

Significance Statement
Although steroid-sensitive nephrotic syndrome (SSNS) is considered an autoimmune disease, its etiology is poorly understood. Genome-wide association studies (GWAS) have provided important insights into other autoimmune diseases, but so far, such studies have reported associations only in the classical HLA region for SSNS. In a GWAS of a large cohort of European ancestry comprising 422 ethnically homogeneous pediatric patients and 5642 ethnically matched controls, the authors found two loci outside the HLA region associated with SSNS at genome-wide significance. The locus with strongest association contains the calcium homeostasis modulator family member 6 gene CALHM6, which has been implicated in the regulation of the immune system. These findings suggest that impaired downregulation of the immune system may be a key mechanism in the pathogenesis of SSNS.
Keywords: GWAS, SSNS, HLA, CALHM6, FAM26F
Visual Abstract

Abstract
Background
Steroid-sensitive nephrotic syndrome (SSNS), the most common form of nephrotic syndrome in childhood, is considered an autoimmune disease with an established classic HLA association. However, the precise etiology of the disease is unclear. In other autoimmune diseases, the identification of loci outside the classic HLA region by genome-wide association studies (GWAS) has provided critical insights into disease pathogenesis. Previously conducted GWAS of SSNS have not identified non-HLA loci achieving genome-wide significance.
Methods
In an attempt to identify additional loci associated with SSNS, we conducted a GWAS of a large cohort of European ancestry comprising 422 ethnically homogeneous pediatric patients and 5642 ethnically matched controls.
Results
The GWAS found three loci that achieved genome-wide significance, which explain approximately 14% of the genetic risk for SSNS. It confirmed the previously reported association with the HLA-DR/DQ region (lead single-nucleotide polymorphism [SNP] rs9273542, P=1.59×10−43; odds ratio [OR], 3.39; 95% confidence interval [95% CI], 2.86 to 4.03) and identified two additional loci outside the HLA region on chromosomes 4q13.3 and 6q22.1. The latter contains the calcium homeostasis modulator family member 6 gene CALHM6 (previously called FAM26F). CALHM6 is implicated in immune response modulation; the lead SNP (rs2637678, P=1.27×10−17; OR, 0.51; 95% CI, 0.44 to 0.60) exhibits strong expression quantitative trait loci effects, the risk allele being associated with lower lymphocytic expression of CALHM6.
Conclusions
Because CALHM6 is implicated in regulating the immune response to infection, this may provide an explanation for the typical triggering of SSNS onset by infections. Our results suggest that a genetically conferred risk of immune dysregulation may be a key component in the pathogenesis of SSNS.
Steroid-sensitive nephrotic syndrome (SNSS) is the most common form of nephrotic syndrome in children, with an incidence of approximately 1–10 per 100,000.1 The majority of affected children experience a chronic relapsing course. The onset of disease manifestations is commonly associated with a preceding activation of the immune system, typically by an upper respiratory tract infection.2 As the name implies, SSNS is characterized by a therapeutic response to glucocorticoids as well as to other immunosuppressants. The apparent triggering of the disease by infection and the therapeutic effect of immunosuppressive treatment have suggested that SSNS is an autoimmune disorder.3
Investigating the genetic architecture of other autoimmune diseases through genome-wide association studies (GWAS) has proven to be successful in providing insight into their etiology and pathogenesis. Unsurprisingly, a common finding in these studies is the identification of association with the HLA region that contains numerous genes critical for the immune system, in particular enabling the distinction between self and foreign.4
Arguably, however, it is the identification of risk loci outside the HLA locus that can provide the most informative mechanistic insights into the pathogenesis of such diseases. Prominent examples from nephrology include membranous nephropathy and IgA nephropathy. In membranous nephropathy, GWAS identified association with PLA2R1, suggesting that the antibody formation against the PLA2R1 receptor observed in membranous nephropathy is a causal disease mechanism.5,6 Similarly, in IgA nephropathy, GWAS have highlighted the important role of the intestinal immune response as well as IgA1 antibody glycosylation in the pathogenesis of the disease.7–9
Three GWAS of SSNS have recently been reported, but in all, association at genome-wide significance was identified within the HLA region only.10–12 We set out to perform a GWAS using the largest number of ethnically homogenous patients and controls studied to date in an attempt to identify additional loci associated with SSNS.
Methods
Full details of all methods can be found in Supplemental Material.
Cohorts
DNA from pediatric patients diagnosed with SSNS and of reported European ethnicity were used in this study. SSNS was defined according to standard clinical criteria.13 DNA was acquired from the Prednisolone in Nephrotic Syndrome (PREDNOS) (EudraCT 2010–022489–29) and PREDNOS2 (EudraCT 2012–003476–39) trials14 as well as from collaborating clinicians at their affiliated institutions. Informed consent was obtained from all participants, and ethical approval was granted by the host institutions. Ethnically matched controls were drawn from publicly available datasets (Supplemental Figure 1). For the replication of our findings, we examined the results of previously published GWAS of SSNS.10–12
Genotyping, Quality Control, and Imputation
Isolation of DNA, genotyping, quality control (QC), and imputation were performed using standard procedures. Patients were genotyped by University College London Genomics (Institute of Child Health, University College London, London, United Kingdom) on the Infinium Multi-Ethnic Global BeadChip v.A1 (Illumina). Controls had been genotyped on a variety of platforms (additional details are in Supplemental Material).15–17 Stringent QC steps for both single-nucleotide polymorphisms (SNPs) and individuals (including missingness, heterozygosity, and deviation from Hardy–Weinberg equilibrium) were carried out on patients and controls separately as well as on the combined cohort (Supplemental Figures 1–3). Principal component analysis was used to identify the subset of patients and controls of European ancestry (Supplemental Figure 4). The genomic inflation factor (λ) was calculated to estimate population stratification. Imputation was performed on the combined patient-control dataset with Beagle 5.0 (https://faculty.washington.edu/browning/beagle/beagle.html) using only markers passing stringent QC and present in all datasets.18 Only SNPs with a dosage R2 of >0.8 were included. Data from the 1000 Genomes Project Phase 3 were used as a reference panel. Golden Helix SNP & Variation Suite version 8.8.1 (SVS; http://goldenhelix.com/products/SNP_Variation/index.html) and PLINK version 1.90β (https://www.cog-genomics.org/plink/1.9) were used for analysis.19
GWAS and Conditional Analyses
The primary association analysis was performed using logistic regression under an additive model with adjustment for the first ten principal components of ancestry.
Conditional analysis of the lead SNPs was performed using a logistic regression model. A genome-wide significance threshold of 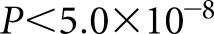 was used. SVS was used for association testing and conditional analysis. A power calculation is detailed in Supplemental Material.
was used. SVS was used for association testing and conditional analysis. A power calculation is detailed in Supplemental Material.
HLA Type Imputing
HLA imputation was performed using SNP2HLA v1.0.3 (http://software.broadinstitute.org/mpg/snp2hla/) with default parameters.20 As input, a subset of 1189 SNPs from those selected for GWAS (post-QC) and overlapping with the SNP2HLA imputation HapMap European reference dataset was used. Logistic regression under an additive model with adjustment for the first ten principal components of ancestry was used to test for association of each HLA allele with SSNS. Conditional analysis of the lead HLA alleles was performed using a logistic regression model as well.
Expression Quantitative Trait Loci Analyses
Publicly available expression quantitative trait loci (eQTLs) databases were queried to ascertain whether variants significantly associated with SSNS were known to influence the expression levels of corresponding gene products in multiple tissues.
Results
Study Cohort
DNA from a total of 712 anonymous patients with SSNS of reported European ethnicity were available to our project. The combined control dataset consisted of 6126 individuals of reported European ethnicity. After stringent QC and selection for European ancestry by principal component analysis, 422 patients and 5642 controls remained (Supplemental Figure 1), with a total of 158,217 overlapping SNPs. These were imputed to 5,216,266 high-quality genome-wide SNPs. The summary statistics are available in Supplemental Material.
GWAS Results
Our GWAS revealed three loci achieving genome-wide significance (Figure 1). Together, these loci explain approximately 14% of the genetic risk for SSNS. The strongest signal corresponded to a broad peak in the classic HLA region on chromosome 6p21.32. The three leading SNPs, in strong linkage disequilibrium with each other, are rs9273542 (P=1.59×10−43; odds ratio [OR], 3.39; 95% confidence interval [95% CI], 2.86 to 4.03) and rs9273529 (P=2.87×10−43; OR, 3.39; 95% CI, 2.85 to 4.03) in the intronic region of the gene HLA-DQB1 and rs9273371 (P=1.64×10−43; OR, 3.29; 95% CI, 2.78 to 3.89) intergenic between HLA-DQA1 and HLA-DQB1 (Figure 2A).
Figure 1.

Manhattan plot. Genome-wide association studies (GWAS) for steroid-sensitive nephrotic syndrome (SSNS) comparing 422 European patients with 5642 ethnically matched controls. The x axis shows the chromosomal position. The y axis shows the log-transformed P value; the horizontal red line indicates the genome-wide significance threshold (P=5×10−8). Three loci surpass this threshold on chromosomes 4 and 6.
Figure 2.

Locus zoom plot of the HLA-DR/DQ (6p21.32) association. The x axis indicates chromosomal position, the left y axis indicates the log-transformed P value, and the right y axis indicates the recombination rate. The purple diamond indicates the single-nucleotide polymorphism (SNP) with the smallest P value within each region. SNPs are colored on the basis of their pairwise linkage disequilibrium to the lead SNP as per 1000 Genomes European reference data according to the key. Recombination hotspots are indicated by blue vertical lines. Data are shown for 200 kb on either side of the lead SNP. (A) Unconditioned analysis. The lead SNP is rs9273542 in the gene HLA-DQB1, with a P value of 1.59×10−43. (B) Postconditioning on rs9273542. The lead SNP is rs2858317 centromeric of HLA-DQB1, with a P value of 4.29×10−31. (C) Postconditioning on rs9273542 and rs2858317. The lead SNP from this analysis is rs3828799 centromeric of HLA-DQB1, with a P value of 2.40×10−8. Note that these results indicate that the association in the HLA-DRB1 region is driven mainly by two independent HLA alleles (indicated by lead SNPs in A and B).
Conditional analysis on rs9273542 decreased the strength of the association such that the minimum P value achieved at this locus changed to P=4.29×10−31 for rs2858317 centromeric of HLA-DQB1 (Figure 2B). Joint conditioning on rs9273542 and rs2858317 significantly reduced the strength of the association to a minimum P value of P=2.40×10−8 at rs3828799 (Figure 2C). These results indicate that the association at this locus is driven by at least two independent signals.
Our two lead SNPs (rs9273542 and rs2858317) are in strong linkage disequilibrium with rs4642516 (identified by Jia et al.12) and two markers rs1063348 and rs28366266 (identified by Debiec et al.11).
The strongest association outside the HLA region was with a locus on chromosome 6q22.1. The three lead SNPs (rs2637678: P=1.27×10−17; OR, 0.51; 95% CI, 0.44 to 0.60; rs2637681: P=3.53×10−17; OR, 0.52; 95% CI, 0.44 to 0.61; and rs2858829: P=1.72×10−16; OR, 0.53; 95% CI, 0.45 to 0.62) are all in close linkage disequilibrium and localize around the gene CALHM6 (previously called FAM26F) (Figure 3A). Genome-wide significance was lost by conditioning on rs2637678 (Figure 3B), indicating that a single signal is responsible for driving this association.
Figure 3.

Locus zoom plot of the CALHM6 (6q22.1) association. The composition of this figure is as per Figure 2. (A) Unconditioned analysis. The lead single-nucleotide polymorphism (SNP) is rs2637678 with a P value of 1.27×10−17. (B) Postconditioning on rs2637678. No additional SNPs reached genome-wide significance level, indicating that the association with CALHM6 is driven by a single haplotype.
The third locus to reach genome-wide significance was on chromosome 4q13.3. The lead SNP, rs10518133, is in the intronic region of the gene PARM1, and it was associated at P=2.50×10−8 with OR of 1.96 (95% CI, 1.57 to 2.45) (Figure 4A). Genome-wide significance was also lost at this locus by conditioning on the lead SNP, indicating that a single signal is driving the association (Figure 4B).
Figure 4.

Locus zoom plot of the PARM1 (4q13.3) association. The composition of this figure is as per Figure 2. (A) Unconditioned analysis. The lead single-nucleotide polymorphism (SNP) is rs10518133 in the gene PARM1, with a P value of 2.50×10−8. (B) Postconditioning on rs10518133. No additional SNPs reached genome wide significance level, indicating that the association with PARM1 is driven by a single haplotype.
A Manhattan plot as well as locus zoom plots of the identical regions using genotyped markers only are provided in Supplemental Figures 5–8, and details for the lead genotyped SNPs at the respective loci are listed in Supplemental Table 1.
Classic HLA Type Analyses
HLA allele imputation was performed for HLA class 1 (HLA-A, -C, and -B) and class 2 (HLA-DRB1 and -DQB1) genes. Genome-wide significance for SSNS was achieved for nine HLA alleles, including high-resolution subtypes at DQA1*01, DQA1*02, DQA1*07, DQA1*13, and B*8 (Supplemental Figure 2, Table 1). The strongest association was observed with HLA-DQA1*02:01, which resides on a haplotype with HLA-DRB1*07:01 (indicated by equal allele frequencies in patients and controls). The HLA-DQA1*01 allele was protective and remained independently associated at genome-wide significance after conditioning on HLA-DQA1*02:01 (P=1.24×10−31; OR, 0.31; 95% CI, 0.25 to 0.38).
Table 1.
Association of imputed classic HLA alleles with steroid-sensitive nephrotic syndrome
| HLA Allele | MAF Patients | MAF Controls | OR | 95% CI | P Value |
|---|---|---|---|---|---|
| HLA_DQA1*02:01 | 0.35 | 0.15 | 3.42 | 2.80 to 4.16 | 1.06×10−32 |
| HLA_DQA1*01 | 0.13 | 0.38 | 0.36 | 0.30 to 0.43 | 1.90×10−31 |
| HLA_DRB1*07:01 | 0.35 | 0.15 | 3.26 | 2.68 to 3.97 | 5.62×10−31 |
| HLA_DQB1*02 | 0.40 | 0.21 | 2.43 | 2.04 to 2.91 | 9.77×10−22 |
| HLA_DQA1*01:03 | 0.02 | 0.09 | 0.24 | 0.15 to 0.38 | 1.79×10−14 |
| HLA_DRB1*13 | 0.04 | 0.11 | 0.31 | 0.22 to 0.44 | 2.41×10−14 |
| HLA_DRB1*13:01 | 0.02 | 0.08 | 0.23 | 0.15 to 0.37 | 3.18×10−14 |
| HLA_DQA1*01:01 | 0.08 | 0.15 | 0.46 | 0.35 to 0.59 | 1.53×10−10 |
| HLA_B*08:01 | 0.20 | 0.13 | 2.95 | 2.05 to 4.23 | 9.17×10−09 |
Minor allele frequencies (MAFs) for patients and controls and odds ratios (ORs) with 95% confidence intervals (95% CIs) for each of the HLA alleles achieving genome-wide significance are shown.
eQTL Analyses
The three leading SNPs at the CALHM6 locus (rs2637678, rs2637681, and rs2858829) exhibit strong cis-eQTL effects in the GTEx database.21 For all three SNPs, the highest normalized effect size (NES) is seen in EBV-transformed lymphocytes (rs2637678: NES=0.66; P=7.2×10−10; rs2637681: NES=0.67; P=1.9×10−9; and rs2858829: NES=0.66; P=3.2×10−9). A strong cis-eQTL effect is also noted in the eQTLgen database from blood (Z score=43.18; P=3.27×10−310), which also involves the neighboring genes DSE, RWDD1, and NT5DC1.22 In both databases, the minor allele is associated with increased expression of CALHM6. Of note, no expression of CALHM6 is noted in whole kidney in the GTEx, NephQTL, and Human Kidney Atlas Expression (HKAE) databases.23,24 The HKAE database suggests specific expression in glomerulus, but this could not be confirmed in the other databases.
No significant eQTLs were found for rs10518133 (tagging the PARM1 locus) in any tissue in the GTEx database, but in eQTLgen, the risk allele is associated with significant downregulation of PARM1.
Discussion
This study has confirmed the association of SSNS with the HLA locus and identified two additional loci on chromosome 6 and chromosome 4 achieving genome-wide significance.
HLA Locus
By far, the most associations identified by GWAS of human diseases are located in the HLA region.25 The vast majority of these phenotypes are autoimmune or infectious diseases. The necessity for an appropriate and ever-evolving response to infection drives variation in HLA peptide binding grooves and some of these variants can lead to inappropriate responses against antigens of the host.26 Thus, unsurprisingly and consistent with previous studies, the strongest signal for association with SSNS that we identified was in the HLA region.10–12 The lead SNP, rs9273542 (Table 2), is located within the HLA-DR/DQ region, specifically HLA-DQB1.
Table 2.
Effect estimates for lead single-nucleotide polymorphism
| Locus | Gene | SNP | DR2 | Minor Allele | MAF Patients | MAF Controls | OR | 95% CI | P Value |
|---|---|---|---|---|---|---|---|---|---|
| 6p21.3 | HLA-DQB1 | rs9273542 | 0.89 | T | 0.51 | 0.24 | 3.39 | 2.86 to 4.03 | 1.59×10−43 |
| 6q22.1 | CALHM6/FAM26F | rs2637678 | 0.96 | C | 0.26 | 0.40 | 0.51 | 0.44 to 0.60 | 1.27×10−17 |
| 4q13.3 | PARM1 | rs10518133 | 0.93 | A | 0.12 | 0.06 | 1.96 | 1.57 to 2.45 | 2.50×10−8 |
Minor allele frequencies (MAFs) and odds ratios (ORs) with 95% confidence intervals (95% CIs) for each of the minor alleles of the lead single-nucleotide polymorphisms (SNPs) from the three loci achieving genome-wide significance. Dosage R2 (DR2) indicates the Beagle imputation quality score.
Imputation of HLA alleles identified that HLA-DQA1*02:01 and HLA-DRB1*07:01 are associated with the strongest risk, whereas HLA-DQA1*01 seems to be independently protective. HLA-DRB1*13 (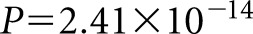 ) is also protective, which is similar to the results of the study by Jia et al.,12 which showed in an Asian population that HLA-DRB1*13:02 and HLA-DQB1*06:04 have protective effects. In the study by Debiec et al.,11 serial conditional analysis of the HLA association revealed an independently associated SNP located within BTNL2 (rs9348883), although this did not reach genome-wide significance. We were unable to confirm an independent association with this gene, which may reflect the different ethnicities analyzed, because the BTNL2 signal was primarily driven by the African cohort in that study.
) is also protective, which is similar to the results of the study by Jia et al.,12 which showed in an Asian population that HLA-DRB1*13:02 and HLA-DQB1*06:04 have protective effects. In the study by Debiec et al.,11 serial conditional analysis of the HLA association revealed an independently associated SNP located within BTNL2 (rs9348883), although this did not reach genome-wide significance. We were unable to confirm an independent association with this gene, which may reflect the different ethnicities analyzed, because the BTNL2 signal was primarily driven by the African cohort in that study.
Associations Outside the HLA Region
The adaptive immune response is triggered by HLA peptide-epitope binding, but it is modulated by regulatory mechanisms, elements of which are encoded in non-HLA genes.4 Arguably, it is the identification of these genes that can provide the most informative insights into the complex architecture of the dysregulated immune response in specific diseases. We here describe the discovery of two loci outside the HLA region achieving genome-wide significant associations with SSNS.
CALHM6 Association (6q22.1)
The strongest signal outside the HLA region is on chromosome 6q22.1, and the lead SNP, rs2637678 (Table 2), is located very near the gene CALHM6 (calcium homeostasis modulator family member 6), previously also annotated as FAM26F or INAM (IRF-3–dependent NK-activating molecule).27 Of note, this locus had been identified as a potential signal in SSNS by Debiec et al.,11 but it did not reach genome-wide significance. The 6q22.1 SNP reported by Debiec et al.11 is identical to one of the lead SNPs identified in this study (rs2858829), and it was associated with a P value of 6.8×10−8. Importantly, the existence of this published suggestive association at rs2858829 provides independent confirmatory evidence for our genome-wide significant finding.
The apparent absence of expression of CALHM6 in the kidney is in contrast to the high expression in lymphocytes and consistent with the notion that impaired immune regulation is a key risk factor for the development of SSNS. Indeed, CALHM6 is thought to have an important role in the regulation of the immune system by facilitating interactions and potential synapses between immune cells.28 It is differentially regulated in response to various immune stimuli, especially IFN-γ, with predicted IFN-stimulated response element and signal transducer and initiator of transcription binding sites in its promoter.29 Members of the CALHM family are thought to belong to the ATP release channel superfamily.30 CALHM6 is highly expressed in lymphocytes, and ATP is a recognized trigger for apoptosis, including of immune cells.31,32 A key effect of glucocorticoids on the immune system is the induction of lymphocyte apoptosis.33 Thus, CALHM6 may play a role in mediating this effect, and impaired CALHM6 function may exacerbate an exaggerated immune response, leading to SSNS, which can be suppressed by pharmacologic doses of glucocorticoids. Of note, the same locus with the same risk allele has been previously associated with another autoimmune disease highly sensitive to steroid treatment, ulcerative colitis, providing additional evidence for the importance of CALHM6 in immune response regulation.34
CALHM6 eQTL Analyses
We performed an eQTL analysis to further assess potential associations between our identified variants and gene expression.35 Of note, for the lead variants in CALHM6, the presence of the minor allele is protective against SSNS so that the major allele is the risk allele and enriched in patients. Interestingly, the lead variant (rs2637678) at the CALHM6 locus is a known eQTL, and the risk allele is associated with decreased CALHM6 expression. This is consistent with the hypothesis that the presence of the risk allele may impair lymphocyte apoptosis and thus, lead to less effective downregulation of an immune response.
However, it is important to note that GWAS identify variants that represent a haploblock rather than a specific gene associated with the disease. This is best illustrated by the initial controversy over the association of MYH9 versus APOL1 with nondiabetic kidney disease in black individuals.36 Thus, it is possible that another gene in the haploblock with CALHM6 may actually be causally associated. Indeed, in the eQTLGen database, rs2637678 also has highly significant eQTL effects on neighboring genes. CALHM6 was prioritized on the basis of its proximity to the GWAS signal and its known function, but additional studies will be required to establish with certainty if this is indeed the gene responsible for driving the observed association.
PARM1 Association (4q13.3)
In addition, we found a genome-wide significant association with a locus on chromosome 4q13.3 (lead SNP rs10518133; 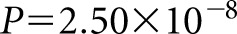 ; OR, 1.96), which is located within the gene PARM1 (prostate androgen-regulated mucin-like protein 1). Interestingly, this locus is near a locus reported by Debiec et al.11 as achieving suggestive evidence of association with SSNS, with their lead SNP located within BTC (Betacellulin), the next gene upstream of PARM1 on chromosome 4. The lead SNP at 4q13 reported by Debiec et al.11 and the PARM1 SNP identified in this study are approximately 250 kb apart, but they are separated by a strong recombination hotspot (>50 cM/Mb) (Supplemental Figure 3). This hot spot is at least equally strong in African populations (Supplemental Tables 2 and 3) so that the different ethnicities cannot explain the separation of this locus between the previous study and our study.
; OR, 1.96), which is located within the gene PARM1 (prostate androgen-regulated mucin-like protein 1). Interestingly, this locus is near a locus reported by Debiec et al.11 as achieving suggestive evidence of association with SSNS, with their lead SNP located within BTC (Betacellulin), the next gene upstream of PARM1 on chromosome 4. The lead SNP at 4q13 reported by Debiec et al.11 and the PARM1 SNP identified in this study are approximately 250 kb apart, but they are separated by a strong recombination hotspot (>50 cM/Mb) (Supplemental Figure 3). This hot spot is at least equally strong in African populations (Supplemental Tables 2 and 3) so that the different ethnicities cannot explain the separation of this locus between the previous study and our study.
Limitations
Our study has several limitations. First, only limited clinical information on our patients is available. However, the majority of patients were obtained through two clinical trials (the PREDNOS and PREDNOS2 trials), which recruited from >100 pediatric units across the United Kingdom, and it is thus highly unlikely that our patient cohort is substantially different from other SSNS cohorts. Indeed, the available data, such as the 2:1 male:female ratio, match perfectly with published data.13
Second, we used publicly available genotype data for controls, and therefore, the data from different genotyping platforms needed to be combined. As detailed in Supplemental Figure 1, this led to a limited set of overlapping genotyped SNPs, and the majority of SNPs used in our final analysis were imputed. However, imputation has become an accepted tool in GWAS, and the fact that the analysis with genotyped markers only (Supplemental Figures 5–9) identifies the same loci, albeit with higher P values, provides strong evidence that these loci are genuinely associated with the phenotype and have not been identified due to imputation artifacts.37
The most important limitation, however, is the lack of a replication cohort. The independent identification of loci at both 6q22.1 and 4q13.3 by Debiec et al.11 provides strong confirmatory evidence for our results. Yet, as detailed above, the lead SNP at 4q13.3 identified in that study is separated from our lead SNP by a recombination hotspot. It, therefore, remains to be determined whether this region is truly associated with SSNS or not and if so, if these loci are independent from each other. Moreover, Debiec et al.11 did not provide detailed information on the risk allele at 6q22.1, and we are thus unable to assess whether the allelic effect is identical in both studies. Additional independent replication is thus needed to confirm the discovery of these loci. Our study identifies two loci outside the HLA region with genome-wide significant association with SSNS and thus, provides important insight into the pathogenesis of SSNS. Because CALHM6 is implicated in regulating the immune response to infection, this may provide an explanation for the typical triggering of disease onset by infections. Additional studies are needed to provide independent replication of our findings and investigate the precise mechanisms and whether these could be amenable to specific treatments.
Disclosures
Dr. N. J. Webb reports employment from Novartis outside the submitted work. Dr. Wetzels reports grants from Sanofi, grants from Pfizer, grants from Amgen, grants from Achillion, other from Shire, and other from Vifor outside the submitted work. Dr. Gale reports personal fees from Alexion Ltd. and personal fees from Otsuka Ltd. outside the submitted work. All of the remaining authors have nothing to disclose.
Funding
Dr. Dufek, Dr. Kleta, and Dr. Bockenhauer are grateful to Dr. Magdi Yaqoob for support through the William Harvey Paediatric Fellowship. Dr. Trompeter, Dr. Kleta, and Dr. Bockenhauer were supported by the Mitchell Charitable Trust, Kids Kidney Research, Kidney Research UK, the Garfield Weston Foundation, and the Grocers’ Charity. Dr. Teeninga and Dr. Nauta were supported by the Dutch Kidney Foundation. Dr. Kari, Dr. Kleta, and Dr. Bockenhauer were supported by the Deanship of Scientific Research, King Abdulaziz University, Jeddah grant 432/003/d for pediatric nephrology research. Dr. Gbadegesin is supported by National Institutes of Health/National Institutes of Diabetes and Digestive and Kidney Diseases grants 5R01DK098135 and 5R01DK094987 and a Doris Duke Charitable Foundation Clinical Scientist Development Award. Dr. Gale, Dr. Stanescu, Dr. Kleta, and Dr. Bockenhauer were supported by St. Peter’s Trust for Kidney, Bladder & Prostate Research. Dr. Gale acknowledges support from the UK Medical Research Council, Kidney Research UK, and the Rosetrees Trust. Dr. Kleta was supported by the David and Elaine Potter Foundation. Dr. Bockenhauer is supported by the NIHR Biomedical Research Centre at GOSH/ICH.
Supplementary Material
Acknowledgments
We are grateful to the team of the Prednisolone in Nephrotic Syndrome (PREDNOS) and PREDNOS2 trials (trial partners are listed in Supplemental Material) and the members of the Mid West Pediatric Nephrology Consortium for their study support. We also thank Gaganjit Madhan Kaur from University College London Genomics for providing expert genotyping support as well as Drs. Fairfax, Makino, and Knight from the University of Oxford for sharing ethnically matched control data.
Footnotes
Published online ahead of print. Publication date available at www.jasn.org.
Supplemental Material
This article contains the following supplemental material online at http://jasn.asnjournals.org/lookup/suppl/doi:10.1681/ASN.2018101054/-/DCSupplemental.
Supplemental Material. Supplementary results, materials and methods, acknowledgments, and summary statistics.
Supplemental Figure 1. Flowchart of quality control steps and GWAS.
Supplemental Figure 2. Results of classical HLA type association analysis.
Supplemental Figure 3. Locus Zoom plot of the association on chromosome 4.
Supplemental Figure 4. Principal component analysis of patients and controls.
Supplemental Figure 5. Manhattan plot with genotyped markers only.
Supplemental Figure 6. Locus zoom plot with genotyped markers only for the HLA locus.
Supplemental Figure 7. Locus zoom plot with genotyped markers only for the CALHM6 locus.
Supplemental Figure 8. Locus zoom plot with genotyped markers only for the chromosome 4q13.3 locus.
Supplemental Figure 9. Linkage disequilibrium of SNP in the HLA region associated with SSNS.
Supplementary Table 1. Effect estimates for the three lead genotyped SNPs at the associated loci.
Supplemental Table 2. Linkage equilibrium at the chromosome 4q13.3 locus.
Supplemental Table 3. List of SNPs with a suggestive association.
References
- 1.Banh TH, Hussain-Shamsy N, Patel V, Vasilevska-Ristovska J, Borges K, Sibbald C, et al.: Ethnic differences in incidence and outcomes of childhood nephrotic syndrome. Clin J Am Soc Nephrol 11: 1760–1768, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Vivarelli M, Massella L, Ruggiero B, Emma F: Minimal change disease. Clin J Am Soc Nephrol 12: 332–345, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Shalhoub RJ: Pathogenesis of lipoid nephrosis: A disorder of T-cell function. Lancet 2: 556–560, 1974 [DOI] [PubMed] [Google Scholar]
- 4.Hu X, Daly M: What have we learned from six years of GWAS in autoimmune diseases, and what is next? Curr Opin Immunol 24: 571–575, 2012 [DOI] [PubMed] [Google Scholar]
- 5.Sekula P, Li Y, Stanescu HC, Wuttke M, Ekici AB, Bockenhauer D, et al.: GCKD Investigators : Genetic risk variants for membranous nephropathy: Extension of and association with other chronic kidney disease aetiologies. Nephrol Dial Transplant 32: 325–332, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Stanescu HC, Arcos-Burgos M, Medlar A, Bockenhauer D, Kottgen A, Dragomirescu L, et al.: Risk HLA-DQA1 and PLA(2)R1 alleles in idiopathic membranous nephropathy. N Engl J Med 364: 616–626, 2011 [DOI] [PubMed] [Google Scholar]
- 7.Wuttke M, Köttgen A: Insights into kidney diseases from genome-wide association studies. Nat Rev Nephrol 12: 549–562, 2016 [DOI] [PubMed] [Google Scholar]
- 8.Kiryluk K, Li Y, Scolari F, Sanna-Cherchi S, Choi M, Verbitsky M, et al.: Discovery of new risk loci for IgA nephropathy implicates genes involved in immunity against intestinal pathogens. Nat Genet 46: 1187–1196, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gale DP, Molyneux K, Wimbury D, Higgins P, Levine AP, Caplin B, et al.: Galactosylation of IgA1 is associated with common variation in C1GALT1. J Am Soc Nephrol 28: 2158–2166, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gbadegesin RA, Adeyemo A, Webb NJ, Greenbaum LA, Abeyagunawardena A, Thalgahagoda S, et al.: Mid-West Pediatric Nephrology Consortium : HLA-DQA1 and PLCG2 are candidate risk loci for childhood-onset steroid-sensitive nephrotic syndrome. J Am Soc Nephrol 26: 1701–1710, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Debiec H, Dossier C, Letouzé E, Gillies CE, Vivarelli M, Putler RK, et al.: Transethnic, genome-wide analysis reveals immune-related risk alleles and phenotypic correlates in pediatric steroid-sensitive nephrotic syndrome. J Am Soc Nephrol 29: 2000–2013, 2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jia X, Horinouchi T, Hitomi Y, Shono A, Khor SS, Omae Y, et al.: Research Consortium on Genetics of Childhood Idiopathic Nephrotic Syndrome in Japan : Strong association of the HLA-DR/DQ locus with childhood steroid-sensitive nephrotic syndrome in the Japanese population. J Am Soc Nephrol 29: 2189–2199, 2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Noone DG, Iijima K, Parekh R: Idiopathic nephrotic syndrome in children. Lancet 392: 61–74, 2018 [DOI] [PubMed] [Google Scholar]
- 14.Webb NJ, Frew E, Brettell EA, Milford DV, Bockenhauer D, Saleem MA, et al.: PREDNOS 2 study group : Short course daily prednisolone therapy during an upper respiratory tract infection in children with relapsing steroid-sensitive nephrotic syndrome (PREDNOS 2): Protocol for a randomised controlled trial. Trials 15: 147, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fairfax BP, Makino S, Radhakrishnan J, Plant K, Leslie S, Dilthey A, et al.: Genetics of gene expression in primary immune cells identifies cell type-specific master regulators and roles of HLA alleles. Nat Genet 44: 502–510, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fairfax BP, Humburg P, Makino S, Naranbhai V, Wong D, Lau E, et al.: Innate immune activity conditions the effect of regulatory variants upon monocyte gene expression. Science 343: 1246949, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wellcome Trust Case Control Consortium : Genome-wide association study of 14,000 cases of seven common diseases and 3,000 shared controls. Nature 447: 661–678, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Browning BL, Zhou Y, Browning SR: A one-penny imputed genome from next-generation reference panels. Am J Hum Genet 103: 338–348, 2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chang CC, Chow CC, Tellier LC, Vattikuti S, Purcell SM, Lee JJ: Second-generation PLINK: Rising to the challenge of larger and richer datasets. Gigascience 4: 7, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jia X, Han B, Onengut-Gumuscu S, Chen WM, Concannon PJ, Rich SS, et al.: Imputing amino acid polymorphisms in human leukocyte antigens. PLoS One 8: e64683, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.GTEx Consortium : The Genotype-Tissue Expression (GTEx) project. Nat Genet 45: 580–585, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Võsa U, Claringbould A, Westra H-J, Bonder MJ, Deelen P, Zeng B, et al. : Unraveling the polygenic architecture of complex traits using blood eQTL meta-analysis [published online ahead of print October 19, 2018]. bioRxiv 10.1101/447367 [Google Scholar]
- 23.Qiu C, Huang S, Park J, Park Y, Ko YA, Seasock MJ, et al.: Renal compartment-specific genetic variation analyses identify new pathways in chronic kidney disease. Nat Med 24: 1721–1731, 2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gillies CE, Putler R, Menon R, Otto E, Yasutake K, Nair V, et al.: Nephrotic Syndrome Study Network (NEPTUNE) : An eQTL landscape of kidney tissue in human nephrotic syndrome. Am J Hum Genet 103: 232–244, 2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lenz TL, Spirin V, Jordan DM, Sunyaev SR: Excess of deleterious mutations around HLA genes reveals evolutionary cost of balancing selection. Mol Biol Evol 33: 2555–2564, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Trowsdale J: The MHC, disease and selection. Immunol Lett 137: 1–8, 2011 [DOI] [PubMed] [Google Scholar]
- 27.Malik U, Javed A, Ali A, Asghar K: Structural and functional annotation of human FAM26F: A multifaceted protein having a critical role in the immune system. Gene 597: 66–75, 2017 [DOI] [PubMed] [Google Scholar]
- 28.Malik U, Javed A: FAM26F: An enigmatic protein having a complex role in the immune system. Int Rev Immunol 19: 1–11, 2016 [DOI] [PubMed] [Google Scholar]
- 29.Chmielewski S, Olejnik A, Sikorski K, Pelisek J, Błaszczyk K, Aoqui C, et al.: STAT1-dependent signal integration between IFNγ and TLR4 in vascular cells reflect pro-atherogenic responses in human atherosclerosis. PLoS One 9: e113318, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ma Z, Tanis JE, Taruno A, Foskett JK: Calcium homeostasis modulator (CALHM) ion channels. Pflugers Arch 468: 395–403, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Di Virgilio F, Pizzo P, Zanovello P, Bronte V, Collavo D: Extracellular ATP as a possible mediator of cell-mediated cytotoxicity. Immunol Today 11: 274–277, 1990 [DOI] [PubMed] [Google Scholar]
- 32.Nagy PV, Fehér T, Morga S, Matkó J: Apoptosis of murine thymocytes induced by extracellular ATP is dose- and cytosolic pH-dependent. Immunol Lett 72: 23–30, 2000 [DOI] [PubMed] [Google Scholar]
- 33.Banuelos J, Lu NZ: A gradient of glucocorticoid sensitivity among helper T cell cytokines. Cytokine Growth Factor Rev 31: 27–35, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Julià A, Domènech E, Chaparro M, García-Sánchez V, Gomollón F, Panés J, et al.: A genome-wide association study identifies a novel locus at 6q22.1 associated with ulcerative colitis. Hum Mol Genet 23: 6927–6934, 2014 [DOI] [PubMed] [Google Scholar]
- 35.Battle A, Montgomery SB: Determining causality and consequence of expression quantitative trait loci. Hum Genet 133: 727–735, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bockenhauer D, Medlar AJ, Ashton E, Kleta R, Lench N: Genetic testing in renal disease. Pediatr Nephrol 27: 873–883, 2012 [DOI] [PubMed] [Google Scholar]
- 37.Marchini J, Howie B: Genotype imputation for genome-wide association studies. Nat Rev Genet 11: 499–511, 2010 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.