

Abstract
Background
Key mechanisms of cell division and its regulation are well understood in model bacteria such as Escherichia coli and Bacillus subtilis. In contrast, current knowledge on the regulation of cell division in Actinobacteria is rather limited. FtsZ is one of the key players in this process, but nothing is known about its transcriptional regulation in Corynebacterium glutamicum, a model organism of the Corynebacteriales.
Results
In this study, we used DNA affinity chromatography to search for transcriptional regulators of ftsZ in C. glutamicum and identified the Cg1631 protein as candidate, which was named FtsR. Both deletion and overexpression of ftsR caused growth defects and an altered cell morphology. Plasmid-based expression of native ftsR or of homologs of the pathogenic relatives Corynebacterium diphtheriae and Mycobacterium tuberculosis in the ΔftsR mutant could at least partially reverse the mutant phenotype. Absence of ftsR caused decreased expression of ftsZ, in line with an activator function of FtsR. In vivo crosslinking followed by affinity purification of FtsR and next generation sequencing of the enriched DNA fragments confirmed the ftsZ promoter as in vivo binding site of FtsR and revealed additional potential target genes and a DNA-binding motif. Analysis of strains expressing ftsZ under control of the gluconate-inducible gntK promoter revealed that the phenotype of the ΔftsR mutant is not solely caused by reduced ftsZ expression, but involves further targets.
Conclusions
In this study, we identified and characterized FtsR as the first transcriptional regulator of FtsZ described for C. glutamicum. Both the absence and the overproduction of FtsR had severe effects on growth and cell morphology, underlining the importance of this regulatory protein. FtsR and its DNA-binding site in the promoter region of ftsZ are highly conserved in Actinobacteria, which suggests that this regulatory mechanism is also relevant for the control of cell division in related Actinobacteria.
Electronic supplementary material
The online version of this article (10.1186/s12866-019-1553-0) contains supplementary material, which is available to authorized users.
Keywords: DNA affinity chromatography, Transcriptional regulation, ChAP-Seq, Corynebacterium diphtheriae, Mycobacterium tuberculosis, Rv1828
Background
Bacterial reproduction is usually characterized by cellular growth followed by binary fission of a mother cell into two daughter cells. During this process, the bacteria need to coordinate several distinct processes such as DNA replication, biogenesis of the new cell wall, and the division process itself. Although the overall process is similar, several different concepts have evolved in distinct bacterial groups. Escherichia coli and Bacillus subtilis represent two of the best studied species in this respect [1, 2]. Corynebacterium glutamicum is a non-pathogenic, facultative anaerobic, Gram-positive soil bacterium frequently used as model organism for the order Corynebacteriales, which includes important pathogens such as Corynebacterium diphtheriae and Mycobacterium tuberculosis causing the fatal infectious diseases diphtheria and tuberculosis [3–6]. C. glutamicum strains are used in industrial biotechnology for the production of l-amino acids and other metabolites, as well as for proteins [7–11].
One of the key players of cell division is the highly conserved and essential FtsZ protein, which is present in most bacteria. It is the first protein that moves to the future division site, polymerizes into a ring structure, and serves as scaffold for the assembly of further cell division proteins [12]. The resulting protein complex is called the divisome and drives the constriction during cell division [13]. Interestingly, Actinobacteria such as C. glutamicum lack homologs of several important cell division-related genes described for E. coli or B. subtilis. One of these is MreB, an actin homolog essential for lateral cell wall elongation in E. coli, Caulobacter crescentus, and B. subtilis [14–16]. In contrast to these species, C. glutamicum inserts new cell wall material at the poles, representing an apical elongation mechanism [17]. Another protein absent in C. glutamicum is FtsN, an essential membrane protein of E. coli with a potential role in coordination of cell division [1, 18]. However, most strikingly is the complete absence of homologs of many known spatial and temporal, positive and negative regulators of cell division such as ftsA, ezrA, noc/slmA, zapA, and minCDE [18, 19]. This implies that cell division in the Corynebacteriales and probably other Actinobacteria is regulated through proteins and mechanisms that differ significantly from previously described ones.
To date, we have only a limited understanding of the regulation of cytokinesis in Corynebacteriales [18]. Accurate expression of ftsZ is critical for normal growth of C. glutamicum and M. tuberculosis and small variations lead to severe morphological changes and affect cell viability [19–21]. Thus, the FtsZ level has to be tightly regulated, which is supported by different studies of the ftsZ promoter region showing a very complex transcription of ftsZ in various actinobacteria [19, 22–24]. Current knowledge on transcriptional regulation of ftsZ expression in C. glutamicum is sparse. A complex of the DNA-binding proteins WhiA (Cg1792) and WhcD (Cg0850) was reported to bind to the ftsZ promoter region [25], however, the effect of this interaction on ftsZ expression remains to be elucidated. We previously showed that the FtsZ protein can be phosphorylated in vitro by the serine/threonine protein kinases PknA, PknB, and PknL and is an in vivo substrate of the phospho-Ser/Thr protein phosphatase Ppp [26], suggesting that the properties and activities of FtsZ can also be influenced by posttranslational regulation [18].
In this study, we identified and characterized the transcriptional regulator Cg1631 of C. glutamicum as an activator of ftsZ expression. Therefore, Cg1631 was renamed FtsR, standing for ftsZ regulator. We show that FtsR is critical for normal growth and cell morphology of C. glutamicum, suggesting an important role of this protein in the regulation of cell division.
Results
Search for transcriptional regulators of ftsZ
To investigate transcriptional control of ftsZ, we searched for potential regulators by DNA affinity chromatography using a 417 bp DNA fragment covering the 3′-end of the ftsQ coding region (36 bp), the intergenic region between ftsQ and ftsZ, and the first 96 bp of the ftsZ coding region. The immobilized DNA fragment was incubated with crude protein extract of C. glutamicum wild type cells. The proteins eluting after several washing steps were separated by SDS-PAGE. Subsequent MALDI-ToF-MS analysis resulted in the identification of eight proteins (Fig. 1, Additional file 1: Table S1). Six of them represent proteins that are often found in DNA affinity purifications with C. glutamicum cell extracts independent of the promoter region used. These are the single-stranded DNA-binding protein Ssb (Cg3307), the restriction endonucleases CglIR (Cg1997) and CglIIR (Cg1998), the ε-subunit of DNA polymerase III (Cg2321), DNA polymerase I (Cg1525), and the transcription termination factor Rho (Cg1354). Most interesting were the remaining two proteins, both of which are annotated as transcriptional regulators, Cg0444 (RamB) and Cg1631. The identity of these two proteins was further verified by MS/MS analysis of selected peptides (Additional file 1: Table S1). The regulator of acetate metabolism RamB (Cg0444) has already been studied quite extensively and represses genes involved in acetate metabolism and alleviates glucose and sucrose uptake [27, 28]. A function in ftsZ regulation has not been described for RamB and a well-conserved RamB-binding site (AA/GAACTTTGCAAA [28]) is absent from the ftsZ promoter region. In contrast to RamB, the function of the second transcriptional regulator enriched with the ftsZ promoter fragment, Cg1631, was completely unknown and Cg1631 was therefore the focus of this study. Due to its role in ftsZ regulation, the protein was named FtsR.
Fig. 1.
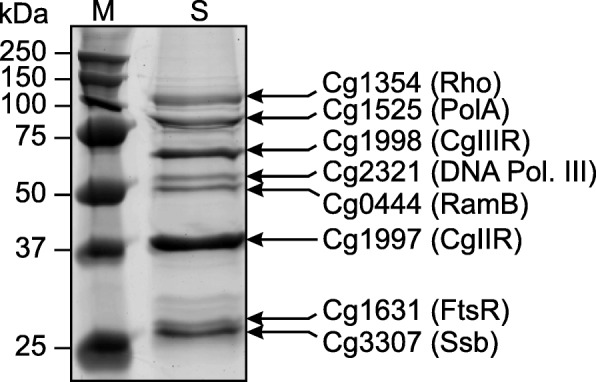
DNA affinity chromatography with the ftsZ promoter region. Crude cell extract of C. glutamicum ATCC13032 cultivated in glucose minimal medium to the mid-exponential growth phase was incubated with an immobilized 417-bp DNA fragment covering the ftsZ promoter region and strongly binding proteins were eluted with a high-salt buffer. Proteins enriched with the ftsZ promoter were separated by SDS-PAGE, stained with colloidal Coomassie, and identified by peptide-mass fingerprinting using MALDI-ToF MS analysis (Additional file 1: Table S1). The band with an apparent mass of about 27 kDa was identified as FtsR. M, marker; S, sample
FtsR, a MerR-type transcriptional regulator conserved in Actinobacteria
The gene ftsR (cg1631) of C. glutamicum ATCC13032 encodes a protein of 252 amino acids (calculated mass 27.23 kDa), which was annotated as a transcriptional regulator of the MerR family due to the sequence similarity of the N-terminal region of FtsR to other members of the MerR family. The ftsR gene and its immediate genomic vicinity including the upstream gene odhI (cg1630) and the two downstream genes (cg1632 and cg1633) were found to be highly conserved within the Actinobacteria (Fig. 2). The odhI gene encodes the 2-oxoglutarate dehydrogenase inhibitor protein OdhI, which acts as a phosphorylation-dependent switch controlling the activity of the 2-oxoglutarate dehydrogenase complex [33–35]. The homologous protein of mycobacteria is GarA [36]. The gene cg1632 encodes a putative bifunctional nuclease having both DNase and RNase activity (PFAM PF02577), and cg1633 encodes another yet uncharacterized transcriptional regulator of the MerR family. Previous RNAseq analysis indicated that ftsR is co-transcribed with these neighboring genes in various combinations: cg1629-cg1633 was annotated as primary operon and cg1630-cg1633 and cg1631-cg1633 as sub-operons [29] (Fig. 2). The genomic region upstream of odhI (cg1630) and downstream of cg1633 varies in different Actinobacteria.
Fig. 2.

Phylogenetic conservation of ftsR (cg1631) and adjacent genes. ftsR and its homologs in different actinobacterial species of various genera are shown in red. Neighboring homologous genes are colored alike, whereas grey arrows indicate genes that are not conserved in the gene cluster analyzed. The black arrows indicate transcriptional start sites determined by RNA-Seq for C. glutamicum [29]. Note that the four-gene-cluster comprising ftsR, its upstream gene odhI/garA, and the two downstream genes encoding a bifunctional nuclease and a MerR-type transcriptional regulator is strongly conserved. The figure was prepared based on data of MicrobesOnline [30] and ERGO [31]. The amino acid sequence identity of the C. glutamicum FtsR homologs is given on the right and was derived from NCBI BLAST [32]. For lack of space, some locus tag prefixes were omitted in short genes. Gene lengths are approximately to scale. The full species names are as following: Corynebacterium glutamicum, Corynebacterium diphtheriae, Mycobacterium tuberculosis, Rhodococcus erythropolis, Nocardia farcinica, Tsukamurella paurometabola, Streptomyces coelicolor, Nakamurella multipartita, Propionibacterium acnes, Acidothermus cellulolyticus, Actinosynnema mirum, Micrococcus luteus
An amino acid sequence alignment of FtsR homologs of different actinobacterial species revealed that especially the N-terminal part is highly conserved (Additional file 1:Figure S1). Two features of the alignment are conspicuous, which are the variable length of the N-termini upstream of the highly conserved region starting with the sequence MSIG and the variable length of the region linking the N-terminal DNA-binding domain and the C-terminal regulatory domain.
Deletion and overexpression of ftsR affects growth behavior and cell shape
To gain insights into the function of FtsR in C. glutamicum, an in-frame ftsR deletion mutant as well as an ftsR-overexpressing strain were constructed (Additional file 1: Tables S2 and S3) and analyzed with respect to growth behavior and cell morphology. Remarkably, the ftsR deletion strain grew significantly slower and to a lower final cell density (backscatter) compared to the wild type in CGXII minimal medium with glucose as carbon source (Fig. 3a). The consequences of ftsR overexpression in the wild-type strain ATCC13032 were tested using plasmid pAN6-ftsR, in which ftsR expression is controlled by the tac promoter. The promoter is known to be leaky in C. glutamicum and allows basal expression of the target gene also in the absence of the inducer IPTG [37]. Without IPTG and with IPTG concentrations up to 10 μM, growth of ATCC13032 pAN6-ftsR was comparable to the reference strain ATCC13032 pAN6. However, when 100 μM IPTG was added, growth of ATCC13032 pAN6-ftsR was strongly inhibited (Fig. 3b). Thus, both the absence and the overexpression of ftsR had negative consequences. We searched our in-house microarray database with more than 1700 experiments for conditions with altered ftsR mRNA levels, but found no evidence for transcriptional regulation of ftsR. Together, these results suggest that ftsR expression in the cell may be constitutive.
Fig. 3.
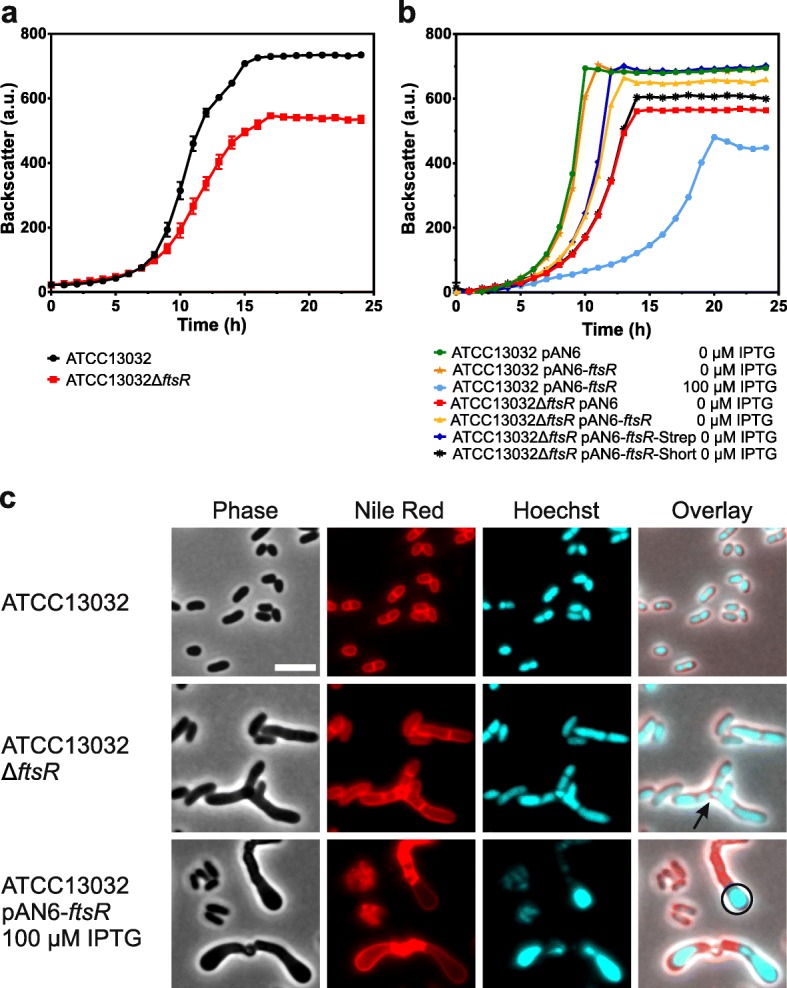
Growth behavior and cell morphology of C. glutamicum ΔftsR and C. glutamicum overexpressing ftsR. a Growth of the ftsR deletion mutant in comparison to the wild type. Mean values and standard deviation of three biological replicates are shown. b Growth of an ftsR overexpressing strain and complementation of the ΔftsR mutant with several FtsR variants including an N-terminally shortened protein and a protein with a C-terminal StrepTag-II. Average values from two biological replicates are shown. For the growth experiments shown in a and b, the strains were pre-cultivated first in BHI medium and then in CGXII medium with 2% (w/v) glucose, followed by the main cultivation in the same medium. For b, all media were supplemented with kanamycin (25 μg/mL) and IPTG as indicated. c Microscopic pictures of cells in the stationary phase. To visualize membranes and DNA, cells were stained with Nile Red and Hoechst 33342, respectively. The arrow points towards a branched cell and the circle indicates a high DNA concentration at the cell pole. The scale bar represents 5 μm
Phase-contrast and fluorescence microscopy with staining of DNA and membranes revealed strong morphological differences to wild-type cells both for the ΔftsR mutant and for the ftsR overexpressing strain (Fig. 3c, Additional file 1: Figure S2). These differences were apparent both in the exponential and stationary growth phases (Additional file 1: Figure S3). While the phenotype of the ΔftsR mutant did not change much along the cultivation, the phenotype of the overexpression strain became more severe over time (Additional file 1: Figure S3). This could be due to an accumulation of FtsR in the cells resulting from the overexpression. The ΔftsR cells were elongated and some of them were branched (Fig. 3c, black arrow). Several cells had multiple septa at different positions in the cell. Overexpression of ftsR led to enlarged cells with DNA accumulated at the poles (Fig. 3c, black circle). These cells also contained additional septa, which were mostly located in the middle of the cells. However, only a fraction of the cells was enlarged, whereas the others remained small and inconspicuous. Please note that the small cells appear DNA-free in some pictures because the brightness was adjusted to the cells with the high DNA concentration. All small cells of the FtsR overexpression strain seem to contain similar DNA amounts like the wild type. The reason for this population heterogeneity is unknown at present. It could result from different FtsR levels in the cells due to heterogeneous induction of pAN6-based ftsR expression, as previously reported for eyfp expression by the pAN6 parent plasmid pEKEx2 [38]. The observed morphological changes caused by ftsR deletion and overexpression, in particular the presence of multiple septa within a single cell that did not lead to a separation of the cells, hint toward a function of FtsR in cell division and to a potential role in the regulation of ftsZ expression. To further evaluate the changes in cell size or rather cell volume, we analyzed the strains using a Coulter counter in the volumetric measurement mode. Additional file 1:Figure S4 shows the size distribution at three different time points during cultivation (3 h, 6 h, 24 h). For the ΔftsR strain in comparison to the wild type, the whole population was shifted to larger cell sizes at all time points and formed a single broad peak. The population of the ftsR overexpressing strain formed two peaks of different sizes at 24 h, differing weakly or strongly in size from wild type carrying pAN6. As mentioned above, this could be due to heterogeneous expression from plasmid pAN6-ftsR. Overall, these measurements confirmed that the ftsR deletion strain comprises a homogeneous population of cells with increased size in different growth phases. In contrast, the strain overexpressing ftsR showed a homogeneous population of enlarged cells in the exponential growth phase, and interestingly two populations with cells of different sizes in the stationary phase, which matches the. These results coincide with the cells size distribution observed by microscopy.
In order to corroborate the results described above, the ftsR gene was also deleted and overexpressed in another C. glutamicum strain termed MB001, which differs from ATCC13032 by the deletion of the three prophage regions CGP1 (13.5 kb), CGP2 (3.9 kb), and CGP3 (187.3 kb) [39]. We previously reported that the MB001 strain behaved like its parent wild type in the majority of conditions tested, but showed improved growth and fitness under SOS response-inducing conditions that trigger CGP3 induction in the wild type. Thus, MB001 is a useful strain background for the analysis of deletions causing stressed cells, as secondary effects due to prophage induction are excluded. Both growth behavior (Additional file 1: Figure S5) and cell morphology of MB001ΔftsR (Additional file 1: Figure S6) were comparable to that of ATCC13032ΔftsR, and also overexpression of ftsR in MB001 caused similar effects as in ATCC13032 (Additional file 1:Figure S5). Thus, the absence of the prophages CGP1, CGP2, and CGP3 had no obvious influence on the effects of ftsR deletion and overexpression on growth and cell morphology.
Complementation of the ΔftsR phenotype with native FtsR and homologs of C. diphtheriae and M. tuberculosis
To confirm that the growth defect and the altered cell morphology of the ΔftsR mutants are caused by the ftsR deletion rather than by secondary mutations that might have occurred during construction of the mutants or polar effects on downstream genes, complementation experiments were performed with plasmid pAN6-ftsR and two variants thereof. Plasmid pAN6-ftsR-Strep encodes an FtsR variant with a carboxyterminal linker sequence (AS) followed by a Streptag-II (WSHPQFEK). Plasmid pAN6-ftsR-short encodes an FtsR variant shortened by 28 amino acids at the N-terminus, which is now MSIGV. An alternative start codon was found here, based on the amino acid sequence alignment reported above (Additional file 1: Figure S1). As shown in Fig. 3b, the growth defect of ATCC13032ΔftsR pAN6 could partially be reversed by pAN6-ftsR and pAN6-ftsR-Strep, but not by pAN6-ftsR-short. This suggests that the longer FtsR variant composed of 256 amino acid residues represents the active protein and that its function is not disturbed by a C-terminal Streptag-II. Furthermore, the morphological changes of ATCC13032ΔftsR could be fully reversed by pAN6-ftsR. When the complementation was performed with strain MB001ΔftsR, a full reversal of the growth defect and of the morphological changes was observed for MB001ΔftsR pAN6-ftsR without IPTG induction (Additional file 1:Figures S5, S6). These results confirm that ftsR deletion causes the growth defect and the morphological changes of the corresponding mutants.
Due to the high conservation of the ftsR locus in different actinobacterial species, we assumed a similar function of the homologous genes and proteins. To test this hypothesis, we expressed ftsR homologs of two pathogenic relatives, CDC7B_1201 of Corynebacterium diphtheriae C7 and Rv1828 of Mycobacterium tuberculosis H37Rv, in the C. glutamicum MB001ΔftsR strain using pAN6-based expression plasmids, and monitored growth and cell morphology. While this manuscript was in preparation, initial biochemical and structural studies of Rv1828 were published [40], which will be discussed alongside our results. With pAN6-CDC7B_1201 almost full reversal of the growth defect and of the morphological changes was achieved by basal expression without IPTG induction (Additional file 1: Figure S6). As previously observed for overexpression of C. glutamicum ftsR, stronger induction of CDC7B_1201 gene expression had a negative effect on growth (data not shown). With pAN6-rv1828, the best complementation was achieved in the presence of 100 μM IPTG (Additional file 1: Figure S6). Whereas a full reversal to wild-type morphology was observed, growth was improved with pAN6-rv1828 but did not reach the wild-type characteristics. These results strongly suggest that the FtsR homologs of Actinobacteria have similar or identical functions and target genes.
Transcriptome comparison of the ATCC13032ΔftsR mutant with its parent wild type
In order to elucidate the effects of ftsR deletion on ftsZ expression, but also on global gene expression as a first step towards definition of the FtsR regulon, transcriptome comparisons of the ATCC13032ΔftsR mutant versus the parent wild type were performed using DNA microarrays and RNA isolated from cells cultivated in glucose minimal medium and harvested in the exponential growth phase. 34 genes showed a ≥ 2-fold increased mRNA level and 18 genes a ≥ 2-fold decreased mRNA level in the ΔftsR mutant (Additional file 1: Table S4). Both the upregulated and the downregulated genes covered a broad and heterogeneous range of cellular functions. Within the group of genes showing a decreased mRNA level, the ftsZ gene with an mRNA ratio of 0.35 was the most prominent one. A reduced expression of ftsZ in the ΔftsR mutant, combined with the fact that FtsR was enriched with the ftsZ promoter region, hints at a possible transcriptional activator function for FtsR. The group of upregulated genes included oppC and oppD encoding components of a putative ABC-type peptide transport system, four prp genes involved in propionate or propionyl-CoA catabolism, seven genes located in the CGP3 prophage region, which in total extends from cg1890 to cg2071, and a cluster of eight neighboring genes (cg0830 – cg0838) including those for an ABC-type trehalose uptake system. Interestingly, genome-resequencing revealed that the genome region comprising cg0830-cg0838 was amplified, which presumably caused the increased mRNA levels of these genes in the transcriptome analysis (Supplementary results, Additional file 1: Table S5, Additional file 1: Figure S7).
Effect of ftsR deletion on ftsZ promoter activity and FtsZ distribution
As an in vivo approach to demonstrate transcriptional activation of ftsZ expression by FtsR, a transcriptional fusion of the ftsZ promoter to a reporter gene encoding the mVenus protein was constructed resulting in plasmid pJC1-PftsZ-venus. The reporter plasmid and, as negative control, the parent plasmid pJC1-venus-term were used to transform strains MB001 and MB001ΔftsR and ftsZ promoter activity was measured as cell-specific fluorescence (defined as ratio of fluorescence and backscatter). As shown in Fig. 4, the lack of ftsR caused a significantly lower specific fluorescence, supporting activation of ftsZ expression by FtsR. Comparable results were obtained with strains ATCC13032 and ATCC13032ΔftsR carrying pJC1-PftsZ-venus (Additional file 1: Figure S8).
Fig. 4.
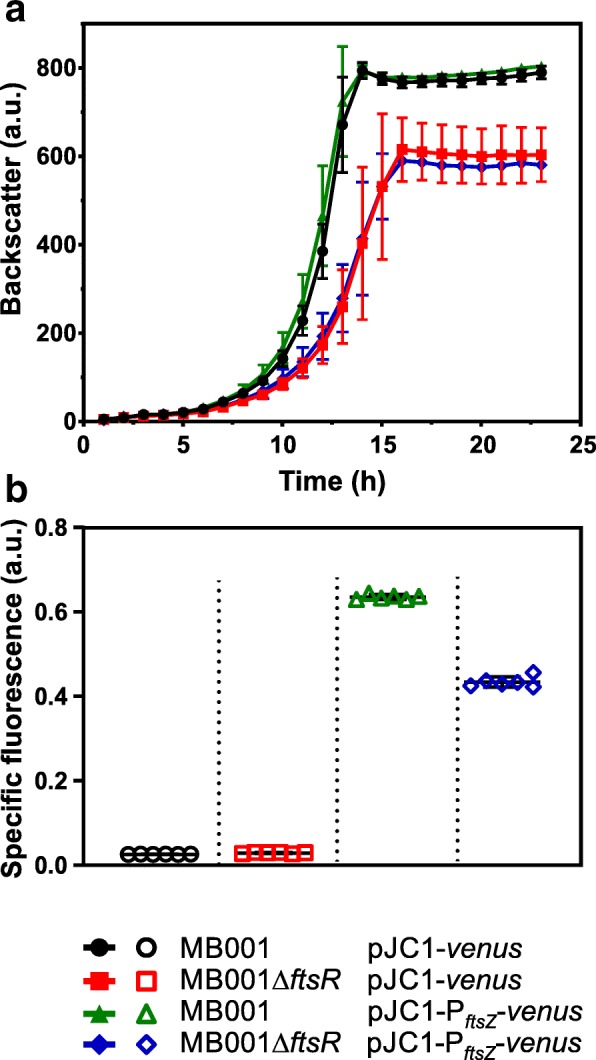
Influence of the ftsR deletion in C. glutamicum MB001 on the expression of the venus reporter gene under control of the ftsZ promoter. The indicated strains were cultivated in CGXII medium with 2% (w/v) glucose and 25 μg/mL kanamycin in a BioLector with automated measurement of Venus fluorescence and of cell density as backscatter (a). Specific fluorescence represents the ratio of fluorescence and backscatter at 20 h cultivation (b). Average values and standard deviations of three biological replicates are shown (a). b shows the result of six biological replicates
To test whether the lack of FtsR influences the distribution of FtsZ within the cell, we generated derivatives of ATCC13032 and ATCC13032ΔftsR with a chromosomal insertion of a second copy of ftsZ fused in frame to the coding sequence of the fluorescent protein mVenus under control of the native ftsZ promoter. Both strains were cultivated in CGXII minimal medium with glucose as carbon source and analyzed by fluorescence microscopy (Additional file 1:Figure S9). In the strain lacking FtsR, the frequency as well as the intensity of the FtsZ rings was reduced. This may be caused by a reduced availability of FtsZ within the cells, which might contribute to the morphological phenotype of the ftsR deletion mutant.
Genome-wide profiling of in vivo FtsR binding sites
With the aim to identify the in vivo binding site of FtsR in the ftsZ promoter region and to identify further target genes of FtsR, a ChAP-Seq experiment was performed. In vivo formaldehyde-crosslinked FtsR-Strep-DNA complexes were purified by StrepTactin-affinity chromatography and the isolated DNA fragments were sequenced. Nine peaks with a sequencing coverage above 2000 were detected, which is at least 50-fold higher than the background noise signal observed for the entire genome with a 40-fold coverage (Table 1, Fig. 5a, Additional file 1: Figure S10). The nine peaks were analyzed with respect to their chromosomal location and the mRNA level of the neighboring genes (Table 1). Based on the mRNA ratios observed in the transcriptome comparison of C. glutamicum ATCC13032ΔftsR vs. ATCC13032, three potentially FtsR-activated genes (ftsZ, cg0852, cg2477) and five potentially FtsR-repressed genes (cop1-cg3181-cg3180, phoC, cg0838) were identified using mRNA ratio (ΔftsR/wild type) cutoffs of 1.5 and 0.75. A repetition of the ChAP-Seq experiment confirmed the results of the first one except that the overall coverage was lower and the order of the peaks varied slightly (Table 1).
Table 1.
Results of ChAP-Seq analysis with C. glutamicum ATCC13032ΔftsR/pAN6-ftsR-Strep. The nine DNA regions that showed a ≥ 50-fold higher coverage than the entire genome (coverage 40) in the ChAP-Seq analysis with FtsR-Strep were analyzed with respect to the location of the peak within the genome and the mRNA levels of the genes in this region. Based on that, a prediction was made which genes could be potential FtsR targets. The arrows indicate whether the genes are encoded on the leading (→) or lagging strand (←) within the genome. Graphical representations of selected peaks as derived from the ChAP-Seq analysis are shown in Additional file 1: Figure S10
| No.a | Coveragea | Peak position | DNA microarray 13032ΔftsR/13032 | Potential FtsR target genes | ||||
|---|---|---|---|---|---|---|---|---|
| in gene | downstream 3′-end of | upstream of | Gene | mRNA ratio | p-value | |||
| 1 (1) | 28,811 (4781) | – | cg2478 (NCgl2178, penicillin binding protein) ← | cg2477 (NCgl2177, conserved hypothetical protein) ← | cg2478 | 0.75 | 0.11 | cg2477 potentially activated by FtsR |
| cg2477 | 0.24 | 0.01 | ||||||
| 2 (5) | 22,757 (934) | – | – | cg3182 (NCgl2777, cop1, trehalose corynemycolyl transferase) ← | cg3182 | 1.70 | 0.20 | cop1-cg3181-cg3180 potentially repressed by FtsR |
| cg3181 | 2.30 | 0.05 | ||||||
| cg3180 | 1.91 | 0.05 | ||||||
|
cg3181 (NCgl2776, put. Secreted protein) ← cg3180 (NCgl2775, put. Secreted protein) ← cg3183 (NCgl2778, put. transposase) → |
cg3183 | n.a. | n.a. | |||||
| 3 (2) | 4437 (1556) | – | cg2367 (NCgl2076, ftsQ, cell division septal protein) ← | cg2366 (NCgl2075, ftsZ, cell division GTPase) ← | cg2367 | 1.20 | 0.02 | ftsZ activated by FtsR |
| cg2366 | 0.35 | 0.02 | ||||||
| 4 (11) | 3899 (373) | cg0470 (NCgl0381, htaB, heme transport-associated protein) → | cg0469 (NCgl0380, hmuV) → | cg0471 (NCgl0382, htaC, heme-transport associated protein) → | cg0469 | 1.41 | < 0.01 | – |
| cg0470 | 1.45 | 0.09 | ||||||
| cg0471 | 1.55 | 0.03 | ||||||
| 5 (12) | 3836 (365) | – | – | cg0852 (NCgl0712, conserv. Hypoth. protein) ← | cg0852 | 0.62 | 0.01 | cg0852 potentially activated by FtsR |
|
cg0853 (NCgl0713, conserv. Hypoth. protein) → cg0854 (NCgl0714, pmmA, phosphomannomutase) → cg0855 (NCgl0715, conserv. Hypoth. protein) → cg0856 (NCgl0716, mannose-6-phosphate isomerase) → |
cg0853 | 1.03 | 0.29 | |||||
| cg0854 | 0.89 | 0.25 | ||||||
| cg0855 | 1.18 | 0.08 | ||||||
| cg0856 | 1.09 | 0.03 | ||||||
| 6 (3) | 3630 (1529) | – |
cg3392 (oxiE, idhA2, NCgl2958) → (myo-inositol 2-dehydrogenase) |
cg3393 (NCgl2959, phoC, secreted cell-wall-associated phosphatase) → | cg3392 | 1.25 | 0.03 | phoC potentially repressed by FtsR |
| cg3393 | 1.60 | 0.03 | ||||||
| 7 (15) | 2871 (315) | cg2695 (NCgl2368, ATPase of ABC transporter) ← | cg2697 (ssb, NCgl2370, single-strand DNA-binding protein) ← |
cg2694 (NCgl2367, put. Phosphodiesterase, nucleotide pyrophosphatase) ← cg2693 (NCgl2366, put. Phosphodiesterase, nucleotide pyrophosphatase) ← |
cg2693 | 1.31 | 0.03 | – |
| cg2694 | 1.29 | 0.05 | ||||||
| cg2695 | 1.06 | 0.42 | ||||||
| cg2697 | 1.04 | 0.24 | ||||||
| 8 (6) | 2355 (858) | – |
cg2518 (NCgl2213, put. Secreted protein) → cg2519 (NCgl2214, conserv. Hypoth. protein) ← cg2520 (NCgl2215, hypoth. protein) ← cg2521 (NCgl2216, fadD15, long chain fatty acid CoA ligase) ← |
– | cg2518 | 0.81 | 0.18 | – |
| cg2519 | 0.49 | 0.03 | ||||||
| cg2520 | 0.92 | 0.45 | ||||||
| cg2521 | 0.91 | 0.43 | ||||||
| 9 (8) | 2248 | cg0839 (NCgl0701) ← (hypoth. protein) | cg0840 (NCgl0702, conserv. Hypoth. protein) ← | cg0838 (NCgl0700, ATP-depend. helicase) ← | cg0838 | 4.61 | 0.03 | cg0838 potentially repressed |
| cg0839 | 2.45 | 0.16 | by FtsR | |||||
| cg0840 | 2.45 | 0.19 | ||||||
a The numbers in brackets represent the result of a second, independent experiment. Please note that the overall coverage was lower in the second experiment
Fig. 5.

Binding of FtsR to the ftsZ promoter. a Enrichment of the ftsZ promoter region in the ChAP-Seq experiment with FtsR-Strep. The negative control is presented below the genes. b FtsR consensus DNA-binding motif identified by the MEME software [41] using the nine DNA regions with the highest coverage in the ChAP-Seq experiment with FtsR-Strep. c DNA sequence of the ftsZ promoter region including parts of the coding regions of ftsQ and ftsZ and the corresponding amino acid sequences. The ribosome binding site of ftsZ is double underlined. The transcriptional start site identified by RNA-Seq [29] in strain ATCC13032 is highlighted in magenta, the transcriptional start sites identified by primer extension and RACE in strain ATCC13689 [19] are indicated in blue. The deduced −10 regions are shown in grey boxes. The FtsR-binding site identified in this work is highlighted in yellow. The 30-bp region used for EMSAs with purified FtsR-Strep is underlined. d In vitro DNA-binding studies with FtsR-Strep. Purified FtsR-Strep was incubated in the indicated concentrations with a constant concentration (1 μM) of a 30-bp double-stranded oligonucleotide covering the predicted FtsR-binding site in the ftsZ-promoter region. The mixture was then analyzed by electrophoresis using a non-denaturing 15% (w/v) polyacrylamide gel. As negative control, a DNA fragment of the promoter region of cg1081 was used (1 μM)
The genes cg0852 and cg2477 encode conserved proteins of unknown function. The putative operon cop1-cg3181-cg3180 encodes three secreted proteins [42]. The Cop1 protein (also termed Csp1) was shown to function as mycolyltransferase involved in the conversion of trehalose monocorynomycolate to trehalose dicorynomycolate [42, 43]. The protein encoded by phoC was proposed to function as cell-wall-associated phosphatase [44, 45] and is induced under phosphate-limiting conditions [46]. The cg0838 gene is part of the DNA region found to be amplified in the ATCC13032ΔftsR mutant but had a higher mRNA ratio than the neighboring genes that were also amplified (Additional file 1: Table S5), pointing to a possible repression by FtsR. The large Cg0838 protein (179 kDa) is proposed to function as an ATP-dependent helicase and contains a unique C-terminal domain including a metal-binding cysteine cluster.
The search for a common DNA sequence motif in the nine sequences with a coverage above 2000 using the MEME software [41] revealed the motif shown in Fig. 5b. The motif was identified in eight of the nine sequences except for the sequence with a coverage of 2871. When looking at the corresponding motif in the ftsZ promoter region, it forms an imperfect 25-bp inverted repeat: AACCCTAAAGTAAAGGTTGAGGGTA. The center of this motif was located at position − 73 with respect to the transcriptional start site of ftsZ as determined by RNA-Seq [29]. This position is compatible with an activating function of FtsR for ftsZ expression (Fig. 5c). It should be mentioned that in C. glutamicum strain ATCC13689, five transcriptional start sites were identified for ftsZ by primer extension and RACE studies [19], none of which corresponds to the one identified by RNA-Seq for strain ATCC13032 [29].
The DNA-binding motif shown above was used to search for similar motifs in the ftsZ promoter regions of other actinobacterial species possessing FtsR homologs using Clustal Omega [47]. All analyzed promoters contained a similar motif (Fig. 6a), supporting the assumption that regulation of ftsZ expression by FtsR homologs is a conserved mechanism in FtsR-containing Actinobacteria. The motif generated from the proposed binding sites also represents a 25 bp inverted repeat (Fig. 6b), which is very similar to the one derived from the C. glutamicum sequences enriched by ChAP-Seq with FtsR (Fig. 5b).
Fig. 6.

Putative FtsR binding sites in the ftsZ promoters of various actinobacteria. a Proposed FtsR binding sites in the ftsZ promoters of several actinobacterial species identified by sequence alignment with the FtsR binding site in the C. glutamicum ftsZ promoter. The respective ftsZ locus tags are given on the right. The proposed binding motifs are shown as colored letters. The annotated start codons of the FtsZ proteins are indicated by bold letters. The distance between the proposed FtsR binding site and the start codon varies from 41 to 193 bp. b Consensus DNA-binding motif generated by MEME from the sequences shown in a using default parameters
In vitro binding of purified FtsR to the proposed binding motif in the ftsZ promoter region
To test whether purified FtsR is able to bind to the proposed binding motif in the ftsZ promoter region, FtsR-Strep was overproduced in C. glutamicum ATCC13032ΔftsR using plasmid pAN6-ftsR-Strep and purified using StrepTactin-Sepharose (Additional file 1: Figure S11A). As described above, FtsR-Strep could complement the growth defect of the ATCC13032ΔftsR mutant to the same extent as native FtsR, indicating that the StrepTag did not negatively influence FtsR activity. Size-exclusion chromatography of affinity-purified FtsR-Strep (calculated mass 28.5 kDa) and comparison to standard proteins indicated that the protein forms a dimer (Additional file 1: Figure S11B). This is in line with the dimeric state of the homologous protein Rv1828 [40]. Purified FtsR-Strep was able to completely shift a 30-bp double-stranded oligonucleotide covering the predicted FtsR-binding site in the ftsZ promoter region at a 2-fold molar excess of the dimeric protein (Fig. 5d). In contrast, a control 30-bp double-strand-oligonucleotide derived from the promoter region of cg1081 was incompletely shifted even at an 8.5-fold molar excess of dimeric FtsR-Strep. The specificity of this interaction was further tested using a competition-EMSA (Additional file 1: Figure S12). A Cy3-labelled 271-bp DNA fragment covering the ftsZ promoter region including the FtsR binding site was incubated with FtsR protein and increasing concentrations of either specific (same fragment as above) or unspecific unlabeled competitor DNA. Only the specific competitor DNA reversed the shift of the labelled DNA fragment by FtsR, supporting its specific binding to the ftsZ promoter region.
FtsR-independent expression of FtsZ
It is known from previous studies that differences in ftsZ expression cause aberrant cell morphology and FtsZ localization in C. glutamicum [19]. In order to test whether the altered cell morphology and the growth defect of the ΔftsR mutant are solely caused by the differences in ftsZ expression, we constructed strains in which the expression of ftsZ is independent of FtsR and controlled by the gluconate-inducible gntK promoter of C. glutamicum [19, 37, 48]. A DNA fragment containing a terminator sequence and the gntK promoter was inserted between the native ftsZ promoter and the ribosome binding site of ftsZ within the chromosomes of C. glutamicum MB001 and MB001ΔftsR by double homologous recombination (Fig. 7a). The promoter exchange strains MB001::PgntK-ftsZ and MB001ΔftsR::PgntK-ftsZ showed growth in CGXII medium containing gluconate, but not in medium containing only glucose or sucrose, confirming gluconate-inducible ftsZ expression (Additional file 1: Figure S13).
Fig. 7.

Promoter exchange of ftsZ and growth of the resulting strains. a Strains with FtsR-independent ftsZ expression were constructed using a DNA fragment with a terminator sequence and the gluconate-inducible gntK promoter, which was inserted between the native ftsZ promoter and the ftsZ start codon in the chromosomes of MB001 and the MB001ΔftsR mutant. b-d Effect of different gluconate concentrations on cell morphology (b) and growth (c, d) of the promoter exchange strains MB001::PgntK-ftsZ and MB001ΔftsR::PgntK-ftsZ. The two strains were first pre-cultivated in BHI medium supplemented with 0.1% (w/v) gluconate to induce ftsZ expression by PgntK. The second pre-cultivation was performed in CGXII medium with 2% (w/v) glucose supplemented with the indicated gluconate concentrations. The main cultures were then performed in media having the same composition as the ones for the second pre-cultivation. b Microscopic pictures of cells from the stationary phase. The scale bar represents 5 μm. c, d The growth experiments show mean values and standard deviations of three biological replicates
In order to analyze the consequences of different ftsZ expression levels on growth and morphology, cells were cultivated in CGXII medium with 2% (w/v) glucose supplemented with 0.005, 0.01, 0.05, and 0.1% (w/v) gluconate. Strain MB001::PgntK-ftsZ showed enlarged cells with thickened poles at the lowest gluconate concentrations and reverted back to wild-type morphology at 0.1% (w/v) gluconate (Fig. 7b). Strain MB001ΔftsR::PgntK-ftsZ displayed strongly branched cells at the two lowest gluconate concentrations and even at 0.1% (w/v) gluconate the cells were still enlarged compared to the ftsR-positive strain (Fig. 7b). With respect to growth, the two strains showed quite similar growth behavior at the lowest gluconate concentration. Increased gluconate concentrations strongly improved growth of the ftsR-positive strain (Fig. 7c), whereas growth of the ftsR-negative strain was only marginally increased (Fig. 7d).
Overall, these results demonstrate the importance of an appropriate FtsZ level for normal cell morphology and growth, but they also show that the branched cells of the ΔftsR mutant are not solely caused by reduced ftsZ expression, but presumably also by altered expression of other genes as a consequence of the ftsR deletion. Similarly, the growth defect of the ΔftsR mutant cannot be rescued simply by increased expression of ftsZ and thus involves further genes. These could either be direct target genes of FtsR or genes whose expression is indirectly influenced by the absence of FtsR. In additional experiments, the promoter exchange strains with FtsR-independent ftsZ expression were also used to confirm transcriptional activation of ftsZ expression by FtsR using the reporter plasmid pJC1-PftsZ-venus (see Supplemental results and Additional file 1: Figure S14).
Discussion
The tubulin-like GTPase FtsZ plays the key role in bacterial cell division [49]. In this study, we identified the transcriptional regulator FtsR that functions as activator of ftsZ expression in C. glutamicum, representing a novel control mechanism of actinobacterial cytokinesis. FtsR was initially enriched by DNA affinity chromatography with the ftsZ promoter region. In vivo binding of FtsR to the ftsZ promoter was confirmed by ChAP-Seq experiments and purified FtsR was shown to bind to a 30 bp-DNA fragment covering the proposed DNA-binding site of FtsR, which represents an imperfect 25-bp inverted repeat. Evidence for the activation of ftsZ expression by FtsR came from transcriptome analysis, where the ftsZ mRNA level was significantly reduced in the ΔftsR mutant, and reporter gene studies showing reduced activity of the ftsZ promoter in the ΔftsR background.
Bioinformatic analyses revealed that FtsR belongs to the MerR superfamily of transcriptional regulators, which mostly function as activators, responding to a vast diversity of stimuli including oxidative stress, xenobiotics, and metal ion stress [50, 51]. MerR-type regulators typically bind as homodimers to inverted repeats in the promoter region of their target genes. The binding sites can be located between the − 10 and − 35 regions, which then show a spacing of 19–20 bp rather than the optimal 16–18 bp. This hampers transcription because the − 10 and − 35 regions are located on opposite sides of the DNA strand [51]. A well-described mechanism of transcriptional activation by regulators of the MerR superfamily is thought to rely on a conformational change of the N-terminal DNA-binding region after recognition of a stimulus by the C-terminal domain, which causes a twist of the bound DNA molecule in a manner that the − 10 and − 35 regions are rearranged, enabling RNA polymerase to bind and initiate transcription [52–55]. FtsR eluted as dimer during gel filtration and the binding site identified in the ftsZ promoter region was found to be an inverted repeat, which was located at position − 61 to − 85 upstream of the ftsZ transcriptional start site identified by RNA-Seq [29] rather than between the − 10 and − 35 regions. This position is compatible with an activating function of FtsR, but might involve a different mechanism of transcriptional activation than the one described above.
It should be mentioned that in C. glutamicum strain ATCC13689 five transcriptional start sites were identified for ftsZ by RACE and primer extension experiments [19], none of which corresponds to the one identified by RNA-Seq for strain ATCC13032 [29]. Further studies are required to test for the presence of additional transcriptional start sites of the ftsZ gene in strain ATCC13032, in which the FtsR binding site might be located between the − 10 and − 35 regions. Multiple promoters for ftsZ expression appear to be the rule in various bacteria such as M. tuberculosis [22, 24], E. coli [23], Streptomyces coelicolor A3 [2] [56], or Streptomyces griseus [57]. They allow a dynamic adjustment of the FtsZ levels according to different stimuli and probably make the cells more robust with respect to spontaneous mutations in the ftsZ promoter region.
FtsR was found to be conserved in many different families of Actinobacteria, including the genus Mycobacterium. In a ChIP-Seq experiment with the FtsR homolog Rv1828 of M. tuberculosis, the promoter region of the ftsZ gene was the top target [58–60]. Using the proposed binding motif of FtsR (AACCCTAAAGTAAAGGTTGAGGGTA) as template, we identified a very similar sequence (AACtCTAAgcctAtGGTTGAGGtTt) in the ftsZ promoter of M. tuberculosis, the region which also showed by far the highest coverage in the ChIP-Seq experiment [59]. Here the motif was located exactly between the − 10 and − 35 regions of the transcriptional start site P1, which is located at position − 43 relative to the ftsZ translational start [24]. The binding motif proposed for Rv1828 in a recent study is different but overlaps partially with our motif [40]. Mutational analysis of this binding motif by EMSAs revealed that binding of Rv1828 is not influenced when the specific part of the motif is altered that does not overlap with our proposed binding motif [40]. This, together with the strong conservation of our motif among many species, suggests that the motif proposed by us is more likely the correct one. Our results obtained for FtsR and Rv1828 together with the recently published characterization of Rv1828 [40] strongly support the notion that FtsR and its actinobacterial homologs are involved in transcriptional regulation of ftsZ. In line with this, the ftsZ promoter regions of various actinobacterial genera were found to contain DNA sequence motifs similar to the ones determined for FtsR and Rv1828 (Fig. 6).
Deletion and overexpression of ftsR led to growth inhibition and an altered cell morphology. The deletion phenotype could be reversed by plasmid-based expression of ftsR or the ftsR homologs of C. diphtheriae and M. tuberculosis (Fig. 3, Additional file 1:Figures. S2, S3, S4). The fact that many ΔftsR cells and also ftsR overexpression cells contained multiple septa indicates that cell division is disturbed. The studies with the promoter exchange strains, in which ftsZ expression was controlled by the gluconate-inducible gntK promoter, revealed that growth behavior and morphology of the ΔftsR mutant are not solely caused by altered ftsZ expression. Despite comparable ftsZ expression, the ΔftsR strain grew much worse than the ftsR-positive reference strain (Fig. 7c, d) and the morphology of the ΔftsR strain differed from the ftsR+ strain, in particular by the formation of branched cells (Fig. 7b). This suggests that i) FtsZ alone is not the cause for the branching phenotype, and ii) there must be some other, FtsR-dependent gene that causes enlarged cells even in the presence of sufficient FtsZ.
Potential further target genes of FtsR were deduced by combining ChAP-Seq and transcriptome results. The target with the most obvious relation to the altered cell morphology of the ΔftsR mutant was the cop1-cg3181-cg3182 gene cluster. These three genes encode secreted proteins and Cop1 (Csp1), a mycolyltransferase converting trehalose monocorynomycolate to trehalose dicorynomycolate. The mycolyl transferase function was located in the N-terminal portion of Cop1 [43]. A C. glutamicum cop1 deletion mutant had an altered cell morphology characterized by enlarged, club-shaped cells and it was speculated that the C-terminal part of Cop1 plays a role in cell shape formation [42]. In the case of cop1, the FtsR-binding site (AAGTCTAAAGTTGAACTTAAGATTG) starts downstream of the − 35 region and ends downstream of the − 10 region (TAAGAT). The transcriptome data suggest a repression of cop1 by FtsR and the morphology of the cells overexpressing ftsR (Fig. 3c, Additional file 1: Figure S3) resemble that of cells lacking functional cop1 [42]. Further studies are required to confirm the influence of FtsR on the expression of the cop1-cg3181-cg3182 gene cluster and to identify further target genes and their function.
Regarding the physiological function of FtsR, a possible role could be to serve as a regulatory mechanism that controls cell division in response to the nutritional status of the cell. In recent years, several studies have revealed metabolic enzymes to directly influence cell division by modulating the activity of FtsZ [61]. Transcriptional regulation, such as shown here for FtsR, is another means to influence FtsZ activity. The fact that FtsR activates ftsZ expression suggests that FtsR signals favorable nutritional conditions with a high cell division rate and thus a high FtsZ demand. As the ftsR gene and its homologs in other Actinobacteria are always located downstream of the odhI/garA gene, a functional link to OdhI/GarA might exist. OdhI/GarA controls the metabolic flux at the 2-oxoglutarate node of metabolism, which is particularly relevant for nitrogen assimilation. Our preliminary attempts to find a potential ligand of FtsR were not successful and the identification of the stimulus controlling FtsR activity is ultimately required to understand the function of this regulator.
Conclusion
In this study, we identified and characterized FtsR as the first transcriptional regulator of FtsZ described for C. glutamicum. Deletion or overexpression of ftsR had severe effects on growth and cell morphology, underlining the importance of FtsR for correct cell division. The fact that FtsR activates ftsZ expression suggests that FtsR might sense favorable nutritional conditions that allow for a high cell division rate and therefore require high FtsZ levels. In summary, our findings provide a first insight into transcriptional regulation of cell division in Actinobacteria.
Methods
Bacterial strains, plasmids and growth conditions
Bacterial strains and plasmids used in this study are listed in Additional file 1: Table S2. The C. glutamicum type strain ATCC13032 or its prophage-free variant C. glutamicum MB001 were used as wild type or reference strain, as indicated. The parental strain ATCC13032 (DSM No. 20300) was obtained from the German Collection of Microorganisms and Cell Cultures GmbH (DSMZ, Braunschweig, Germany). The prophage-free strain MB001 is from in-house stock and can also be obtained from the DSMZ (DSM No. 102070). For growth experiments, C. glutamicum was precultivated for 6–8 h at 30 °C and 170 rpm in 5 mL BHI medium (BD Bacto™ Brain Heart Infusion, Becton Dickinson and Company, Heidelberg, Germany). The cells were harvested by centrifugation, washed with phosphate-buffered saline (PBS, 137 mM NaCl, 2.7 mM KCl, 4.3 mM Na2HPO4, 1.4 mM KH2PO4, pH 7.3) and used as inoculum for a second preculture in 20 mL CGXII minimal medium [62] containing 30 mg/L 3,4-dihydroxybenzoate as iron chelator and, if not stated otherwise, 2% (w/v) glucose as carbon source. This preculture was incubated overnight at 30 °C and 120 rpm and after harvesting the cells were washed in PBS and used for inoculation of the main culture, using again CGXII medium with 30 mg/L 3,4-dihydroxybenzoate and 2% (w/v) glucose. Growth experiments were either performed in 100 mL shake flasks with 20 mL medium (initial optical density at 600 nm (OD600) of 1.0) that were shaken at 30 °C and 120 rpm or in 48-well FlowerPlates® (m2p-labs GmbH, Baesweiler, Germany) with a culture volume of 800 μL (initial OD600 of 0.5) that were shaken in a BioLector (m2p-labs GmbH, Baesweiler, Germany) at 30 °C and 1200 rpm. Growth was monitored as cell density by determining either OD600 (shake flasks) or as scattered light at 620 nm in the BioLector [63], which is termed “backscatter” throughout this study. For growth experiments with promoter exchange strains where expression of the ftsZ gene was under control of the gntK promoter, the first pre-culture was supplemented with 0.1% (w/v) gluconate and the second pre-culture were supplemented with 0.01% (w/v) gluconate and 1.99% (w/v) glucose as carbon source. The gluconate concentration of the main culture is indicated for each experiment. For cloning purposes, Escherichia coli DH5α was used and routinely cultivated at 37 °C in lysogeny broth (LB) [64]. When required, the media were supplemented with 25 μg/mL kanamycin or 10 μg/mL chloramphenicol for C. glutamicum or with 50 μg/mL kanamycin or 34 μg/mL chloramphenicol for E. coli.
Recombinant DNA work and construction of insertion and deletion mutants
Routine methods such as PCR, DNA restriction and ligation, Gibson assembly and transformation were performed using standard protocols [64–67]. Phusion Green High Fidelity DNA Polymerase (Thermo Fisher Scientific Inc., Rockford, IL, USA) was used for cloning purposes. For all other PCRs, either the KAPA2G Fast ReadyMix PCR Kit (Kapa Biosystems, Wilmington, USA) or DreamTaq DNA Polymerase (Thermo Fisher Scientific Inc., Rockford, IL, USA) were used. The oligonucleotides used in this study are listed in Additional file 1: Table S3 and were purchased from Eurofins Genomics GmbH (Ebersberg, Germany). DNA sequencing was performed by Eurofins Genomics GmbH. The ΔftsR mutants of C. glutamicum and the promoter exchange strains carrying a DNA fragment with a transcription terminator and the gntK promoter inserted into the chromosome between the native ftsZ promoter and the ftsZ coding region were constructed via a two-step homologous recombination protocol as described [37, 68, 69] using plasmids pK19mobsacB-ΔftsR and pK19mobsacB-PgntK-ftsZ. The strains ATCC13032::ftsZ-venus and ATCC13032ΔftsR::ftsZ-venus were constructed by chromosomal insertion of pK18mob-ftsZ-venus by single homologous recombination. In all tested clones of both strains, the plasmid did not insert into the intergenic region between cg1121 and cg1122, but in the ftsZ region. As the insertion plasmid was constructed in such a way that these mutants also carry one native copy of ftsZ and one fused to venus, these strains were used for microscopy, anyway.
Fluorescence microscopy
C. glutamicum cells were centrifuged and suspended in PBS containing 100 ng/mL Hoechst 33342 dye to stain DNA and 300 ng/mL Nile red to stain membranes (Sigma-Aldrich Chemie GmbH, Taufkirchen, Germany). After 10 min of incubation in the dark at room temperature, samples were spotted onto a glass slide covered with a thin agarose layer (1% (w/v) in TAE buffer (40 mM Tris, 1 mM Na2EDTA, 20 mM glacial acetic acid, pH 8)) and analyzed using a Zeiss Axio Imager M2 microscope equipped with a Zeiss AxioCam MRm camera (Carl Zeiss AG, Oberkochen, Germany). Images were acquired using a Plan-Apochromat 100×/1.40-numerical aperture phase contrast oil immersion objective and AxioVision 4.8 software (Carl Zeiss Microscopy GmbH, Jena, Germany). Hoechst 33342 fluorescence was visualized with filter set 49 and Nile red with filter set 63 HE (both Carl Zeiss AG). C. glutamicum strains producing the FtsZ-Venus fusion protein were directly used for microscopy without further staining.
Purification of FtsR
FtsR-Strep was purified using C. glutamicum ATCC13032ΔftsR carrying plasmid pAN6-FtsR-Strep. The preculture was prepared using a single colony from a fresh BHI agar plate to inoculate 20 mL BHI with 2% (w/v) glucose and 25 μg/mL kanamycin and incubated at 30 °C and 120 rpm for 6–8 h. The main culture (500 mL of the same medium in a 2 L baffled flask) was inoculated with 1 mL of the preculture and incubated overnight at 30 °C and 90 rpm. On the following morning, 50 μM isopropyl β-D-1-thiogalactopyranoside (IPTG) was added and the culture was incubated for additional 4 h at 30 °C and 120 rpm. Subsequently, the cells were harvested by centrifugation (4 °C, 30 min, 3399 g) and resuspended in 25 mL buffer A (100 mM Tris-HCl, 100 mM NaCl, pH 7.5) supplemented with cOmplete Mini EDTA-free protease inhibitor cocktail tablets (Roche Diagnostics GmbH, Mannheim, Germany). Cell disruption was performed by five passages through a French pressure cell using a HTU-Digi-F-Press (G. Heinemann Ultraschall- und Labortechnik, Schwaebisch Gmuend, Germany) at a pressure of 103.4 MPa (15,000 psi). Cell debris was removed by centrifugation for 20 min at 5300 g and 4 °C, followed by ultracentrifugation for 1 h at 84,000 g and 4 °C. The supernatant was used for affinity chromatography using a Strep-Tactin-Sepharose column (IBA, Göttingen, Germany) with 1 mL bed volume equilibrated with buffer W (50 mM Tris-HCl, 250 mM NaCl, pH 7.5). After the protein extract had passed, the column was washed three times with 15 mL buffer W (50 mM Tris-HCl, 250 mM NaCl, pH 7.5) and FtsR-Strep was eluted with 10 × 1 mL buffer E (buffer W with 15 mM d-desthiobiotin obtained from Sigma-Aldrich Chemie GmbH, Steinheim, Germany). Aliquots of the elution fractions were analyzed by SDS-PAGE [70] and the fractions containing FtsR-Strep were pooled and concentrated using Amicon Ultra-4 Centrifugal Filter Devices with a 10 kDa cut-off (Merck Millipore Ltd., Carrigtwohill, Cork, Ireland). FtsR-Strep was further purified by gel filtration using a Superdex™ 200 Increase 10/300 GL column attached to an ÄKTA™ pure 25 system (GE Healthcare Bio-Sciences AB, Uppsala, Sweden). The column was equilibrated with buffer W and elution was performed with a flow rate of 0.6 mL/min.
Electrophoretic mobility shift assays (EMSAs)
Two complementary single-stranded 30-bp oligonucleotides (see Additional file 1: Table S3) covering the putative FtsR-binding motif in the ftsZ promoter region were annealed by heating the samples containing 10 μM Tris-HCl pH 8.0, 50 mM NaCl, 1 mM EDTA and 10 μM of each oligonucleotide to 95 °C for 5 min, followed by slow cooling to room temperature. The double-stranded oligonucleotide (final concentration 1 μM) was incubated at room temperature for 30 min with increasing concentrations (0–8.5 μM) of freshly purified dimeric FtsR protein (Additional file 1: Figure S11) in binding buffer (10 mM Tris-HCl pH 7.5, 30 mM NaCl, 1.5 mM Na2EDTA). A 30 bp fragment located in the cg1081 promoter region was used as negative control (Additional file 1: Table S3). After incubation, a suitable volume of the 5-fold concentrated sample buffer (0.1% (w/v) xylene cyanol, 0.1% (w/v) bromophenol blue, 20% (v/v) glycerol in 1x TBE (89 mM Tris base, 89 mM boric acid, 2 mM Na2EDTA)) was added and the samples were separated by native PAGE using 15% (w/v) polyacrylamide gels in 1x TBE buffer. Subsequently, the DNA was stained with SYBR® Green I Nucleic Acid Gel Stain (Invitrogen, Ltd., Paisley, UK) and photographed. The competition-EMSA was performed as described previously [71]. In brief, a 271 bp DNA-fragment covering the ftsZ promoter region including the binding site was amplified using oligonucleotides ftsZ_prom250_fw andCy3-ftsZ_prom_rv to generate Cy3-labelled DNA or oligonucleotides ftsZ_prom250_fw/ftsZ_prom_rv to generate unlabeled specific competitor DNA. A 260 bp DNA-fragment further upstream in the ftsZ promoter was amplified using the oligonucleotide-pair ftsZ_prom500_fw/ftsZ_prom250up_rv and used as unspecific competitor DNA. Purified FtsR protein was incubated with the DNA fragment(s) in a total volume of 10 μL binding buffer. After addition of loading buffer and separation by native PAGE on a 10% polyacrylamide gel, the gel was scanned using a Typhoon TrioTM scanner (GE Healthcare).
Promoter activity studies with PftsZ
In order to analyze transcriptional regulation of ftsZ by FtsR in vivo, a DNA fragment covering the ftsZ promoter region and the native ribosomal binding site extending from position − 494 to + 3 with respect to the ftsZ translational start site was fused to a DNA sequence coding for the fluorescent protein mVenus and cloned into the pJC1 vector backbone (for details see Additional file 1: Tables S4 and S5). Strains carrying their chromosomal ftsZ gene either under control of its native promoter or under control of the gntK promoter were transformed with the resulting plasmid pJC1-PftsZ-venus. For complementation studies, the strains were additionally transformed with pEC-ftsR or pEC-XC99E as control. Growth experiments were performed in the BioLector® as described above, with additional monitoring of the ftsZ promoter activity by online measurement of mVenus fluorescence at an excitation wavelength of 508 nm and an emission wavelength of 532 nm.
Chromatin affinity purification with subsequent sequencing (ChAP-Seq)
For ChAP-Seq experiments, C. glutamicum ATCC13032ΔftsR/pAN6-FtsR-Strep and as negative control C. glutamicum ATCC13032ΔftsR/pAN6 were cultivated as described above for FtsR purification with 10 μM instead of 50 μM IPTG. FtsR-Strep was purified as described above with the following alterations: after harvesting (15 min, 6371 g, 4 °C), washing with 40 mL PBS, and a second centrifugation step (15 min, 5525 g, 4 °C), the supernatant was discarded and the cells were suspended in 20 mL PBS containing 1% (w/v) formaldehyde and incubated at room temperature for 20 min, followed by addition of glycine to a final concentration of 125 mM and incubation for another 5 min at room temperature. The cells were washed twice with 40 mL buffer A and suspended in 20 mL buffer A with a suitable amount of cOmplete Mini EDTA-free protease inhibitor cocktail tablets and 5 mg of RNAse A (both Roche Diagnostics GmbH, Mannheim, Germany). After cell disruption using a French press as described above, the crude extract was transferred to a sonication vessel, placed in an ice bath and sonified 3 × 30 s using a Branson Sonifier 250 (G. Heinemann Ultraschall- und Labortechnik, Schwaebisch Gmuend, Germany) with a pulse length of 40% and an intensity of one to shear the genomic DNA. The sonified crude extract was used for StrepTactin affinity chromatography as described above. The FtsR-containing elution fractions were pooled, 1% (w/v) SDS was added, and the samples were incubated overnight at 65 °C, followed by proteinase K treatment (final concentration 400 μg/mL) for 3 h at 55 °C. The DNA in the samples was purified by phenol-chloroform-isoamyl alcohol extraction [72], precipitated with ethanol and sodium acetate, washed with 70% (v/v) ethanol, dried and dissolved in 50 μL deionized water. Sequencing of the DNA fragments and data evaluation were performed as described previously [73]. Genomic binding sites of FtsR were defined to have a sequence coverage that is at least 50-fold above the average genome coverage determined for the negative control strain C. glutamicum ATCC13032 pAN6.
DNA microarrays
DNA microarray analysis was performed to compare the mRNA levels of the ATCC13032ΔftsR mutant with the wild type. Cells were cultivated in 50 mL CGXII medium with 2% (w/v) glucose and harvested on ice in the exponential growth phase. RNA sample preparation, labelling, hybridization, and comparative transcriptome analysis were performed as described previously [74]. The full data set of this experiment has been deposited in the NCBI Gene Expression Omnibus and can be found under the GEO accession number GSE107921.
DNA affinity purification and MALDI-ToF-MS analysis
To identify putative regulatory proteins binding to the ftsZ promoter region, DNA affinity chromatography was performed. A 404-bp DNA fragment, extending from position − 285 to + 96 with respect to the translational start site of ftsZ and including a 23 bp overhang for the addition of the biotin-tag, was amplified by PCR using the oligonucleotides AP_PftsZ_fw and AP_PftsZ_rv_bio. The resulting fragment was tagged with biotin by a second PCR using the biotinylated oligonucleotide biotin_oligo and AP_PftsZ_fw. The PCR products were purified by size exclusion chromatography using a column with 8 mL Sephacryl™ S-400 High Resolution Chromatography Media (GE Healthcare Bio-Sciences AB, Uppsala, Sweden) as bed material. Fractions containing the desired DNA fragment were pooled, concentrated to about 500 μL using an Eppendorf Concentrator plus (Eppendorf AG, Hamburg, Germany) and precipitated with ethanol. The precipitate was dissolved in 50 μL TE buffer and the DNA concentration was measured with a Colibri Microvolume Spectrophotometer (Berthold Detection Systems GmbH, Pforzheim, Germany) to assure a minimal amount of 220 pmol biotinylated DNA, which was required for the following steps. The DNA affinity chromatography was performed using Dynabeads® M-280 Streptavidin (Life Technologies AS, Oslo, Norway) as described [73]. In brief, the biotinylated DNA was coupled to the beads and incubated with crude cell extract of C. glutamicum ATCC13032 cells that had been cultivated in glucose minimal medium and harvested in the exponential growth phase. After several washing steps, the proteins were eluted using buffer containing 2 M NaCl. The elution fractions were subjected to protein precipitation with trichloroacetic acid (TCA, 10% (w/v) final concentration) and washed once with acetone. The precipitated protein was dissolved in 20 μL Tris-HCl buffer pH 7.5 and analyzed by SDS-PAGE [70] using a 12% Mini-PROTEAN® TGX™ gel (Bio-Rad Laboratories, Inc., Hercules, CA, USA). The gel was stained with GelCode® Blue Stain Reagent (Thermo Fisher Scientific Inc., Rockford, IL, USA). Proteins enriched with the promoter region of ftsZ were identified by peptide mass fingerprinting after tryptic in-gel digestion of the excised bands of the polyacrylamide gel followed by MALDI-ToF-MS using an Ultraflex III TOF/TOF mass spectrometer (Bruker Daltonics, Bremen, Germany) as described [75, 76].
Additional file
Contains additional methods, results, figures and tables. (PDF 3699 kb)
Acknowledgements
We would like to thank Susana Matamouros and Jannick Kappelmann for their help with Coulter counter measurements and data interpretation and Christina Mack and Doris Rittmann for excellent technical assistance.
Abbreviations
- BHI
Brain Heart Infusion (complex medium for C. glutamicum)
- CGP1, CGP2, CGP3
C. glutamicum prophages 1, 2 and 3
- CGXII
minimal medium for C. glutamicum
- IPTG
Isopropyl β-D-1-thiogalactopyranoside
- LB
Lysogeny broth
- OD600
Optical density at 600 nm
- PBS
Phosphate buffered saline
- TCA
Trichloroacetic acid
Authors’ contributions
MBa and MBo designed and coordinated the study. KJK performed most of the experiments. TP generated and analyzed the NGS data for genome resequencing and the ChAP-Seq experiment. KJK, MBa, and MBo analyzed the data and wrote the manuscript. All authors read and approved the final manuscript.
Funding
No Funding was obtained for this study.
Availability of data and materials
The microarray data has been deposited in the GEO database under GSE107921 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE107921). All other data generated or analysed during this study are included in this published article and its supplementary information files. All plasmids and strains used in this study are available from the corresponding author.
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Footnotes
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Meike Baumgart, Phone: +49 2461 615515, Email: m.baumgart@fz-juelich.de.
Michael Bott, Phone: +49 2461 613294, Email: m.bott@fz-juelich.de.
References
- 1.Du S, Lutkenhaus J. Assembly and activation of the Escherichia coli divisome. Mol Microbiol. 2017;105(2):177–187. doi: 10.1111/mmi.13696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hajduk IV, Rodrigues CD, Harry EJ. Connecting the dots of the bacterial cell cycle: coordinating chromosome replication and segregation with cell division. Semin Cell Dev Biol. 2016;53:2–9. doi: 10.1016/j.semcdb.2015.11.012. [DOI] [PubMed] [Google Scholar]
- 3.Wagner KS, White JM, Lucenko I, Mercer D, Crowcroft NS, Neal S, et al. Diphtheria in the postepidemic period, Europe, 2000-2009. Emerg Infect Dis. 2012;18(2):217–225. doi: 10.3201/eid1802.110987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lawn SD, Zumla AI, et al. Lancet. 2011;378(9785):57–72. doi: 10.1016/S0140-6736(10)62173-3. [DOI] [PubMed] [Google Scholar]
- 5.WHO . Tuberculosis fact sheet. 2015. [Google Scholar]
- 6.CDC . Diphtheria. 2014. [Google Scholar]
- 7.Eggeling L, Bott M. Editors. Handbook of Corynebacterium glutamicum. Boca Raton: CRC Press, Taylor & Francis Group; 2005. [Google Scholar]
- 8.Eggeling L, Bott M. A giant market and a powerful metabolism: L-lysine provided by Corynebacterium glutamicum. Appl Microbiol Biotechnol. 2015;99(8):3387–3394. doi: 10.1007/s00253-015-6508-2. [DOI] [PubMed] [Google Scholar]
- 9.Wendisch VF, Jorge JM, Perez-Garcia F, Sgobba E. Updates on industrial production of amino acids using Corynebacterium glutamicum. World J Microbiol Biotechnol. 2016;32(6):105. doi: 10.1007/s11274-016-2060-1. [DOI] [PubMed] [Google Scholar]
- 10.Becker J, Wittmann C. Bio-based production of chemicals, materials and fuels -Corynebacterium glutamicum as versatile cell factory. Curr Opin Biotechnol. 2012;23(4):631–640. doi: 10.1016/j.copbio.2011.11.012. [DOI] [PubMed] [Google Scholar]
- 11.Freudl R. Beyond amino acids: use of the Corynebacterium glutamicum cell factory for the secretion of heterologous proteins. J Biotechnol. 2017;258:101–109. doi: 10.1016/j.jbiotec.2017.02.023. [DOI] [PubMed] [Google Scholar]
- 12.Margolin W. FtsZ and the division of prokaryotic cells and organelles. Nat Rev Mol Cell Biol. 2005;6(11):862–871. doi: 10.1038/nrm1745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ortiz C, Natale P, Cueto L, Vicente M. The keepers of the ring: regulators of FtsZ assembly. FEMS Microbiol Rev. 2016;40(1):57–67. doi: 10.1093/femsre/fuv040. [DOI] [PubMed] [Google Scholar]
- 14.Kruse T, Bork-Jensen J, Gerdes K. The morphogenetic MreBCD proteins of Escherichia coli form an essential membrane-bound complex. Mol Microbiol. 2005;55(1):78–89. doi: 10.1111/j.1365-2958.2004.04367.x. [DOI] [PubMed] [Google Scholar]
- 15.Figge RM, Divakaruni AV, Gober JW. MreB, the cell shape-determining bacterial actin homologue, co-ordinates cell wall morphogenesis in Caulobacter crescentus. Mol Microbiol. 2004;51(5):1321–1332. doi: 10.1111/j.1365-2958.2003.03936.x. [DOI] [PubMed] [Google Scholar]
- 16.Jones LJ, Carballido-Lopez R, Errington J. Control of cell shape in bacteria: helical, actin-like filaments in Bacillus subtilis. Cell. 2001;104(6):913–922. doi: 10.1016/s0092-8674(01)00287-2. [DOI] [PubMed] [Google Scholar]
- 17.Daniel RA, Errington J. Control of cell morphogenesis in bacteria: two distinct ways to make a rod-shaped cell. Cell. 2003;113(6):767–776. doi: 10.1016/s0092-8674(03)00421-5. [DOI] [PubMed] [Google Scholar]
- 18.Donovan C, Bramkamp M. Cell division in Corynebacterineae. Front Microbiol. 2014;5:132. doi: 10.3389/fmicb.2014.00132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Letek M, Ordonez E, Fiuza M, Honrubia-Marcos P, Vaquera J, Gil JA, et al. Characterization of the promoter region of ftsZ from Corynebacterium glutamicum and controlled overexpression of FtsZ. Int Microbiol. 2007;10(4):271–282. [PubMed] [Google Scholar]
- 20.Ramos A, Letek M, Campelo AB, Vaquera J, Mateos LM, Gil JA. Altered morphology produced by ftsZ expression in Corynebacterium glutamicum ATCC 13869. Microbiology. 2005;151(Pt 8):2563–2572. doi: 10.1099/mic.0.28036-0. [DOI] [PubMed] [Google Scholar]
- 21.Dziadek J, Rutherford SA, Madiraju MV, Atkinson MA, Rajagopalan M. Conditional expression of Mycobacterium smegmatis ftsZ, an essential cell division gene. Microbiology. 2003;149(Pt 6):1593–1603. doi: 10.1099/mic.0.26023-0. [DOI] [PubMed] [Google Scholar]
- 22.Roy S, Ajitkumar P. Transcriptional analysis of the principal cell division gene, ftsZ, of Mycobacterium tuberculosis. J Bacteriol. 2005;187(7):2540–2550. doi: 10.1128/JB.187.7.2540-2550.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Flardh K, Garrido T, Vicente M. Contribution of individual promoters in the ddlB-ftsZ region to the transcription of the essential cell-division gene ftsZ in Escherichia coli. Mol Microbiol. 1997;24(5):927–936. doi: 10.1046/j.1365-2958.1997.4001762.x. [DOI] [PubMed] [Google Scholar]
- 24.Kiran M, Maloney E, Lofton H, Chauhan A, Jensen R, Dziedzic R, et al. Mycobacterium tuberculosis ftsZ expression and minimal promoter activity. Tuberculosis (Edinb). 2009;89(Suppl 1):S60–S64. doi: 10.1016/S1472-9792(09)70014-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lee DS, Kim P, Kim ES, Kim Y, Lee HS. Corynebacterium glutamicum WhcD interacts with WhiA to exert a regulatory effect on cell division genes. Antonie Van Leeuwenhoek. 2018;111(5):641–648. doi: 10.1007/s10482-017-0953-0. [DOI] [PubMed] [Google Scholar]
- 26.Schultz C, Niebisch A, Schwaiger A, Viets U, Metzger S, Bramkamp M, et al. Genetic and biochemical analysis of the serine/threonine protein kinases PknA, PknB, PknG and PknL of Corynebacterium glutamicum: evidence for non-essentiality and for phosphorylation of OdhI and FtsZ by multiple kinases. Mol Microbiol. 2009;74(3):724–741. doi: 10.1111/j.1365-2958.2009.06897.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gerstmeir R, Cramer A, Dangel P, Schaffer S, Eikmanns BJ. RamB, a novel transcriptional regulator of genes involved in acetate metabolism of Corynebacterium glutamicum. J Bacteriol. 2004;186(9):2798–2809. doi: 10.1128/JB.186.9.2798-2809.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Auchter M, Cramer A, Hüser A, Rückert C, Emer D, Schwarz P, et al. RamA and RamB are global transcriptional regulators in Corynebacterium glutamicum and control genes for enzymes of the central metabolism. J Biotechnol. 2011;154(2–3):126–139. doi: 10.1016/j.jbiotec.2010.07.001. [DOI] [PubMed] [Google Scholar]
- 29.Pfeifer-Sancar K, Mentz A, Rückert C, Kalinowski J. Comprehensive analysis of the Corynebacterium glutamicum transcriptome using an improved RNAseq technique. BMC Genomics. 2013;14:888. doi: 10.1186/1471-2164-14-888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Alm EJ, Huang KH, Price MN, Koche RP, Keller K, Dubchak IL, et al. The MicrobesOnline web site for comparative genomics. Genome Res. 2005;15(7):1015–1022. doi: 10.1101/gr.3844805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Overbeek R, Larsen N, Walunas T, D'Souza M, Pusch G, Selkov E, Jr, et al. The ERGO genome analysis and discovery system. Nucleic Acids Res. 2003;31(1):164–171. doi: 10.1093/nar/gkg148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. Basic local alignment search tool. J Mol Biol. 1990;215(3):403–410. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- 33.Niebisch A, Kabus A, Schultz C, Weil B, Bott M. Corynebacterial protein kinase G controls 2-oxoglutarate dehydrogenase activity via the phosphorylation status of the OdhI protein. J Biol Chem. 2006;281:12300–12307. doi: 10.1074/jbc.M512515200. [DOI] [PubMed] [Google Scholar]
- 34.Krawczyk S, Raasch K, Schultz C, Hoffelder M, Eggeling L, Bott M. The FHA domain of OdhI interacts with the carboxyterminal 2-oxoglutarate dehydrogenase domain of OdhA in Corynebacterium glutamicum. FEBS Lett. 2010;584(8):1463–1468. doi: 10.1016/j.febslet.2010.03.028. [DOI] [PubMed] [Google Scholar]
- 35.Raasch K, Bocola M, Labahn J, Leitner A, Eggeling L, Bott M. Interaction of 2-oxoglutarate dehydrogenase OdhA with its inhibitor OdhI in Corynebacterium glutamicum: mutants and a model. J Biotechnol. 2014;191:99–105. doi: 10.1016/j.jbiotec.2014.05.023. [DOI] [PubMed] [Google Scholar]
- 36.Ventura M, Rieck B, Boldrin F, Degiacomi G, Bellinzoni M, Barilone N, et al. GarA is an essential regulator of metabolism in Mycobacterium tuberculosis. Mol Microbiol. 2013;90(2):356–366. doi: 10.1111/mmi.12368. [DOI] [PubMed] [Google Scholar]
- 37.Baumgart M, Schubert K, Bramkamp M, Frunzke J. Impact of LytR-CpsA-Psr proteins on cell wall biosynthesis in Corynebacterium glutamicum. J Bacteriol. 2016;198(22):3045–3059. doi: 10.1128/JB.00406-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kortmann M, Kuhl V, Klaffl S, Bott M. A chromosomally encoded T7 RNA polymerase-dependent gene expression system for Corynebacterium glutamicum: construction and comparative evaluation at the single-cell level. Microb Biotechnol. 2015;8(2):253–265. doi: 10.1111/1751-7915.12236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Baumgart M, Unthan S, Rückert C, Sivalingam J, Grünberger A, Kalinowski J, et al. Construction of a prophage-free variant of Corynebacterium glutamicum ATCC 13032 for use as a platform strain for basic research and industrial biotechnology. Appl Environ Microbiol. 2013;79(19):6006–6015. doi: 10.1128/AEM.01634-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Singh S, Sevalkar RR, Sarkar D, Karthikeyan S. Characteristics of the essential pathogenicity factor Rv1828, a MerR family transcription regulator from Mycobacterium tuberculosis. FEBS J. 2018;285(23):4424–4444. doi: 10.1111/febs.14676. [DOI] [PubMed] [Google Scholar]
- 41.Bailey TL, Boden M, Buske FA, Frith M, Grant CE, Clementi L, et al. MEME SUITE: tools for motif discovery and searching. Nucleic Acids Res. 2009;37(Web Server issue):W202–W208. doi: 10.1093/nar/gkp335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Brand S, Niehaus K, Pühler A, Kalinowski J. Identification and functional analysis of six mycolyltransferase genes of Corynebacterium glutamicum ATCC 13032: the genes cop1, cmt1, and cmt2 can replace each other in the synthesis of trehalose dicorynomycolate, a component of the mycolic acid layer of the cell envelope. Arch Microbiol. 2003;180(1):33–44. doi: 10.1007/s00203-003-0556-1. [DOI] [PubMed] [Google Scholar]
- 43.Puech V, Bayan N, Salim K, Leblon G, Daffe M. Characterization of the in vivo acceptors of the mycoloyl residues transferred by the corynebacterial PS1 and the related mycobacterial antigens 85. Mol Microbiol. 2000;35(5):1026–1041. doi: 10.1046/j.1365-2958.2000.01738.x. [DOI] [PubMed] [Google Scholar]
- 44.Schaaf S, Bott M. Target genes and DNA-binding sites of the response regulator PhoR from Corynebacterium glutamicum. J Bacteriol. 2007;189(14):5002–5011. doi: 10.1128/JB.00121-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wendisch VF, Bott M. Phosphorus metabolism of Corynebacterium glutamicum. In: Eggeling L, Bott M, editors. Handbook of Corynebacterium glutamicum. Boca Raton: CRC Press; 2005. pp. 377–396. [Google Scholar]
- 46.Ishige T, Krause M, Bott M, Wendisch VF, Sahm H. The phosphate starvation stimulon of Corynebacterium glutamicum determined by DNA microarray analyses. J Bacteriol. 2003;185(15):4519–4529. doi: 10.1128/JB.185.15.4519-4529.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sievers F., Wilm A., Dineen D., Gibson T. J., Karplus K., Li W., Lopez R., McWilliam H., Remmert M., Soding J., Thompson J. D., Higgins D. G. Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Molecular Systems Biology. 2014;7(1):539–539. doi: 10.1038/msb.2011.75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Frunzke J, Engels V, Hasenbein S, Gätgens C, Bott M. Co-ordinated regulation of gluconate catabolism and glucose uptake in Corynebacterium glutamicum by two functionally equivalent transcriptional regulators, GntR1 and GntR2. Mol Microbiol. 2008;67(2):305–322. doi: 10.1111/j.1365-2958.2007.06020.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Adams DW, Errington J. Bacterial cell division: assembly, maintenance and disassembly of the Z ring. Nat Rev Microbiol. 2009;7(9):642–653. doi: 10.1038/nrmicro2198. [DOI] [PubMed] [Google Scholar]
- 50.McEwan AG, Djoko KY, Chen NH, Couñago RLM, Kidd SP, Potter AJ, et al. Novel bacterial MerR-like regulators: their role in the response to carbonyl and nitrosative stress. Adv Microb Physiol. 2011;Volume 58:1–22. doi: 10.1016/B978-0-12-381043-4.00001-5. [DOI] [PubMed] [Google Scholar]
- 51.Brown NL, Stoyanov JV, Kidd SP, Hobman JL. The MerR family of transcriptional regulators. FEMS Microbiol Rev. 2003;27(2–3):145–163. doi: 10.1016/S0168-6445(03)00051-2. [DOI] [PubMed] [Google Scholar]
- 52.Ansari AZ, Chael ML, O'Halloran TV. Allosteric underwinding of DNA is a critical step in positive control of transcription by hg-MerR. Nature. 1992;355(6355):87–89. doi: 10.1038/355087a0. [DOI] [PubMed] [Google Scholar]
- 53.Frantz B, O'Halloran TV. DNA distortion accompanies transcriptional activation by the metal-responsive gene-regulatory protein MerR. Biochemistry. 1990;29(20):4747–4751. doi: 10.1021/bi00472a001. [DOI] [PubMed] [Google Scholar]
- 54.Newberry KJ, Brennan RG. The structural mechanism for transcription activation by MerR family member multidrug transporter activation. N terminus J Biol Chem. 2004;279(19):20356–20362. doi: 10.1074/jbc.M400960200. [DOI] [PubMed] [Google Scholar]
- 55.Philips SJ, Canalizo-Hernandez M, Yildirim I, Schatz GC, Mondragon A, O'Halloran TV. Allosteric transcriptional regulation via changes in the overall topology of the core promoter. Science. 2015;349(6250):877–881. doi: 10.1126/science.aaa9809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Flardh K, Leibovitz E, Buttner MJ, Chater KF. Generation of a non-sporulating strain of Streptomyces coelicolor A3(2) by the manipulation of a developmentally controlled ftsZ promoter. Mol Microbiol. 2000;38(4):737–749. doi: 10.1046/j.1365-2958.2000.02177.x. [DOI] [PubMed] [Google Scholar]
- 57.Kwak J, Dharmatilake AJ, Jiang H, Kendrick KE. Differential regulation of ftsZ transcription during septation of Streptomyces griseus. J Bacteriol. 2001;183(17):5092–5101. doi: 10.1128/JB.183.17.5092-5101.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Galagan JE, Sisk P, Stolte C, Weiner B, Koehrsen M, Wymore F, et al. TB database 2010: overview and update. Tuberculosis (Edinb) 2010;90(4):225–235. doi: 10.1016/j.tube.2010.03.010. [DOI] [PubMed] [Google Scholar]
- 59.Minch KJ, Rustad TR, Peterson EJ, Winkler J, Reiss DJ, Ma S, et al. The DNA-binding network of Mycobacterium tuberculosis. Nat Commun. 2015;6:5829. doi: 10.1038/ncomms6829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Reddy TB, Riley R, Wymore F, Montgomery P, DeCaprio D, Engels R, et al. TB database: an integrated platform for tuberculosis research. Nucleic Acids Res. 2009;37(Database issue):D499–D508. doi: 10.1093/nar/gkn652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Monahan LG, Harry EJ. You are what you eat: metabolic control of bacterial division. Trends Microbiol. 2016;24(3):181–189. doi: 10.1016/j.tim.2015.11.007. [DOI] [PubMed] [Google Scholar]
- 62.Keilhauer C, Eggeling L, Sahm H. Isoleucine synthesis in Corynebacterium glutamicum - molecular analysis of the IlvB-IlvN-IlvC operon. J Bacteriol. 1993;175(17):5595–5603. doi: 10.1128/jb.175.17.5595-5603.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Kensy F, Zang E, Faulhammer C, Tan RK, Büchs J. Validation of a high-throughput fermentation system based on online monitoring of biomass and fluorescence in continuously shaken microtiter plates. Microb Cell Factories. 2009;8:31. doi: 10.1186/1475-2859-8-31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Sambrook J, Russell D. Molecular cloning. A laboratory manual. 3rd ed. Cold Spring Harbor: Cold Spring Harbor Laboratory Press; 2001. [Google Scholar]
- 65.Hanahan D. Studies on transformation of Escherichia coli with plasmids. J Mol Biol. 1983;166(4):557–580. doi: 10.1016/s0022-2836(83)80284-8. [DOI] [PubMed] [Google Scholar]
- 66.van der Rest ME, Lange C, Molenaar D. A heat shock following electroporation induces highly efficient transformation of Corynebacterium glutamicum with xenogeneic plasmid DNA. Appl Microbiol Biotechnol. 1999;52(4):541–545. doi: 10.1007/s002530051557. [DOI] [PubMed] [Google Scholar]
- 67.Gibson DG, Young L, Chuang RY, Venter JC, Hutchison CA, 3rd, Smith HO. Enzymatic assembly of DNA molecules up to several hundred kilobases. Nat Methods. 2009;6(5):343–345. doi: 10.1038/nmeth.1318. [DOI] [PubMed] [Google Scholar]
- 68.Schäfer A, Tauch A, Jäger W, Kalinowski J, Thierbach G, Pühler A. Small mobilizable multi-purpose cloning vectors derived from the Escherichia coli plasmids pK18 and pK19: selection of defined deletions in the chromosome of Corynebacterium glutamicum. Gene. 1994;145(1):69–73. doi: 10.1016/0378-1119(94)90324-7. [DOI] [PubMed] [Google Scholar]
- 69.Niebisch A, Bott M. Molecular analysis of the cytochrome bc1-aa3 branch of the Corynebacterium glutamicum respiratory chain containing an unusual diheme cytochrome c1. Arch Microbiol. 2001;175(4):282–294. doi: 10.1007/s002030100262. [DOI] [PubMed] [Google Scholar]
- 70.Laemmli UK. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature. 1970;227:680–685. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- 71.Garcia-Nafria J, Baumgart M, Turkenburg JP, Wilkinson AJ, Bott M, Wilson KS. Crystal and solution studies reveal that the transcriptional regulator AcnR of Corynebacterium glutamicum is regulated by citrate-Mg2+ binding to a non-canonical pocket. J Biol Chem. 2013;288(22):15800–15812. doi: 10.1074/jbc.M113.462440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Chomczynski P, Sacchi N. The single-step method of RNA isolation by acid guanidinium thiocyanate-phenol-chloroform extraction: twenty-something years on. Nat Protoc. 2006;1(2):581–585. doi: 10.1038/nprot.2006.83. [DOI] [PubMed] [Google Scholar]
- 73.Pfeifer E, Hünnefeld M, Popa O, Polen T, Kohlheyer D, Baumgart M, et al. Silencing of cryptic prophages in Corynebacterium glutamicum. Nucleic Acids Res. 2016;44(21):10117–10131. doi: 10.1093/nar/gkw692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Vogt M, Haas S, Klaffl S, Polen T, Eggeling L, van Ooyen J, et al. Pushing product formation to its limit: metabolic engineering of Corynebacterium glutamicum for L-leucine overproduction. Metab Eng. 2014;22:40–52. doi: 10.1016/j.ymben.2013.12.001. [DOI] [PubMed] [Google Scholar]
- 75.Schaffer S, Weil B, Nguyen VD, Dongmann G, Günther K, Nickolaus M, et al. A high-resolution reference map for cytoplasmic and membrane-associated proteins of Corynebacterium glutamicum. Electrophoresis. 2001;22(20):4404–4422. doi: 10.1002/1522-2683(200112)22:20<4404::AID-ELPS4404>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]
- 76.Koch-Koerfges A, Kabus A, Ochrombel I, Marin K, Bott M. Physiology and global gene expression of a Corynebacterium glutamicum ΔF1FO-ATP synthase mutant devoid of oxidative phosphorylation. Biochim Biophys Acta. 2012;1817(2):370–380. doi: 10.1016/j.bbabio.2011.10.006. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Contains additional methods, results, figures and tables. (PDF 3699 kb)
Data Availability Statement
The microarray data has been deposited in the GEO database under GSE107921 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE107921). All other data generated or analysed during this study are included in this published article and its supplementary information files. All plasmids and strains used in this study are available from the corresponding author.