PubMed] [Google Scholar]
- 27. Landgraf B. J., Arcinas A. J., Lee K.-H., and Booker S. J. (2013) Identification of an intermediate methyl carrier in the radical S-adenosylmethionine methylthiotransferases RimO and MiaB. J. Am. Chem. Soc. 135, 15404–15416 10.1021/ja4048448 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Grove T. L., Ahlum J. H., Sharma P., Krebs C., and Booker S. J. (2010) A consensus mechanism for radical SAM-dependent dehydrogenation? BtrN contains two [4Fe-4S] clusters. Biochemistry 49, 3783–3785 10.1021/bi9022126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Grove T. L., Benner J. S., Radle M. I., Ahlum J. H., Landgraf B. J., Krebs C., and Booker S. J. (2011) A radically different mechanism for S-adenosylmethionine–dependent methyltransferases. Science 332, 604–607 10.1126/science.1200877 [DOI] [PubMed] [Google Scholar]
- 30. Blaszczyk A. J., Wang B., Silakov A., Ho J. V., and Booker S. J. (2017) Efficient methylation of C2 in l-tryptophan by the cobalamin-dependent radical S-adenosylmethionine methylase TsrM requires an unmodified N1 amine. J. Biol. Chem. 292, 15456–15467 10.1074/jbc.M117.778548 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Blaszczyk A. J., Wang R. X., and Booker S. J. (2017) TsrM as a model for purifying and characterizing cobalamin-dependent radical S-adenosylmethionine methylases. Methods Enzymol. 595, 303–329 10.1016/bs.mie.2017.07.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Lanz N. D., Blaszczyk A. J., McCarthy E. L., Wang B., Wang R. X., Jones B. S., and Booker S. J. (2018) Enhanced solubilization of Class B radical S-adenosylmethionine methylases by improved cobalamin uptake in Escherichia coli. Biochemistry 57, 1475–1490 10.1021/acs.biochem.7b01205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Rüdiger H. (1971) The vitamin B12-dependent methionine synthetase: the cycle of transmethylation. Eur. J. Biochem. 21, 264–268 10.1111/j.1432-1033.1971.tb01465.x [DOI] [PubMed] [Google Scholar]
- 34. LaMattina J. W., Wang B., Badding E. D., Gadsby L. K., Grove T. L., and Booker S. J. (2017) NosN, a radical S-adenosylmethionine methylase, catalyzes both C1 transfer and formation of the ester linkage of the side-ring system during the biosynthesis of nosiheptide. J. Am. Chem. Soc. 139, 17438–17445 10.1021/jacs.7b08492 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Zhang Z., Mahanta N., Hudson G. A., Mitchell D. A., and van der Donk W. A. (2017) Mechanism of a Class C radical S-adenosyl-l-methionine thiazole methyl transferase. J. Am. Chem. Soc. 139, 18623–18631 10.1021/jacs.7b10203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Stupperich E., and Kräutler B. (1988) Pseudo vitamin B12 or 5-hydroxybenzimidazolyl-cobamide are the corrinoids found in methanogenic bacteria. Arch. Microbiol. 149, 268–271 10.1007/BF00422016 [DOI] [Google Scholar]
- 37. Scherer P., Höllriegel V., Krug C., Bokel M., and Renz P. (1984) On the biosynthesis of 5-hydroxybenzimidazolylcobamide (vitamin B12-factor III) in Methanosarcina barkeri. Arch. Microbiol. 138, 354–359 10.1007/BF00410903 [DOI] [Google Scholar]
- 38. DiMarco A. A., Bobik T. A., and Wolfe R. S. (1990) Unusual coenzymes of methanogenesis. Annu. Rev. Biochem. 59, 355–394 10.1146/annurev.bi.59.070190.002035 [DOI] [PubMed] [Google Scholar]
- 39. Sato S., Kudo F., Kuzuyama T., Hammerschmidt F., and Eguchi T. (2018) C-methylation catalyzed by Fom3, a cobalamin-dependent radical SAM enzyme in fosfomycin biosynthesis, proceeds with inversion of configuration. Biochemistry 57, 4963–4966 10.1021/acs.biochem.8b00614 [DOI] [PubMed] [Google Scholar]
- 40. Wang Y., Schnell B., Baumann S., Müller R., and Begley T. P. (2017) Biosynthesis of branched alkoxy groups: iterative methyl group alkylation by a cobalamin-dependent radical SAM enzyme. J. Am. Chem. Soc. 139, 1742–1745 10.1021/jacs.6b10901 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Benjdia A., Pierre S., Gherasim C., Guillot A., Carmona M., Amara P., Banerjee R., and Berteau O. (2015) The thiostrepton A tryptophan methyltransferase TsrM catalyses a cob(II)alamin-dependent methyl transfer reaction. Nat. Commun. 6, 8377 10.1038/ncomms9377 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Allen K. D., and Wang S. C. (2014) Spectroscopic characterization and mechanistic investigation of P-methyl transfer by a radical SAM enzyme from the marine bacterium Shewanella denitrificans OS217. Biochim. Biophys. Acta 1844, 2135–2144 10.1016/j.bbapap.2014.09.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Kim H. J., McCarty R. M., Ogasawara Y., Liu Y. N., Mansoorabadi S. O., LeVieux J., and Liu H. W. (2013) GenK-catalyzed C-6′ methylation in the biosynthesis of gentamicin: isolation and characterization of a cobalamin-dependent radical SAM enzyme. J. Am. Chem. Soc. 135, 8093–8096 10.1021/ja312641f [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Sato S., Kudo F., Kim S.-Y., Kuzuyama T., and Eguchi T. (2017) Methylcobalamin-dependent radical SAM C-methyltransferase Fom3 recognizes cytidylyl-2-hydroxyethylphosphonate and catalyzes the nonstereoselective C-methylation in fosfomycin biosynthesis. Biochemistry 56, 3519–3522 10.1021/acs.biochem.7b00472 [DOI] [PubMed] [Google Scholar]
- 45. Gerlt J. A., Bouvier J. T., Davidson D. B., Imker H. J., Sadkhin B., Slater D. R., and Whalen K. L. (2015) Enzyme function initiative-enzyme similarity tool (EFI-EST): a web tool for generating protein sequence similarity networks. Biochim. Biophys. Acta 1854, 1019–1037 10.1016/j.bbapap.2015.04.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Bridwell-Rabb J., Zhong A., Sun H. G., Drennan C. L., and Liu H. (2017) A B12-dependent radical SAM enzyme involved in oxetanocin A biosynthesis. Nature 544, 322–326 10.1038/nature21689 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Markowitz V. M., Chen I.-M., Palaniappan K., Chu K., Szeto E., Pillay M., Ratner A., Huang J., Woyke T., Huntemann M., Anderson I., Billis K., Varghese N., Mavromatis K., Pati A., et al. (2014) IMG 4 version of the integrated microbial genomes comparative analysis system. Nucleic Acids Res. 42, D560–D567 10.1093/nar/gkt963 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Mitchell A., Chang H.-Y., Daugherty L., Fraser M., Hunter S., Lopez R., McAnulla C., McMenamin C., Nuka G., Pesseat S., Sangrador-Vegas A., Scheremetjew M., Rato C., Yong S.-Y., Bateman A., et al. (2015) The InterPro protein families database: the classification resource after 15 years. Nucleic Acids Res. 43, D213–D221 10.1093/nar/gku1243 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Finn R. D., Bateman A., Clements J., Coggill P., Eberhardt R. Y., Eddy S. R., Heger A., Hetherington K., Holm L., Mistry J., Sonnhammer E. L., Tate J., and Punta M. (2014) Pfam: the protein families database. Nucleic Acids Res. 42, D222–D230 10.1093/nar/gkt1223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Iwig D. F., and Booker S. J. (2004) Insight into the polar reactivity of the onium chalcogen analogues of S-adenosyl-l-methionine. Biochemistry 43, 13496–13509 10.1021/bi048693+ [DOI] [PubMed] [Google Scholar]
- 51. Zehnder A. J., and Wuhrmann K. (1976) Titanium (III) citrate as a nontoxic oxidation-reduction buffering system for the culture of obligate anaerobes. Science 194, 1165–1166 10.1126/science.793008 [DOI] [PubMed] [Google Scholar]
- 52. Lanz N. D., Grove T. L., Gogonea C. B., Lee K.-H., Krebs C., and Booker S. J. (2012) RlmN and AtsB as models for the overproduction and characterization of radical SAM proteins. Methods Enzymol. 516, 125–152 10.1016/B978-0-12-394291-3.00030-7 [DOI] [PubMed] [Google Scholar]
- 53. Altschul S. F., Madden T. L., Schäffer A. A., Zhang J., Zhang Z., Miller W., and Lipman D. J. (1997) Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 25, 3389–3402 10.1093/nar/25.17.3389 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Smoot M. E., Ono K., Ruscheinski J., Wang P.-L., and Ideker T. (2011) Cytoscape 2.8: new features for data integration and network visualization. Bioinformatics 27, 431–432 10.1093/bioinformatics/btq675 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. UniProt Consortium (2014) UniProt: a hub for protein information. Nucleic Acids Res. 43, 204–212 10.1093/nar/gku989 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Sievers F., Wilm A., Dineen D., Gibson T. J., Karplus K., Li W., Lopez R., McWilliam H., Remmert M., Söding J., Thompson J. D., and Higgins D. G. (2011) Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol. Syst. Biol. 7, 539 10.1038/msb.2011.75 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.