

A spatiotemporal microproteomics analysis was applied to an experimental model of TBI within the first 10-day window post injury. Proteomics profiles linked to systems biology analysis revealed that the brain protein profile is restored to noninjured conditions at 10 days after injury. Analyzing the substantia nigra after 3 days of injury, the ipsilateral hemisphere substantia nigra revealed an upregulation of several proteins previously reported in Parkinson's disease, thus further suggesting a link between TBI and Parkinson's disease at 3 days.
Keywords: Mass Spectrometry, Parkinson's disease, Lipidomics, Systems biology*, Imaging, Label-free quantification, Long chain Acylcarnitines, MALDI Mass spectrometry imaging, Spatially resolved Microproteomics, Spatiotemporal, Traumatic Brain Injury
Graphical Abstract
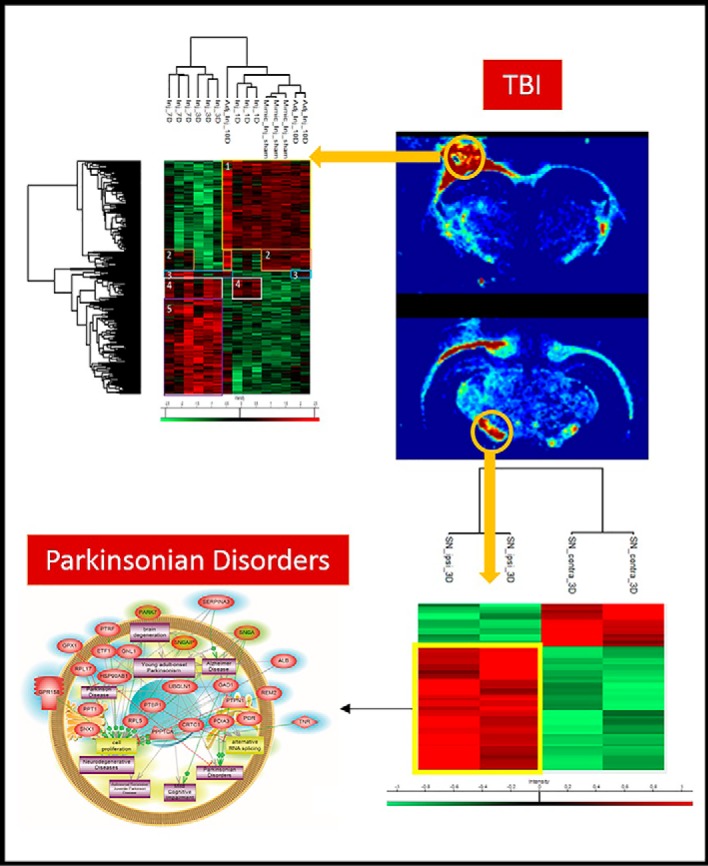
Highlights
Spatiotemporal microproteomics analysis of TBI.
Injury site microproteomics reveal distinct phases in 10-day frame post TBI.
Uninjured proteomic profile is restored in TBI at 10 days post injury.
Substantia nigra protein post 3 days suggest link to Parkinson's disease.
Abstract
Traumatic brain injury (TBI) represents a major health concerns with no clinically-approved FDA drug available for therapeutic intervention. Several genomics and neuroproteomics studies have been employed to decipher the underlying pathological mechanisms involved that can serve as potential neurotherapeutic targets and unveil a possible underlying relation of TBI to other secondary neurological disorders. In this work, we present a novel high throughput systems biology approach using a spatially resolved microproteomics platform conducted on different brain regions in an experimental rat model of moderate of controlled cortical injury (CCI) at a temporal pattern postinjury (1 day, 3 days, 7 days, and 10 days). Mapping the spatiotemporal landscape of signature markers in TBI revealed an overexpression of major protein families known to be implicated in Parkinson's disease (PD) such as GPR158, HGMB1, synaptotagmin and glutamate decarboxylase in the ipsilateral substantia nigra. In silico bioinformatics docking experiments indicated the potential correlation between TBI and PD through alpha-synuclein. In an in vitro model, stimulation with palmitoylcarnitine triggered an inflammatory response in macrophages and a regeneration processes in astrocytes which also further confirmed the in vivo TBI proteomics data. Taken together, this is the first study to assess the microproteomics landscape in TBI, mainly in the substantia nigra, thus revealing a potential predisposition for PD or Parkinsonism post-TBI.
Traumatic brain injury (TBI)1 is a leading cause of mortality and morbidity worldwide. TBI is characterized by the transfer of a force from an external object to the head, leading to neuropathologic damage and dysfunction (1). According to the Center of Disease Control and Prevention, and based on data between the years of 2002 and 2006, ∼1.7 million people sustain a TBI annually (2). Several neurotherapeutic strategies have been proposed and studied in TBI, mainly: neuroprotection, neurovascular regeneration, and neurorestoration (3). Although several efforts over the decades have been put to develop treatment strategies, TBI still lacks an effective therapy. This major weakness in establishing an optimal TBI treatment may be because of a deficiency in the full understanding of molecular mechanisms occurring within the injured microenvironment considering different biomolecule partners, including lipids, proteins, and metabolites along with their potential interactions. Such integrative biology studies involving proteomics and the possible influence of other biomolecular partners such as lipids on the proteomic signature within the injury microenvironment is still missing. In the context of TBI, a combined focus on injury-related molecules, including proteins and lipids will help find new therapeutic targets.
Moreover, a better understanding of the molecular mechanisms occurring in brain regions after remote injury is also required to develop innovative strategies for the long-term consequences of TBI. Indeed, it has been suggested that TBI is implicated in several other neurodegenerative diseases such as Parkinson's disease (PD) (4) and Alzheimer's disease (AD) (5) (6). TBI is notably associated with a 44% increased risk of developing PD over 5 to 7 years (7). In fact, Acosta et al. showed that the expression of α-synuclein-positive dopaminergic neurons in the substantia nigra pars compacta was elevated in the ipsilateral hemisphere of a rat TBI model at 60 days postinjury, when compared with the contralateral hemisphere of the same injured brain region, and either hemispheres of sham noninjured rats (8). The aggregation and accumulation of truncated forms of α-synuclein in the substantia nigra, along with the continuous loss of dopaminergic neurons is a hallmark of PD (9). Also, the expression α-synuclein was shown to be regulated by microglia in the substantia nigra and not astrocytes at 30 days postinjury in a mouse CCI model (10) with a marked neuropathological-regenerative role of microglia and astrocytes post -TBI (11).
In the present study, we applied a high-resolution, state-of-the-art, “field microproteomics” approach on CCI brain sections within the acute and subacute phase post impact (1 day, 3 days, 7 days, and 10 days) to study proteomic changes within the injured microenvironment (1 mm2 surface area). This was achieved via a spatially resolved microproteomics technique (12), which provided a more accurate, intricate and detailed brain region proteomic characterization compared with previously performed TBI proteomic studies using the whole cortex or hippocampus tissue for analysis (13, 14). In addition, in vitro secrotome experiments along with systems biology and in silico bioinformatics and docking experiments, assessed the spatially resolved microproteomics data to generate a novel depiction of TBI pathophysiological changes that indicate a strong correlation among the TBI molecular changes and predisposition for PD physiopathology.
EXPERIMENTAL PROCEDURES
Reagents
Absolute methanol (MeOH), water (H2O), formic acid (FA), acetonitrile (ACN), chloroform, and trifluoroacetic acid (TFA) were obtained from Biosolve B. V. (Dieuze, France). dl-dithiothreitol (DTT), thiourea, and iodoacetamide (IAA) were purchased from Sigma-Aldrich (Saint-Quentin Fallavier, France). LysC/Trypsin was purchased from Promega (Charbonnieres, France). Polylysine-coated slides were purchased from Thermo-Scientific (Braunschweig, Germany). NR8383 is a rat alveolar macrophage cell line (CRL-2192) obtained from ATCC (USA). Rat brain cell line DITNC1 (astrocyte) was purchased from Sigma Aldrich. Dulbecco's Modified Eagle's Medium (DMEM), Ham's F12K, fetal bovine serum (FBS) and phosphate buffer saline (PBS) were obtained from Invitrogen Life Technologies (Milan, Italy). Penicillin and streptomycin were purchased from Invitrogen, Thermo Fisher Scientific.
Animals
Experiments were conducted on adult male Sprague-Dawley rats (225–250g, 7–8 weeks old) in accordance with the National Institute of Health Guidelines for Animal Research (Guide for the Care and Use of Laboratory Animals) and approved by the Institutional Animal Care and Use Committee (IACUC) at the American University of Beirut (AUB). The rats were maintained and housed under pathogen-free conditions at room temperature in a 12h/12h (light/dark) cycles with 24 h access to food and water, and each 3 rats of same day group were housed together in one cage. Animals were provided with constant temperature and humidity control at the AUB Animal Care Facility (ACF). All surgical procedures were conducted under deep anesthesia.
Experimental Design and Statistical Rational
All experiments were conducted with biological replicates (n = 3). Our study included 5 cohorts of animals including the sham cohort and 4 experimental moderate injury cohorts (1 day, 3 days, 7 days, and 10 days). A total of 15 rats were included in the study each cohort involved 3 animals for the proteomics studies for each of the conditions. The statistical analysis carried out for the spatially resolved microproteomics injury site data was multiple sample ANOVA test with a p value = 0.01. For cell line experiments (macrophages and astrocytes), and microproteomic analysis of the substantia nigra, Student's t test was applied with a p value = 0.05. Normalization was achieved by using the Z-score. Data analysis was carried out on Perseus Software.
For the ClueGo analysis, only pathways with a p value ≤ 0.05 were displayed. For the Scatter Plot analysis using GraphPad Prism 5, the LFQ values were subjected to ANOVA using Tukey's Multiple Comparison Test with a p value < 0.05.
Experimental Cell Lines
NR8383 were cultured in Ham's F12 K medium supplemented with 15% FBS, 100 U/ml penicillin and 100 μg/ml Streptomycin, at 37 °C in a humidified atmosphere (5% CO2). DITNC1 cell line was grown in DMEM medium supplemented with 10% FBS, 2% l-glutamate, 100 U/ml penicillin, 100 μg/ml Streptomycin and 1% sodium pyruvate, at 37 °C in a humidified atmosphere (5% CO2). ND7/23 cell line was grown in DMEM medium supplemented with 10% FBS, 1% l-glutamate, 100 U/ml penicillin and 100 μg/ml Streptomycin, at 37 °C in a humidified atmosphere (5% CO2).
Experimental Controlled Cortical Impact (CCI), Study Design, Brain Harvesting, and Tissue Preparation
After anesthesia, the cortex of rats was exposed by craniotomy, followed by controlled cortical impact (CCI) to induce mechanical damage to the cortex tissue (15). Briefly, after anesthesia using a ketamine (10 mg/kg)-xylazine (100 mg/kg) mix, rats were fixed on a stereotactic frame, and ear bars were used to secure the position of the rat head. A midline incision was then performed using a sterilized blade to expose the underlying tissue and the skin was retracted on the right hemisphere (ipsilateral). A target was set half-way between the bregma and lambda using the machine software (ipsilateral), and a craniotomy (5–7 mm diameter) at the target site was performed. An impact was induced using an impactor tip of 2 mm diameter, 2 mm depth, 4 m/s velocity and impact duration of 0.8 s. Each animal was considered as an experimental unit. Postinjury, the rats' heads were sutured and maintained until selected time points for the study, including: 1 day, 3 days, 7 days, and 10 days. The sham (noninjured) rats received the same procedure except with no craniotomy nor impact to the underlying tissue. At the corresponding time points postinjury, rats were anesthetized using 5% isoflurane and sacrificed by decapitation. The complete brain was then collected and snap-frozen on isopentane which was previously cooled on dry ice. The frozen brains were then stored at −80 °C until the experiments were carried out. At the time of experimentation, the samples were then transferred from −80 °C to −20 °C and kept for 2 h, prevent possible cracking within the tissue samples. Cryostat (Leica Microsystems, Nanterre, France) was used to cut the brain samples in coronal view with a thickness of 20 μm per tissue section. The sections where then thaw-mounted on Polylysine-coated slides.
Spatially Resolved Microproteomics
In-situ LysC Tryptic Digestion
Lesion sections from all injured time points (1 day, 3 days, 7 days, and 10 days) and a corresponding area from the sham tissue, were all chosen directly consecutive to the MALDI-MSI imaged sections and subjected to in situ tryptic digestion. Briefly, these tissue sections were dried in the vacuum desiccator for 15 min before experimentation. Several washes were performed in a delipidation approach as follows: 30 s in 70% ethanol, 30 s in 100% ethanol, and 2 × 30 s in chloroform. The tissue was subjected to drying in the desiccator between all steps. Post washing steps, LysC trypsin at 40 μg/ml resuspended in Tris-HCl (50 mm, pH 8.0) was spotted on the injured cortical tissue using a piezoelectric microspotter Chemikal Inkjet Printer (CHIP-1000, Shimadzu, CO, Kyoto, Japan). The digested area was controlled by assigning a 4 × 4 grid of microspots to obtain a final digested area of 1 mm2. Each spot was 200 μm in diameter and contained a droplet volume of 150 pL/cycle. The digestion was carried out for 2 h, and 25 cycles of 0.1%TFA were spotted postdigestion.
Liquid Microjunction Microextraction
Using the TriVersa Nanomate platform (Advion Biosciences Inc., Ithaca, NY) with the installed option of Liquid Extraction Surface Analysis (LESA), the digested peptides within the injured cortical tissue were collected. Briefly, three prepared solvents were used in the extraction process: a) 0.1% TFA, b) ACN/0.1% TFA (8:2 v/v ratio), and c) MeOH/0.1% TFA (7:3 v/v ratio). After the tissue is scanned and digested, the cortical tissue is designated as a target for extraction. An automatically controlled tip deposits a volume of each solvent onto the digested area and performs several aspiration-deposition actions, before finally aspirating all the volume and depositing in small tubes. Two extraction cycles per mentioned solvent type are performed to ensure the maximum collection of peptides as possible. The content of the tubes is then dried using speedvac, and the tubes are stored in −80 °C until subjected to LC-MS/MS.
Shotgun Proteomics Analysis after Stimulation of Cell lines with Palmitoylcarnitine
Cell Line Stimulation with Palmitoylcarnitine
Using a 6-well plate, ∼1.8 million cells were applied to each well (300,000 cells/well) for macrophages and 2.7 million cells for astrocytes (450,000 cells/well). The following day, cells were then starved overnight at 37 °C in their corresponding medium (F12 for NR8383 and DMEM for astrocytes) with a lower concentration of serum (2% FBS, 1% penicillin-streptomycin and sodium pyruvate). Finally, the cells were then stimulated by adding palmitoyl-l-carnitine (0.1 μm) to the medium in the absence of serum, thus 0% FBS in medium for all cells. After stimulation for 24 h, cells were collected and centrifuged at 150 RCF/5 min for the macrophages, and at 1000 RPM/5 min for the astrocytes. The controls underwent the same experimental procedure except for no stimulation with palmitoyl-l-carnitine.
Protein Extraction
Protein extraction was performed using sodium dodecyl sulfate buffer (SDS). Precisely, 50 μls of extraction buffer (4% SDS, Tris 0.1 m, pH 7.8) were added to each tube containing the previously stimulated cells, or their corresponding controls. Samples were then heated for 15 min at 95 °C and sonicated for another 15 min. After, a centrifugation step was performed at 16,000 × g for 10 min at 20 °C. Postcentrifugation, the supernatant containing the extracted proteins were collected. Using Bradford quantification method, all samples were quantified and stored at −80 °C until further experimentation.
FASP Protocol
For protein identification, we performed a shotgun bottom-up proteomic approach. All sample concentrations were normalized to obtain a final concentration of 1.5 μg/μl per sample. With a normalized concentration, 30 μl volume each sample was prepared to obtain a final protein concentration of 45 μg for digestion. An equal volume, 30 μl, or reduction solution (Dithiothreitol- DTT 0.1 m) was added to each of the samples and followed by incubation for 40 min at 56 °C. Samples were then transferred, and experimentation carried out using filter-aided sample preparation (FASP) method (16). Briefly, this method uses a filter with a nominal molecular weight limit of 30,000 kDa. (Amicon Ultra-0.5 30K, Millipore). After transferring the samples into the FASP filters, an alkylation step was performed in the dark by adding iodoacetamide (IAA) solution (0.05 m) for 20 min at room temperature. The samples were then digested overnight at an incubation temperature of 37 °C by adding LysC/trypsin at a concentration of 40 μg/ml prepared in 50 mm Tris-HCL solution at pH 8. The following day, the digested proteins in the filter were eluted with 50 μl of saline solution (NaCl 0.5 m) and the digestion reaction was stopped by adding an acidic solution (10 μl of TFA 5%) to each of the filters. Desalting of the samples, along with their enrichment, was performed with ZipTip C-18 (Millipore) just before processing using LC-MS/MS.
LC-MS and MS/MS Data Acquisition for Shotgun Proteomics
MS analysis was performed by a nanoAcquity UPLC system (Waters) coupled with a Q-Exactive Orbitrap mass spectrometer (Thermo Scientific) containing a nano-electrospray ionization source. The analysis was carried out in reverse phase, and all samples were loaded into a preconcentration column (nanoAcquity Symmetry C18, 5 μm, 180 μm × 20 mm). The peptides were separated using an analytical column (nanoAcquity BEH C18, 1.7 μm, 75 μm × 250 mm) by applying a linear gradient of acetonitrile in 0.1% formic acid (5–35%, for 2 h) at a flow rate of 300 nL/min. Within the Orbitrap mass analyzer, the MS analysis was performed with a resolution of 70,000 FWHM, a m/z mass range between 300 and 1600, an AGC of 3e6 ions and a maximum injection time of 120 ms. The MS/MS was performed in a data-dependent acquisition mode defined to analyze the 10 most intense ions within the primary MS analysis (Top 10). Regarding the MS/MS fragmentation parameters, the resolution was set at 17,500 FWHM, an m/z range between 200 and 2000, an AGC of 5e4 ions, and a maximum injection time of 60 ms. The isolation window was set at 4.0 m/z.
Protein Identification and Data Processing
All raw data were analyzed by MaxQuant software version 1.5.8.3 (17). Proteins were identified by comparing all raw spectra with a proteome reference database of Rattus norvegicus (Uniprot, release 20180712, 8027 entries for the palmitoylcarnitine stimulated protein identifications, and release 20180302, 8022 entries for the in situ spatially resolved microproteomics identifications). The parameters chosen for the identification include: digestion enzyme used was trypsin LysC, and the maximum number allowed of missed cleavages was two. The oxidation of methionine and N-terminal protein acetylation was chosen as variable modifications. As for fixed modifications, carbamidomethylation of cysteine was set for the identification of proteins in the palmitoylcarnitine stimulation case, and this modification was removed for the micro-proteomics approach. Proteins were identified based on a minimum of 2 peptides per protein, in which 1 was unique. As for initial mass tolerance, 6 ppm was selected for MS mode, and 20 ppm was set for fragmentation data with regard to MS/MS tolerance. The false discovery rate (FDR) was specified to 1% for both protein and peptide. The label-free quantification (LFQ) was performed by keeping the default parameters of MaxQuant. After data treatment using MaxQuant, statistical analysis was done using Perseus software (version 1.6.0.7). Each cell line was processed alone in its own Perseus file. The beginning matrix was filtered by removing the potential contaminants, reverse and only identified by site. Then, the LFQ intensity was logarithmized (log2[x]). The principal component analysis (PCA) was then applied using the default parameters of the software. Regarding the spatially resolved microproteomics analysis, the samples were annotated based on the corresponding time point (sham, 1 day, 3 days, 7 days, and 10 days) resulting in 5 different groups, each containing 3 replicates of data corresponding to lesion sections. The hierarchical clustering and profile plot of only the statistically significant proteins were all performed and visualized by Perseus using default parameters.
Subnetwork Enrichment Pathway Analysis
Using Elseviers's Pathway Studio (version 11.0//Elsevier), all relationships between the differentially expressed proteins among all conditions were depicted based on the Ariadne ResNet database (18, 19). For proteins identified in the spatially resolved microproteomics analysis and the ones identified in the shotgun analysis poststimulation of cell lines with palmitoylcarnitine, Subnetwork Enrichment Analysis (SNEA) algorithm was used to detect the statistically significant altered biological pathways in which the identified proteins are involved. This algorithm uses Fisher's statistical test to detect any nonrandom associations between two categorical variables organized by a specific relationship. Also, this algorithm starts by creating a central “seed” from all the relevant identities in the database and builds connections with associated entities based on their relationship with the seed. SNEA compares the sub-network distribution to the background distribution using one-sided Mann-Whitney U-Test, and calculates a p value, thus representing a statistical significance between different distributions. In all analysis that we performed, the GenBank ID was used to form experimental groups based on the different conditions present for analysis. The pathway networks were reconstructed based on biological processes and molecular functions for each single protein, along with its associated targets (20). In addition, we performed targeted protein pathway analysis for PD using the protein list identified in the ipsilateral substantia nigra at 3 days postinjury.
Western Blot Analysis
Extracts from liquid microjunction microextraction samples were used for Western blotting assays. First, proteins were separated by SDS-PAGE electrophoresis and then transferred onto a nitrocellulose membrane. Membranes were blocked for 1 h at room temperature in TBS-Tween 0.1% + milk 5% and incubated overnight at 4 °C with primary antibodies directed against rabbit anti-GPR158 (1:500, from AbCam (ab121388)), rabbit anti-HMGB1 (1:500, from AbCam (Cambridge, UK) (ab77302)). Horseradish peroxidase-coupled goat anti-rabbit secondaries (Jackson ImmunoResearch) were used at 1:20,000 respectively. The proteins were visualized with the enhanced chemiluminescence kit (West Dura from Pierce) according to the manufacturer's instructions. ImageJ software was used to quantify the bands. Experiments were done in triplicate (n = 3).
Modeling and Protein Interactions Prediction
All models have been realized with the software I-Tasser (21) using the complete structure of the proteins reconstitute the complete amino acid sequence of the proteins that are realized in-silico from the closest analogs. The most stable model, with a C-Score between −5 and 2 is retained. The HMGB1 model was generated from its amino acid sequence. This rat protein has never been described by 3D modeling and has no structural data, but it can be predicted by sequence homology and the nature of these amino acids. I-Tasser generated this model with a C-Score of −3.66, which complies with standard recommendations [-5; 2] based on the structural human analog HMGB1 model obtained by NMR (PDB (22) model n°2YRQ) (23). In a similar approach as applied to HMGB1, alpha-synuclein protein has no experimental model in the rat database and, thus we were carried out the analysis using I-Tasser. The generated model has a C-Score of 1.06 in agreement with the recommendations. This model is mainly based on the structural analog, human micelle-bound alpha-synuclein (PDB n°1XQ8) (24). The modeling of GPR158 had the most stable model with a C-Score of −0.27 and does not respect the biological morphology described in Uniprot. Indeed GPR158 is described as having an extracellular domain, a transmembrane domain, and an intracellular domain. The most stable used model complying with this architecture had a C-score of −2.98, still in agreement with the C-score values described. Finally, osteocalcin was also modeled on I-Tasser, with a C-score of −2.92 and having as its main structural analog nitric oxide reductase (PDB model n°3AYF) described as a transmembrane structure.
The prediction of protein-protein interactions was performed with ClusPro (25) software. The HMGB1 is identified as a receptor and the alpha-synuclein as a ligand. The GPR158 receptor has been used complete without distinction of architecture, to find all the possible fixation sites of HMGB1 or osteocalcin. The interaction model is carried out by docking the ligand on the receptor. ClusPro then generates multiple interaction models ranked in order of stability. The selected models are still part of the Top5 “balanced” models considering the best stability structure.
The selected interactions are then recreated with Chimera (26). For that the file “.PDB” generated by ClusPro is loaded on Chimera and the model is split between the ligand and the receptor to form two independent chains, then the surface of the model is represented for manually controlling the position and possibility of protein interaction. Chimera allowed the graphical representation of the interactions, and thanks to the “MatchMaker” tool, the interaction between HMGB1 and alpha-synuclein could be compared with the GPR158-HMGB1 interaction to represent the positioning of alpha-synuclein in the GPR158-HMGB1 interaction. In the same manner, Chimera allowed the observation of colocalization between osteocalcin and HMGB1 on GPR158, after superposition of models GPR158-HMGB1 and GPR158-osteocalcin.
ClueGo Interrelation Analysis
ClueGO analysis was used to visualize the possible interactions of the obtained clusters in biological networks. These networks were obtained by interrogation against all of the following ontologies/pathways:BiologicalProcess-GOA, CellularComponent-GOA, ImmueSystemProcess-GOA, MolecularFunction-GOA, KEGG, and REACTOME, and the GO Term Fusion option was selected. Only pathways with a p value ≤ 0.05 were displayed. The GO tree Interval had a minimum value of 5 and a maximum value of 11. The GO Term/Pathway selection was based on a minimum number of 3 genes and a Kappa score = 0.6.
Scatter Plot Statistical Analysis
GraphPad Prism 5 software was used to obtain the scatter plot based on the LFQ intensity between the substantia nigra ipsilateral and contralateral of the following proteins: Syn11, Hmgb1, Gad1, and GPR158. The entered LFQ values were subjected to ANOVA using Tukey's Multiple Comparison Test with a p value < 0.05.
RESULTS
Spatially Resolved Microproteomics Analysis Reveals Time Point-specific Proteomic Phases
With respect to time, the injured cortical tissue was subjected to on-spot tryptic digestion followed by liquid micro-junction extraction within a total surface area of 1 mm2 as seen in the magnified picture of the optical scan corresponding to lesion tissue section at 3 days postinjury (supplemental Fig. S1A). The extracted peptides were subjected to shotgun analysis followed by label-free quantification. 1950 proteins (see supplemental Spreadsheet S1) were identified across all samples, and 306 proteins (see supplemental Spreadsheet S2) showed a significant difference in expression based on LFQ values after subjecting the data to multiple ANOVA tests with a p value = 0.01. Principal component analysis (PCA) was applied to the 306 quantified proteins comparing all injured time points and the noninjured sham, to describe the variance across all samples of different conditions. The first component explains 63.2% of the variance of samples, whereas the second component explains 12.8%, thus yielding a sum of 76% of the variance observed. Plot of the first two PCs shows that for each time point, all three replicates clustered together with no interference from the other conditions (supplemental Fig. S1B). Time points 3 and 7 days cluster separately from the rest of the time points and are positively correlated to the first component. Hierarchal clustering performed on the 306 proteins shows 5 main clusters (Fig. 1A), from which we can deduce 4 main phases of protein processes that progress throughout the first 10 days of TBI. Phase 1 is characterized by the proteins that are upregulated in the sham condition and at 10 days postinjury, thus showing a possible restoration of function at this time point. This phase is divided into two parts: phase 1a corresponding to cluster 1 and phase 1b corresponding to cluster 2 in Fig. 1A. Phase 1a corresponds to a group of proteins that are elevated at sham and surprisingly at 1 day postinjury (see supplemental Spreadsheet S3). This expression decreases at 3 and 7 days postinjury and then re-elevates again at 10 days. This cluster includes all proteins which are found in normal cell functions. Detected proteins such as: Atp1a2, Brsk1, Napa, Ppp1r9a and Slca3 are all implicated in neurotransmitter transport as seen in the ClueGo analysis (supplemental Fig. S2A). Several proteins involved in normal cellular respiration were also upregulated at the mentioned time points including: Aldh5a1, Cox4i1, and Dlat. Phase 1b (cluster 2) shows elevation in expression of 22 proteins at sham condition. This expression decreases at directly after injury, i.e. 1 and 3 days, and then restores its upregulated expression at 7 and 10 days. Several proteins in this cluster such as Slc27a1, Acsf2, and Acsbg1 are associated with lipid processes within the cells such as lipid transport and energy production (see supplemental Spreadsheet S4). Phase 2 is represented by cluster 4 in Fig. 1A, and mainly is characterized by the set of proteins that are upregulated just after injury (1 day postinjury) and continue their upregulation throughout 3 and 7 days postimpact, were after that, the expression decreases again at 10 days to similar conditions as the sham. Phase 2 is mainly characterized by blood-related processes including platelet activation and aggregation (supplemental Fig. S2B), resulting in a platelet plug formation because of the increase in expression of the several fibrinogen proteins including: F2, Fga, Fgb, Fgg, and Hrg (see supplemental Spreadsheet S5). In this phase also, many proteins involved in acute inflammation are overexpressed including complement protein C3, Mug1, and Serpina1. Phase 3, corresponding to cluster 5, in the first 10 days is also injury related, but is not present until 3 days postinjury and only stays elevated until 7 days postinjury were the expression levels of these proteins return to the sham levels at 10 days postinjury. In this phase, several proteins involved in apoptotic execution phase are detected, including: Gsn, Lmnb1, and Vim. Further, as Hspb1, Pak2, Psme1, and Psme2 involved in MAPK6/MAPK4 signaling pathway are increased (see supplemental Spreadsheet S6). Finally, and within the same phase, several DNA-related proteins, especially ones involved in translation have increased expression (supplemental Fig. S2C). These mainly include elongation factor proteins such as Eef1d and Eef2 along with a major amount of ribosomal proteins including: Fau, Rpl10a, Rpl13, Rpl15, Rpl18, Rpl4, Rpl6, Rplp0, Rps13, Rps3, and Rps8. Phase 4 within the first 10 days corresponds to cluster 3 which contains a small group of proteins including GFAP and Heat shock-related protein (Hspa2), and FABP7 that have increased expression at 3, 7, and 10 days post impact (see supplemental Spreadsheet S7). These proteins are mainly injury related but are not expressed until 3 days after impact and remain with high expression even after 10 days of impact. The proteins found in this cluster are mainly implicated in the initiation of regeneration, astrocytosis, and remyelinization. A representative scheme summing up all described phases is shown in Fig. 1B.
Fig. 1.

Spatially resolved microproteomics analysis reveals time point-specific proteomic phases. A, Heat map based on the hierarchical clustering analysis of the LFQ value corresponding to the detected proteins within the 1 mm2 microenvironment of the following conditions: sham, 1 day, 3 days, 7 days, and 10 days postinjury. Distinct clusters are highlighted and assigned numbers from 1 to 5. B, Representative scheme of all 4 phases concluded from the spatially resolved microproteomic data.
Microproteomics Applied On the Substantia Nigra Reveals Upregulation in PD Related Proteins
We have characterized the involvement of palmitoylcarnitine, and other acylcarntine lipid family members, by applying a spatiotemporal lipid MALDI MSI study of TBI. Palmitoylcarnitine showed maximum expression at 3 days post impact with a spatial distribution surrounding the injury core and colocalizing with the resident microglia of the brain. Surprisingly, palmitoylcarnitine also showed an elevated expression in the ipsilateral substantia nigra at 3 days postinjury, with a similar expression intensity to that of the injury site. Thus, the same spatially resolved microproteomics approach applied to the injured cortical tissue was now applied on tissue sections containing substantia nigra of both ipsilateral (hemisphere of injury) and contralateral hemispheres (hemisphere opposite to injury) at 3 days postinjury. The areas of microdigestion are illustrated on the corresponding figure from the rat brain atlas (27) presented in Fig. 2. The shotgun proteomics yielded the identification of 1513 (see supplemental Spreadsheet S8) proteins in which 55 (see supplemental Spreadsheet S9) were statistically significant between the ipsilateral substantia nigra and the contralateral based on the LFQ expression with a p value = 0.05. The generated heat map showed that 41 proteins are upregulated within the ipsilateral substantia nigra when compared with the contralateral side (Fig. 2 and supplemental Spreadsheet S10). Interestingly, among these ipsilateral overexpressed proteins, synaptotagmins, high-mobility group protein 1 (HMGB-1), glutamate decarboxylase (GAD1), neurotensin and GPR158, have been previously reported to be involved in PD. Quantitative expression studies of Syt11, HMGB1, GAD1, and GPR158, in substantia nigra ipsilateral and contralateral confirmed their upregulation in substantia nigra ipsilateral (supplemental Fig. S3). Targeted pathway analysis revealed that several of our identified proteins upregulated in the ipsilateral substantia nigra (including GAD1 and SERPINA3) have been reported to be involved in PD (Fig. 2 and supplemental Spreadsheet S11). Based on the literature, and as depicted in the figure, several other proteins such as PPT1, ALB, and UBQNL1 have been previously reported in neurodegenerative diseases and cognitive impairment. To validate the data obtained for GPR158 and HMGB1, liquid microjunction microextraction procedures as previously described (12) were realized in substantia nigra ipsilateral and contralateral areas (Fig. 3A). The collected samples were then subjected to Western blotting analyses performed with anti-GPR158 (Fig. 3B) and anti-HMGB (Fig. 3C). Results confirmed the overexpression of a protein of 70kDa corresponding to GPR158 proteolytic form and at 25k Da corresponding to HMGB1 in the substantia nigra ipsilateral area compared with the contralateral ones (Fig. 3B), thus confirming the proteomic data. We also focused our attention on GPR158 and HGMB1, along with their possible interaction proteins known to be involved in PD such as α -synuclein, or cognitive impairment such as osteocalcin.. Docking generated by ClusPro 2.0 reflects the possibility of interaction in ipsilateral and contralateral between HMGB1 and GPR158 from one side, and between GPR158 and osteocalcin from another. Docking all three proteins together allowed us to observe that same docking site is registered between osteocalcin and GPR158, and between HMGB1 and GPR158, which can occur in the 7TM region. Interestingly to note, a competition between HMGB1 and osteocalcin can be suggested as presented in (Fig. 4). Similar in silico analysis was performed, but this time between HMGB1, GPR158, and α-synuclein to better understand how such interactions may affect the synaptic transmission and hint in possible PD like pathology. Docking results show the possibility of α-synuclein to structurally roll up with HMGB1, and to be captured by GPR158, thus hinting to a possible inhibition of α-synuclein function because of its binding to the mentioned proteins (Fig. 5).
Fig. 2.

Microproteomic analysis of the substantia nigra 3 days post TBI reveals elevation in proteins related to PD. Atlas figure with the designated area that were microdigested. The heat map based on the hierarchical clustering analysis of the LFQ value corresponding to the detected proteins in the spatially resolved microproteomic experiments performed on the substantia nigra in both ipsilateral and contralateral hemispheres at 3 days post impact is also shown. Distinct clusters are highlighted. Targeted protein pathway analysis for PD. The figure shows proteins identified (highlighted in blue) in the ipsilateral substantia nigra along with their interaction with proteins reported to be involved in PD (highlighted in green).
Fig. 3.

Western blotting analyses of proteins extracts using liquid microjunnction extraction collected in ipsilateral and contralateral S. nigra area. A, Picture of a slides on which the liquid microjunction microextraction was performed. B, Western blotting analyses performed with the collected samples with the anti-GPR158 (n = 3) and C, Western blotting analyses performed with the collected samples with the anti-HMGB1 (n = 3).
Fig. 4.
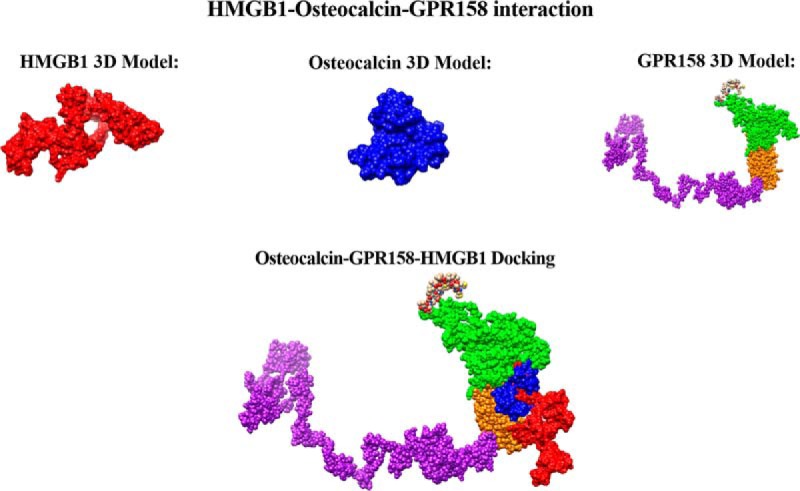
Interaction of HMGB1-Osteocalcin-GPR158. Docking generated by Cluspro 2.0 of osteocalcin (blue), GPR158 (green is the extracellular outside part of the receptor, yellow the transmembrane domain, and purple the intracellular domain), and HMGB1 (red). The interaction of all three proteins is also shown and occurs at the transmembrane region of the GPR158.
Fig. 5.
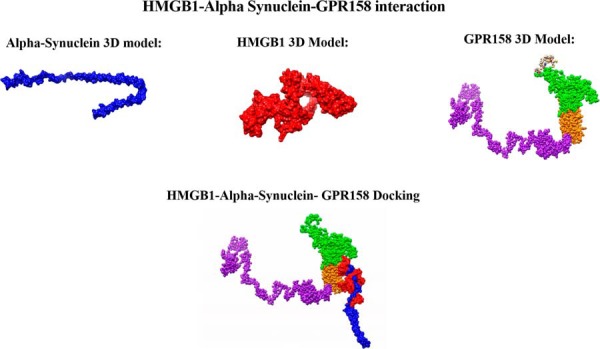
Interaction HMGB1-Alpha Synuclein-GPR158. Docking generated by ClusPro 2.0 of HMGB1 alone (red), α-synuclein alone (blue), and GPR158 (green is the extracellular outside part of the receptor, yellow the transmembrane domain, and purple the intracellular domain) alone along with HMGB1-GPR158-Synuclein_ITasser.
Palmitoylcarnitine Induces Inflammatory Action in Macrophages and Regeneration in Astrocytes
To understand the biological effect of palmitoylcarnitine on different cells present in injured environment, in vitro experiments were carried out by stimulating macrophage and astrocyte cell lines with palmitoylcarnitine. Poststimulation, a shotgun bottom-up proteomics approach was performed on the protein extracts from the mentioned cell lines, along with label-free quantification (LFQ). Proteomic results were subjected to principal component analysis and (PCA) and results show that the proteomic profile upon palmitoylcarnitine stimulation of both cell lines differs from their corresponding control nonstimulated condition (supplemental Fig. S4).
Shotgun bottom-up proteomics analysis of astrocytes yielded 1744 (see supplemental Spreadsheet S12) protein identifications across all samples, 74 (see supplemental Spreadsheet S13) of which showed a significant difference in LFQ expression between palmitoylcarnitine-stimulated astrocytes and control (supplemental Fig. S5). Twenty-eight proteins were upregulated in the control when compared with the palmitoylcarnitine-stimulated astrocytes including TFRC, CDK2, SKP1, and HSP90AA1, proteins which are implicated in cell homeostasis, meiosis, cell cycle, DNA repair and DNA replication (see supplemental Spreadsheet S14 andS15). In addition, these same proteins are involved in signaling mechanisms, mainly NF-kB signaling, along with other proteins such as BST2 and PABPC1. Finally, upregulated control proteins such as ICAM1, CDK2, and PSMA5 all have functions in immunity-related proteins. In contrast, 46 proteins were found to be upregulated upon the stimulation with palmitoylcarnitine when compared with the control. Such upregulated proteins include SPARC, CTNNB1, VIM, and LOX proteins which are implicated in axonogenesis and regeneration (see supplemental Spreadsheet S16 and S15). At the same time, these same proteins, along with others such as PLEC, CAPG, PTPNS, and SERPINE1 are all involved in cellular processes like invasion, cell adhesion, migration, and spreading processes. At the mitochondrial level, proteins involved in fatty acid oxidation and lipid metabolism such as PC, ALDH6A1, ACAA1, and ATP5B were all overexpressed after palimtoylcarnitine stimulation. Thus, stimulation of astrocytes by palmitoylcarnitine leads to altered lipid metabolism, along with an increase in the aggressiveness of such cells by performing actions such as cellular invasion and spreading but also trigger the neurogenesis.
The same approach of palmitoylcarnitine stimulation was then applied on WT macrophage cell line. The proteomic analysis yielded a total of 1099 proteins (see supplemental Spreadsheet S17) identified across all samples, of which 783 (See supplemental Spreadsheet S18) were statistically significant between the stimulated and control conditions with p value = 0.05. Surprisingly, only 19 proteins were upregulated in the control condition when compared with stimulated one, whereas 764 proteins were significantly upregulated after palmitoylcarnitine stimulation (Fig. 6). In the control condition, proteins involved in synaptogenesis, PLC and phospholipase D signaling such as TXN, CFL1, and MARCKS were identified (see supplemental Spreadsheet S19 and S20). However, upon stimulation, major inflammation reactions and inflammatory signaling pathways were overexpressed. Examples of proteins such as LGALS3, PLAA, ASCL1, and RAF1 which are implicated in inflammation were all over expressed (see supplemental Spreadsheet S21). These proteins, along with others such as STAT5B, TMED7, CCDC22, MSN, VAV1, and RPL27A are precisely implicated in NF-kB signaling, a signaling pathway known for involvement in inflammation (28). Besides, a large amount of proteins involved in processes such as immune response and phagocytosis were overexpressed (see supplemental Spreadsheet S20). These include: STAT3, HMGB2, GLS, CD48, GAPDH, PARP1, FBL, PDIA3, and FTH1. Along with the previously mentioned processes observed post palmitoylcarnitine stimulation, others such as ROS generation, cell damage, neuronal death, and injuries were all enhanced by overexpression of several proteins including: RAF1, PLA2G4A, HSPA9, RHOA, PRKCD, and ANXA1. Taken altogether, stimulation of macrophages with palmitoylcarnitine led to an increase in pro-inflammatory response and enhancement of the phagocytic role of these cells. This stimulation led to an activation of such cells toward a full inflammatory phenotype with the ability to induce damage and clear cellular debris, thus possibly explaining one mode of activation of macrophages postinjury as a pro-inflammatory stimulator.
Fig. 6.

Palmitoylcarnitine induces inflammatory action in macrophages. Heat map based on the hierarchical clustering analysis of the LFQ value corresponding to the detected proteins extracted from macrophages stimulated with palmitoylcarnitine or not. Distinct clusters are highlighted. System biology proteomic pathway analysis for network identification for the highlighted clusters is also shown.
DISCUSSION
In the present study, we performed a proteomic analysis on an experimental model of moderate rat CCI using spatially resolved microproteomics to assessed the protein profile at four injury time points (1 day, 3 days, 7 days, and 10 days) depicting the acute, subacute and subchronic phases of clinical TBI. Our microproteomic results collected from within the injury core allowed us to characterize 4 phases of biological processes occurring within the first 10 days post impact. Although phase 1 (a and b) was characterized by proteomic pathways that are usually depicted in uninjured conditions, phases 2, 3, and 4 explained better the injury-related processes in a time course. Phase 2 is characterized by proteins involved in acute inflammation and infiltration of blood-related proteins, which is expected as our CCI model induces breach within the blood-brain barrier. Thus blood related proteins from the circulatory system can enter the injury site. Surprisingly, phase 3, which corresponds to the proteins elevated at 3 and 7 days postimpact, showed a dual effect with regard to survival and death of cells in a synchronized manner. Although the dying cells are undergoing programmed cell death execution such as apoptosis, the surviving cells within the same spatial distribution initiate cellular repair at the DNA level by upregulating several ribosomal proteins involved in the translation process. Thus, in TBI, the initiation of repair at the DNA level occurs as early as 3 days after impact. Compared with spinal cord injury (SCI), these proteomic phases are not depicted, as we have previously shown that the proteomic profile postinjury to the spinal cord is follows an extensive inflammatory pattern that becomes fully chronicle without any repair with the progression of time (29, 30). In SCI, there is now detection of regeneration-related processes at the 3 day time point that was detected in the case of TBI. Moreover, the proteomic profile of the brain tends to be restored to a similar profile of sham condition, as we have shown in this study. These findings clearly demonstrate the high plasticity factor of the brain by possessing the capability to try and restore normal protein profile after 10 days of mechanical impact.
As previously mentioned, we have characterized an injury related lipid marker, which had a major expression at 3 days postimpact. Long chain acylcarnitines (LC AC) and even the medium chain acylcarnitines (MC AC) were proposed to have a pro-inflammatory effect on immune cells. Treatment of bone marrow-derived macrophages with lauroylcarnitine induced an “M1” pro-inflammatory phenotype in these cells (31). In addition, treatment of RAW 264.7 cells, a murine monocytic cell, by acylcarnitine with a 14-carbon side chain led to an increase in COX-2 expression, an important toll-like receptor (TLR) target gene product, along with the increase in reactive oxygen species (ROS) production (32). This same treatment also induced the phosphorylation of JNK and ERK via MyD88 signaling pathway. By going deeper into our overexpressed proteomic data from macrophages stimulated with palmitoylcarnitine, we have depicted several proteins that can either stimulate or are directly involved in the MyD88 inflammatory pathway. One protein, high mobility group box 1 protein (HMGB1), can activate macrophages by acting as a ligand for toll-like receptor (TLR) 2 and 4, resulting in the activation of the MyD88 signaling pathway (33). Another protein upregulated upon palmitoylcarnitine treatment, tyrosine protein kinase (SYK), plays a critical role as a regulator of MyD88 posttranslational modifications, precisely by phosphorylating MyD88 (34). Signal transducer and activator of transcription 3 (STAT3) was also elevated post stimulation with palmitoylcarnitine. In the hypothalamus of mice, and in the state of infection and inflammation, MyD88 has an important role in the activation of STAT3 (35). Two other major proteins of importance, which were also increased post stimulation with palmitoylcarnitine are Galectin-3 (Gal-3) and Heat shock Protein (HSPD1). HSPD1, also known as 60-KDa heat shock protein (HSP60), mediates neurodegeneration via the TLR4-MyD88 pathway in the cerebral cortex of a mouse model (36). Gal-3 can induce the proliferation and survival of cancer cells by signaling through TLR4 pathway, as the case of lung adenocarcinoma by activating the TLR4/MyD88/p-p65 pathway expression (37). Gal-3 is known to bind to TLR-4, and administration of a neutralizing antibody against Gal-3 decreases the expression of IL-1β, IL-6, TNFα and NOS2 and promotes neuroprotection in cortical and hippocampal cell populations after head injury (38). Gal-3 is constitutively expressed in the subventricular zone (SVZ) and the rostral migratory stream (RMS) (39) and does not affect neurogenesis, but, instead, regulates neuronal migration. Gal-3-null mice exhibit aberrant SVZ astrocyte morphology and reduced SVZ neuroblast migration and explants treated with Gal-3-blocking antibody show decreased neuronal migration, whereas expression of recombinant Gal-3 increases migration distances.
By comparing with our tissue microproteomic data, we were also able to detect the overexpression of Gal-3 and HSPD1 in cluster 5 of the heatmap in Fig. 1, i.e. elevated expression at 3 and 7 days postinjury. Gal-3 protein has been previously described in the context of TBI (40) but our work suggests a possible explanation for one mode of activation mechanism for Gal-3 postinjury. The peak of Gal-3 expression at 3 days postinjury in the spatially resolved microproteomic data is in coherence with the maximum expression pattern of palmitoylcarnitine, and several other acylcarnitine members, which is also at 3 days postinjury. Gal-3 is a chimeric galectin that possesses both a C-terminal carbohydrate-recognition domain (CRD), and an N-terminal aggregating domain that can interact with noncarbohydrate ligand partners. It has been suggested to act as a pathogen pattern recognition receptor (PRR) that can detect pathogen-associated molecular patterns (PAMPs) and endogenous ligands from injured tissue and has been reported to bind to LPS via both C' and N′ terminals (41). Saturated medium and long-chain fatty acids have been reported to mimic the pro-inflammatory effect of the Lipid A moiety of LPS, by activating NF-KB via TLR mechanisms. As such, acylcarnitines containing these long chain fatty acids could likewise act in a similar manner by serving as ligands for Gal-3 binding. This needs to be further confirmed by PRR reporter gene assays as both can act as alarmins in this process. The upregulation of Gal-3 and enhanced presence of acylcarnitines within the lesion site at specific time points lead us to the mentioned proposal of the cycle of events that occur within the 10-day timeframe after injury.
Our spatially resolved microproteomic experiments performed on the substantia nigra established the over-expression of several proteins, including: GAD1, Syt11, HMGB1, PPT1 and GPR158 of which most have been linked to PD. Glutamate decarboxylase enzyme is one key enzyme involved in Parkinson's disease (42). The bilateral delivery of GAD1 by an adeno-associated viral vector into the subthalamic nucleus of patients with Parkinson disease resulted in significant improvement during a six-month study (43). Synaptotagmin proteins are involved in neurodegeneration (44). HMGB1 has been reported to induce neuroinflammation, and interact with α-synuclein, in addition to participating in pathogenesis of PD (45). Recently, it has been demonstrated that HMGB1 could be beneficial by promoting the autophagy dependent degradation of wild type or mutated α-synuclein to limit the α-synuclein associated degeneration in SH-SY5Y cells (46). Our docking analysis confirmed that α-synuclein and HMGB1 can directly interact, and α-synuclein can roll up with HMGB1. Similarly, GPR158 can also bind to α-synuclein and the synchronized binding of both GPR158 and HMGB1 can capture α-synuclein. It is known that in PD, alpha-synuclein lose their monomer structure and form fibrils, which thus inhibit its monomer function in aiding synaptic function (47). We further suggest a new deleterious mechanism by which α-synuclein cannot play its biological function i.e. synaptic transmission and thus lead to PD. We propose that the binding of both HMGB1 and GPR158 to α-synuclein after TBI, restrict the ability of α-synuclein monomers to aid synaptic transmission. Indeed, α-syn modulate endocytic import of iron and vesicle transport of dopamine during normal physiology. Disregulated α-syn function may mediate iron and dopamine-dependent oxidative stress through impaired cellular location and increase propensity for α-syn aggregation (48). Gpr158 is abundant in neurons of the CA3 region of the hippocampus, the brain's memory center. Recently GPR158 has been shown to be involved in depression following chronic stress by affecting key signaling pathways involved in mood regulation in the region of the brain called prefrontal cortex (49). Recently it has been shown that Gpr158 mediates osteocalcin's regulation of cognition (50). Docking generated between osteocalcin and GPR158, and between HMGB1 and GPR158 are similar and thus leading to the idea of competition between HMGB1 and osteocalcin for GPR158. Thus, if HMGB1 can bind to GPR158 and inhibit the binding of osteocalcin, it may lead to a decrease in the osteocalcin related cognition which is mainly mediated by GPR158. HMGB1 is also known to be involved in depression, and thus, such competition could contribute to our observation (51).
Taken altogether, our full proteomic results lead us to hypothesize a link between acylcarnitines produced by microglia in substantia nigra and the initiation of changes associated to PD 3 days after TBI. This is mainly because of the idea that we have found several PD implicated proteins overexpressed directly after TBI. It is also known that boxers are developing PD in later stages of life after chronicle TBI (52). Acylcarnitine expression in substantia nigra, produced by microglia can be an inducer of the early stage of the disease. Chronic stress will produce much more of such lipids in this region, leading to a local inflammatory environment in which GPR158, synaptotagmin, GAD1, HMGB1 will be over-expressed and will be the precursors of the disease. The last question in the case of an open head injury is about the proximal relation of the injury site in the cortex and the acylcarnitine localization in the substantia nigra. We have previously shown that there is no continuous expression of acylcarnitine between the injured cortical tissue and the substantia nigra, as both are distant from each other and possess the same intensity of expression for acylcarnitines. We hypothesize that extracellular vesicles (EVs) containing acylcarnitines produced by microglia at the lesion site, will impact microglia cells in substantia nigra or inflammation propagation from the injury site to the other brain regions including substantia nigra. Inflammation is restricted to the lesion site and inflammation linked lipids do not go to the substantia nigra post-TBI as we have already published (53). EVs containing carnitine transporter OCTN2 have been recently demonstrated (54) where immune cells are known to produce EVs containing acylcarnitines (55). Also, there is growing evidence indicating the role of EVs in PD (56). We can thus speculate as we present in Fig. 7, that EVs from lesion site can target microglia in substantia nigra leading to a local inflammatory environment stimulating expression of proteins involved in stress.
Fig. 7.

Schematic representation of molecular mechanisms associated with acylcarnitine expression in substantia nigra 3 days post-TBI. Acylcarnitines were found to be associated with microglia and are specifically found by MALD MSI, 3 days post-TBI, in the subtantia nigra. Spatially resolved proteomics reveals the expression of proteins known to be involved in PD. Acylcarnitines are known to activate microglial through TLR4/MyD88 pathway leading to inflammatory cytokines and alarmins (Galectin-3). GAL-3 is a pro-inflammatory protein known to promote neuron inflammation leading to the expression of proteins which were recently shown to be involved in PD (GPR158, HMGB1, GAD1). Docking models between HMGB1 and α -synuclein as well as between HMGB1-osteocalcin and GPR158 are also presented. This putative physiological pathway could make a link between chronic TBI and PD.
In conclusion, our spatiotemporal study of mild-TBI conducted by applying a spatially resolved microproteomics analysis revealed the plasticity factor of the brain. At the 10 days after injury, the brain tries to recompensate to the noninjured state at the proteomic level. These integrative data lead to unveiling the central role of acylcarnitines lipid family in the inflammation and associated molecular mechanisms. Very interestingly, such study gives also new insights on how TBI can affect at longer spatial distance other areas of the brain such as substantia nigra giving molecular support to clinical observation relating repetitive TBI events and emergence of PD.
Data Availability
Spatially resolved microproteomics, astrocytes and macrophage proteomic datasets including MaxQuant files and annotated MS/MS datasets, as well as the MALDI imaging datasets, were uploaded to ProteomeXchange Consortium via the PRIDE database, and was assigned the dataset identifier PXD011262.
Supplementary Material
Acknowledgments
We would like to acknowledge Ministere del'Enseignement Superieur, de la Recherche et de l'Innovation (MESRI), Institut National de la Santé et de la Recherche Mé dicale (Inserm) and Université de Lille, and the Association of Scientific Orientation and Specialization (ASOS Scholarship).
Footnotes
* This research was supported by funding from Ministère de l'Enseignement Supérieur, de la Recherche et de l'Innovation (MESRI), Institut National de la Santé et de la Recherche Médicale (Inserm) and Université de Lille. K.M. acknowledges funding from the Association of Scientific Orientation and Specialization (ASOS Scholarship).
 This article contains supplemental Figures and Spreadsheets. We declare no competing interests.
This article contains supplemental Figures and Spreadsheets. We declare no competing interests.
1 The abbreviations used are:
- TBI
- traumatic brain injury
- AD
- Alzheimer's disease
- CCI
- controlled cortical impact
- LFQ
- label free quantification
- MALDI
- matrix assisted laser desorption/ionization
- MSI
- mass spectrometry imaging
- PD
- Parkinson's disease.
REFERENCES
- 1. Mckee A. C. and Daneshvar D. H. (2015) in Handbook of clinical neurology, pp 45–66 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Faul M., Xu L., Wald M., and Coronado V. (2010) Traumatic brain injury in the United States: Emergency department visits, hospitalizations and deaths, 2002–2006 [Google Scholar]
- 3. Galgano M., Toshkezi G., Qiu X., Russell T., Chin L., and Zhao L. R. (2017) Traumatic brain injury. Cell Transplant. 26, 1118–1130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Gardner R. C., Byers A. L., Barnes D. E., Li Y., Boscardin J., and Yaffe K. (2018) Mild TBI and risk of Parkinson disease. Neurology 90, e1771–e1779 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Johnson V. E., Stewart W., and Smith D. H. (2010) Traumatic brain injury and amyloid-β pathology: a link to Alzheimer's disease? Nat. Rev. Neurosci. 11, 361–370 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Roberts G. W., Gentleman S. M., Lynch A., Murray L., Landon M., and Graham D. I. (1994) Beta amyloid protein deposition in the brain after severe head injury: implications for the pathogenesis of Alzheimer's disease. J. Neurol. Neurosurg. Psychiatry 57, 419–425 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Gardner R. C., Burke J. F., Nettiksimmons J., Goldman S., Tanner C. M., and Yaffe K. (2015) Traumatic brain injury in later life increases risk for Parkinson disease. Ann. Neurol. 77, 987–995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Acosta S. A., Tajiri N., de la Pena I., Bastawrous M., Sanberg P. R., Kaneko Y., and Borlongan C. V. (2015) Alpha-synuclein as a pathological link between chronic traumatic brain injury and Parkinson's disease. J. Cell. Physiol. 230, 1024–1032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Xu L. and Pu J. (2016) Alpha-synuclein in Parkinson's disease: from pathogenetic dysfunction to potential clinical application. Parkinsons. Dis. 2016, 1–10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Impellizzeri D., Campolo M., Bruschetta G., Crupi R., Cordaro M., Paterniti I., Cuzzocrea S., and Esposito E. (2016) Traumatic brain injury leads to development of Parkinson's disease related pathology in mice. Front. Neurosci. 10, 458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Karve I. P., Taylor J. M., and Crack P. J. (2016) The contribution of astrocytes and microglia to traumatic brain injury. Br. J. Pharmacol. 173, 692–702 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Quanico J., Franck J., Dauly C., Strupat K., Dupuy J., Day R., Salzet M., Fournier I., and Wisztorski M. (2013) Development of liquid microjunction extraction strategy for improving protein identification from tissue sections. J. Proteomics 79, 200–218 [DOI] [PubMed] [Google Scholar]
- 13. Kobeissy F. H., Ottens A. K., Zhang Z., Liu M. C., Denslow N. D., Dave J. R., Tortella F. C., Hayes R. L., and Wang K. K. W. (2006) Novel differential neuroproteomics analysis of traumatic brain injury in rats. Mol. Cell. Proteomics 5, 1887–1898 [DOI] [PubMed] [Google Scholar]
- 14. Ding J., Ding Z., Yuan F., Guo J., Chen H., Gao W., Wang R., Gu Y., Chen J., Guo Y., and Tian H. (2015) Proteomics analysis after traumatic brain injury in rats: the search for potential biomarkers. Arq. Neuropsiquiatr. 73, 342–349 [DOI] [PubMed] [Google Scholar]
- 15. Osier N. D. and Dixon C. E. (2016) The controlled cortical impact model: applications, considerations for researchers, and future directions. Front. Neurol. 7, 134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Wiśniewski J. R., Zougman A., Nagaraj N., and Mann M. (2009) Universal sample preparation method for proteome analysis. Nat. Methods 6, 359–362 [DOI] [PubMed] [Google Scholar]
- 17. Cox J. and Mann M. (2008) MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat. Biotechnol. 26, 1367–1372 [DOI] [PubMed] [Google Scholar]
- 18. Yuryev A., Kotelnikova E., and Daraselia N. (2009) Ariadne's ChemEffect and Pathway Studio knowledge base. Expert Opin. Drug Discov. 4, 1307–1318 [DOI] [PubMed] [Google Scholar]
- 19. Bonnet A., Lagarrigue S., Liaubet L., Robert-Granié C., SanCristobal M., and Tosser-Klopp G. (2009) Pathway results from the chicken data set using GOTM, Pathway Studio and Ingenuity softwares. BMC Proc. 3, S11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Pyatnitskiy M., Mazo I., Shkrob M., Schwartz E., and Kotelnikova E. (2014) Clustering gene expression regulators: new approach to disease subtyping. PLoS ONE 9, e84955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Zhang Y. (2008) I-TASSER server for protein 3D structure prediction. BMC Bioinformatics 9, 40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Berman H. M., Westbrook J., Feng Z., Gilliland G., Bhat T. N., Weissig H., Shindyalov I. N., and Bourne P. E. (2000) The protein data bank. Nucleic Acids Res. 28, 235–242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Xu Y., Yang W., Wu J., and Shi Y. (2002) Solution structure of the first HMG box domain in human upstream binding factor. Biochemistry 41, 5415–5420 [DOI] [PubMed] [Google Scholar]
- 24. Ulmer T. S., Bax A., Cole N. B., and Nussbaum R. L. (2005) Structure and dynamics of micelle-bound human synuclein. J. Biol. Chem. 280, 9595–9603 [DOI] [PubMed] [Google Scholar]
- 25. Comeau S. R., Gatchell D. W., Vajda S., and Camacho C. J. (2004) ClusPro: a fully automated algorithm for protein-protein docking. Nucleic Acids Res. 32, W96–W99 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Pettersen E. F., Goddard T. D., Huang C. C., Couch G. S., Greenblatt D. M., Meng E. C., and Ferrin T. E. (2004) UCSF Chimera? A visualization system for exploratory research and analysis. J. Comput. Chem. 25, 1605–1612 [DOI] [PubMed] [Google Scholar]
- 27. Paxinos G. and Watson C. (1998) The rat brain in stereotaxic coordinates. Academic Press, pp.1–474 [DOI] [PubMed] [Google Scholar]
- 28. Liu T., Zhang L., Joo D., and Sun S.-C. (2017) NF-κB signaling in inflammation. Signal Transduct. Target. Ther. 2, 17023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Cizkova D., Le Marrec-Croq F., Franck J., Slovinska L., Grulova I., Devaux S., Lefebvre C., Fournier I., and Salzet M. (2014) Alterations of protein composition along the rostro-caudal axis after spinal cord injury: proteomic, in vitro and in vivo analyses. Front. Cell. Neurosci. 8, [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Devaux S., Cizkova D., Quanico J., Franck J., Nataf S., Pays L., Hauberg-Lotte L., Maass P., Kobarg J. H., Kobeissy F., Mériaux C., Wisztorski M., Slovinska L., Blasko J., Cigankova V., Fournier I., and Salzet M. (2016) Proteomic analysis of the spatio-temporal based molecular kinetics of acute spinal cord injury identifies a time- and segment-specific window for effective tissue repair. Mol. Cell. Proteomics 15, 2641–2670 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Sampey B. P., Freemerman A. J., Zhang J., Kuan P.-F., Galanko J. A., O'Connell T. M., Ilkayeva O. R., Muehlbauer M. J., Stevens R. D., Newgard C. B., Brauer H. A., Troester M. A., and Makowski L. (2012) Metabolomic profiling reveals mitochondrial-derived lipid biomarkers that drive obesity-associated inflammation. PLoS ONE 7, e38812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Rutkowsky J. M., Knotts T. A., Ono-Moore K. D., McCoin C. S., Huang S., Schneider D., Singh S., Adams S. H., and Hwang D. H. (2014) Acylcarnitines activate proinflammatory signaling pathways. Am. J. Physiol. Metab. 306, E1378–E1387 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Peng H.-H., Liu Y.-J., Ojcius D. M., Lee C.-M., Chen R.-H., Huang P.-R., Martel J., and Young J. D. (2017) Mineral particles stimulate innate immunity through neutrophil extracellular traps containing HMGB1. Sci. Rep. 7, 16628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Gurung P., Fan G., Lukens J. R., Vogel P., Tonks N. K., and Kanneganti T.-D. (2017) Tyrosine Kinase SYK Licenses MyD88 Adaptor Protein to Instigate IL-1α-Mediated Inflammatory Disease. Immunity 46, 635–648 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Yamawaki Y., Kimura H., Hosoi T., and Ozawa K. (2010) MyD88 plays a key role in LPS-induced Stat3 activation in the hypothalamus. Am. J. Physiol. Integr. Comp. Physiol. 298, R403–R410 [DOI] [PubMed] [Google Scholar]
- 36. Rosenberger K., Dembny P., Derkow K., Engel O., Krüger C., Wolf S. A., Kettenmann H., Schott E., Meisel A., and Lehnardt S. (2015) Intrathecal heat shock protein 60 mediates neurodegeneration and demyelination in the CNS through a TLR4- and MyD88-dependent pathway. Mol. Neurodegener. 10, 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Zhou W., Chen X., Hu Q., Chen X., Chen Y., and Huang L. (2018) Galectin-3 activates TLR4/NF-κB signaling to promote lung adenocarcinoma cell proliferation through activating lncRNA-NEAT1 expression. BMC Cancer 18, 580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Yip P. K., Carrillo-Jimenez A., King P., Vilalta A., Nomura K., Chau C. C., Egerton A. M. S., Liu Z.-H., Shetty A. J., Tremoleda J. L., Davies M., Deierborg T., Priestley J. V., Brown G. C., Michael-Titus A. T., Venero J. L., and Burguillos M. A. (2017) Galectin-3 released in response to traumatic brain injury acts as an alarmin orchestrating brain immune response and promoting neurodegeneration. Sci. Rep. 7, 41689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Comte I., Kim Y., Young C. C., van der Harg J. M., Hockberger P., Bolam P. J., Poirier F., and Szele F. G. (2011) Galectin-3 maintains cell motility from the subventricular zone to the olfactory bulb. J. Cell Sci. 124, 2438–2447 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Nishikawa H. and Suzuki H. (2018) Possible role of inflammation and galectin-3 in brain injury after subarachnoid hemorrhage. Brain Sci. 8, 30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Rutkowsky J. M., Knotts T. A., Ono-Moore K. D., McCoin C. S., Huang S., Schneider D., Singh S., Adams S. H., and Hwang D. H. (2014) Acylcarnitines activate proinflammatory signaling pathways. AJP Endocrinol. Metab. 306, E1378–E1387 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Lloyd K. G. and Hornykiewicz O. (1973) L-glutamic acid decarboxylase in Parkinson's disease: effect of L-Dopa therapy. Nature 243, 521–523 [DOI] [PubMed] [Google Scholar]
- 43. Lewitt P. A., Rezai A. R., Leehey M. A., Ojemann S. G., Flaherty A. W., Kostyk S. K., Thomas K., Sarkar A., Siddiqui M. S., Tatter S. B., Schwalb J. M., Poston K. L., Henderson J. M., Kurlan R. M., Richard I. H., Van Meter L., Sapan C. V., During M. J., Kaplitt M. G., and Feigin A. (2011) Articles AAV2-GAD gene therapy for advanced Parkinson's disease: a double-blind, sham-surgery controlled, randomised trial. Lancet Neurol. 10, 309–319 [DOI] [PubMed] [Google Scholar]
- 44. Glavan G., Schliebs R., and Zivin M. (2009) Synaptotagmins in neurodegeneration. Anat. Rec. 292, 1849–1862 [DOI] [PubMed] [Google Scholar]
- 45. Ko E. A., Min H. J., and Shin J.-S. (2012) Interaction of high mobility group box-1 (HMGB1) with synuclein and its aggregation (172.28). J. Immunol. 188, (1 Supplement) 172.28 [Google Scholar]
- 46. Guan Y., Li Y., Zhao G., and Li Y. (2018) HMGB1 promotes the starvation-induced autophagic degradation of α-synuclein in SH-SY5Y cells Atg 5-dependently. Life Sci. 202, 1–10 [DOI] [PubMed] [Google Scholar]
- 47. Ruipérez V., Darios F., and Davletov B. (2010) Alpha-synuclein, lipids and Parkinson's disease. Prog. Lipid Res. 49, 420–428 [DOI] [PubMed] [Google Scholar]
- 48. Duce J. A., Wong B. X., Durham H., Devedjian J. C., Smith D. P., and Devos D. (2017) Post translational changes to α-synuclein control iron and dopamine trafficking; a concept for neuron vulnerability in Parkinson's disease. Mol. Neurodegener. 12, 45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Sutton L. P., Orlandi C., Song C., Oh W. C., Muntean B. S., Xie K., Filippini A., Xie X., Satterfield R., Yaeger J. D. W., Renner K. J., Young S. M., Xu B., Kwon H., and Martemyanov K. A. (2018) Orphan receptor GPR158 controls stress-induced depression. Elife 7, [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Khrimian L., Obri A., Ramos-Brossier M., Rousseaud A., Moriceau S., Nicot A. S., Mera P., Kosmidis S., Karnavas T., Saudou F., Gao X. B., Oury F., Kandel E., and Karsenty G. (2017) Gpr158 mediates osteocalcin's regulation of cognition. J. Exp. Med. 214, 2859–2873 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Wang B., Lian Y. J., Su W.-J., Peng W., Dong X., Liu L. L., Gong H., Zhang T., Jiang C. L., and Wang Y. X. (2018) HMGB1 mediates depressive behavior induced by chronic stress through activating the kynurenine pathway. Brain. Behav. Immun. 72, 51–60 [DOI] [PubMed] [Google Scholar]
- 52. Lolekha P., Phanthumchinda K., and Bhidayasiri R. (2010) Prevalence and risk factors of Parkinson's disease in retired Thai traditional boxers. Mov. Disord. 25, 1895–1901 [DOI] [PubMed] [Google Scholar]
- 53. Mallah K., Quanico J., Trede D., Kobeissy F., Zibara K., Salzet M., and Fournier I. (2018) Lipid changes associated with traumatic brain injury revealed by 3D MALDI-MSI. Anal. Chem. 90, 10568–10576 [DOI] [PubMed] [Google Scholar]
- 54. Console L., Scalise M., Tonazzi A., Giangregorio N., and Indiveri C. (2018) Characterization of Exosomal SLC22A5 (OCTN2) carnitine transporter. Sci. Rep. 8, 3758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Colombo M., Raposo G., and Théry C. (2014) Biogenesis, secretion, and intercellular interactions of exosomes and other extracellular vesicles. Annu. Rev. Cell Dev. Biol. 30, 255–289 [DOI] [PubMed] [Google Scholar]
- 56. Russo I., Bubacco L., and Greggio E. (2012) Exosomes-associated neurodegeneration and progression of Parkinson's disease. Am. J. Neurodegener. Dis. 1, 217–225 [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Spatially resolved microproteomics, astrocytes and macrophage proteomic datasets including MaxQuant files and annotated MS/MS datasets, as well as the MALDI imaging datasets, were uploaded to ProteomeXchange Consortium via the PRIDE database, and was assigned the dataset identifier PXD011262.