

Sir,
We hereby describe the case of a 21-year-old gentleman who had presented with features of delayed puberty and sexual infantilism. He was born as the first child of a non-consanguineous marriage. There was no cryptorchidism, microphallus or dysmorphic features at birth. His developmental milestones were normal. However, he did not experience a normal pubertal growth spurt, but con'tinued to have a steady but slow growth until the time of presentation. He had history suggestive of anosmia. He had no hearing loss, dental agenesis, or cleft lip/palate. There was no significant family history.
On physical examination, his height was 173 cm, with an arm span of 177 cm, an upper to lower segment ratio of 0.94 and weight of 63 kg (body mass index, 21 kg/m2). He had poor development of secondary sexual characteristics in the form of absent facial hair, sparse axillary and pubic hair (Tanner stage 2), testicular volume of 2 mL bilaterally and a stretched penile length (SPL) of 4 cm. Baseline investigations had revealed serum LH and FSH to be less than 0.1 mIU/mL and serum testosterone that was undetectable. The Magnetic Resonance Imaging (MRI) brain showed absent olfactory bulb and sulci bilaterally [Figure 1].
Figure 1.
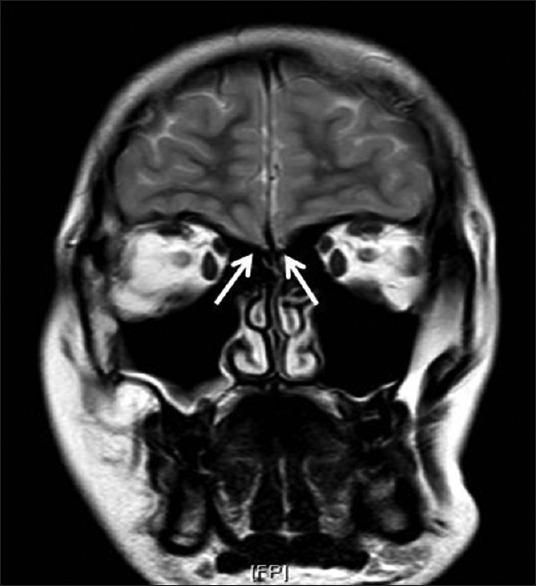
MRI Brain showing bilateral absence of olfactory bulbs
Based on these findings, he was diagnosed with Kallmann syndrome and initiated on Testosterone replacement therapy (100 mg every 4 weeks with a planned increase after 3 months to 250 mg every four weeks). He visited our clinic after 8 months of the initial presentation, having discontinued Testosterone therapy 2 months prior to his review. A repeat clinical assessment had revealed significant improvement in his secondary sexual characteristics with SPL of 5 cm, testicular volume of 6 mL bilaterally and Tanner stage 3 pubic hair. Repeat biochemical evaluation showed serum Testosterone level of 178 ng/dL and LH level of 3.8 mIU/mL. Thus, within 6 months of initiating testosterone therapy, he attained considerable improvement in pubertal features with spontaneous increase in testicular volume, which was strongly indicative of a reversal of Kallmann syndrome.[1]
Typically, patients diagnosed with Kallmann syndrome cannot complete puberty and achieve fertility unless exposed to physiological doses of gonadotropins or pulsatile GnRH. They require lifelong replacement therapy with testosterone to maintain male secondary sexual characteristics. A few subjects, however, have been described who present with a rare variant of Kallmann syndrome in which gonadotropin and testosterone secretion recovers spontaneously.
Many mechanisms are described to explain the reversibility in Kallmann syndrome. One hypothesis is that the GnRH neuronal migratory defects may not be complete and variable degrees of endogenous GnRH secretion may occur. The final maturation of the GnRH neuronal pathway is triggered by exogenous sex steroids.[2] Epigenetic influences may also be involved in reversible cases.[3] Some cases of reversible hypogonadotropic hypogonadism have shown a dramatic response to exogenous kisspeptin.[4] Mutations identified in reversible cases include FGFR1, GnRHR, TAC3, TAC3R and HS6ST1. The reversal could also be due to an androgen driven up-regulation of genes involved in the maturation of the GnRH neurons.[3]
Declaration of patient consent
The authors certify that they have obtained all appropriate patient consent forms. In the form the patient(s) has/have given his/her/their consent for his/her/their images and other clinical information to be reported in the journal. The patients understand that their names and initials will not be published and due efforts will be made to conceal their identity, but anonymity cannot be guaranteed.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
REFERENCES
- 1.Laitinen EM, Tommiska J, Sane T, Vaaralahti K, Toppari J, Raivio T. Reversible congenital hypogonadotropic hypogonadism in patients with CHD7, FGFR1 or GNRHR mutations. PLoS One. 2012;7:e39450. doi: 10.1371/journal.pone.0039450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Pitteloud N, Acierno JS, Meysing AU, Dwyer AA, Hayes FJ, Crowley WF. Reversible kallmann syndrome, delayed puberty, and isolated anosmia occurring in a single family with a mutation in the fibroblast growth factor receptor 1 gene. J Clin Endocrinol Metab. 2005;90:1317–22. doi: 10.1210/jc.2004-1361. [DOI] [PubMed] [Google Scholar]
- 3.Dwyer AA, Raivio T, Pitteloud N. Management of endocrine disease: Reversible hypogonadotropic hypogonadism. Eur J Endocrinol. 2016;174:R267–274. doi: 10.1530/EJE-15-1033. [DOI] [PubMed] [Google Scholar]
- 4.Boehm U, Bouloux PM, Dattani MT, de Roux N, Dodé C, Dunkel L, et al. Expert consensus document: European consensus statement on congenital hypogonadotropic hypogonadism--pathogenesis, diagnosis and treatment. Nat Rev Endocrinol. 2015;11:547–64. doi: 10.1038/nrendo.2015.112. [DOI] [PubMed] [Google Scholar]