

Fusion proteoforms represent all protein products that can be generated by translation of fusion transcripts. FusionPro is developed as a sensitive tool for detecting and annotating fusion transcripts and proteoform by analyzing RNA-Seq data. In this study, we found the evidence of fusion proteoforms present in MS/MS data and analyzed their translational patterns based on the sequence information which is obtained from FusionPro. Our pipeline will facilitate the proteogenomic identification of fusion-derived peptides and characterization of various oncogenic fusion proteoforms.
Keywords: Proteogenomics, Bioinformatics, Ovarian cancer, Mass Spectrometry, Translation, Customized database, Fusion proteoform, Fusion transcript
Graphical Abstract

Highlights
FusionPro, a versatile tool for studying fusion proteoforms, has been developed.
Fusion peptides were identified against a customized database built by FusionPro.
Types and features of fusion proteoforms were efficiently predicted by FusionPro.
Abstract
Fusion proteoforms are translation products derived from gene fusion. Although very rare, the fusion proteoforms play important roles in biomedical science. For example, fusion proteoforms influence the development of tumors by serving as cancer markers or cell cycle regulators. Although numerous studies have reported bioinformatics tools that can predict fusion transcripts, few proteogenomic tools are available that can predict and identify proteoforms. In this study, we develop a versatile proteogenomic tool “FusionPro,” which facilitates the identification of fusion transcripts and their potential translatable peptides. FusionPro provides an independent gene fusion prediction module and can build sequence databases for annotated fusion proteoforms. FusionPro shows greater sensitivity than the available fusion finders when analyzing simulated or real RNA sequencing data sets. We use FusionPro to identify 18 fusion junction peptides and three potential fusion-derived peptides by MS/MS-based analysis of leukemia cell lines (Jurkat and K562) and ovarian cancer tissues from the Clinical Proteomic Tumor Analysis Consortium. Among the identified fusion proteins, we molecularly validate two fusion junction isoforms and a translation product of FAM133B:CDK6. Moreover, sequence analysis suggests that the fusion protein participates in the cell cycle progression. In addition, our prediction results indicate that fusion transcripts often have multiple fusion junctions and that these fusion junctions tend to be distributed in a nonrandom pattern at both the chromosome and gene levels. Thus, FusionPro allows users to detect various types of fusion translation products using a transcriptome-informed approach and to gain a comprehensive understanding of the formation and biological roles of fusion proteoforms.
The term “proteoform” encompasses all the various forms of proteins that result from molecular or cellular modifications (1). Given their important biological roles, proteoforms have been included in the list of proteome parts examined in the global Chromosome-centric Human Proteome Project (C-HPP)1 (2–4). Fusion proteoforms are protein products that are translated from the fusion transcripts that form when two different transcripts join because of chromosomal translocation, structural variation, or trans-splicing. Fusion genes and their proteoforms are valuable in biomedical research because they can serve as new drug targets or disease biomarkers (5). The function of fusion proteoforms may be continually activated by deletion of the regulatory site or permanently repressed by loss of the active site, both of which can affect tumorigenesis. However, it is difficult to infer the functionality of fusion proteoforms without a clear understanding of their composition (5–7).
An example of a well-studied fusion proteoform is BCR:ABL1 (8), which is a fusion protein of BCR and ABL1 gene fusion. BCR:ABL1 causes chronic myeloid leukemia by continuous activation of the tyrosine kinase activity of ABL1 caused by the loss of its regulatory site. It is well known that the drugs that inhibit the tyrosine kinase activity of this fusion protein are effective in treating chronic myeloid leukemia (9). Another example is the ESR1:CCDC170 fusion proteoform, which has an N-terminal-truncated form of wild-type CCDC170 and has been recurrently found in an aggressive subset of ER-positive breast cancers (10). We were interested to find that both of these fusion proteoforms produce several fusion products that result from different fusion sites: 11 or more breakpoints in BCR:ABL1 and four in ESR1:CCDC170. These findings suggest that a fusion gene can be transcribed and translated into diverse forms and does not always form the same fusion proteoform. Therefore, it is important to develop a bioinformatics tool that can accurately screen potential fusion junction isoforms and characterize the diverse forms of their fusion proteoforms (6).
Proteogenomics involves the comprehensive and integrative study of proteomes by annotation and identification of novel peptides based on genomic or transcriptomic data (11). Several proteogenomic approaches have been proposed to identify or predict unannotated fusion proteoforms. For example, Frenkel-Morgenstern et al. (12) identified 12 fusion proteins from mass spectrometry data sets of normal human tissues using six- or three-frame translated sequences of chimeric transcripts that comprise expressed sequence tag (EST) and mRNA. Similarly, Sun et al. (13) identified 11 fusion proteins in nonsmall cell lung cancer samples using chimeric transcript data. However, both studies used only fusion sequences that had been deposited in the database (DB) because of the lack of efficient bioinformatic tools for the study of sample-specific fusion proteoforms. Therefore, the research on fusion events emphasizes the importance of developing a proteogenomic tool that has two specific functions: (1) the detection of sample-specific fusion transcripts and (2) the construction of a customized sequence database of fusion proteoforms for tandem mass spectrometry (MS/MS)–based fusion identification.
Recent advances in RNA sequencing (RNA-Seq) technology have enabled investigators to analyze several sample-specific novel fusion genes in silico (14). These sample- or cell-specific fusion genes can be identified using fusion prediction programs called “fusion finders” that map distant gapped reads from RNA-Seq data. However, none of the fusion finders have shown satisfactory performance in the evaluation of simulated or biological data sets (15). As in the case of BCR:ABL1, fusion genes often have multiple junctions, and in general, it is difficult to detect every fusion site simultaneously using a single fusion finder because of their limited sensitivity. Thus, improving the fusion prediction power is the first step toward the identification of more fusion proteoform-derived peptides. The next step is to construct a full-sequence DB to identify and characterize the fusion proteoforms. Only two tools have yet been developed to annotate and identify sequences of fusion transcripts and fusion proteoforms: INTEGRATE-neo (16) and ProTIE (17). INTEGRATE-neo was developed to predict fusion peptides for identification of gene fusion neoantigens. Although the pipeline enables the prediction of possible fusion peptide neoantigens, it does not provide the full sequences of all possible proteoforms necessary for general annotation and characterization. ProTIE is a proteogenomic pipeline used to identify microstructural variant peptides. However, this framework can only generate fusion peptide sequences from RNA-Seq-based analyses, not from user-specified fusion transcripts. Thus, we believe it necessary to develop an efficient proteogenomic tool that offers more sensitive prediction and versatile functions than the currently available tools for determination of interesting fusion junctions and their potential translation products as peptides from MS/MS data.
In this study, we develop a versatile proteogenomic tool, named FusionPro, which aims to facilitate the accurate prediction and annotation of fusion transcripts and the construction of “customized DBs” that contain full-length sequences of fusion proteoforms or the sequences of the tryptic fusion junction peptides. FusionPro is a modular pipeline that allows all the constituting modules to be easily run with a single installation of references and a single configuration file. Here we present the key features of FusionPro with some case studies in which our pipeline efficiently identifies the fusion transcripts and their potential translation products in leukemia cell lines (Jurkat and K562) and ovarian cancer data sets from the Clinical Proteomic Tumor Analysis Consortium (CPTAC) (18, 19).
FusionPro was developed in Python and Perl and is freely available for academic purposes at the Bitbucket repository (https://bitbucket.org/chaeyeon/fusionpro).
EXPERIMENTAL PROCEDURES
Details of the Modules Included in FusionPro
FusionPro comprises the following eight modules, which are used to detect the putative fusion transcripts and create a customized DB of fusion proteoforms: CreateReference, RunSoapfuse, RunTophatfusionS, RunMapspliceS, RunCufflinks, ProcessResults, FindFusionProteoform, and MergeMultipleResults. The CreateLibrary module generates all the reference data sets required in the FusionPro processes. The Run[Program Name] modules execute certain fusion finders to determine the fusion transcript candidates. In particular, the last “S” of RunTophatfusionS and RunMapspliceS represents tools that are run using the processed intermediate output of the RunSoapfuse module. Filtration and integration of the output of the three fusion finders are performed by two modules, RunCufflinks and ProcessResults. In the final step, a customized DB is built using the FindFusionProteoform module, and the results of multiple samples are merged using the MergeMutipleResults module. These modules can also be run using a module called RunPipeline with a single configuration setting. In addition, a customized DB can be generated by analyzing user-specified gene fusion information using another standalone Perl script, FusionPro-C.pl. The internal workflow of FusionPro is delineated in supplemental Fig. S1, and the detailed processes are described in the following subsections. Note that all of the programs used in FusioPro require a moderate amount of memory (up to 8 GB).
Fusion Gene Calling
The SOAPfuse (v1.27) (20) algorithm aligns whole RNA-Seq reads, first against the whole genome (WG) and then against the annotated transcripts. During these steps, discordantly mapped reads are collected after the WG mapping, and unmapped reads are collected after both the WG and transcriptome alignment to create fastq-format data for further analysis using TopHat-Fusion (v2.0.14) (21) and MapSplice2 (v2.1.8) (22). Discordant read pairs that encompass a fusion junction indicate the existence of a fusion transcript. Moreover, unpaired and unmapped reads can span a fusion junction and thus provide direct evidence of such an element. FusionPro then skips the redundant WG mapping of TopHat-Fusion and MapSplice2, which has already been performed by SOAPfuse, and thus significantly reduces the running time while exercising all the junction alignment strategies. Instead, TopHat-Fusion is executed with both discordant and unmapped reads. In contrast, MapSplice2 is executed with only unmapped reads because the algorithm does not require paired-end read alignment and instead splits all of the reads to detect the fusion junctions. Finally, the fusion gene candidates are acquired by the above workflow using the three fusion finders (SOAPfuse, TopHat-Fusion, and MapSplice2), which use different strategies for discordant read mapping (supplemental Fig. S2).
Filtration of False Gene Fusion Predictions
To prevent increasing erroneous discoveries by combining the results, FusionPro removes fusion genes that are matched with the following criteria, represented by the IPX (Intronic junction, Paralog/Pseudogene, no eXpression) filtration module: (1) at least one of the fusion junction coordinates falls into the intron region; (2) two gene fusion partners have a paralog/pseudogene relationship; and (3) at least one of the two partners has no significant fragments per kilobase of exon model per million reads mapped (FPKM), that is normalized estimation of gene expression, calculated by Cufflinks (v2.2.1) (23) (default FPKM cut-off is set to 0). Overall, 97,204 paralog gene pairs were obtained using Ensembl biomart (https://www.ensembl.org/biomart) by searching the paralog list of protein-coding genes (downloaded on July 21, 2017). A list of 10,371 pseudogene-wild-type gene pairs was downloaded from gencode version 10 of the psiCube database (24).
Splicing Site Selection
We assumed that canonical transcripts with no expression were not involved in the formation of the fusion transcripts. Thus, FusionPro only selected the fusion transcripts that had Cufflinks FPKM values above the user-defined cut-off (default = 0) used in the fusion gene-level filtration step. A fusion transcript annotated as exon or exon boundary for its fusion junction was then selected when applying the intron filtration (supplemental Fig. S3). A fusion transcript is commonly defined as a putative transcript if it has both fusion junctions located on the exon boundary or within an exon region. Therefore, performing the intron filtration step is strongly recommended.
Customized DB Generation
A fusion proteoform sequence was generated by combining the Ensembl cDNA sequence of each fusion transcript partner. Candidate transcripts represented by every start to stop codon and having at least 30 nt were three-frame translated. In addition, we categorized the fusion proteoform sequences according to their sequence origin: a truncated or frameshift sequence of each partner sequence or a combination of two partner sequences. This annotation information was also included with its sequence in the FASTA-formatted customized DB.
Fabrication of the 50-Fusion Transcript-Simulated RNA-Seq Data Sets
We randomly extracted 50 genes from 19,805 protein-coding genes from Ensembl (GRCh38.p10; https://www.ensembl.org) and prepared their genome models to create synthetic fusion genes. Theoretically, a single simulation could have generated a maximum of 2450 distinct fusion genes from a combination of the 50 genes. However, it was still necessary to perform multiple simulations to create fusion junction isoforms with the same gene pairs but different junction positions because fusion genes often have multiple fusion breakpoints and recurrent partners in real situations (7). Thus, based on these 50 genes, we simulated 200 fusion genes per simulation process using the ChimSim program introduced in the ChimePipe study (25) and produced 978 different fusion transcripts by repeating this process five times. Briefly, in each repetition, 50 fusion genes were simulated for four of five classes except the readthrough in the five classes that ChimSim could produce (i.e. read-through, intrachromosomal, inverted, interstrand, interchromosomal). Among the 978 different fusion transcripts, we randomly selected 50 fusion transcripts, including 21 fusion gene pairs with one junction, 10 fusion gene pairs with two junctions, two fusion gene pairs with three junctions, and one fusion gene pair with three junctions. The details of the simulated fusion transcripts are presented in supplemental Tables S1 and S4. The 102,149 nonchimeric sequences provided by the ChimPipe study were then downloaded and merged with the 600-bp fusion junctional cDNA sequences of the 50 fusion transcripts. The Art-illumina program (v2.5.8) (26) was used to build three synthetic RNA-Seq data sets, based on the concatenated sequences of the chimeric and canonical cDNAs with read lengths of 50, 75, and 100 bp. Each simulated RNA-Seq data set is fabricated for both 20X and 5X coverage, and the mean fragment lengths were set to 200, 250, and 300 bp according to the 50-, 75-, and 100-bp sequences in the data sets, with standard deviations of 20, 25, and 30 bp, respectively.
Performance Evaluation of the Fusion Finders by Analyzing Gold Standard Data Sets
To test the improved prediction power of FusionPro's integrative fusion calling module, we analyzed the 50-fusion transcript-simulated data and the fusion gene-validated biological data. For the biological data set, we downloaded RNA-Seq data on four breast cancer cell lines (BT474, MCF7, SKBR3, and KPL4) from the TopHat-Fusion website (https://ccb.jhu.edu/software/tophat/fusion_tutorial.shtml) and RNA-Seq data on 13 glioma samples. The breast cancer cell line data set was originally produced by Edgren et al. (27), and the replicates of BT474 and SKBR3 in the data were merged by Kim and Salzberg (21). The 13 glioma samples that have two or more validated fusion genes were selected and downloaded from the SRA archive by searching the accession number [SRP027383] (28). All of the reference files for the FusionPro analysis were downloaded simultaneously by FusionPro using Ensembl version GRCh38.82 and UCSC genome version hg38. The parameters used for each program and the version information of the programs are provided in supplemental Table S2.
Experimental Design and Statistical Rational
Cell Culture and Cell Preparation
Jurkat clone E6-1 and HEK293 cell lines were purchased from the Korean Cell Line Bank (Seoul, Republic of Korea). The cell lines were cultured in RPMI 1640 (Thermo Fisher Scientific) and DMEM (Thermo Fisher Scientific), respectively, containing 10% (v/v) FBS, 100 units/ml of penicillin, and 100 μg/ml of streptomycin. The cells were washed with phosphate-buffered saline solution (PBS) and harvested via centrifugation.
Total RNA Preparation and Validation of the Fusion Transcripts
Total RNA was extracted using TRIzol reagent (Thermo Fisher Scientific) and purified using the RNeasy kit (Qiagen) according to the manufacturer's instructions. The extracted RNA was reverse transcribed using a Transcriptor First Strand cDNA synthesis kit (Roche) with oligo-dT priming. The fusion transcript of FAM133B and CDK6 was amplified from Jurkat clone E6–1 cell cDNA by polymerase chain reaction using Phusion High-Fidelity DNA Polymerase (NEB). The long and short isoforms of the amplified fusion transcripts were separated by gel electrophoresis on 1% agarose gel, and the purified fusion transcripts were then ligated into pTOP TA V2 (TOP clonerTM TA core kit, Enzynomics). The sequences of the primers used in this experiment are provided in supplemental Table S3. The sequences of the constructed plasmids were validated by sequence analysis (Macrogen, Republic of Korea).
Protein Preparation and Western Blot Analysis
For the whole cell protein, the cell pellets were homogenized in modified RIPA buffer (50 mm Tris-HCl, 150 mm NaCl, 1% NP-40, and 0.25% sodium deoxycholate, pH 7.4) with protease inhibitor (Roche) and centrifuged at 14,000 rpm for 20 min, and the supernatants were then collected. For the subcellular fractionation, we modified the previous method (29). Briefly, Jurkat cells were disrupted with a glass homogenizer (Type B) with hypotonic buffer (10 mm HEPES, 1.5 mm MgCl2, 10 mm KCl, and 1 mm DTT (Dithiothreitol), pH 7.9, 4 °C) containing protease inhibitor (Roche). The cell lysates were centrifuged, and the supernatants were collected by low-speed centrifugation (4000 rpm for 15 min) (cytosolic fraction). The pellets were homogenized with a gradient high-salt hypotonic buffer (0.2∼1.6 M KCl), and the supernatants were then collected by high-speed centrifuge (12,000 rpm for 45 min) (nucleus fraction). The cytosolic and nucleus fractions were mixed (intracellular fraction, IC) and the mixture was used for the CDK6 detection. Cell debris was extracted in a modified RIPA buffer with a protease inhibitor and centrifuged, and the supernatants were then collected (membrane fraction). All supernatants were treated with 10% trichloroacetic acid (10%, v/v), the precipitates were dissolved in urea buffer (7 m urea, 2 m thiourea, 4% CHAPS, 50 mm DTT and 30 mm Tris, pH 8.5), and the protein concentration was determined using a 2D Quant kit (GE Healthcare). Fifty micrograms of protein were loaded onto a 10% SDS-PAGE gel and transferred to a nitrocellulose membrane. Blocking was carried out with PBS containing 5% skim milk and 0.2% Tween-20 for 1 h at room temperature. The membrane was incubated with primary antibodies overnight at 4 °C. After washing, the membrane was incubated with secondary antibodies for 1 h at room temperature. The band signal was developed by ECL plus reagents (GE Healthcare) and scanned using a Typhoon 9400 scanner (GE Healthcare). The anti-CDK6 (C-terminal epitope 300–326) and anti-β-tubulin were purchased from Abcam and Santacruz.Bioinformatics Analysis for Identifying Fusion-Derived Peptides in the MS/MS Data (Jurkat and K562).
First, raw MS/MS data of Jurkat (PeptideAtlas I.D.: PASS00215) (30) and K562 (ProteomeXchange I.D.: PXD002395) (31) were downloaded and converted to mgf format using MSConvert of the ProteoWizard software (v3.0.10462) (32). Next, the mgf files were searched by Mascot (v2.6.0, Matrix Science) against the customized DB from FusionPro concatenated with the neXtProt database (released on January 17, 2018; 42,241 entries) (33) to identify the fusion-derived peptides in the Jurkat and K562 cell lines. To analyze these MS/MS data sets of trypsin-digested peptides, semi-trypsin and two missed cleavages were set for the peptide digestion information, and carbamidomethylation on cysteine and oxidation on methionine were set for the fixed and variable modification. Further parameters were set as follows: the fragment tolerance was set to 0.8 Da, peptide tolerance was set to 40 ppm, and peptide charge was specified as 2+, 3+, and 4+. The Mascot taget-decoy function was checked to calculate the false discovery rate (FDR) of the peptide-spectrum matches, and only the fusion-derived peptides in the 1% FDR at the peptide-spectrum match (PSM) level were considered.
CPTAC Ovarian Cancer Samples and Bioinformatics Analysis for Fusion Peptide Identification
The CPTAC has produced comprehensive proteome data on ovarian cancer using the isobaric tags for relative and absolute quantification (iTRAQ) method for absolute and relative quantification (19). Sixty-two sets of global proteomic data sets on trypsin-digested peptides containing 158 clinical samples were downloaded from the CPTAC data portal. Thirty-four experiments were analyzed by Johns Hopkins University and 28 by Pacific Northwest National Laboratory. To identify the fusion junction peptides, mzML-formatted data of the ovarian cancer samples were searched by Mascot (v2.6.0) against the neXtProt protein sequences (January 17, 2018; 42,241 entries) together with the customized DB, which was built by FusionPro based on the data from two public repositories of a fusion gene: TumorFusions (34) and ChimerDB 3.0 (35). TumorFusions is a TCGA fusion gene database that stores PRADA (36) predicted data on the fusion genes of TCGA tumor samples. ChimerDB 3.0 also provides possible fusion transcripts based on the analysis of the TCGA RNA-Seq and EST data. In the data analysis of CPTAC MS/MS data sets, the quantification option was set to iTRAQ 4plex, the fragment tolerance was set to 0.4 Da, and the peptide tolerance was set to 20 ppm. The other parameters and the FDR cut-off were set as the same as those in the aforementioned method for the cell line analysis.
Identification of Antibody-Captured Protein by LC-MS/MS
The target band of the preparative gel was sliced, destained, reduced with 50 mm DTT, alkylated with 25 mm IAA (Iodoacetamide), and then digested with 1/10-fold trypsin per protein containing 50 mm ammonium bicarbonate buffer. The peptide solution was collected, evaporated, and dissolved with 3% formic acid. The peptide separation and identification were analyzed using liquid chromatography (LC) and MS with a LTQ-Orbitrap XL MS (Thermo Fisher Scientific, CA) coupled to an EASY-nLC 1000 system (Thermo Fisher Scientific). A C18 silica-packed nanobore column (ZORBAX 300SB-C18, 150 mm × 0.1 mm, 3-μm pore size, Agilent) was used for peptide separation. The mobile phase A for the LC separation comprised 3% acetonitrile and 0.1% formic acid in deionized water, and the mobile phase B was 0.1% formic acid in acetonitrile. The chromatography gradient was designed to increase linearly from 0% B to 32% B in 40 min, 32% B to 60% B in 4 min, 60% B to 95 B in 4min, 95% B in 4 min, and 0% B in 6 min. The flow rate was maintained at 1500 nL/min. The settings used for the MS analyses for identification of the antibody-captured proteins were mass resolution setting for full MS scan: 30,000; mass range for MS1: 300 to 1500; isolation window for precursor ion selection: 5; number or precursors selected for tandem MS in each scan event: 10; intensity threshold for MS2: 100,000.0; charge-state screening parameters: 2 or more; relative collision energy: 35.0 in CID (collision-induced dissociation) and 40.0 in HCD (higher-energy collisional dissociation); mass analyzer for MS2: ion trap; dynamic exclusion settings: enable (repeat count: 1; repeat duration: 20.00, exclusion list size: 20.00, exclusion mass width relative to mass; exclusion list size: 20.00, exclusion mass width relative to low (ppm): 10.00; and exclusion mass width relative to high (ppm): 10.00. The MS data files were processed using Proteome Discoverer (v1.4; Thermo Fisher Scientific). The spectra were searched against the sequences of neXtProt database (January 17, 2018), and FAM133B:CDK6 fusion junction tryptic peptide (PTW:LFDVCTVSR) by Mascot (v2.6.0) with global 1% FDR cut-off. The other parameters were set as follows: fixed modification was set to carbamidomethylation at cysteine, fragment tolerance was set to 0.8 Da, peptide tolerance was set to 40 ppm, enzyme was set to trypsin with 1 missed cleavage, and peptide charge was set to 2+, 3+. The raw mass spectrometry data were deposited in a MassIVE database (MassIVE I.D.: MSV000083822).
RESULTS
Overview of the FusionPro Functionalities
General Features of FusionPro
One of goals of FusionPro is to provide a FASTA-formatted customized DB that contains full-length sequences of all possible fusion proteoforms. The customized DB is mainly used for further MS/MS-based identification of fusion proteoforms. To provide in-depth annotations of the fusion products and make the customized DB, our pipeline is implemented either by using the custom input of the fusion transcripts or by predicting sample-specific fusion transcripts from RNA-Seq data sets (.fastq) using our own FusionPro modules (Fig. 1 and supplemental Fig. S1).
Fig. 1.

Data analysis workflow of FusionPro. FusionPro contains modules that provide predictions of the fusion transcript and fusion proteoform sequences. First, the gene fusion prediction module provides a list of candidate fusion transcripts annotated by selective integration of the workflow of the three fusion finders (SOAPfuse, TopHat-fusion, and MapSplice2). From the predicted fusion genes or user-specified fusion genes, the “IPX” filtration module filters out the false positives and integrates the results into a single data format. Finally, the fusion cDNA sequences are three-frame translated and annotated to construct a customized DB for the identification of the fusion proteoforms.
Description of the FusionPro Modules and Strategies
FusionPro contains some unique features that can enhance the data processing modules for the prediction, construction, and annotation of fusion proteoforms. FusionPro also introduces an integrative fusion gene prediction module that selectively integrates the workflow of three popular fusion finders: SOAPfuse (20), TopHat-Fusion (21), and MapSplice2 (22) (Fig. 1). These fusion finders were selected for two technical reasons. First, because the programs can share the same Ensembl reference to consistently output the fusion junction coordinates, the results can be combined as a single output. Accordingly, FusionPro can automatically generate a comprehensive reference and execute modified codes of the three fusion detectors using the same version of the Ensembl references. This feature makes it easy to set up and use the fusion-calling module of FusionPro. Second, the fusion finders use different strategies for fusion junction inference (supplemental Fig. S2) (37). We hypothesized that integration of the results from various fusion finders that use different strategies would allow the detection of more fusion genes than with a single tool because each mapping method might provide unique predictions of the fusion junctions. We used three typical strategies to align the discordant reads that can support fusion junctions. The first strategy allows the paired-end reads to be aligned against the genome and transcriptome data and constitutively trims the unmapped reads to map against the annotated transcripts. For example, the SOAPfuse program uses this first strategy and shows good prediction power. The second strategy, which is used in TopHat-Fusion, enables prediction of the fusion junctions by fragmenting the unmapped reads instead of trimming them. The third strategy is the “direct fragmentation” approach used in MapSplice2, which splits the RNA-Seq reads in the first step to directly detect the fusion junctions from pieces of the whole reads.
However, rather than simply integrating the fusion finders in a parallel manner, our FusionPro offers a selective integration function by using SOAPfuse to perform the initial alignment of the raw RNA-Seq data and provides unmapped and discordant reads as inputs to TopHat-fusion and MapSplice2. This selective integration of multiple fusion finders eliminates the redundant and time-consuming tasks of whole-genome or transcriptome mapping (Fig. 1) and can improve the prediction sensitivity while efficiently avoiding an increase in the running time.
We used a heuristic filtration step to remove possible false candidates with intronic fusion junctions, no expression, or highly similar fusion partners from the predicted or specified fusion transcripts. We used Cufflinks (v2.2.1) (23) to filter out the unexpressed parent transcripts by calculating the FPKM values based on the supporting reads of the fusion transcripts (supplemental Fig. S3). This process used the reads (.bam) that were initially unmapped or discordantly mapped by SOAPfuse but consequently mapped by TopHat-fusion. Given that fusion genes tend to have an exonic fusion junction generated by an intronic fusion breakpoint (38) and that fusion finders often make inaccurate predictions because of the similar sequences of fusion partners, which are paralogs or have wildtype-pseudogene relationships, this process enabled the elimination of fusion partners with intronic fusion junctions and homologous sequences (supplemental Fig. S3). Thus, this filtration strategy is one of the key functions of FusionPro, which is designed to maximally reduce the number of redundant and irrelevant transcripts that will be used to make the final fusion proteoform sequences.
FusionPro is also able to annotate fusion transcripts and their translation products using the genome model (.gtf) and Mitelman Database of Chromosome Aberrations and Gene Fusions in Cancer (http://cgap.nci.nih.gov/Chromosomes/Mitelman). The annotations of the predicted fusion transcripts and fusion proteoforms provide not only fundamental information on the parent genes (i.e. Ensembl transcript symbol, chromosome number, and regional coordinates), but also detailed information on the fusion events, such as the fusion junction coordinates, region, and Mitelman database references. We suggest that the integrative fusion detector, heuristic filter, and fusion annotator modules make FusionPro a more versatile and powerful tool for the detection of potential fusion transcripts and fusion proteoforms.
Evaluation of FusionPro Using Simulated and Real Data Sets
The prediction performance of the integrative fusion gene calling module of FusionPro was estimated and compared with five well-known benchmarked fusion finders: SOAPfuse (20), TopHat-Fusion (21), MapSplice2 (22), FusionCatcher (39), and JAFFA (40). FusionCatcher and JAFFA were selected because their performance exceeds those of other currently available programs (15). We used these fusion finders to analyze both simulated RNA-Seq data (supplemental Tables S1 and S4) and biological RNA-Seq data with validated fusion genes (supplemental Table S5) and to compare their performance to that of FusionPro.
Benchmark Test Using the Simulated Data Sets
To more accurately evaluate the performance of the fusion finders, we processed newly simulated RNA-Seq data sets, which are the first case of mimicking real gene fusion events with multiple junctions and recurrent partners. Fifty fusion transcripts from 33 random pairs of fusion genes were simulated by reusing the gene partners or by creating two or more junction positions (supplemental Tables S1 and S4). These simulated RNA-Seq data were generated in 50-, 75-, and 100-bp read lengths with 20× coverage using ChimSim (25). The preparation process for the simulated data sets is described in the Experimental Procedures. These data sets are suitable for evaluation of the fusion junction-level sensitivity, which is calculated by the number of fusion junctions, rather than the fusion gene pair-level sensitivity that is usually evaluated in other studies. They can be used as a gold standard data set for the accurate evaluation of the sensitivity of any available fusion finders. The average of the performance measures (Recall, also referred to as sensitivity, Precision, and F-score; Equation 1) for the three simulated RNA-Seq data sets (50, 75, and 100 bp) indicates that FusionPro not only predicted more fusion junctions but also recorded the highest F-score, an indicator of the comprehensive prediction power that considers both precision and recall (Fig. 2A and supplemental Table S6). FusionPro also showed a higher recall value than the other tools when the recall value was calculated as the number of predicted fusion junctions rather than as the predicted fusion gene pairs (Fig. 2A). Similarly, FusionPro usually produced the highest recall values at the fusion transcript-level for the simulated data with different read lengths and low coverage (5×) (supplemental Fig. S4).
Fig. 2.

Performance evaluation of FusionPro and the other fusion finders in the analyses of simulated and real data sets. A, The average of the precision, recall, and F-score values of the six fusion finders and the combined results of S, T, and M (SOAPfuse, TopHat-fusion, and MapSplice2) were obtained from the analysis of 50-, 75-, and 100-bp simulated RNA-Seq data sets containing 50 simulated fusion transcripts. The fusion-transcript-level recall value considers not only the accurate gene fusion pairs but also the precise junction locations. Based on the F-score and transcript-level recall value obtained from this evaluation, of the six fusion finders, FusionPro was found to be the most reliable tool with the highest sensitivity in detecting fusion junctions. B–C, Comparison of the prediction power of five currently available fusion finders and FusionPro estimated by analyzing real data sets ((B) breast cancer cell lines and (C) gliomas). The numbers on the bar indicate the number of true and false predictions. B, FusionPro predicted 116 fusion genes and 46 validated fusion genes with the highest recall values when analyzing the four breast cancer cell lines (BT474, MCF7, SKBR3, and KPL4). C, FusionPro predicted all the 31 validated fusion genes in the glioma samples. The fusion gene prediction results for the (B) breast cancer cell lines and (C) gliomas show that the selectively integrative approach and heuristic filtration module of FusionPro removes a significant number of false predictions when comparing with the simple union of SOAPfuse, Tophat-fusion and MapSplice2 results (S+T+M). *S+T+M, the merged prediction results of SOAPfuse, TopHat-fusion, and MapSplice2.
Benchmark Test Using the Real Data Sets
Further, we evaluated the prediction power of FusionPro and the other fusion finders using real data sets on breast cancer cell lines and gliomas. We first analyzed RNA-Seq data sets of breast cancer cell lines. The experiment was based on a list of 120 RT-PCR-validated fusion genes obtained from six validation studies of fusion genes in four breast cancer cell lines (BT474, MCF7, SKBR3, and KPL4) (27, 41–45) (supplemental Table S5). Because we used a larger number of validated fusion genes than in other studies, we expected that the performance of each fusion finder would be measured more accurately. Indeed, FusionPro predicted the truest fusion genes among the evaluated fusion finders, based on the prediction of 116 fusion genes and 46 verified fusion genes (Fig. 2B and supplemental Table S7). We next evaluated the performance of FusionPro by analyzing RNA-Seq data sets obtained from gliomas. We chose 13 samples that contained at least two RT-PCR-validated fusion genes from 274 glioma samples analyzed in a previous fusion transcript study (28). The 31 true fusion genes contained in the 13 samples are listed in supplemental Table S5. The results show that FusionPro identified all 31 true positives from a total 635 predictions, whereas the other tools predicted a maximum of 27 true fusion genes (Fig. 2C and supplemental Table S8).
Assessing the Power of the Filtration Module
To estimate the effectiveness of IPX filtration module of FusionPro, we compared the prediction result of FusionPro and the combined prediction results obtained from fusion finders making up FusionPro (SOAPfuse, TopHat-fusion, MapSplice2). Assuming that all predictions except for the validated fusion genes are false predictions, the false prediction of the real data set was greatly reduced to 15.7% for breast cancer and to 38.9% for glioma as in the simulation data analysis, although few true predictions were lost (Fig. 2B–2C). The results indicate that selective integration of the fusion finders and three heuristic filtration strategies of FusionPro efficiently removes the erroneous predictions that increased during the integration phase.
Application of FusionPro in Identifying Fusion Peptides from Leukemia Cell Lines
Prediction of Fusion Transcripts in RNA-Seq Data Using FusionPro
We used FusionPro to detect putative fusion transcripts and build a customized DB to identify fusion transcript-derived translated peptides in two leukemia cell lines, Jurkat and K562. We downloaded paired-end RNA-Seq data and MS/MS data of the Jurkat cell line of T-cell acute lymphoblastic leukemia, which Sheynkman et al. (30) generated to identify splice variants. Further, the proteogenomics data sets of K562 cells were acquired from different studies which produced RNA-Seq data (46, 47) and MS/MS data (31). Hereafter, the two K562 RNA-seq data sets are referred to as K562_Slavoff and K562_Berger for convenience. FusionPro detected 82 fusion gene pairs and 101 fusion transcripts in the Jurkat cell line and 281 and 95 fusion gene pairs with 363 and 296 fusion transcripts in the K562_Slavoff and K562_Berger data sets, respectively (Table I). Most of the fusion genes detected by FusionPro contained one fusion junction, but about 20% of the predicted fusion genes had two to eight fusion junctions (Fig. 3A). The K562_Berger cells contained 24 fusion genes with two or more breakpoints, resulting in approximately three times more fusion transcripts than the number of fusion gene pairs (Fig. 3A). Moreover, the detected fusion transcripts were not randomly distributed, but were concentrated regions, with asterisks indicating the top five regions of link density (10-Mbp window) (Fig. 3B). As indicated by asterisks, the top five regions of link density in the Jurkat cell line were observed on chr 2, chr 10 and chr 15. However, both K562 cell lines harbored marked regions on chr 3 and chr 11. Additionally, regions on chr1 and chr6, which were marked in the K562_Berger and K562_Slavoff lines, respectively, were also in top 10 in both data sets but were not observed in Jurkat cells. Overall, the link densities exhibit a cell line-specific junction pattern. These phenomena can be explained by the finding that the regions that involve copy number variations or tandem duplications are closely related to fusion gene formation (44, 48).
Table I. Numerical summary of fusion events in leukemia cell lines predicted by FusionPro.
| Fusion gene | Fusion junction isoform | Fusion proteoform | Fusion junction spanning sequence | Upstream protein truncated sequence | Downstream protein truncated sequence | Possible fusion-derived peptide (PSM FDR<1%) | |
|---|---|---|---|---|---|---|---|
| Jurkat | 82 | 101 | 3121 | 125 | 891 | 1809 | 2 |
| K562 (Slavoff) | 281 | 363 | 4461 | 269 | 1289 | 2343 | 2 |
| K562 (Berger) | 95 | 296 | 3964 | 191 | 842 | 1371 | 2 |
Fig. 3.

Characterization of the fusion junction formation on fusion genes and chromosomes in leukemia cells. A, Percentage of fusion junctions on the predicted fusion gene in leukemia cell lines. The numbers of fusion sites on each fusion gene are shown in different colors. The ratios of the numbers of fusion junctions are represented by a bar for each sample data set. B, The distribution of the predicted fusion transcripts is visualized as links on the chromosomes by a Circos plot. In the prediction results from each RNA-Seq data set, the top five regions of link density (10-Mbp window), which were determined by the numbers of fusion junctions, are marked with colored asterisks (*). Six asterisks are present in the Jurkat results because this cell line contained two equally ranked regions.
Construction of a Customized DB for Fusion Proteoforms
Based on the prediction results, we created a putative sequence of a fusion transcript by combining the cDNA sequence of each splice isoform that passed the filtration steps of our tool. This “filtering out first and combining next” approach of FusionPro helped to reduce the numbers of possible false sequences before conducting further MS/MS data analysis. These fusion cDNA sequences were then three-frame translated, and the resulting sequences of the fusion proteoforms were deposited in the customized DBs built for the Jurkat, K562_Slavoff, and K562_Berger cell lines (Table I). The identified fusion peptides were categorized into three types according to their sequence origin and frame: (1) C- or N-terminal truncated protein sequence, (2) frameshift sequence, and (3) combined sequence of two partner proteins. Among these types, only the last two can be discriminated from canonical protein sequences (supplemental Fig. S5). Note that the sum of the numbers of different forms may not have agreed with the total number of fusion proteoforms because some of the fusion proteoforms shared the same fusion junction sequence, although they had different splicing site.
Identification of Fusion-Derived Peptides from MS/MS Data
To identify the fusion-derived peptides, 28 MS/MS mass spectra raw files on Jurkat cells and 18 MS/MS spectral data on K562 cells were searched with MASCOT software (v2.6.0) against a neXtProt (January 17, 2018; 42,241 entries) database concatenated with each cell's customized DB built from the RNA-Seq data sets. This is so-called “all-together strategy” which maximizes the identification of novel peptides by merging the canonical proteome DB with the variant proteome DB and calculating the combined FDR value of both the wild-type and variant proteins (49, 50). During the identification process, the spectrum of the variant peptide does not incorrectly match the canonical peptide by using this strategy (50). Details of the bioinformatics analysis are presented in supplemental Fig. S5. Using the “all-together strategy,” MASCOT identified 151,287 (Jurkat), 119,383 (K562_Slavoff), and 119,650 (K562_Berger) PSMs when the PSM-level FDR cut-off was set as 1%. The FDR values can be considered reliable because despite a slight reduction in the number of canonical peptide identifications, the decoy identification was nearly identical when compared with a data analysis based only on neXtProt DB (supplemental Fig. S6). The only junction-spanning peptide that spanned at least two amino acids on either side of the junction was selected as direct evidence of fusion protein. This MS/MS ion search resulted in the matching of three spectra with two fusion-derived peptides in the Jurkat cells and eight spectra with four fusion-derived peptides in the K562 cells (Table II and supplemental Table S9). Of these six peptides, two were the frameshift peptides of the downstream parental protein and three were the junction-spanning peptides of the fusion protein. The last peptide, predicted from the K562_Slavoff data set, was a translation product of the upstream parent, which was a pseudogene. Although a stringent FDR cut-off of <1% was applied, all peptides that passed this threshold could not be described as confidently identified, given the imperfect assumption of the error types in the target-decoy approach. In addition, although frameshift peptides and pseudogene products were identified, further experimental analysis may be required to ensure that they came from the fusion events.
Table II. Potential fusion-derived peptides identified in leukemia cell lines.
| Sample | Fusion gene | Chr. | Fusion junction | Junction boundary | Peptide type | Peptide sequence | # of spectra |
|---|---|---|---|---|---|---|---|
| Jurkat | FAM133B:CDK6 | 7–7 | 92581506–92774831 | Exon-Exon | Fusion junction peptide | PTW:LFDVCTVSR | 2 |
| Jurkat | FAM24B:CUZD1 | 10–10 | 122855745–122839231 | Exon-Exon | Frameshift peptide | KGLKVTHRPAC | 1 |
| K562 (Slavoff) | HNRNPA1P13:HNRNPA2B1 | 5–7 | 136429638–26193641 | Exon-Exon | Fusion junction peptide | ALVVVDMVAVGMAIMDMV:VEDLDMATR | 1 |
| K562 (Slavoff) | HNRNPA1P58:HNRNPA2B1 | 6–7 | 24002209–26195899 | Exon-Exon | Peptide of pseudogene | QEMVSASSSQRGR | 3 |
| K562 (Berger) | RPL14:NCOR2 | 3–12 | 40462013–124402538 | Exon-Exon | Fusion junction peptide | ASPKKA:AAAAAAAH | 2 |
| K562 (Berger) | C20orf203:NOL4L | 20–20 | 32632893–32527913 | Exon-5'UTR | Frameshift peptide | LGEDLGV | 2 |
Case Study: Identification of FAM133B:CDK6 Fusion Transcripts and Protein in Leukemia Cell Lines by FusionPro
In the series of trials with the new FusionPro workflow, we were able to identify the fusion junction peptide of FAM133B:CDK6 in the Jurkat cell line. Our FusionPro analysis revealed, with a enough supporting reads, that this FAM133B:CDK6 fusion gene had two junction isoforms (supplemental Fig. S7). The two isoforms shared the same junction location in CDK6, but the junction locations of FAM133B differed in exon 1 and exon 2. For convenience of description, the fusion junction location of FAM133B can be divided into two types of isoforms: a “long isoform” with a fusion junction at exon 2, and a “short isoform” with a fusion junction at exon 1. The long isoform has been reported to be formed specifically in Jurkat cells and not in other leukemia cells and has been validated at the transcription level (51). However, the short isoform was a novel form discovered by FusionPro. For the RT-PCR validation and DNA sequencing of these two fusion transcripts, we designed a primer that captures two isoforms and another primer that is specific to the long isoform (supplemental Table S3). We found that the long and short fusion isoforms were separately detected in the Jurkat cell line and that each isoform had the same sequence previously predicted by FusionPro (Fig. 4). We also confirmed the presence of the long isoform using the long isoform-specific primer, which captured both sides of the fusion junction of the isoform (supplemental Fig. S8).
Fig. 4.
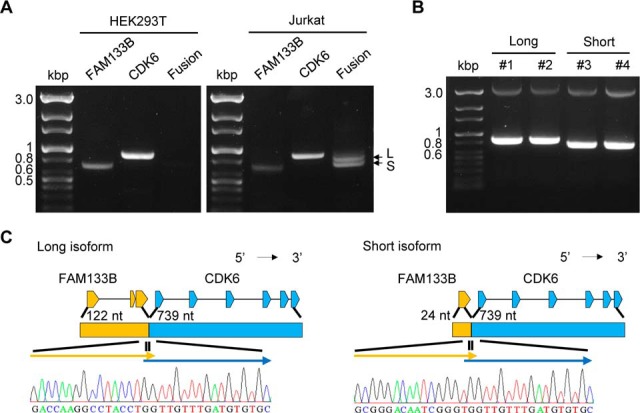
Molecular validation of two FAM133B:CDK6 isoforms and their protein product by RT-PCR amplification and DNA sequencing. A, FAM133B (740 bp), CDK6 (906 bp), and FAM133B:CDK6 (a long isoform (L, 870 bp) and a short isoform (S, 772 bp)) were amplified by RT-PCR in the HEK293 (negative control) and Jurkat cell lines. B, PCR amplification of the long (870 bp, Lane 1 and 2) and short isoforms (772 bp, Lane 3 and 4) after cloning each isoform. C, DNA sequencing results for each junction region of the two isoforms, which are well aligned with the predicted sequences obtained from FusionPro (refer to supplemental Fig. S7).
Although the short isoform did not have a translatable open reading frame, the long isoform was translated into a combined sequence of the FAM133B and CDK6 proteins that were confirmed by MS/MS-based identification of the fusion junction peptide with a stringent 1% FDR cut-off. Further, the FAM133B:CDK6 fusion protein was validated by comparing the spectrum of the identified fusion junction peptide with the spectrum of the synthetic peptide with the same sequence. These spectra showed a high cosine similarity score (0.911), which was calculated by the peak intensity and m/z value of the two spectra of the matched ions, representing the validation of the identified endogenous peptide (Fig. 5A). For molecular validation of the existence of fusion protein, a CDK6 antibody- and MS-based assay was performed in cellular fractions. Unlike the HEK293 cells, two bands representing the wildtype CDK6 (37 kDa) and the predicted fusion protein of long isoform (33 kDa) were detected in the intracellular fraction (IC) of the Jurkat cells (Fig. 5B). Using preparative gel, the antibody-captured bands were analyzed with tandem MS. The band at 37 kDa was identified as CDK6 with three unique peptides, and the “chimeric” band was identified as CDK6 with a unique peptide of CDK6, which is also included in fusion protein (Fig. 5C). This peptide was identified with a global FDR 1% cut-off, and its expectation value of 0.0043 indicates identity or extensive homology (p < 0.02687). Although a specific peptide (PTW:LFDVCTVSR) of the fusion protein was not detected, the data indicate that the band at 33 kDa is highly likely to be a FAM133B:CDK6 fusion protein for two reasons. First, the CDK6 antibody captures the C-terminal epitope of CDK6 (300–326 amino acids) and a fusion protein with a molecular mass of 33 kDa, and the chimera band is also located at about 33 kDa in our result (Fig. 5B). Also, CDK6 has not been reported to be cleaved or broken. Therefore, the identification of the common peptide of CDK6 and FAM133B:CDK6 indirectly demonstrated the presence of the fusion protein in this case.
Fig. 5.

Experimental validation and characterization of the FAM133B:CDK6 fusion protein. A, Validation of the identified fusion junction peptide (PTW:LFDVCTVSR) of the FAM133B:CDK6 fusion protein. The m/z value of the matched peaks of the synthetic peptide, which was synthesized based on the identified fusion junction sequence, is consistent with the m/z value of the original matched peaks of the endogenous fusion peptide. B, Dual position of CDK6 detected by antibody-based assay in the Jurkat and HEK293 cells. Fifty micrograms of each fraction of the Jurkat and HEK293 cell lysates were loaded onto a gel, and CDK6 (top panel) and β-tubulin as an intracellular marker (middle panel) were detected by Western blot analysis. The preparative gel was stained using CBB as a loading control (bottom panel). C, The spectrum shows the antibody-captured “chimeric” band (33 kDa) identified as the common peptide (ILDVIGLPGEEDWPR) of FAM133B:CDK6 and wildtype CDK6 by LTQ-Orbitrap-MS. D, The sequences of FAM133B, CDK6, and FAM133B:CDK6 fusion protein with domains from InterPro (https://www.ebi.ac.uk/interpro/) and p16-binding residues are shown. *WC, whole cell lysates; MB, membrane fraction; IC, intracellular fraction; CBB, Coomassie brilliant blue; Mr (calc), relative molecular mass of the matched sequence.
In this fusion protein, the N-terminal sequence of FAM133B (41-amino acid length) was linked to the sequence of the CDK6 protein that lost the N-terminal regulatory region (249-amino acid length), resulting in loss of ATP and p16 binding site (Fig. 5D) (52, 53). Reportedly, the CDK6 expression was higher in J.RT3-T3.5, a Jurkat-derivative cell line, than in the other leukemia cell lines (51). Further, the CDK6 protein regulates the G1 phase of the cell cycle (54) and is found in various types of leukemia to form fusion genes recurrently with other fusion partners, such as IGK (55, 56) and TLX3 (57). In summary, we validated and characterized a potential oncogenic FAM133B:CDK6 fusion protein that is translated from the longer one of two fusion junction isoforms of FAM133B:CDK6 through a proteogenomic workflow of FusionPro.
Application of FusionPro in Identifying Fusion Peptides from CPTAC Ovarian Cancer Data
CPTAC generated the MS/MS data set of The Cancer Genome Atlas (TCGA) samples, which contains qualitative and quantitative information on the proteins present in various clinical samples (18). To leverage the CPTAC data set for our proteogenomic work on fusion proteoforms, we used an independent script FusionPro-C that creates a customized DB using the user-specified information on the fusion transcripts. In this application study, we obtained the information on fusion events from TumorFusions (34) and ChimerDB 3.0 (35), which have predicted fusion transcripts from TCGA ovarian cancer data sets and other transcriptomic data (supplemental Table S10, see also Experimental Procedures). Using these data, we parsed the name, chromosome, strand, and junction coordinate of each fusion gene partner and used them as inputs to construct a customized DB using FusionPro.
Overall, we analyzed 687 distinct fusion junction isoforms (supplemental Table S10) using our tool and reference data sets corresponding to Ensembl GRCh37.72, of which 115 were filtered out for their intronic junctions, highly similar sequences of gene pairs, and obsolete annotations. Finally, the 16,159 fusion proteoform sequences, which were three-frame translated from 498 fusion transcripts, were deposited in the customized sequence DB, which contained 1809 unique fusion junction sequences. Using the abovementioned “all-together search strategy,” we identified total 5,216,091 PSMs and 15 fusion junction-spanning peptides that were well matched with 76 spectra in the 62 CPTAC ovarian cancer samples downloaded from the CPTAC Data Portal (58) (PSM-level FDR < 1%) (Table III and see supplemental Table S9, 11). In this process, six peptides were excluded because they did not properly cover fusion junctions (at least two amino acids on each side of the fusion junction) or were not the top-ranking peptide match on the spectrum. Also, we did not count 210 identified frameshift peptides as possible fusion-derived peptides in this analysis because the customized DB is not patient-specific and because frameshift events could occur for various reasons, such as insertion, deletion, or alternative translation initiation sites.
Table III. Fusion junction peptides identified in CPTAC ovarian samples.
| Fusion gene | Chr. | Fusion junction | Peptide sequence | # of spectra | Source DBa |
|---|---|---|---|---|---|
| ANXA2P2:ANXA2 | 9–15 | 33625242–60639739 | DTN:AERDALNIETAIK | 1 | C |
| **ANXA2P2:ANXA2 | 9–15 | 33625242–60639739 | SLYYYIQQDTN:A | 1 | C |
| *SPAG9:MBTD1 | 17–17 | 49156945–49258055 | DQ:MTTLQLK | 1 | C, T |
| EMID1:CBY1 | 22–22 | 29630338–39069164 | QGSAGQR:AGGRR | 18 | C, T |
| PPFIBP1:SMCO2 | 12–12 | 27677298–27654856 | EVCELCTL:MALTPTNLNNK | 15 | C, T |
| *CCDC6:ANK3 | 10–10 | 61665880–61802517 | ASVTI:QGEGFK | 3 | C, T |
| *CCDC6:ANK3 | 10–10 | 61665880–61802514 | ASVTI:GEGFK | 1 | C, T |
| MAPK9:UBL7 | 5–15 | 179718848–74749012 | IPAI:GAGALGSAGSMCV | 1 | C, T |
| OBSL1:STK11IP | 2–2 | 220430127–220467497 | PPPHVP:PSGTEHP | 2 | C |
| ACTN4:CAPN12 | 19–19 | 39217667–39220940 | VGWEQLLTTIA:LG | 23 | C |
| ACTN4:CAPN12 | 19–19 | 39219963–39220936 | ATCEAPD:YVFIMRAGETAEI | 1 | C |
| FBXO34:LGALS3 | 14–14 | 55738255–55604088 | RAGAAPGQAAAGAGAQ:WKPKP | 5 | C, T |
| SLC25A32:SMYD3 | 8–1 | 104419862–246518396 | FILGTLLFL:MEP | 1 | T |
| H19:COL1A1 | 11–17 | 2018215–48264480 | GSSGTAFSGLCRSLEG:PLALLVLLA | 1 | C |
| C2CD3:SYTL1 | 11–1 | 73849765–27671788 | LLS:SSVCPAGAQPQLMPQR | 1 | C, T |
| SH3BP5:METTL6 | 3–3 | 15372011–15425694 | MISTDGRLNLR:SQT | 2 | C, T |
*This fusion protein is also detected in other tissues according to the Mitelman Database.
**This fusion protein does not span junction properly, but another junction peptide (DTN:AERDALNIETAIK) supports this same fusion junction.
aC: ChimerDB 3.0; T: TumorFusions.
Among the identified fusion proteins, SPAG9:MBTD1 (59) and CCDC6:ANK3 (42, 43) have been found in the large intestine and breast cancer samples, according to the Mitelman database. CCDC6 was found to be involved in various fusion events as an upstream partner, and two protein isoforms of CCDC6:ANK3 were identified. This suggests that the fusion event occurs recurrently and can be stably translated to proteins in a cell. The remaining identified fusion partners have also been shown to produce fusion genes with other partners, which is consistent with studies in which the features of the DNA sequence resulted in recurrent formation of fusion genes regardless of the tissue source (7, 60).
Exploring the Pattern of the Fusion Junction Locations on Fusion Parental Genes
In our application studies, FusionPro detected 899 upstream transcript parents and 911 downstream transcript parents that formed distinct fusion transcripts as predicted from the transcriptome data on the Jurkat, K562, and ovarian cancer samples. Using the prediction results we attempted to explore the pattern of the fusion junction locations because this would help to predict the forms in which the fusion transcripts would mainly be translated. To this end, we created a model for detecting the pattern of fusion junction locations by establishing a working equation that calculated the relative position of a fusion junction on the fusion parents' longest cDNA, the length of which was considered as 1:
The upstream and downstream parental genes both showed a pattern of fusion junctions at the 5′ untranslated region (UTR) or 5′ side of the cDNA, as shown in Fig. 6A. This finding suggests that the fusion junctions of both parents often appeared near the 5′ UTR, and this trend could explain the research that showed that in general, downstream parents usually maintain their functional domains, whereas upstream parents tend to lose their domains or functions (7). However, in the upstream parent, the occurrence of fusion junctions increased again near the 3′ end (Fig. 6A, left panel), whereas in the downstream parent, the number of fusion junctions decreased toward the 3′ end (Fig. 6A, right panel). Based on this finding, we can also predict the major forms of a translatable fusion proteoform (Fig. 6B) and their expected functions. For example, upstream parents with a fusion breakpoint near the 3′ UTR should have an intact start codon and may be transcribed without miRNA-mediated perturbation while being translated into a canonical form (61). In addition, the protein product generated by the in-frame alternative TIS of the downstream parental protein can serve as an oncoprotein or a tumor-suppressor because of the loss of a regulatory site resulting from the N-terminal truncation. Additionally, alternative TIS products that were frameshifted or originated from an upstream parental protein, might still be translated. All those fusion proteoforms can be verified with immunoprecipitation-based technology. Validation of the various forms of the fusion proteoform would contribute to the characterization of its unknown functions. As mentioned above, fusion proteoforms may have unknown functions or effects depending on their translated form. For example, the biological role of fusion proteoforms may affect cellular translocation (62), increase the expression of the truncated parent gene (61), or suppress the tumor by acting as an immunopeptidome (16, 63). Future studies should focus on the various forms of fusion proteoforms and their oncogenic properties.
Fig. 6.

The proteogenomics study explains the expected forms of the fusion proteoform and the limited identification of the fusion peptide. A, The trend of the fusion junction locations on the longest cDNA of the 899 upstream parent of the fusion transcripts and 911 downstream parent of the fusion transcripts. The bar indicates the fusion site count in the window of relative length 0.05 when the total length of the cDNA is 1. B, Three major forms of a translational product of a fusion transcript deduced from the patterns of the fusion junction locations on the upstream and downstream parents. C, Tryptic peptide coverage on the predicted fusion junctions in the Jurkat, K562, and ovarian cancer tissues when zero or one missed cleavage is allowed. D, The amino acid composition of the fusion proteoform junction detected in the Jurkat, K562, and ovarian cancer samples. Approximately 30% of the fusion sites have trypsin cleavage sites (Lys or Arg).
Tryptic Peptide Coverage on Fusion Junctions
To investigate the number of fusion junctions that can be covered by a tryptic peptide, we performed in silico trypsin digestion of fusion proteins by Proteogest (http://sites.utoronto.ca/emililab/proteogest.htm) (64). Of the 2381 fusion junction sequences obtained from the K562, Jurkat, and ovarian cancer samples, nearly half of the fusion proteoform junctions could not be covered by a tryptic peptide with no missed cleavage (Fig. 6C). Although one missed cleavage was allowed, 458 fusion junctions (19%) still could not be detected by the MS/MS analysis of the trypsin-digested peptides (Fig. 6C). This poor coverage can be explained by the amino acid composition at fusion junction. Interestingly, 711 (30%) fusion junctions had at least one trypsin cleavage site of lysine (Lys, K) or arginine (Arg, R) (Fig. 6D). Thus, we suggest here that one of obstacles in identifying novel proteins by MS/MS analysis is the lack of unique tryptic peptides spanning fusion junctions. For example, although the BCR:ABL1 fusion transcript and its protein sequences were predicted in K562 cells, the protein product could not be identified by MS/MS analysis because of the trypsin cleavage site (K) at the fusion junction (QSS:KALQR).
Further, many fusion junctions have been reported to lie on the exon boundaries (65). In line with this finding, 46% (590 of 1276) of the fusion transcripts predicted in our study had a fusion junction on the exon boundary of either fusion parent. Studies have shown that the percentages of Lys and Arg that constitute the exon boundary are relatively high and that chymotrypsin and Asp-N greatly improves the peptide coverage of the exon-exon splice junction containing K/R residue (66). These findings corroborate our result that high levels of Lys and Arg are found at the fusion junction (Fig. 6D).
DISCUSSION
Because the proteoform has become one of the major targets of the C-HPP (3, 4), we attempted to create a versatile proteogenomic pipeline, named FusionPro, for identification and characterization of various fusion proteoforms, including the fusion protein, truncated protein, and frameshift protein. FusionPro was developed to make the most of the available resources by running with either RNA-Seq data or a list of interesting user-specified fusion transcripts.
Our comparative performance evaluation using two types of RNA-Seq data sets (simulated data sets and biological data sets) produced three noteworthy findings. First, we created simulated RNA-Seq data sets containing fusion junction isoforms and recurrent fusion partners to precisely evaluate the prediction capability of the fusion finders. Second, we gathered RNA-Seq data on biological samples (breast cancer cell lines and gliomas) and used a larger number of validated fusion genes in the corresponding samples than in the existing evaluation studies. Third, the fusion-detecting module of FusionPro outperformed the currently available five fusion finders in analyzing both types of RNA-Seq data sets. To identify novel fusion proteoforms it is essential to detect as many fusion transcripts as possible by increasing the coverage of the customized DBs. Our results indicate that the selectively integrated fusion caller and heuristic filter, which were designed to improve the sensitivity of the fusion finder, make FusionPro a fine proteogenomic tool for finding various types of fusion proteoforms.
However, the performance tests on the biological data sets may have underestimated the power of FusionPro for two reasons. First, although the breast cancer cell lines and gliomas used in the tests have been well studied for fusion genes, more novel fusion events may remain unexplored. For example, the SULF2-ZNF217 fusion gene, previously reported in breast cancer tissue (67) but not included in the list of validated fusion genes we referenced, was predicted by FusionPro, but was not predicted by SOAPfuse, which was the second most sensitive fusion finder in the analysis. The estimated sensitivity of FusionPro can be increased and the false predictions reduced by further validation of the novel fusion events. Second, a precise junction location was not specified in the validation set of breast cancer cells. Thus, although FusionPro predicted 160 fusion junction isoforms from 116 fusion genes in four breast cancer cell lines, we considered only the correct fusion gene pairs and ignored the various junction positions when calculating the sensitivity of the fusion finders. Because fusion genes tend to have multiple junctions, evaluation of accuracy based on the correct fusion junctions would have provided a more direct indicator of the performance power of the fusion finders. Because of these limitations of the validated data set, the precision, which can be biased by the number of not-yet-validated true events, was not considered in the benchmarking tests of the biological data sets.
In the application studies, FusionPro enabled the identification of 18 novel fusion proteins from two leukemia cell lines and CPTAC ovarian cancer tissues. This identification of fusion peptides led us to explore the potential oncogenic function of these proteins. For example, we were able to molecularly validate two predicted isoforms and a translation product of the FAM133B:CDK6 long isoform. This fusion protein was expected to function as an N-terminal-modified CDK6, which is likely involved in cell cycle progression by escaping the diverse regulation of suppressors or DNA methylation. The N-terminal region of CDK6, which was truncated during the generation of the fusion gene, contained a p16-binding site and a DNA methylation site (Fig. 5D) (68, 69). Thus, CDK6 down-regulation by methylation did not occur in this fusion event because the 5′ methylation site was removed. In addition, although the cell cycle progression mediated by CDK6 was inhibited by p16 binding, CDK6 Lys29 and Arg31 mutations have been reported to disrupt the binding of CDK6 and p16 (53, 54). However, this inhibition might not have occurred because the p16-binding site was removed from the FAM133B:CDK6 fusion protein (supplemental Fig. S9). Therefore, the resulting fusion protein might promote cell proliferation in Jurkat cells because the p16 could not bind to the N-terminal-truncated CDK6 for cell growth inhibition and the modified CDK6 could not be downregulated by 5′ DNA methylation.
We also showed that fusion sites appear at multiple positions with a certain pattern on the parent gene and are also not distributed uniformly on the chromosomes, which were discovered by analyzing the global fusion transcripts data obtained from our application studies. This emphasizes the important role that proteogenomics research has played in unveiling a variety of translation products that have nonrandom forms, which can be listed as a new family of the human proteome (1, 3). Although there are diverse fusion proteoforms, only some of them can be identified by trypsin digestion because of the poor tryptic peptide coverage on the fusion junction regions. Thus, semi-tryptic and maximum two missed cleavages were used as search options in our study, resulting in an increase of fusion junction coverage of tryptic peptide and the identification of more fusion junction peptides. Also, our pipeline provides in silico digestions of trypsin, chymotrypsin and Asp-N, which have been reported to enhance the detection of exon boundary junctions (66). With this inclusion, we aim to accelerate MS/MS experiments involving these enzymes and identify more fusion junction peptides.
In conclusion, our study has demonstrated that the FusionPro tool, which bridges genomics and proteomics, facilitated the collation of a variety of evidence and the characterization of fusion events from both RNA-Seq and MS/MS data sets. Our proteogenomic tool is capable of detecting more supporting reads for fusion events, which will allow users to discover more fusion junction isoforms and build sample-specific customized DBs for further MS/MS analysis. With the customized DB, users can identify more fusion-derived peptides obtained from MS/MS data and perform functional studies. This proteogenomic pipeline also helps characterize the potential roles of the fusion junction isoforms (e.g. FAM133B:CDK6) in Jurkat T cells. As the scope of the global C-HPP is extended to mapping human proteoforms (3, 4), FusionPro may contribute to this global project by deepening the pool of proteomes, which could provide potential biomarkers and drug targets for human cancers.
Data Access
FusionPro was developed using Python and Perl, and the open source program is available freely for academic purposes at the Bitbucket repository: https://bitbucket.org/chaeyeon/fusionpro.
The fusion-transcript-simulated RNA-Seq data sets can be downloaded at https://goo.gl/kvZWLm
DATA AVAILABILITY
Data supporting the findings of this study include raw MS/MS data and RNA-Seq data, and sources of data can be available within the Experimental Procedures section of the paper. RNA-Seq data of Jurkat (SRA project I.D.: SRP019939), K562 (SRA project I.D.: SRP000931 and SRP010061), and Glioma (SRA project I.D.: SRP027383) can be downloaded from the SRA archive (https://www.ncbi.nlm.nih.gov/sra). Raw MS/MS data of Jurkat (http://www.peptideatlas.org/, PeptideAtlas I.D.: PASS00215) and K562 (http://www.proteomexchange.org/, ProteomeXchange I.D.: PXD002395) were previously published and can be downloaded in each data repository. Also, the raw MS/MS data of CDK6 antibody-captured chimeric band were deposited in a MassIVE database (https://massive.ucsd.edu/ProteoSAFe/static/massive.jsp, MassIVE I.D.: MSV000083822).
Supplementary Material
Acknowledgments
The data used in this study were generated by the Clinical Proteomic Tumor Analysis Consortium (NCI/NIH).
Footnotes
* This study was supported by grants from the Korean Ministry of Health and Welfare: [HI13C2098] International C-HPP Consortium Project and [HI16C0257] (to Y.-K. Paik).
 This article contains supplemental material.
This article contains supplemental material.
1 The abbreviations used are:
- C-HPP
- Chromosome-centric Human Proteome Project
- EST
- expressed sequence tag
- DB
- database
- MS/MS
- tandem mass spectrometry
- CPTAC
- Clinical Proteomic Tumor Analysis Consortium
- WG
- whole genome
- FPKM
- fragments per kilobase of exon model per million reads mapped
- PBS
- phosphate-buffered saline solution
- DTT
- dithiothreitol
- IAA
- iodoacetamide
- FDR
- false discovery rate
- PSM
- peptide-spectrum match
- iTRAQ
- isobaric tags for relative and absolute quantification
- LC
- liquid chromatography
- CID
- collision-induced dissociation
- HCD
- higher-energy collisional dissociation
- WC
- whole cell lysates
- MB
- membrane fraction
- IC
- intracellular fraction
- CBB
- Coomassie brilliant blue
- Mr (calc)
- relative molecular mass of the matched sequence
- TCGA
- The Cancer Genome Atlas.
REFERENCES
- 1. Smith L. M., Kelleher N. L., and Consortium for Top Down, P. (2013) Proteoform: a single term describing protein complexity. Nat. Methods 10, 186–187 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Paik Y. K., Jeong S. K., Omenn G. S., Uhlen M., and Hanash S. (2012) The Chromosome-Centric Human Proteome Project for cataloging proteins encoded in the genome. Nat. Biotechnol. 30, 221. [DOI] [PubMed] [Google Scholar]
- 3. Paik Y. K., Omenn G. S., Hancock W. S., Lane L., and Overall C. M. (2017) Advances in the Chromosome-Centric Human Proteome Project: looking to the future. Expert Rev. Proteomics 14, 1059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Paik Y.-K., Lane L., Kawamura T., Chen Y.-J., Cho J.-Y., LaBaer J., Yoo J. S., Domont G., Corrales F., Omenn G. S., Archakov A., Encarnación-Guevara S., Lui S., Salekdeh G. H., Cho J.-Y., Kim C.-Y., and Overall C. M. (2018) Launching the C-HPP neXt-CP50 Pilot Project for Functional Characterization of Identified Proteins with No Known Function. J. Proteome Res. 17, 4042–4050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Mitelman F., Johansson B., and Mertens F. (2007) The impact of translocations and gene fusions on cancer causation. Nat. Rev. Cancer 7, 233–245 [DOI] [PubMed] [Google Scholar]
- 6. Casado-Vela J., Lacal J. C., and Elortza F. (2013) Protein chimerism: novel source of protein diversity in humans adds complexity to bottom-up proteomics. Proteomics 13, 5–11 [DOI] [PubMed] [Google Scholar]
- 7. Latysheva N. S. and Babu M. M. (2016) Discovering and understanding oncogenic gene fusions through data intensive computational approaches. Nucleic Acids Res. 44, 4487–4503 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Laurent E., Talpaz M., Kantarjian H., and Kurzrock R. (2001) The BCR gene and philadelphia chromosome-positive leukemogenesis. Cancer Res. 61, 2343–2355 [PubMed] [Google Scholar]
- 9. Druker B. J. and Lydon N. B. (2000) Lessons learned from the development of an abl tyrosine kinase inhibitor for chronic myelogenous leukemia. J. Clin. Invest. 105, 3–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Veeraraghavan J., Tan Y., Cao X. X., Kim J. A., Wang X., Chamness G. C., Maiti S. N., Cooper L. J., Edwards D. P., Contreras A., Hilsenbeck S. G., Chang E. C., Schiff R., and Wang X. S. (2014) Recurrent ESR1-CCDC170 rearrangements in an aggressive subset of oestrogen receptor-positive breast cancers. Nat. Commun. 5, 4577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Nesvizhskii A. I. (2014) Proteogenomics: concepts, applications and computational strategies. Nat. Methods 11, 1114–1125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Frenkel-Morgenstern M., Lacroix V., Ezkurdia I., Levin Y., Gabashvili A., Prilusky J., Del Pozo A., Tress M., Johnson R., Guigo R., and Valencia A. (2012) Chimeras taking shape: potential functions of proteins encoded by chimeric RNA transcripts. Genome Res. 22, 1231–1242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Sun H., Xing X., Li J., Zhou F., Chen Y., He Y., Li W., Wei G., Chang X., Jia J., Li Y., and Xie L. (2013) Identification of gene fusions from human lung cancer mass spectrometry data. BMC Genomics 14, S5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Wang Q., Xia J., Jia P., Pao W., and Zhao Z. (2013) Application of next generation sequencing to human gene fusion detection: computational tools, features and perspectives. Brief Bioinform. 14, 506–519 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Liu S., Tsai W. H., Ding Y., Chen R., Fang Z., Huo Z., Kim S., Ma T., Chang T. Y., Priedigkeit N. M., Lee A. V., Luo J., Wang H. W., Chung I. F., and Tseng G. C. (2016) Comprehensive evaluation of fusion transcript detection algorithms and a meta-caller to combine top performing methods in paired-end RNA-seq data. Nucleic Acids Res. 44, e47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Zhang J., Mardis E. R., and Maher C. A. (2017) INTEGRATE-neo: a pipeline for personalized gene fusion neoantigen discovery. Bioinformatics 33, 555–557 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Lin Y. Y., Gawronski A., Hach F., Li S., Numanagic I., Sarrafi I., Mishra S., McPherson A., Collins C. C., Radovich M., Tang H., and Sahinalp S. C. (2018) Computational identification of micro-structural variations and their proteogenomic consequences in cancer. Bioinformatics 34, 1672–1681 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Ellis M. J., Gillette M., Carr S. A., Paulovich A. G., Smith R. D., Rodland K. K., Townsend R. R., Kinsinger C., Mesri M., Rodriguez H., Liebler D. C., and Clinical Proteomic Tumor Analysis, C. (2013) Connecting genomic alterations to cancer biology with proteomics: the NCI Clinical Proteomic Tumor Analysis Consortium. Cancer Discov. 3, 1108–1112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Zhang H., Liu T., Zhang Z., Payne S. H., Zhang B., McDermott J. E., Zhou J. Y., Petyuk V. A., Chen L., Ray D., Sun S., Yang F., Chen L., Wang J., Shah P., Cha S. W., Aiyetan P., Woo S., Tian Y., Gritsenko M. A., Clauss T. R., Choi C., Monroe M. E., Thomas S., Nie S., Wu C., Moore R. J., Yu K. H., Tabb D. L., Fenyo D., Bafna V., Wang Y., Rodriguez H., Boja E. S., Hiltke T., Rivers R. C., Sokoll L., Zhu H., Shih I. M., Cope L., Pandey A., Zhang B., Snyder M. P., Levine D. A., Smith R. D., Chan D. W., Rodland K. D., and Investigators C. (2016) Integrated proteogenomic characterization of human high-grade serous ovarian cancer. Cell 166, 755–765 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Jia W., Qiu K., He M., Song P., Zhou Q., Zhou F., Yu Y., Zhu D., Nickerson M. L., Wan S., Liao X., Zhu X., Peng S., Li Y., Wang J., and Guo G. (2013) SOAPfuse: an algorithm for identifying fusion transcripts from paired-end RNA-Seq data. Genome Biol. 14, R12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Kim D. and Salzberg S. L. (2011) TopHat-Fusion: an algorithm for discovery of novel fusion transcripts. Genome Biol. 12, R72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Wang K., Singh D., Zeng Z., Coleman S. J., Huang Y., Savich G. L., He X., Mieczkowski P., Grimm S. A., Perou C. M., MacLeod J. N., Chiang D. Y., Prins J. F., and Liu J. (2010) MapSplice: accurate mapping of RNA-seq reads for splice junction discovery. Nucleic Acids Res. 38, e178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Trapnell C., Williams B. A., Pertea G., Mortazavi A., Kwan G., van Baren M. J., Salzberg S. L., Wold B. J., and Pachter L. (2010) Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat. Biotechnol. 28, 511–515 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Sisu C., Pei B., Leng J., Frankish A., Zhang Y., Balasubramanian S., Harte R., Wang D., Rutenberg-Schoenberg M., Clark W., Diekhans M., Rozowsky J., Hubbard T., Harrow J., and Gerstein M. B. (2014) Comparative analysis of pseudogenes across three phyla. Proc. Natl. Acad. Sci. U.S.A. 111, 13361–13366 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Rodriguez-Martin B., Palumbo E., Marco-Sola S., Griebel T., Ribeca P., Alonso G., Rastrojo A., Aguado B., Guigo R., and Djebali S. (2017) ChimPipe: accurate detection of fusion genes and transcription-induced chimeras from RNA-seq data. BMC Genomics 18, 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Huang W., Li L., Myers J. R., and Marth G. T. (2012) ART: a next-generation sequencing read simulator. Bioinformatics 28, 593–594 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Edgren H., Murumagi A., Kangaspeska S., Nicorici D., Hongisto V., Kleivi K., Rye I. H., Nyberg S., Wolf M., Borresen-Dale A. L., and Kallioniemi O. (2011) Identification of fusion genes in breast cancer by paired-end RNA-sequencing. Genome Biol. 12, R6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Bao Z. S., Chen H. M., Yang M. Y., Zhang C. B., Yu K., Ye W. L., Hu B. Q., Yan W., Zhang W., Akers J., Ramakrishnan V., Li J., Carter B., Liu Y. W., Hu H. M., Wang Z., Li M. Y., Yao K., Qiu X. G., Kang C. S., You Y. P., Fan X. L., Song W. S., Li R. Q., Su X. D., Chen C. C., and Jiang T. (2014) RNA-seq of 272 gliomas revealed a novel, recurrent PTPRZ1-MET fusion transcript in secondary glioblastomas. Genome Res. 24, 1765–1773 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Abmayr S. M., Yao T., Parmely T., and Workman J. L. (2006) Preparation of nuclear and cytoplasmic extracts from mammalian cells. Curr. Protoc. Mol. Biol. Chapter 12, Unit 12 11 [DOI] [PubMed] [Google Scholar]
- 30. Sheynkman G. M., Shortreed M. R., Frey B. L., and Smith L. M. (2013) Discovery and mass spectrometric analysis of novel splice-junction peptides using RNA-Seq. Mol. Cell. Proteomics 12, 2341–2353 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Geiger T., Wehner A., Schaab C., Cox J., and Mann M. (2012) Comparative proteomic analysis of eleven common cell lines reveals ubiquitous but varying expression of most proteins. Mol. Cell. Proteomics 11, M111 014050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Adusumilli R. and Mallick P. (2017) Data conversion with ProteoWizard msConvert. Methods Mol. Biol. 1550, 339–368 [DOI] [PubMed] [Google Scholar]
- 33. Lane L., Argoud-Puy G., Britan A., Cusin I., Duek P. D., Evalet O., Gateau A., Gaudet P., Gleizes A., Masselot A., Zwahlen C., and Bairoch A. (2012) neXtProt: a knowledge platform for human proteins. Nucleic Acids Res. 40, D76–D83 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Hu X., Wang Q., Tang M., Barthel F., Amin S., Yoshihara K., Lang F. M., Martinez-Ledesma E., Lee S. H., Zheng S., and Verhaak R. G. W. (2018) TumorFusions: an integrative resource for cancer-associated transcript fusions. Nucleic Acids Res. 46, D1144–D1149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Lee M., Lee K., Yu N., Jang I., Choi I., Kim P., Jang Y. E., Kim B., Kim S., Lee B., Kang J., and Lee S. (2017) ChimerDB 3.0: an enhanced database for fusion genes from cancer transcriptome and literature data mining. Nucleic Acids Res. 45, D784–D789 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Torres-Garcia W., Zheng S., Sivachenko A., Vegesna R., Wang Q., Yao R., Berger M. F., Weinstein J. N., Getz G., and Verhaak R. G. (2014) PRADA: pipeline for RNA sequencing data analysis. Bioinformatics 30, 2224–2226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Carrara M., Beccuti M., Lazzarato F., Cavallo F., Cordero F., Donatelli S., and Calogero R. A. (2013) State-of-the-art fusion-finder algorithms sensitivity and specificity. Biomed. Res. Int. 2013, 340620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Annala M. J., Parker B. C., Zhang W., and Nykter M. (2013) Fusion genes and their discovery using high throughput sequencing. Cancer Lett. 340, 192–200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Nicorici D., Satalan M., Edgren H., Kangaspeska S., Murumagi A., Kallioniemi O., Virtanen S., and Kilkku O. (2014) FusionCatcher - a tool for finding somatic fusion genes in paired-end RNA-sequencing data. bioRxiv. DOI: 10.1101/011650 [DOI] [Google Scholar]
- 40. Davidson N. M., Majewski I. J., and Oshlack A. (2015) JAFFA: High sensitivity transcriptome-focused fusion gene detection. Genome Med 7, 43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Inaki K., Hillmer A. M., Ukil L., Yao F., Woo X. Y., Vardy L. A., Zawack K. F., Lee C. W., Ariyaratne P. N., Chan Y. S., Desai K. V., Bergh J., Hall P., Putti T. C., Ong W. L., Shahab A., Cacheux-Rataboul V., Karuturi R. K., Sung W. K., Ruan X., Bourque G., Ruan Y., and Liu E. T. (2011) Transcriptional consequences of genomic structural aberrations in breast cancer. Genome Res. 21, 676–687 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Robinson D. R., Kalyana-Sundaram S., Wu Y. M., Shankar S., Cao X., Ateeq B., Asangani I. A., Iyer M., Maher C. A., Grasso C. S., Lonigro R. J., Quist M., Siddiqui J., Mehra R., Jing X., Giordano T. J., Sabel M. S., Kleer C. G., Palanisamy N., Natrajan R., Lambros M. B., Reis-Filho J. S., Kumar-Sinha C., and Chinnaiyan A. M. (2011) Functionally recurrent rearrangements of the MAST kinase and Notch gene families in breast cancer. Nat. Med. 17, 1646–1651 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Kalyana-Sundaram S., Shankar S., Deroo S., Iyer M. K., Palanisamy N., Chinnaiyan A. M., and Kumar-Sinha C. (2012) Gene fusions associated with recurrent amplicons represent a class of passenger aberrations in breast cancer. Neoplasia 14, 702–708 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Kangaspeska S., Hultsch S., Edgren H., Nicorici D., Murumagi A., and Kallioniemi O. (2012) Reanalysis of RNA-sequencing data reveals several additional fusion genes with multiple isoforms. PLoS ONE 7, e48745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Sakarya O., Breu H., Radovich M., Chen Y., Wang Y. N., Barbacioru C., Utiramerur S., Whitley P. P., Brockman J. P., Vatta P., Zhang Z., Popescu L., Muller M. W., Kudlingar V., Garg N., Li C. Y., Kong B. S., Bodeau J. P., Nutter R. C., Gu J., Bramlett K. S., Ichikawa J. K., Hyland F. C., and Siddiqui A. S. (2012) RNA-Seq mapping and detection of gene fusions with a suffix array algorithm. PLoS Comput. Biol. 8, e1002464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Berger M. F., Levin J. Z., Vijayendran K., Sivachenko A., Adiconis X., Maguire J., Johnson L. A., Robinson J., Verhaak R. G., Sougnez C., Onofrio R. C., Ziaugra L., Cibulskis K., Laine E., Barretina J., Winckler W., Fisher D. E., Getz G., Meyerson M., Jaffe D. B., Gabriel S. B., Lander E. S., Dummer R., Gnirke A., Nusbaum C., and Garraway L. A. (2010) Integrative analysis of the melanoma transcriptome. Genome Res. 20, 413–427 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Slavoff S. A., Mitchell A. J., Schwaid A. G., Cabili M. N., Ma J., Levin J. Z., Karger A. D., Budnik B. A., Rinn J. L., and Saghatelian A. (2013) Peptidomic discovery of short open reading frame-encoded peptides in human cells. Nat. Chem. Biol. 9, 59–64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Hampton O. A., Den Hollander P., Miller C. A., Delgado D. A., Li J., Coarfa C., Harris R. A., Richards S., Scherer S. E., Muzny D. M., Gibbs R. A., Lee A. V., and Milosavljevic A. (2009) A sequence-level map of chromosomal breakpoints in the MCF-7 breast cancer cell line yields insights into the evolution of a cancer genome. Genome Res. 19, 167–177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Woo S., Cha S. W., Na S., Guest C., Liu T., Smith R. D., Rodland K. D., Payne S., and Bafna V. (2014) Proteogenomic strategies for identification of aberrant cancer peptides using large-scale next-generation sequencing data. Proteomics 14, 2719–2730 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Ivanov M. V., Lobas A. A., Karpov D. S., Moshkovskii S. A., and Gorshkov M. V. (2017) Comparison of false discovery rate control strategies for variant peptide identifications in shotgun proteogenomics. J. Proteome Res. 16, 1936–1943 [DOI] [PubMed] [Google Scholar]
- 51. Giacomini C. P., Sun S., Varma S., Shain A. H., Giacomini M. M., Balagtas J., Sweeney R. T., Lai E., Del Vecchio C. A., Forster A. D., Clarke N., Montgomery K. D., Zhu S., Wong A. J., van de Rijn M., West R. B., and Pollack J. R. (2013) Breakpoint analysis of transcriptional and genomic profiles uncovers novel gene fusions spanning multiple human cancer types. PLoS Genet. 9, e1003464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Finn R. D., Attwood T. K., Babbitt P. C., Bateman A., Bork P., Bridge A. J., Chang H. Y., Dosztanyi Z., El-Gebali S., Fraser M., Gough J., Haft D., Holliday G. L., Huang H., Huang X., Letunic I., Lopez R., Lu S., Marchler-Bauer A., Mi H., Mistry J., Natale D. A., Necci M., Nuka G., Orengo C. A., Park Y., Pesseat S., Piovesan D., Potter S. C., Rawlings N. D., Redaschi N., Richardson L., Rivoire C., Sangrador-Vegas A., Sigrist C., Sillitoe I., Smithers B., Squizzato S., Sutton G., Thanki N., Thomas P. D., Tosatto S. C., Wu C. H., Xenarios I., Yeh L. S., Young S. Y., and Mitchell A. L. (2017) InterPro in 2017-beyond protein family and domain annotations. Nucleic Acids Res. 45, D190–D199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Russo A. A., Tong L., Lee J. O., Jeffrey P. D., and Pavletich N. P. (1998) Structural basis for inhibition of the cyclin-dependent kinase Cdk6 by the tumour suppressor p16INK4a. Nature 395, 237–243 [DOI] [PubMed] [Google Scholar]
- 54. Kollmann K., Heller G., Schneckenleithner C., Warsch W., Scheicher R., Ott R. G., Schafer M., Fajmann S., Schlederer M., Schiefer A. I., Reichart U., Mayerhofer M., Hoeller C., Zochbauer-Muller S., Kerjaschki D., Bock C., Kenner L., Hoefler G., Freissmuth M., Green A. R., Moriggl R., Busslinger M., Malumbres M., and Sexl V. (2013) A kinase-independent function of CDK6 links the cell cycle to tumor angiogenesis. Cancer Cell 24, 167–181 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Hayette S., Tigaud I., Callet-Bauchu E., Ffrench M., Gazzo S., Wahbi K., Callanan M., Felman P., Dumontet C., Magaud J. P., and Rimokh R. (2003) In B-cell chronic lymphocytic leukemias, 7q21 translocations lead to overexpression of the CDK6 gene. Blood 102, 1549–1550 [DOI] [PubMed] [Google Scholar]
- 56. Parker E. P., Siebert R., Oo T. H., Schneider D., Hayette S., and Wang C. (2013) Sequencing of t(2;7) translocations reveals a consistent breakpoint linking CDK6 to the IGK locus in indolent B-cell neoplasia. J. Mol. Diagn. 15, 101–109 [DOI] [PubMed] [Google Scholar]
- 57. Liu Y., Easton J., Shao Y., Maciaszek J., Wang Z., Wilkinson M. R., McCastlain K., Edmonson M., Pounds S. B., Shi L., Zhou X., Ma X., Sioson E., Li Y., Rusch M., Gupta P., Pei D., Cheng C., Smith M. A., Auvil J. G., Gerhard D. S., Relling M. V., Winick N. J., Carroll A. J., Heerema N. A., Raetz E., Devidas M., Willman C. L., Harvey R. C., Carroll W. L., Dunsmore K. P., Winter S. S., Wood B. L., Sorrentino B. P., Downing J. R., Loh M. L., Hunger S. P., Zhang J., and Mullighan C. G. (2017) The genomic landscape of pediatric and young adult T-lineage acute lymphoblastic leukemia. Nat. Genet. 49, 1211–1218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Edwards N. J., Oberti M., Thangudu R. R., Cai S., McGarvey P. B., Jacob S., Madhavan S., and Ketchum K. A. (2015) The CPTAC data portal: a resource for cancer proteomics research. J. Proteome Res. 14, 2707–2713 [DOI] [PubMed] [Google Scholar]
- 59. Nome T., Thomassen G. O., Bruun J., Ahlquist T., Bakken A. C., Hoff A. M., Rognum T., Nesbakken A., Lorenz S., Sun J., Barros-Silva J. D., Lind G. E., Myklebost O., Teixeira M. R., Meza-Zepeda L. A., Lothe R. A., and Skotheim R. I. (2013) Common fusion transcripts identified in colorectal cancer cell lines by high-throughput RNA sequencing. Transl. Oncol. 6, 546–553 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Yoshihara K., Wang Q., Torres-Garcia W., Zheng S., Vegesna R., Kim H., and Verhaak R. G. (2015) The landscape and therapeutic relevance of cancer-associated transcript fusions. Oncogene 34, 4845–4854 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Pathak E., Bhavya Mishra D., Atri N., and Mishra R. (2017) Deciphering the role of microRNAs in BRD4-NUT fusion gene induced NUT midline carcinoma. Bioinformation 13, 209–213 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. French C. A., Miyoshi I., Kubonishi I., Grier H. E., Perez-Atayde A. R., and Fletcher J. A. (2003) BRD4-NUT fusion oncogene: a novel mechanism in aggressive carcinoma. Cancer Res. 63, 304–307 [PubMed] [Google Scholar]
- 63. Bassani-Sternberg M., Braunlein E., Klar R., Engleitner T., Sinitcyn P., Audehm S., Straub M., Weber J., Slotta-Huspenina J., Specht K., Martignoni M. E., Werner A., Hein R., Peschel D. H. B. C., Rad R., Cox J., Mann M., and Krackhardt A. M. (2016) Direct identification of clinically relevant neoepitopes presented on native human melanoma tissue by mass spectrometry. Nat. Commun. 7, 13404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Cagney G., Amiri S., Premawaradena T., Lindo M., and Emili A. (2003) In silico proteome analysis to facilitate proteomics experiments using mass spectrometry. Proteome Sci. 1, 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Lai J., An J., Seim I., Walpole C., Hoffman A., Moya L., Srinivasan S., Perry-Keene J. L., Australian Prostate Cancer, B., Wang C., Lehman M. L., Nelson C. C., Clements J. A., and Batra J. (2015) Fusion transcript loci share many genomic features with non-fusion loci. BMC Genomics 16, 1021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Wang X., Codreanu S. G., Wen B., Li K., Chambers M. C., Liebler D. C., and Zhang B. (2018) Detection of Proteome Diversity Resulted from Alternative Splicing is Limited by Trypsin Cleavage Specificity. Mol. Cell. Proteomics 17, 422–430 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Nik-Zainal S., Davies H., Staaf J., Ramakrishna M., Glodzik D., Zou X., Martincorena I., Alexandrov L. B., Martin S., Wedge D. C., Van Loo P., Ju Y. S., Smid M., Brinkman A. B., Morganella S., Aure M. R., Lingjaerde O. C., Langerod A., Ringner M., Ahn S. M., Boyault S., Brock J. E., Broeks A., Butler A., Desmedt C., Dirix L., Dronov S., Fatima A., Foekens J. A., Gerstung M., Hooijer G. K., Jang S. J., Jones D. R., Kim H. Y., King T. A., Krishnamurthy S., Lee H. J., Lee J. Y., Li Y., McLaren S., Menzies A., Mustonen V., O'Meara S., Pauporte I., Pivot X., Purdie C. A., Raine K., Ramakrishnan K., Rodriguez-Gonzalez F. G., Romieu G., Sieuwerts A. M., Simpson P. T., Shepherd R., Stebbings L., Stefansson O. A., Teague J., Tommasi S., Treilleux I., Van den Eynden G. G., Vermeulen P., Vincent-Salomon A., Yates L., Caldas C., van't Veer L., Tutt A., Knappskog S., Tan B. K., Jonkers J., Borg A., Ueno N. T., Sotiriou C., Viari A., Futreal P. A., Campbell P. J., Span P. N., Van Laere S., Lakhani S. R., Eyfjord J. E., Thompson A. M., Birney E., Stunnenberg H. G., van de Vijver M. J., Martens J. W., Borresen-Dale A. L., Richardson A. L., Kong G., Thomas G., and Stratton M. R. (2016) Landscape of somatic mutations in 560 breast cancer whole-genome sequences. Nature 534, 47–54 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Kollmann K., Heller G., Ott R. G., Scheicher R., Zebedin-Brandl E., Schneckenleithner C., Simma O., Warsch W., Eckelhart E., Hoelbl A., Bilban M., Zochbauer-Muller S., Malumbres M., and Sexl V. (2011) c-JUN promotes BCR-ABL-induced lymphoid leukemia by inhibiting methylation of the 5′ region of Cdk6. Blood 117, 4065–4075 [DOI] [PubMed] [Google Scholar]
- 69. Kollmann K., Heller G., and Sexl V. (2011) c-JUN prevents methylation of p16(INK4a) (and Cdk6): the villain turned bodyguard. Oncotarget 2, 422–427 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Data supporting the findings of this study include raw MS/MS data and RNA-Seq data, and sources of data can be available within the Experimental Procedures section of the paper. RNA-Seq data of Jurkat (SRA project I.D.: SRP019939), K562 (SRA project I.D.: SRP000931 and SRP010061), and Glioma (SRA project I.D.: SRP027383) can be downloaded from the SRA archive (https://www.ncbi.nlm.nih.gov/sra). Raw MS/MS data of Jurkat (http://www.peptideatlas.org/, PeptideAtlas I.D.: PASS00215) and K562 (http://www.proteomexchange.org/, ProteomeXchange I.D.: PXD002395) were previously published and can be downloaded in each data repository. Also, the raw MS/MS data of CDK6 antibody-captured chimeric band were deposited in a MassIVE database (https://massive.ucsd.edu/ProteoSAFe/static/massive.jsp, MassIVE I.D.: MSV000083822).