

Abstract
P450 119 peroxygenase was found to catalyze the sulfoxidation of thioanisole and the sulfonation of sulfoxide in the presence of tert‐butyl hydroperoxide (TBHP) for the first time with turnover rates of 1549 min−1 and 196 min−1 respectively. Several mutants were designed to improve the peroxygenation activity and thioanisole specificity by site‐directed mutagenesis. The F153G/T213G mutant gave an increase of sulfoxide yield and a decrease of sulfone yield. Moreover the S148P/I161T/K199E/T214V mutant and the K199E mutant with acidic Glu residue contributed to improving the product ratio of sulfoxide to sulfone. Addition of short‐alkyl‐chain organic acids to the P450 119 peroxygenase‐catalyzed sulfur oxidation of thioanisole was investigated. Octanoic acid was found to induce a preferred sulfoxidation of thioanisole catalyzed by the F153G/T213G mutant to give approximately 2.4‐fold increase in turnover rate with a k cat value of 3687 min−1 relative to that of the wild‐type, and by the F153G mutant to give the R‐sulfoxide up to 30 % ee. The experimental control and the proposed mechanism for the P450 119 peroxygenase‐catalyzed sulfoxidation of thioanisole in the presence of octanoic acid suggested that octanoic acid could partially occupy the substrate pocket; meanwhile the F153G mutation could enhance the substrate specificity, which could lead to efficiently regulate the spatial orientation of thioanisole and facilitate the formation of Compound I. This is the most effective catalytic system for the P450 119 peroxygenase‐catalyzed sulfoxidation of thioanisole.
Keywords: P450 peroxygenase, site-directed mutagenesis, sulfoxidation reactions, turnover rate, catalytic mechanism
1. Introduction
Cytochrome P450 enzymes (P450s or CYPs) are a family of hemoproteins that catalyze a wide range of oxidative reactions including aliphatic and aromatic hydroxylation, olefin epoxidation and sulfur oxidation in a regio‐ and stereoselective manner.1, 2, 3 This makes P450s highly promising candidates for the production of fine chemicals that are difficult to synthesize by standard chemical means. For many oxidative reactions, P450s typically catalyze the insertion of one atom of molecular oxygen (monooxygenation) into exogenous and endogenous substrates through a classical two‐electron oxotransfer pathway.3 The mechanism of P450 monooxygenases involves a putative Compound I species, which is a known intermediate formed in oxidative reactions of other heme‐containing enzymes.1 One of the limitations to apply P450 monooxygenases to biocatalysts is that the process requires expensive cofactors and complicated electron‐transfer systems such as NADH or NADPH in synthetic chemistry. In contrast to P450 monooxygenases, P450 peroxygenases employ hydroperoxide or other peroxides as surrogate oxygen donors to catalyze the oxygenation of substrates through a so‐called shunt pathway, and in principle make reaction much simpler in the absence of NAD(P)H and redox protein partners.4, 5 However, excess peroxides may deactivate P450s by heme destruction and oxidative modification of amino residues, which makes the use of the shunt pathway not productive. Furthermore, formation of Compound I by the peroxide shunt pathway is restricted by the size and shape of the substrate‐binding pocket of the P450s, which allows other competitive radical species to generate secondary substrate oxidation.
P450 119 cloning from archaebacteria Sulfolobus acidocaldarius contains the typical P450‐fold with a relatively compact structure.6, 7 The available crystal structure indicated that CYP119 undergoes a significant conformational change in the F/G region upon binding of medium substrates, which is helpful to define the determinants of substrate specificity for the subsequent modification of the protein to the desired properties.7 It was suggested that the extensive aromatic cluster and entropic contribution could lead to the enhanced thermal stability of CYP119. [8] It is an attractive system for the formation of the Compound I species by the shunt pathway in mechanistic investigations of the P450 catalytic cycle.9 Generally, the thermal stability of CYP119 does not depend on the iron spin state or the active site of the substrate‐binding pocket.6 Though both the native electron donor partners and the endogenous substrate for CYP119 are still unknown, its site‐directed mutant can hydroxylate lauric acid by putidaredoxin (Pd) and putidaredoxin reductase (PdR) with NADH as surrogate redox partners.10 The T214 V/D77R mutant with a kcat value of 8.8 min−1 showed 15‐fold enhanced activity over the wild‐type. [10] It was suggested that the T214 V mutation should increase the rate of hydroxylation by modifying the size and shape for the substrate binding, whereas the D77R mutation should improve the affinity between CYP119 and the redox protein partner. CYP119 was initially isolated from S. solfataricus, which is closely related to S. acidocaldarius, so the electron transfer proteins or proliferating cell nuclear antigen from this thermophilic organism were reconstituted into P450 119 monooxygenation, in which the homologous recombination from sulfur‐containing microorganism makes hydroxylation of lauric acid work.11, 12, 13
P450 119 was also found to catalyze the peroxygenation of styrene and substituted styrenes in the presence of H2O2 and other peroxides. Styrene was peroxygenated to the corresponding epoxides by using the wild‐type P450 119 at 30 °C and low catalytic activity was observed with a rate of 0.6 min−1, while the P450 119 T214 V mutant was found to increase the H2O2‐dependent epoxidation rate by about 3‐fold under the same reaction conditions.6 Niemeyer et al reported that P450 119 catalyzed the epoxidation of styrene with an increased turnover rate of 78 min−1 by using the optimized reaction conditions at 70 °C and pH 8.5 in the presence of tert‐butyl hydroperoxide (TBHP)13. We reported that introduction of the T213G mutation into the wild‐type P450 119 enhanced the turnover rate for the epoxidation of styrene with k cat value of 51.2 min−1 and 346.2 min−1 respectively at 35 °C and 70 °C in the presence of TBHP at pH 7.5.14 The increased turnover rate might be mainly due to the reduced steric hindrance of the T213G mutation, which could improve the access of styrene and TBHP to the catalytic site. The T213 M mutant can improve the enantioselectivity for the epoxidation of cis‐β‐methylstyrenes, but it gave low turnover number with a rate of 32.9 min−1 at 35 °C under the same reaction condition.15 Our recent study showed that the quadruple mutant S148P/I161T/K199E/T214 V (No.3‐39) obtained by using the directed evolution can significantly improve the turnover number for the epoxidation of styrene and its derivatives with up to 308.8 min−1 at 35 °C and pH 7.5 in the presence of TBHP.16 The proposed mechanism suggested that the improved access of the substrate channel and the increased affinity of the acid‐base controlling in the catalytic pocket of P450 119 peroxygenase might result in high efficiency for the formation of Compound I and the subsequent peroxygenation.
Chiral sulfoxides are important building blocks in medicinal chemistry and pharmaceutical industry. Among several sulfur oxidations catalyzed by cytochrome P450 enzymes,17, 18 an auxiliary flavin containing cofactor and NADH‐NADPH dependent flavoenzymes need to be considered. For example, P450 monooxygenase CYP116B4 from Labrenzia aggregata exhibits sulfoxidation activity towards alkyl‐aryl sulfides with a distinctive CYP‐redox system. [19] The extensively studied P450BM3 naturally fused with an FAD/FMN redox partner was also mutated for application in the sulfoxidation of 1‐thiochroman‐4‐one by directed evolution. [20] There are only few cases reported for the sulfoxidation of thioether by using P450 peroxygenase. For example, P450BSβ peroxygenase isolated from Bacillus subtilis was reported with an enhanced turnover rate for the sulfoxidation of thioanisole in the presence of carboxylic acids by utilizing hydrogen peroxide.21 The P450BM3 F87 A mutant peroxygenase also worked well for the sulfoxidation of thioanisole by using dual‐functional small molecules in the presence of H2O2.22 To our knowledge, no sulfoxidation of thioanisole has been achieved by using P450 119 peroxygenase.
In the present work, we report that for the first time the thermophilic P450 119 peroxygenase can catalyze the sulfur oxidation of thioanisole in the presence of TBHP. Site‐directed mutations of CYP119 and screening assays of the reaction conditions have been conducted to enhance the peroxygenation activity and substrate specificity for the sulfoxidation of thioanisole.
2. Results and Discussion
We first examined the sulfur oxidation of thioanisole 1 catalyzed by the wild‐type P450 119 peroxygenase at 35 °C and pH 7.5 in the presence of TBHP according to our previously optimized reaction conditions.16 Trace products were found in 10 min under these conditions (Table 1, Entry 1). Since TBHP was reported to increase the stability of P450 119 under the peroxidation conditions,13 we prolonged the incubation time. Within 60 min, sufficient amounts of the products were observed in this reaction. An incomplete conversion of 1 with formation of the sulfoxide 2 (36 % yield) was determined by GC‐MS analyses (Scheme 1 and Table 1, Entry 2), with the aid of an internal standard calibration curve prepared by using acetophenone (Figure S1 in Supporting Information). Since a high concentration of TBHP relative to the concentrations of the substrate and P450 119 can also be attributed to the improved turnover rate of a peroxidation in an earlier activity assay, [13] we investigated the sulfoxidation of 1 catalyzed by P450 119 with various concentrations of TBHP. As shown in entries 3–6 of Table 1, the yield of the sulfoxide 2 increased from 46 % to 59 % with the increase of the TBHP concentration. Meanwhile formation of the sulfone 3 was also observed in 23–31 % yield, and the ratio of the sulfoxide product to the sulfone did not show significant change under these conditions. Complete conversion of 1 by using P450 119 gave maximum yields of the sulfoxide 2 and the sulfone 3 in 61 % and 33 % respectively in the presence of 20 mM TBHP (Entry 4). An enantioselectivity of 16.9 % ee was also observed for the sulfoxidation of 1, and the configuration of the sulfoxide 2 was determined to be R by HPLC (Daicel Chiralpak OD−H column) in comparison with the data reported in the literature.21 Neither 2 nor 3 was observed without P450 119 or TBHP in the control assay for the sulfur oxidation of 1.
Table 1.
The results of the sulfur oxidation of thioanisole for P450 119 at 35 °C and pH 7.5 in presence of various concentrations of TBHP.
| Entry | Conc. of TBHP | Yield (%) 2 [a] | Yield (%) 3 [a] | Conf. 2 [b] |
|---|---|---|---|---|
| 1[c] | 2.5 mM | trace | – | – |
| 2 | 2.5 mM | 36.0 | 0.3 | R |
| 3 | 8.0 mM | 46.2 | 26.0 | R |
| 4 | 15.0 mM | 48.0 | 31.3 | R |
| 5 | 20.0 mM | 59.3 | 30.3 | R |
| 6 | 30.0 mM | 54.8 | 23.8 | R |
[a] yield of 2 and 3 determined by GC‐MS. [b] enantioselectivity determined by HPLC and configuration determined by comparing the HPLC data the literature 21. [c] reaction conditions from reference 16.
Scheme 1.
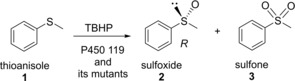
Cytochrome P450 119‐catalyzed sulfur oxidation of thioanisole in the presence of TBHP.
The sulfoxidation of 1 by using P450 119 gave only trace amount of the sulfone 3 (0.3 % yield) in 2.5 mM TBHP (Table 1, Entry 2). The initial rate for the formation of the sulfoxide 2 was investigated in the oxidation of 1 catalyzed by P450 119 in the presence of 20 mM TBHP. The kinetic constant for the sulfoxidation of thioanisole was calculated from the resulting regression equation. The wild‐type P450 119 converted 1 into the sulfoxide 2 with a k cat value of 1549 min−1 and a K m value of 4.3 mM on the basis of the double‐reciprocal plot (Figure S2a in SI). The sulfoxide 2 was also used as the substrate for the oxidation catalyzed by P450 119, and formation of the sulfone 3 was observed under the same reaction conditions (Scheme 2). The kinetic constants k cat and K m for the oxidation of the sulfoxide 2 catalyzed by P450 119 were 196 min−1 and 6.3 mM (see Figure S2b in SI). So, there were 8 times difference in the turnover rate for the sulfoxidation of thioanisole and the sulfonation of the sulfoxide. This is the first time that P450 119 was found to catalyze the sulfur oxidation of thioanisole in the presence of TBHP, in which P450 119 maintained its peroxygenation activity with the formation of a sulfoxide 2 and a sulfone 3, and moreover P450 119 showed its substrate specificity between 1 and 2 with a different yield of 2 and 3.
Scheme 2.
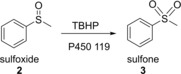
Cytochrome P450 119‐catalyzed the oxidation of the sulfoxide in the presence of TBHP.
In order to improve the peroxygenation activity and substrate specificity of P450 119 for the sulfur oxidation of thioanisole, we designed several new site‐directed mutants on the basis of our previously reported mutants.14, 15 Earlier, we found that introduction of glycine and methionine into the Thr213 residue of the wild‐type P450 119, giving the T213G and T213 M mutants respectively, can enhance the turnover rate and enantioselectivity for the peroxide‐dependent styrene epoxidation.14 A histidine residue as a proximal or distal ligand plays a vital role in high peroxidase and peroxygenase activity.5, 23 We speculate that the site‐directed mutation of Thr213 to histidine as a distal side of P450 119 might enhance P450 peroxygenase activity, in which the distal His213 plays general acid‐base function while the proximal Cys317 is still maintained to ligate heme. In order to enhance the substrate specificity of P450 119 for the sulfur oxidation of thioanisole, molecular dynamics (MD) simulations were carried out and the root‐mean‐square fluctuation (RMSF) of the whole protein and residues was analyzed (Figure S3 in SI). We found that the F/G loop from 148 to 163 residues constitute the significant binding region to recognize substrate with more conformational flexibility (Figure S3 in SI), while the Phe153 residue with phenyl side chain is located into the region with large steric hindrance and rigidity. We propose that the site‐directed mutation of the Phe153 residue in P450 119 to a small and flexible residue such as glycine, alanine or valine might contribute to the special recognition of thioanisole.
Molecular docking was used to investigate the sulfoxidation of 1 with the wild‐type P450 119 and the F153G/T213G mutant. We modeled the docking of the thioanisole into the wild‐type P450 119 and the F153G/T213G mutant respectively. As shown in Figure 1, the distances between the sulfur atom and the iron‐oxo species of the heme are 2.4 Å and 1.9 Å respectively (Figure 1a and 1b), which suggests that the shorter the distance of the sulfur atom from Compound I, the higher the peroxygenation activity of the P450s for the sulfoxidation of 1. It should be noted that when F153 was mutated into G153, the F/G helix of the F153G residue became shorter, and the region near the substrate center formed a loop structure to squeeze the substrate closer to the active center (Figure 1b).
Figure 1.
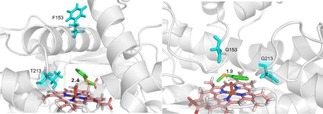
The molecular docking of the thioanisole 1 into (a) the wild‐type P450 119 and (b) the F153G/T213G mutant.
Recently, we also reported that the quadruple mutant S148P/I161T/K199E/T214V (No.3‐39) obtained by using the directed evolution can significantly improve the turnover number for the peroxide‐dependent epoxidation.16 The modelling structures revealed that introduction of the acidic Glu residue into the basic Lys199 residue located within the I helix of the quadruple mutant could lead to the reconstruction of the whole catalytic pocket,16 which might be crucial for both the peroxygenation activity and the substrate specificity. Therefore, we decided to examine whether a single K199E mutant could influence the sulfoxidation of thioanisole in comparison with the quadruple mutant. Similar to the construction, expression, and purification of the above‐mentioned mutants, the K199E mutant was obtained with a typical P450s character in the UV/Vis absorption spectrum (Figure S5 in SI). The sulfoxidation of 1 catalyzed by the No.3‐39 mutant and the K199E mutant was investigated. As shown in Table 2 (Entries 9 and 10), the sulfoxidation of 1 catalyzed by the No.3‐39 mutant gave 76.2 % yield of the 2 and 6.9 % yield of the 3, and the K199E mutant retained similar yields of 2 and 3 with 70.2 % and 18.5 % respectively. Thus, an acidic surrounding of the catalytic pocket in the quadruple mutant and the K199E mutant might have enhanced both the peroxygenation activity and substrate specificity for the sulfoxidation of 1.
Table 2.
The results of the sulfur oxidation of thioanisole for P450 119 and its mutants at 35 °C and pH 7.5 in presence of 20 mM TBHP.
| Entry | Enzyme | Yield (%) 2/3 [a] | ee (%) 2 [b] | Conf. 2 [c] |
|---|---|---|---|---|
| 1 | Wild‐Type | 59.3/30.3 | 16.9 | R |
| 2 | T213G | 72.0/14.5 | 0.2 | R |
| 3 | T213M | 10.3/– | – | – |
| 4 | T213H | 15.4/– | – | – |
| 5 | F153G | 71.3/22.2 | 22.9 | R |
| 6 | F153A | 53.4/29.5 | 19.9 | R |
| 7 | F153V | 60.6/21.9 | 17.5 | R |
| 8 | F153G/T213G | 75.1/11.3 | 6.8 | R |
| 9 | No. 3–39 | 76.2/6.9 | 14.8 | R |
| 10 | K199E | 70.2/18.5 | 12.3 | R |
P450 119 originated from the organism S. acidocaldarius, therefor its optimal growth conditions were initially attached to thermophilic and acidophilic sulfur‐containing surrounding. [6] It was found that several natural P450 peroxygenases can catalyze non‐native substrates in the presence of short‐alkyl‐chain carboxylic acids. [24] We thus investigated the sulfoxidation of 1 catalyzed by P450 119 in presence of TBHP and short‐alkyl‐chain carboxylic acids. As shown in Table 3, acetic acid was found to induce almost equivalent sulfoxidation and sulfonation catalyzed by P450 119 with the yield of 45.5 % and 49.4 % respectively (Entry 2), while heptanoic acid gave the yields of 2 and 3 in 53.6 % and 28.9 % respectively (Entry 3) similar to the results of the sulfur oxidation of 1 catalyzed by the wild‐type without carboxylic acid (Entry 1). A great difference in the 2/3 yield ratio was obtained with 79.1 % versus 15.4 % in the presence of octanoic acid, indicating significant enhancement in the peroxygenation activity and the substrate specificity (Entry 4). Carboxylic acids with a phenyl group gave smaller differences in the yields of the 2 and 3 with 47.3–47.8 % and 37.9–40.9 % respectively for the sulfur oxidation of 1 catalyzed by P450 119 (Entries 5–6). These carboxylic acids examined gave the R‐sulfoxide 2 with increased enantioselectivity ranging from 21.4 % to 28.7 % ee. Thus, P450 119 peroxygenases in the sulfoxidation of 1 exhibit significant dependence on the alkyl‐chain length of the carboxylic acid additives.
Table 3.
The results of the sulfur oxidation of thioanisole catalyzed by P450 119 in presence of 20 mM TBHP and various carboxylic acids.
| Entry | Carboxylic acid | Yield (%) 2/3 [a] | ee (%) 2 [b] | Conf. 2 [c] |
|---|---|---|---|---|
| 1 | none | 59.3/30.3 | 16.9 | R |
| 2 | CH3COOH[d] | 45.5/49.4 | 23.6 | R |
| 3 | CH3(CH2)5COOH[d] | 53.6/28.9 | 28.4 | R |
| 4 | CH3(CH2)6COOH[d] | 79.1/15.4 | 21.4 | R |
| 5 | Ph(CH2)2COOH[d] | 47.8/37.9 | 28.7 | R |
| 6 | Ph(CH2)3COOH[d] | 47.3/40.9 | 22.1 | R |
[a] yield of 2 and 3 determined by GC‐MS . [b] enantioselectivity determined by HPLC. [c] configuration determined by comparing the HPLC data with the literature 21. [d] concentration of carboxylic acid was 20 mM.
We studied the octanoic acid‐attached P450 119 system with an adjusted pH value of 6.5 in potassium phosphate buffer (50 mM). The optimized conditions were applied to the sulfur oxidation of 1 catalyzed by the P450 119 mutants at pH 6.5 in presence of TBHP and octanoic acid, and the results are summarized in Table 4. As shown in Table 4, octanoic acid had no effect on the sulfur oxidation of 1 catalyzed by the T213G, T213 M and T213H mutants, in which the 2 and 3 yields were maintained in comparison with that without any carboxylic acid (Entries 2–4). The F153G mutation increased the peroxygenation activity and substrate specificity with a 2/3 yield ratio of 80.2/13.5 (Entry 5), while the F153 A and F153 V mutants significantly increased the 2/3 yield ratio to 72.8/10.1 and 87.7/10.3 in the presence of octanoic acid (Entries 6–7). Especially, the F153G/T213G mutant showed excellent peroxygenation activity and substrate specificity for the sulfoxidation of 1 in the presence of octanoic acid to give the 2/3 yield ratio up to 90.1 % versus 9.4 % (Entry 8). Octanoic acid was also found to induce a preferred sulfoxidation of 1 catalyzed by the quadruple mutant and the K199E mutant with large 2/3 yield ratios of 88.9/8.2 and 80.4/14.6 respectively (Entries 9–10). The sulfoxidation of 1 catalyzed by the P450 119 and its mutants gave R‐sulfoxide 2 in presence of octanoic acid, and the F153G mutant showed enantioselectivity up to 30 % ee (Table S1 in SI). Kinetic constants were calculated from the resulting regression equation for the sulfoxidation of 1 catalyzed by the F153G/T213G mutant in the presence of TBHP and octanoic acid. The T213G/F153G mutant converted 1 into the sulfoxide 2 with a k cat value of 3687 min−1 on the basis of the double‐reciprocal plot in the presence of TBHP with octanoic acid (Figure S6 in SI), which exhibited approximately 2.4‐fold increase in turnover rate relative to that of the wild‐type without any carboxylic acid with a k cat value of 1549 min−1.
Table 4.
The results of the sulfur oxidation of thioanisole for P450 119 and its mutants at pH 6.5 with octanoic acid and pH 8.5 without octanoic acid.
| Entry | Enzyme | pH 6.5[b] | pH 8.5 |
|---|---|---|---|
| Yield of 2/3 (%)[a] | Yield of 2/3 (%)[a] | ||
| 1 | Wild‐Type | 79.1/15.4 | 65.6/38.2 |
| 2 | T213G | 74.4/13.0 | 55.4/35.8 |
| 3 | T213M | 10.9/– | 39.2/0.36 |
| 4 | T213H | 14.1/– | 36.9/0.4 |
| 5 | F153G | 80.2[c]/13.5 | 63.3/23.8 |
| 6 | F153A | 72.8/10.1 | 59.8/27.3 |
| 7 | F153V | 87.7/10.3 | 58.7/32.9 |
| 8 | F153G/T213G | 90.1/9.4 | 79.2/9.3 |
| 9 | No. 3–39 | 88.9/8.2 | 82.8/6.5 |
| 10 | K199E | 80.4/14.6 | 79.3/16.0 |
[a] yield of 2 and 3 determined by GC‐MS, [b] concentration of octanoic acid was 20 mM, [c] enantioselectivity 30 % ee determined by HPLC and R‐configuration determined by comparing the HPLC data with the literature 21.
To further understand the enhanced activity and stereochemistry in the peroxygenation of P450 119, we also modelled the docking pose of thioanisole into the active pocket of the F153G mutant without and with octanoic acid. As shown in Figure 2a and 2 b, the distances between the sulfur atom and the iron‐oxo species of the heme are 2.5 Å and 1.8 Å without and with octanoic acid respectively, which suggests that the thioanisole molecule corresponding to the formation of the R‐enantiomer of sulfoxide should be closer to the iron‐oxo species of the heme in the active center with octanoic acid than without octanoic acid.
Figure 2.
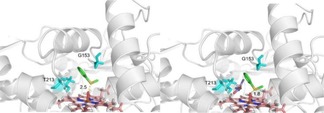
The molecular docking poses of thioanisole 1 into the F153G mutant (a) without octanoic acid (b) with octanoic acid.
It was reported that the P450 119 peroxidase activity was pH dependent in the presence of peroxides, and the peroxidase activity was highest at pH 8.5. [13] We further investigated the sulfur oxidation of 1 catalyzed by P450 119 and its mutants at pH 8.5 in presence of TBHP without octanoic acid in order to confirm the role of the carboxylic acid. As shown in Table 4, the 2/3 yield ratios by using the wild‐type (65.6/38.2) and the T213G mutant (55.4/35.8) at pH 8.5 in the control group were apparently lower than that at pH 6.5 in the octanoic acid‐attached P450 119 group (Entries 1–2). The T213 M and T213H mutants gave an enhanced activity for the sulfoxidation of 1 with the yield of the 2 in 39.2 % and 36.9 % respectively at pH 8.5, which was in agreement with the P450 119 peroxidase activity at pH 8.5 reported earlier (Entries 3–4).13 The F153G, F153 A and F153H mutations decreased the peroxygenation activity and substrate specificity with the 2/3 yield ratios of 63.3/23.8, 59.8/27.3 and 58.7/32.9 respectively (Entrie 5–7). The F153G/T213G mutant also gave an obvious decrease in the 2/3 yield ratio with 79.2/9.3 at pH 8.5 in comparison with that at pH 6.5 in presence of octanoic acid. The No.3–39 mutant and the K199E mutant almost maintained the peroxygenation activity and substrate specificity with more tolerance of the basic surrounding at pH 8.5, and gave the 2/3 yield ratios of 82.8/6.5 and 79.3/16.0 respectively, in which the acidic Glu199 residue with a carboxylic side chain in the mutational K199E position might play a very important role as a substitution of octanoic acid. Thus, octanoic acid might enter the catalytic pocket of P450 119, participate in the formation of Compound I, and regulate the spatial orientation of thioanisole.
We have proposed a mechanism for the sulfoxidation of 1 catalyzed by the P450 119 F153G peroxygeanse in the presence of TBHP and octanoic acid according to the studies reported earlier.17, 18, 24, 25 As shown in Figure 3, after octanoic acid and thioanisole enter the catalytic pocket of the P450 119 F153G peroxygenase, thioanisole could be oriented properly by the mutant and octanoic acid. The mutant, thioanisole and octanoic acid together had great influence on the stability of the FeIII(tBuOOH) intermediate (II) by the hydrogen‐bonding interactions in the active site, in which the orientation of thioanisole could also prevent tBuOOH release. The hydrogen‐bonding network activated by octanoic acid could lock the tBuO⋅ radical formed by the O−O homolysis (III), then the radical abstracts the hydrogen atom from Fe−O−H to generate tBuOH. Compound I could be formed thereby, which could subsequently oxidize thioanisole to form the methyl‐phenyl sulfoxide 2 with R‐enantioselectivity.
Figure 3.
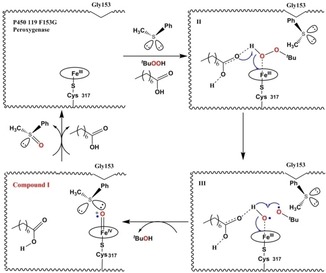
A proposed mechanism for the sulfoxidation of thioanisole 1 catalyzed by the P450 119 F153G peroxygenase.
3. Conclusions
We found for the first time that P450 119 peroxygenase catalyzes the sulfoxidation of thioanisole 1 and the subsequent sulfonation in the presence of TBHP with turnover rates of 1549 min−1 and 196 min−1 respectively. We rationally designed several new site‐directed P450 mutants, and found that the F153G/T213G mutant can enhance the peroxygenation activity and substrate specificity with an increase of the sulfoxide 2 yield and a decrease of the sulfone 3 yield, and the No.3–39 mutant and the K199E mutant with an acidic Glu residue can improve the 2/3 yield ratio. We investigated the addition of short‐alkyl‐chain carboxylic acids into the P450 119 peroxygenase‐catalyzed sulfur oxidation of 1, and found that octanoic acid can induce a preferred sulfoxidation of 1 catalyzed by the F153G/T213G mutant to give approximately 2.4‐fold increase in turnover rate with a k cat value of 3687 min−1 relative to that of the wild‐type, and by the F153G mutant to give the R‐sulfoxide 2 up to 30 % ee. The mechanistic study for the P450 119 peroxygenase‐catalyzed sulfoxidation of 1 in the presence of octanoic acid suggests that introduction of the Gly153 residue into the active pocket of the P450 119 peroxygenase should contribute to improving the substrate specificity, and octanoic acid could occupy the catalytic pocket to efficiently participate in the formation of Compound I by hydrogen‐bonding interactions which in combination with the mutant could regulate the spatial orientation of thioanisole. This is the most effective catalytic system for the P450 119 peroxygenase‐catalyzed sulfoxidation of thioanisole.
Experimental Section
Materials
Escherichia coli strain BL21 (DE3) plysS and pET30a vector were obtained from Novagen (La Jolla, CA). All restriction enzymes and ExTaq Polymerase were purchased from TaKaRa Biotechnology (Liaoning China). Water was generated by using a Milli‐Q‐Gradient purification system (Millipore). Medium components tryptone and yeast extract were purchased from Oxoid. Isopropylb‐d‐1‐thiogalactopyranoside (ITPG, >99 %), ampicillin sodium salt (>99 %), kanamycin sulfate (>99 %), and meta‐chloroperbenzoic acid were purchased from Sigma‐Aldrich. Thioanisole was purchased from J&K Scientific and used without further purification. Acetic acid, hexanoic acid, heptanoic acid, octanoic acid, 2‐phenylpropionic acid, n‐butanoicacid, oxone, hydrogen peroxide were purchased form Adamas‐beta. All other chemicals and reagents were purchased from J&K Scientific, China.
Construction, Expression and Purification of CYP119 Mutants
Mutants of CYP119 were constructed by using the Quickchange Lighting Site‐directed Mutagenesis Kit (Agilent Techologies). Mutagenic PCR was applied on pET30a‐CYP 119 plasmid constructed previously as the template DNA.15 For the T213H mutant, the forward and reverse primers were 5’‐TTCTCATAGCGGGTAATGAGCATACAACTAACTTA ATATC‐3′, 5′‐TTTGATATTAAGTTAGTTGTATGCTCATTACCCGCTATGA G −3′. For the F153 A mutant, the primers were 5’‐TAGTCGCAGCAAGGTTGGGTA AGCCTGGAG‐3’, 5’‐CAACCTTGCTGCGACTAAGTCTGACCACTC‐3’. For the F153 V mutant, the primers were 5’‐AGTCGCAGTTAGG TTGGGTAAGCCTGGAG‐3’, 5’‐CCAACCTAACTGCGACTAAGTCTGA CCACTC‐3’. For the F153G mutant, the primers were 5’‐AGTCGCA GGTAGGTTGGGTAAGCCTGGAG‐3’, 5’‐CCAACCTACC TGCGACTAA GTCTGACCACTC‐3’. For the K199E mutant, the primers were 5’‐ CATAGAGGAACTCGGATACATTATTTTACTTCTCA‐3’, 5’‐TGTATC CGAGTTCCTCTATGTCTGAGAGGTTTGAG‐3’ (Bold and lined face indicates the positions of the mutations). All enzymes were overexpressed in E.coli BL21 (DE3) plysS cells, induced by isopropyl‐β‐D‐thiogalactopyranoside as previous study. [15] The purity of the CYP119 mutants was determined by SDS‐polyacryl‐amide gel electrophoresis with single 45‐kDa band eluted from the Ni‐NTA column. Protein concentration was calculated with use of the molar extinction coefficient of ϵ 415=104 mM.cm−1. The reduced‐CO complex formation of CYP 119 mutants was monitored by UV‐visible spectroscopy.
Synthesis of Standard Samples of the Phenyl Methyl Sulfoxide 2
Hydrogen peroxide (30 % by mass, 23.3 mmol) was added dropwise via addition funnel to a stirred solution of methyl phenyl sulfide (7.76 mmol) in ethanol (15 mL) on cooled in an ice‐water bath. After the mixture was stirred at RT for 24 h, the reaction mixture was transferred to CH2Cl2 (50 mL) and cooled in an ice‐water bath, and aqueous sodium bisulfite (15 %) was added to decompose the unreacted H2O2 until the oxidant was no longer visualized by using the potassium iodide‐starch paper. The organic phase was separated and dried over Na2SO4 overnight. After filtration, the solvent was removed by rotary evaporation. After purified by flash chromatography on a silica gel column, the sulfoxide product was identified by 1H NMR analysis: 1H NMR (400 MHz, Chloroform‐d) δ 7.66–7.64 (t, 2H), 7.53–7.50 (m, 3H), 2.72 (s, 3H). The sulfoxide product was used as the standards in the HPLC and GC‐MS analysis for identification of the enzymatic reaction product.
Synthesis of Standard Sample of Phenyl Methyl Sulfone 3
The reaction was performed in de‐ionized H2O (10 mL) containing methyl phenyl sulfide (2 mmol) and Oxone (3 mmol). The reaction was initiated by vigorous stirring at 60 °C for 12 h. Samples were then extracted with CH2Cl2 (2×2.5 mL) and the combined CH2Cl2 layers were separated and dried over with Na2SO4.The crude product was purified by flash chromatography on a silica gel column and characterized by 1H NMR analysis: 1H NMR (400 MHz, Chloroform‐d) δ 7.91–7.88 (m, 2H), 7.62–7.59 (t, 1H), 7.54–7.50 (m, 2H), 3.00 (s, 3H). The sulfone product was used as standards in the HPLC and GC‐MS analysis for identification of the enzymatic reaction product.
Catalytic Activity Assay
The reaction was carried out in closed glass vials in total volumes of 200 μL containing 7 μM CYP 119 enzyme, 2 mM thioanisole, and variable TBHP concentrations (for the determination of the optimum concentration of TBHP) in 50 mM potassium phosphate buffer, pH 7.5 or in 50 mM glycine buffer, pH 8.5 at 35 °C for 1 h. For the determination of the short‐alkyl‐chain carboxylic acids effect, a concentration of carboxylic acid was 20 mM in 200uL reaction mixture. Acetophenone was added as an internal standard after the reaction was quenched with 200 μL CH2Cl2 or hexane. For the determination of product yield, the reaction mixture was extracted with 200 μL CH2Cl2 for three times. The CH2Cl2 layer was analyzed with gas chromatography (Thermo Scientific ITQ900) on a DB‐1MS column (30 m×0.25 mm inner diameter, 90 °C for 2 min, followed by 10 °C/min rise to 210 °C, and finally 210 °C for 1 min). For the determination of sulfoxide enantioselectivity, the reaction mixture was then extracted with 200 μL of hexane for three times and the products were analyzed by using chiral HPLC on a Waters1525 Breeze™ with a UV detector at 254 nm using Daicel Chiralpak OD−H column (hexane/2‐propanol=92/8, tR1=10.8 min, tR2=12.5 min). The configuration of sulfoxide was determined by comparing the HPLC data with the literature. [21] All the experiments were carried out in triplicate at least. One control experiment was done without CYP119 or TBHP. Another experimental control was conducted for the oxidation of sulfoxide as substrate catalyzed by P450 119 and TBHP respectively.
Kinetics Studies
The kinetics constants of P450 119 and its mutants were determined with thioanisole 1 and sulfoxide 2 as substrate. The reaction mixture consisted of 0.5 μM CYP119 mutant, variable concentrations of substrate and TBHP (10‐fold concentration of substrate). Phosphate buffer (pH 7.5) was added to the final volume at 100 μL. The reaction mixtures were incubated at 35 °C for 15 second (for the substrate thioanisole) or 30 second (for the substrate phenyl methyl sulfoxide) in a closed glass vials and stopped by addition of CH2Cl2 (100 μL). The products were analyzed by gas chromatography (Thermo Scientific ITQ900) on a DB‐1MS column (30 m ×0.25 mm inner diameter). The purified sulfoxide 2 and sulfone 3 were used as authentic standards to identify the retention time and to prepare calibration curve. The retention times found with this condition were ca. 11.2 min for sulfoxide and ca. 4.5 min for sulfone. The initial rates of formation of sulfoxide and sulfone were estimated at various concentrations of the substrate and analyzed by double‐reciprocal plot method to evaluate the kinetic parameters K m and k cat.
Molecular Dynamics Simulations
The crystallographic structure of CYP119 complexed with 4‐Phenylimidazole (PDB code: 1F4T) was used as the starting point for this work. The simulation was set up by removing the 4‐Phenylimidazole and water molecules. The site directed mutations on the site 153 were constructed by MOE v2014.0901. All MD simulations were performed using the Gromacs package v.5.0.5 and the CHARMM27 all‐atom force field. The SPC/E model was used for water molecules. The protein model was solvated in a rhombic dodecahedral box, considering a minimum distance between the protein and box walls of 1 nm. The water molecules and five extra Na+ ions were added to neutralize the system. The system was then minimized for 1000 steps, using the steepest descent algorithm to relieve bad crystal contacts and equilibrated for 400 ps at 308 K using the NVT ensemble and 105 Pa using the NPT ensemble. MD simulations were performed for 100 ns with periodic boundary conditions. The flexibility of the protein was analyzed by inspecting the root‐mean‐square fluctuation (RMSF) of each residue from its time‐average position.
Molecular Docking Studies
The crystallographic structure of CYP119 with a resolution of 1.93 Å was obtained from Protein Data Bank database (PDB code: 1F4T). The structures of the F135G and F135G/T213G mutants were built with Swiss‐Model (https://www.swissmodel.expasy.org/) by using the structure of CYP119 as template. Molecular docking study was carried out with AutoDockTools. The structure of thioanisole was downloaded from PubChemCompound database. Before docking simulation, solvent was removed from the protein structures. Heme and oxygen atom were retained and hydrogen atoms were added. The substrate thioanisole was treated as a flexible ligand and allowed to rotate freely. Autogrid (4.2) was used to generate grid maps for the ligand and the grid box was fixed to cover the catalytic site containing heme moiety. Local Search Parameters were selected and Docking Parameters were set by default in the docking process with Autodock (4.2) program. The docking results were analyzed by using Pymol.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
This project is supported by Sichuan Science and Technology Program (2019JDTD0016) and Program of Education Department of Sichuan Province (14TD0017). We are thankful to Dr. Qin Xu (School of Life Sciences and Biotechnology, Shanghai Jiao Tong University, Shanghai, China) for providing Gromacs package v.5.0.5 for molecular dynamics simulation.
X. Wei, C. Zhang, X. Gao, Y. Gao, Y. Yang, K. Guo, X. Du, L. Pu, Q. Wang, ChemistryOpen 2019, 8, 1076.
Contributor Information
Lin Pu, Email: lp6n@virginia.edu.
Prof. Dr. Qin Wang, Email: wq_ring@hotmail.com.
References
- 1. Groves J. T. in Cytochrome P450 Structure, Mechanism, and Biochemistry, 3rd ed (Eds.: P. R. Ortiz de Montellano), Kluwer: New York, 2005, pp. 1–44. [Google Scholar]
- 2. Shaik S., De Visser S. P. in Cytochrome P450 Structure, Mechanism, and Biochemistry, 3rd ed (Eds.: P. R. Ortiz de Montellano), Kluwer: New York, 2005, pp. 45–86. [Google Scholar]
- 3. de Montellano P. R. Ortiz, De Voss J. J. in Cytochrome P450 Structure, Mechanism, and Biochemistry, 3rd ed (Eds.: P. R. Ortiz de Montellano), Kluwer: New York, 2005, pp. 183–245. [Google Scholar]
- 4.
- 4a. Hrycay E. G., Bandiera S. M., Arch. Biochem. Biophys. 2012, 522, 71–89; [DOI] [PubMed] [Google Scholar]
- 4b. Hayakawa S., Matsumura H., Nakamura N., Yohda M., Ohno H., FEBS J. 2014, 281, 1409–1416; [DOI] [PubMed] [Google Scholar]
- 4c. Faponle A. S., Quesne M. G., De Visser S. P., Chem. Eur. J. 2016, 22, 5478–5483. [DOI] [PubMed] [Google Scholar]
- 5. Shoji O., Watanabe Y., J. Biol. Inorg. Chem. 2014, 19, 529–539. [DOI] [PubMed] [Google Scholar]
- 6. Koo L. S., Tschirret-Guth R. A., Straub W. E., Moënne-Loccoz P., Loehr T. M., de Montellano P. R. Ortiz, J. Biol. Chem. 2000, 275, 14112–14123. [DOI] [PubMed] [Google Scholar]
- 7. Yano J. K., Koo L. S., Schuller D. J., Li H., Ortiz de Montellano P. R., Poulos T. L., J. Biol. Chem. 2000, 275, 31086–31092. [DOI] [PubMed] [Google Scholar]
- 8.
- 8a. Puchkaev A. V., Koo L. S., de Montellano P. R. Ortiz, Arch. Biochem. Biophys. 2003, 409, 52–58; [DOI] [PubMed] [Google Scholar]
- 8b. Nishida C. R., de Montellano P. R. Ortiz, Biochem. Biophys. Res. Commun. 2005, 338, 437–445; [DOI] [PubMed] [Google Scholar]
- 8c. Liu Z., Lemmonds S., Huang J., Tyagi M., Hong L., Jain N., Proc. Natl. Acad. Sci. USA. 2018, 115, 10049–10058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.
- 9a. Kellner D. G., Hung S.-C., Weiss K. E., Sligar S. G., J. Biol. Chem. 2002, 277, 9641–9644; [DOI] [PubMed] [Google Scholar]
- 9b. Sheng X., Horner J. H., Newcomb M., J. Am. Chem. Soc. 2008, 130, 13310–13320; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9c. Wang Q., Sheng X., Horner J. H., Newcomb M., J. Am. Chem. Soc. 2009, 131, 10629–10636; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9d. Rittle J., Green M. T., Science 2010, 330, 933–937. [DOI] [PubMed] [Google Scholar]
- 10. Koo L. S., Immoos C. E., Cohen M. S., Farmer P. J., de Montellano P. R. Ortiz, J. Am. Chem. Soc. 2002, 124, 5684–5691. [DOI] [PubMed] [Google Scholar]
- 11. Puchkaev A. V., de Montellano Y P. R. Ortiz, Arch. Biochem. Biophys. 2005, 434, 169–177. [DOI] [PubMed] [Google Scholar]
- 12. Suzuki R., Hirakawa H., Nagamune T., Biotechnol. J. 2014, 9, 1573–1581. [DOI] [PubMed] [Google Scholar]
- 13. Rabe K. S., Kiko K., Niemeyer C. M., ChemBioChem 2008, 9, 420–425. [DOI] [PubMed] [Google Scholar]
- 14. Zhang C., Li J., Yang B., He F., Yang S.-Y., Yu X.-Q., Wang Q., RSC Adv. 2014, 4, 27526–27531. [Google Scholar]
- 15. Zhang C., Liu P.-X., Huang L.-Y., Wei S.-P., Wang L., Yang S.-Y., Yu X.-Q., Pu L., Wang Q., Chem. Eur. J. 2016, 22, 10969–10975. [DOI] [PubMed] [Google Scholar]
- 16. Wang L., Wei S., Pan X., Liu P., Du X., Zhang C., Pu L., Wang Q., Chem. Eur. J. 2018, 24, 2741–2749. [DOI] [PubMed] [Google Scholar]
- 17. Cryle M. J., De Voss J. J., Angew. Chem. 2006, 118, 8401–8403; [Google Scholar]; Angew. Chem. Int. Ed. 2006, 45, 8221–8223. [DOI] [PubMed] [Google Scholar]
- 18. Li C., Zhang L., Zhang C., Hirao H., Wu W., Shaik S., Angew. Chem. 2007, 119, 8316–8318; [DOI] [PubMed] [Google Scholar]; Angew. Chem. Int. Ed. 2007, 46, 8168–8170. [DOI] [PubMed] [Google Scholar]
- 19. Yin Y.-C., Yu H.-L., Luan Z.-J., Li R.-J., Ouyang P.-F., Liu J., Xu J.-H, ChemBioChem 2014, 15, 2443–2449. [DOI] [PubMed] [Google Scholar]
- 20. Wang J., Ilie A., Reetz M. T., Adv. Synth. Catal. 2017, 359, 2056–2060. [Google Scholar]
- 21. Fujishiro T., Shoji O., Watanabe Y., Tetrahedron Lett. 2010, 52, 395–397. [Google Scholar]
- 22. Ma N., Chen Z., Chen J., Chen J., Wang C., Zhou H., Yao L., Shoji O., Watanabe Y., Cong Z., Angew. Chem. Int. Ed. 2018, 57, 7628–7633; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2018, 130, 7754–7759. [Google Scholar]
- 23. McIntosh J. A., Heel T., Buller A. R., Chio L., Arnold F. H., J. Am. Chem. Soc. 2015, 137, 13861–13865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Shoji O., Fujishiro T., Nakajima H., Kim M., Nagano S., Shiro Y., Watanabe Y., Angew. Chem. Int. Ed. 2007, 46, 3656–3659; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2007, 119, 3730–3733. [Google Scholar]
- 25.
- 25a. Kumar D., Karamzadeh B., Sastry G. N., De Visser S. P., J. Am. Chem. Soc. 2010, 132, 7656–7667. [DOI] [PubMed] [Google Scholar]
- 25b. Sainna M. A., Kumar S., Kumar D., Fornarini S., Crestoni M. E., De Visser S. P., Chem. Sci., 2015, 6, 1516–1529; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25c. Wang B., Li C., Dubey K. D., Shaik S., J. Am. Chem. Soc. 2015, 137, 7379–7390; [DOI] [PubMed] [Google Scholar]
- 25d. Ramanan R., Dubey K. D., Wang B., Mandal D., Shaik S., J. Am. Chem. Soc. 2016, 138, 6786–6797. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary