

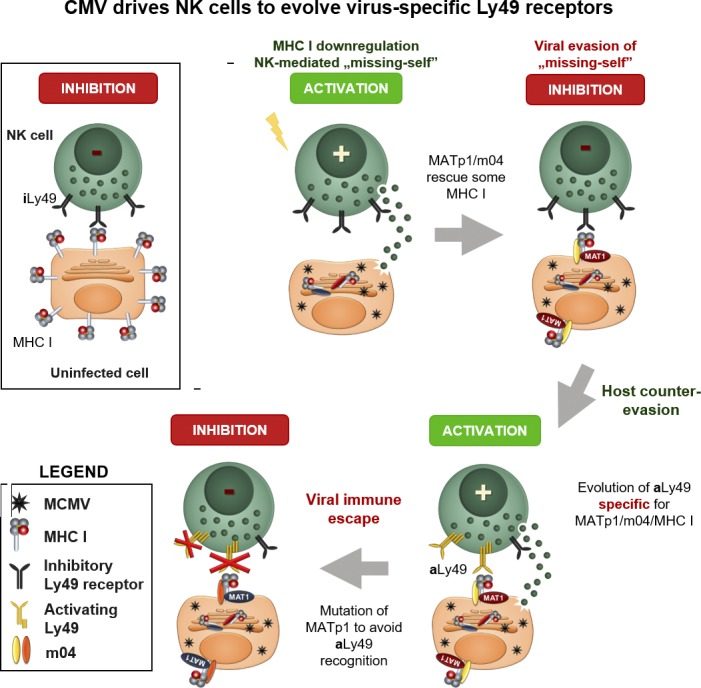
Železnjak et al. demonstrate that two MCMV-encoded proteins interact with MHC I molecules, forming an altered-self complex that prevents missing self recognition by increasing specificity for inhibitory Ly49 receptors. This led to the evolution of CMV-specific activating Ly49s.
Abstract
CMVs efficiently target MHC I molecules to avoid recognition by cytotoxic T cells. However, the lack of MHC I on the cell surface renders the infected cell susceptible to NK cell killing upon missing self recognition. To counter this, mouse CMV (MCMV) rescues some MHC I molecules to engage inhibitory Ly49 receptors. Here we identify a new viral protein, MATp1, that is essential for MHC I surface rescue. Rescued altered-self MHC I molecules show increased affinity to inhibitory Ly49 receptors, resulting in inhibition of NK cells despite substantially reduced MHC I surface levels. This enables the virus to evade recognition by licensed NK cells. During evolution, this novel viral immune evasion mechanism could have prompted the development of activating NK cell receptors that are specific for MATp1-modified altered-self MHC I molecules. Our study solves a long-standing conundrum of how MCMV avoids recognition by NK cells, unravels a fundamental new viral immune evasion mechanism, and demonstrates how this forced the evolution of virus-specific activating MHC I–restricted Ly49 receptors.
Graphical Abstract
Introduction
CMVs use a plethora of mechanisms to efficiently evade immune control. With a prevalence of >90% in many mammalian species, they have been an important driving force in the evolution of their hosts’ immune systems. This is best demonstrated in the mouse cytomegalovirus (MCMV) animal model (Brune, 2013; Lisnić et al., 2015), where viral immune evasion strategies to suppress natural killer (NK) cell activation by engaging inhibitory NK cell receptors have driven the evolution of activating NK cell receptors (Arase and Lanier, 2002; Carrillo-Bustamante et al., 2013; Rahim and Makrigiannis, 2015). The latest example of this is MCMV-encoded m12, which can be recognized by both inhibitory NKR-P1B and activating NKR-P1C (NK1.1) receptors (Aguilar et al., 2015, 2017; Rahim et al., 2016). Similarly, Smith MCMV–encoded m157 can be directly recognized either by inhibitory Ly49I129/J, leading to poor NK cell–mediated control, or by activating Ly49HC57BL/6 receptor, resulting in strong NK cell activation and successful virus control (Arase et al., 2002; Corbett et al., 2011; Pyzik et al., 2014). MCMV is thus the prototype of a virus that prompted the evolution of its own dedicated activating NK cell receptors.
MHC I molecules display peptide fragments of proteins from within the cell to CTLs. To evade recognition by CTLs, many viruses interfere with antigen presentation and remove MHC I from the cell surface. During evolution, NK cells evolved inhibitory receptors (Ly49 receptors in mice and Killer-cell immunoglobulin-like receptor [KIR] receptors in humans; Carlyle et al., 2008) that recognize and monitor surface MHC I levels. Cells unable to display MHC I trigger NK cell activation due to a lack of inhibitory signals, a process termed “missing self recognition” (Kärre et al., 1986). NK cells thereby restrict the ability of viruses to target MHC I for CTL evasion. In addition, inhibitory NK cell receptors play an important role in NK cell education and licensing (Fernandez et al., 2005; Kim et al., 2005; Brodin et al., 2009; Chalifour et al., 2009).
MCMV evades CTL recognition by down-modulation of surface MHC I expression via two viral proteins, m06 and m152 (Ziegler et al., 1997; Hengel et al., 1999; Reusch et al., 1999). We have previously shown that this triggers NK cell activation via missing self recognition (Babić et al., 2010). However, MCMV utilizes a third viral protein (m04) to bypass MHC I targeting via m06 and m152. m04 binds a small portion of properly folded, β2-microglobulin (β2m)-associated MHC I molecules in the ER and escorts them to the cell surface, where they engage inhibitory Ly49 receptors and inhibit NK cell activation (Kleijnen et al., 1997; Babić et al., 2010). Interestingly, while m04 is highly abundant in the cell, only a minor fraction of MHC I is rescued and leaves the ER (Kleijnen et al., 1997). Moreover, while m04 can form a complex with MHC I after transfection into uninfected cells, MCMV infection is required for such complexes to be efficiently exported from the ER to the cell surface (Kavanagh et al., 2001a; Lu et al., 2006). This implies the existence of another MCMV-encoded factor necessary for the efficient transport of m04/MHC I complex to the cell surface.
In various mouse strains, a number of activating Ly49 receptors (Ly49PMA/My, Ly49LBALB, Ly49P1NOD/Ltj, and Ly49D2PWK/Pas) have evolved to specifically recognize virus-altered MHC I molecules. We have previously demonstrated that m04 is necessary but insufficient for recognition by these virus-specific activating Ly49 receptors (Kielczewska et al., 2009; Pyzik et al., 2011). Here, we identify the missing viral factor (MATp1) required for this recognition as the product of a novel viral short open reading frame (ORF). We show that MATp1 is required for the efficient formation of m04/MHC I complexes and their escort to the cell surface. This facilitates the engagement of inhibitory Ly49A receptors with increased affinity, thereby efficiently preventing missing self recognition by NK cells despite substantially reduced MHC I surface levels. Furthermore, our data highlight how this novel immune evasion mechanism may have driven the evolution of activating Ly49 receptors capable of recognizing infected cells through MATp1/m04-modified altered-self MHC I molecules. Our study is thus the first to demonstrate several novel concepts. First, we show and explain the necessary components for MHC I–restricted altered-self recognition of CMV-infected cells by inhibitory and activating Ly49 receptors. Second, we provide a mechanism explaining the longstanding question why licensed NK cells are unable to control MCMV infection despite marked down-regulation of MHC I molecules in the infected cells. Finally, we show that this selective pressure from the virus could have caused the evolution of at least three activating Ly49 receptors specific exclusively for MCMV-modified MHC I molecules. Altogether, our studies demonstrate a fundamentally new paradigm in virus–host interactions where the virus exploits self molecules to evade the immune system rather than encoding self-like decoy ligands or peptides.
Results
MCMV-encoded protein MATp1 is essential in prevention of missing self–dependent NK cell recognition
Our previous studies demonstrated that inhibitory Ly49 receptors strongly recognize MCMV-infected cells (Babić et al., 2010). To investigate this further, we have employed Ly49 reporter cells (RCs) that express the ectodomain of a single Ly49 receptor and, upon ligand engagement, express GFP (Arase et al., 2002; Babić et al., 2010; Pyzik et al., 2011). Using Ly49A-RCs cocultured with WT MCMV–infected or mock-infected BALB/c primary murine embryonic fibroblasts (MEFs), we have confirmed our previous observation that WT MCMV–infected MEFs induce significantly stronger activation of Ly49A-RC compared with mock-infected cells (Fig. 1 A). This phenotype was present despite substantially lower levels of surface H-2Dd MHC I molecules on WT MCMV–infected cells, which serve as the ligand for the Ly49A receptor (Fig. 1 B). In accordance with our previous data, MCMV lacking the m04 gene (Δm04 MCMV) was unable to engage Ly49A due to strong down-regulation of MHC I from the surface of infected cells (Fig. 1 A; Babić et al., 2010). We therefore postulated that MCMV increases the engagement of MHC I with inhibitory Ly49 receptors to prevent missing self recognition and that an additional viral factor is required for this.
Figure 1.

MCMV-encoded protein MATp1 is essential for prevention of missing self recognition. (A) Primary BALB/c MEF cells were infected with 1.5 PFU/cell of indicated viruses or left uninfected. At 16 h p.i., Ly49A-RCs were added and cocultured for 24 h. Following cocultivation, RCs were washed from MEFs, and GFP expression was analyzed on flow cytometer. (B) BALB/c MEFs were either infected with 1.5 PFU/cell of WT MCMV or left uninfected, collected at indicated times p.i., and stained for surface H-2Dd using 34-5-8S antibody. Irrelevant isotype-matched antibody was used as a negative control. (C) Scheme depicting the structure of the MAT locus. White boxes depict ORFs from Rawlinson’s annotation. As previously described (Marcinowski et al., 2012; Juranic Lisnic et al., 2013), a single, 1.7-kb-long and spliced transcript is generated from this locus and encodes one protein that is mostly colinear with m169 (MATp2) ORF and a binding site for cellular miR-27. In addition, we detected an upstream ORF encoding a small, 11-kD protein (MATp1). (D and E) Expression of MATp1 and MATp2 in MCMV mutants harboring various deletions within the MAT locus. As deletion of MATp1 from the MCMV genome results in significantly stronger translation of MATp2 protein, we generated a recombinant virus scMATp1 MCMV in which MATp1 ORF was replaced with an ORF of the same length, codon-usage, propensity for forming secondary structures but different amino acid sequence: in short, an ORF with scrambled codons (scMATp1 MCMV). scMATp1 MCMV generated amounts of MATp2 protein comparable with WT MCMV. Western blot analysis was performed three times on lysates of BALB/c MEFs infected with 1.5 PFU/cell at 48 h p.i. and reacted with indicated antibodies. (F–H) Primary BALB/c MEFs were infected and cocultured with Ly49A-RCs as described in A. (G and H) Level of Ly49A-RC GFP activation expressed as either percentage or median fluorescence intensity (MFI) of GFP+ cells from live population. Reporter cell assays as well as MHC I analyses were performed three times or more, each time using different virus stocks and Ly49A-RC and BALB/c MEF stocks. One-way ANOVA with post hoc test was used for statistical analysis. Graphs show mean with SEM as error bars. ****, P ≤ 0.0001; ***, P ≤ 0.001; **, P ≤ 0.01; *, P ≤ 0.05.
To that aim, we analyzed Ly49A-RC binding to MEF cells infected with several MCMV-deletion mutants lacking genes or regions of unknown function. Among various deletion mutants tested, only a virus lacking the m169–m170 region, known to encode large, highly abundant transcript (MAT; ΔMAT MCMV; Juranic Lisnic et al., 2013), resulted in the loss of stronger activation of Ly49A-RCs observed in WT-infected cells (Fig. 1 A). In fact, with the virus lacking MAT, Ly49A-RC activation was comparable to that of uninfected cells that express MHC I molecules alone. Thus, in addition to m04, efficient engagement of inhibitory Ly49A receptor by MCMV-infected cells requires a factor encoded by the MAT transcript. MAT is a 1.7-kb-long transcript that encodes at least one protein (Juranic Lisnic et al., 2013) and contains a binding site for two cellular microRNAs, miR-27a and miR-27b (Fig. 1 C; Marcinowski et al., 2012). Interestingly, ribosomal profiling (Ribo-seq) from MCMV-infected NIH-3T3 fibroblasts at 48 h after infection (p.i.) demonstrated translation of a novel small upstream ORF (MATp1 for MAT protein 1) at much higher levels than the previously identified m169 ORF (here called MATp2 for MAT protein 2) downstream (Fig. S1 A). We thus generated MCMV mutants lacking either MATp1 or MATp2 proteins (ΔMATp1 MCMV and ΔMATp2 MCMV, respectively). Using these viruses and a virus with mutated binding site for miR-27 (miR-27 mut; Marcinowski et al., 2012), plus our antibodies against MATp1 and MATp2, we demonstrate that MAT indeed encodes two proteins; MATp1 of ∼11 kD (Fig. 1 D), and MATp2 (previously known as m169) of ∼17 kD (Fig. 1 E). Translation of MATp1 strongly interfered with translation of the downstream MATp2, which was relieved upon deletion of the MATp1 ORF (Fig. 1 E). Since ΔMATp1 MCMV–infected cells had more MATp2 protein, which could interfere with interpretation of results, we generated a scrambled (sc) virus (scMATp1 MCMV). In scMATp1 MCMV, we replaced the MATp1 ORF sequence with a different sequence of the same length that contains the same ATGC content; has the same codon usage, codon adaptation index, and stability of secondary RNA structures; and displays the same in vitro growth curve as WT MCMV (Figs. S1 and S2, A–F). Therefore, scMATp1 MCMV lacks the MATp1 protein but retains the same levels of MATp2 (Fig. 1 E).
We then analyzed the ability of MEFs infected with these mutant viruses to activate Ly49A-RCs. Absence of MATp1 in ΔMATp1 and scMATp1 MCMV abrogated the enhanced activation of Ly49A-RCs, whereas cells infected with viruses lacking either MATp2 ORF or with the mutated miR-27 binding site behaved like WT MCMV–infected MEFs (Fig. 1, F–H). We conclude that MAT-encoded MATp1 protein is responsible for the observed stronger activation of Ly49A-RCs in WT-infected cells compared with mock-infected cells. Moreover, this stronger activation resulted in both increased frequency of GFP-expressing Ly49A-RCs (Fig. 1 G) and greater median fluorescence intensity (Fig. 1 H). This implies not only that a greater proportion of Ly49A-RCs is activated, but also that these cells are activated to a greater extent. Altogether, these results introduce MATp1 as an essential new player in Ly49A–MHC I interaction during infection, which contributes to the enhanced engagement of this inhibitory receptor.
MATp1 is required for surface expression and maturation of MHC I molecules
To understand the molecular mechanism of how MATp1 affects recognition by Ly49A receptors, we analyzed the impact of m04 and MATp1 on different MHC I alleles. Both viral proteins were required to increase surface levels of H-2Dd, the only ligand for the Ly49A receptor in BALB/c mice. In contrast, no such increase in surface expression was observed for H-2Kd and H-2Ld (Fig. 2 A). Likewise, restoration of H-2Kb expression, which acts as a crucial self-ligand for the inhibitory Ly49C receptor in C57BL/6 mice, was also dependent on the presence of MATp1 and m04 (Fig. 2 B). To explain the mechanism by which MATp1 rescues H-2Dd on MCMV-infected cells, we radioactively labeled cells, mock treated or infected with WT, Δm04, and ΔMAT MCMV, and performed immunoprecipitation with mAbs against peptide-loaded H-2Dd MHC I molecules (34-5-8S). Immunoprecipitated MHC I molecules were then treated with endoglycosidase H (EndoH) or left untreated. Absence of m04 or MAT resulted in the loss of the mature, EndoH-resistant MHC I form (Fig. 2 C). To confirm this finding, we performed a pulse-chase experiment and followed the glycosylation pattern of H-2Dd molecules for 90 min. In cells lacking MAT, we observed the complete absence of fully matured H-2Dd. Instead, accumulation of only partly EndoH-resistant molecules, indicative of retention of H-2Dd in a premedial-Golgi compartment by m152 (Janßen et al., 2016), was observed (Fig. 2 D). We conclude that MATp1 rescues the maturation of H-2Dd molecules.
Figure 2.

MATp1 is required for proper surface expression and maturation of H-2Dd MHC I molecules. (A) Surface expression of classic MHC I molecules in BALB/c (H-2d) MEF cells infected with 1.5 PFU/cell at 24 h p.i. with indicated viruses or during mock infection. Isotype-matched control or staining with secondary antibody only was used as a negative control. Representative histograms are shown as well as bar charts showing all performed experiments. In bar charts, the results are shown as MFI as follows: MFIsample – MFInegative control normalized (norm.) to MFIWT – MFIWT negative control. Data are representative of at least three independent experiments. Unpaired two-tailed Student’s test was used for statistical analysis. Graphs show mean with SEM as error bars. ****, P ≤ 0.0001; ***, P ≤ 0.001; **, P ≤ 0.01; *, P ≤ 0.05. (B) Surface expression of classic MHC I molecules in C57BL/6 (H-2b) MEFs 24 h p.i. with indicated viruses or left uninfected. Infection, MHC I staining, and statistics were performed as described in A. (C) Immunoprecipitation of H-2Dd molecules from BALB/c MEFs infected with WT MCMV. Cells were infected for 24 h with 1.5 PFU/cell of indicated viruses. Mock-infected cells were treated with rIFN-γ (0.1 µg/ml) for 24 h to induce MHC I expression or left untreated. H-2Dd molecules were immunoprecipitated with 34-5-8S antibody. Some samples were treated with EndoH, as indicated. The positions of bands corresponding to EndoH-resistant and -sensitive molecules are indicated on the right. Red asterisks (*) mark EndoH-resistant bands present in WT MCMV but absent in viruses lacking m04 or MATp1. (D) Pulse-chase analysis of H-2Dd molecules. BALB/c MEF cells were mock treated or rIFN-γ treated (0.1 µg/ml) or infected with 1.5 PFU/cell of WT or ΔMAT MCMV for 24 h. Cells were pulse labeled for 20 min, chased for the indicated times, and lysed. H-2Dd molecules were immunoprecipitated with 34-5-8S antibody, digested with EndoH as indicated, and separated on SDS-PAGE. The positions of bands corresponding to EndoH-resistant and -sensitive molecules are indicated on the right. Red asterisk (*) marks EndoH-resistant bands present in WT MCMV but absent in viruses lacking m04 or MATp1. Data shown in C and D are representative of at least two independent experiments.
MATp1 is necessary for m04/MHC I complex to reach the cell surface
After showing the impact of MATp1 on the surface levels and maturation of H-2Dd molecules, we next analyzed whether MATp1 also affects the function of m04, known for its role as an MHC I rescue protein. Previous data demonstrated that m04 can bind to MHC I and form a complex in uninfected cells. However, viral infection is needed for such a complex to be efficiently exported to the cell surface, i.e., another viral component is required (Kavanagh et al., 2001b; Lu et al., 2006). For this reason, we generated a new m04-specific antibody that recognizes the extracellular domain of m04 in native conditions such as flow cytometry and immunoprecipitation (Fig. S3 A) and followed the kinetics of surface H-2Dd and m04 levels in MCMV-infected primary BALB/c MEFs (Fig. 3 A). The kinetics of surface levels of m04 and H-2Dd coincided, with the biggest expression levels being visible at ∼24 h p.i. In the absence of MATp1, surface levels of both m04 and H-2Dd MHC I were reduced. In agreement with previous data, the absence of m04 also precluded surface expression of MHC I (Fig. 3 A; Wagner et al., 2002; Babić et al., 2010). Moreover, the majority of analyzed cells were double positive for MHC I and m04, especially ∼24 h p.i., when surface levels of both proteins reach their peak (Fig. 3 B). As expected, m04 was unable to reach the cell surface in MCMV-infected cells lacking stabile MHC I (TAP1 KO or β2m KO MEFs; Figs. 3 C and S3 B). In addition, surface levels of m04 were dramatically reduced in the absence of MATp1, even in BALB.K (H-2k) and BALB.B (H-2b) MEFs (Fig. S3, C and D). Finally, expression of other MHC I regulators m152 and m06 was not changed in scMATp1 MCMV–infected cells compared with WT MCMV–infected cells (Fig. S2, G and H). Taken together, these data demonstrate that MATp1 is required for m04/MHC I complex egress to the cell surface. Since the m04 protein is abundant in MCMV-infected cells whereas MATp1 is poorly expressed, this suggests that the amount of MHC I molecules that will reach the cell surface is exclusively dictated by the level of MATp1.
Figure 3.

MATp1 is required for m04/MHC I complex to reach the cell surface and engage the inhibitory Ly49A receptor. (A) Total surface levels of H-2Dd and m04 at various times p.i. BALB/c MEFs were infected with 1.5 PFU of WT, Δm04, and scMATp1 MCMV or left uninfected, followed by surface staining with α-H-2Dd (34-5-8S) and α-m04 (m04.17) antibodies and their respective isotype controls. Shown is a representative experiment (of four), and MHC I expression is presented as MFI as follows: MFIsample – MFInegative control. Gray areas denote times p.i. with the highest expression of m04 and MHC I. ctrl, control (isotype). (B) Representative dot plots of results at 24 h p.i. depicted in A (left) and frequency of cells coexpressing MHC I and m04 at indicated time points (right). (C) Surface level of m04 in TAP1 or β2m KO MEF. MHC I–deficient MEFs isolated from β2m KO and TAP1 KO BALB/c mice were infected with 1.5 PFU/cell of indicated viruses for 24 h p.i. and stained for surface m04. Experiments were repeated three times. Representative histograms are shown. (D) Ly49A receptor specifically recognizes both normal and altered-self H-2Dd. Primary BALB/c MEFs were infected with indicated viruses and then incubated with 10 µg of indicated antibody for 1 h at 4°C before the addition of RCs. Ly49A-RCs were cocultivated with targets for 24 h, as described previously. Addition of α-H-2Dd antibody (34-5-8S) completely abrogated the recognition of either infected or mock-infected cells by Ly49A-RCs, demonstrating that they indeed recognize H-2Dd. Incubation with α-m04.16 antibody partially blocked Ly49A-RC activation, reducing it to the level of activation seen when Ly49-RCs are incubated with mock-infected MEFs. α-MATp1 antibody was unable to block interaction between WT MCMV–infected cells and Ly49A-RCs. Perpendicular dashed lines denote MFI of Ly49A-RCs after incubation with WT MCMV–infected MEFs. Representative histograms (of three independent experiments) are shown. norm., normalized.
We next set out to demonstrate that Ly49A-RCs activate in an MHC I–dependent manner and that m04 and MATp1 altered-self MHC I molecules are responsible for stronger recognition by Ly49A-RCs. To that aim, we performed the reporter assay as in Fig. 1. 1 h before the addition of Ly49A-RCs, we added 10 µg of α-H-2Dd (clone 34-5-8S), α-m04.16, α-MATp1, or irrelevant antibody to target cells. Antibodies were incubated with cells on ice for 1 h, and then RCs were added and left for an additional 24 h at 37°C in a CO2 incubator. We observed no activation of Ly49A-RCs when they were incubated with target cells and α-H-2Dd antibody, indicating that Ly49A-RCs activate in a H-2Dd-dependent manner (Fig. 3 D). When WT MCMV–infected target cells were incubated with α-m04 antibody, the level of Ly49A-RC activation was diminished to the level of activation seen in mock-infected cells. Treatment of cells with α-MATp1 antibody had no effect, which could indicate either that MATp1 is inaccessible to the antibody while on the cell surface, that the antibody is not blocking, or that MATp1 does not reach the cell surface.
MATp1 and m04 bind to MHC I cooperatively
To verify the interactions between MATp1, m04, and MHC I, we performed a series of coimmunoprecipitation studies. We immunoprecipitated MHC I using epoxy Dynabeads coupled with 34-5-8S antibody that recognizes the α1–α2 domain of MHC I (Fig. S3 E). The success of 34-5-8S coupling to Dynabeads was confirmed by staining with α-mouse Ig antibody (Fig. S3 F). Beads were incubated with cell lysates of WT, Δm04, or scMATp1 MCMV–infected cells, and precipitated MHC I was detected by staining with antibody that recognizes the α3 domain of MHC I (34-2-12). Equal amounts of MHC I were coprecipitated from lysates of WT, Δm04, or scMATp1 MCMV–infected cells (Fig. 4 A). Despite this, m04 could not be coprecipitated with MHC I in the absence of MATp1 in either FACS or Western blot analysis (Fig. 4, B and C). We conclude that MATp1 is required for m04/MHC I complex formation in MCMV-infected cells, thereby enabling its surface expression required for ligation of inhibitory Ly49 receptors.
Figure 4.

MATp1 and m04 bind to MHC I molecules cooperatively. (A–C) Coimmunoprecipitation of m04 with MHC I in the presence or absence of MATp1. (A) Epoxy Dynabeads were coupled with 34-5-8S antibody (recognizes α1–α2 domain of H-2Dd molecules) or irrelevant antibody of the same isotype (irrelevant IgG2a DyB). The beads were then incubated for 1 h at 37°C with lysates of BALB/c MEFs infected with WT, Δm04, or scMATp1 MCMV (indicated in the top right corner of histograms). Following incubation, the beads were washed, and an aliquot was stained with 34.2.12-PE antibody (recognizes α3 domain of H-2Dd molecules, indicated on the x axis; A) or with m04.17-biotin antibody (B) followed by streptavidin-PE and detected in flow cytometry. The scheme of the experiment is depicted in Fig. S3 E. (C) Following coimmunoprecipitation described in A, an aliquot of Dynabeads incubated with WT, Δm04, or scMATp1 MCMV–infected cell lysates was washed according to the manufacturer’s protocol, and coprecipitated molecules were eluted and analyzed on 12% SDS-PAGE. m04 was successfully coimmunoprecipitated with H-2Dd only in WT MCMV–infected lysates. m04 was not immunoprecipitated with isotype control antibody coupled (IC IP) or uncoupled (EB IP) beads, indicating that m04 bands eluted from 34-5-8S–coupled beads are coprecipitated m04 and not a consequence of unspecific binding of free m04 from the lysate to the beads. (D and E) PLA (In Situ Duolink for flow and fluorescence; Sigma-Aldrich) demonstrating interactions between m04, MHC I, and MATp1. Primary BALB/c MEFs were infected with 1.5 PFU of indicated viruses and then seeded on glass slides in 12-well plates (D) or seeded on regular cell-culture Petri dishes (E). 24 h p.i., the cells were stained on glass slides (D) or detached using trypsin (E) and then stained with α-MHC I (34-5-8S), α-m04.16, α-MATp1, and secondary antibodies according to the manufacturer’s instructions and described in Materials and methods. In agreement with our coimmunoprecipitation and previous observations (Kleijnen et al., 1997; Lu et al., 2006), m04 and MHC I interacted strongly only in the presence of MATp1 (first row in D and also quantified by FACS in E). MATp1 was shown to interact with both MHC I and m04. There was less interaction between MATp1 and MHC I in virus lacking m04. We thus conclude that MATp1 and m04 interact with MHC I cooperatively. All experiments were performed at least two times. White horizontal bar located at the lower right corner of each image denotes 50 µm. norm., normalized.
Since we have not coprecipitated MATp1 with MHC I and m04, we next performed a proximity ligation assay (PLA; Duolink; Sigma-Aldrich) using 34-5-8S and our α-m04.16 and α-MATp1 antibodies (Fig. 4, D and E). In PLA, potential interacting partners are detected with antibodies labeled with complementary oligonucleotides. If the two potential interactors are close enough (∼40 nm), DNA is amplified in a rolling-circle manner, and this amplified DNA is detected via DNA-binding dyes. Thus, signal is generated only if two potential interactors are close enough (Fredriksson et al., 2002; Bagchi et al., 2015). As before, interaction between MHC I and m04 was apparent only in WT MCMV–infected MEFs. In the absence of m04, no signal was generated (Fig. 4, D and E), while in scMATp1 MCMV–infected MEFs, the signal was very poor. Furthermore, when PLA was performed using antibodies against MHC I and MATp1 or m04 and MATp1, we could also detect signals in WT MCMV–infected cells but none in Δm04 MCMV– or scMATp1 MCMV–infected cells. Finally, we also could not detect any interactions between MATp1 and MHC I in the absence of m04. We thus conclude that binding of m04 and MATp1 to MHC I is cooperative.
Virus lacking MATp1 is attenuated in vivo in an NK- and MHC I–dependent manner
Since MATp1 modulates the expression of H-2Dd, we asked whether MATp1 deletion would be relevant for virus control in vivo by NK cells. We infected BALB/c mice with either WT or MAT MCMV mutants and analyzed virus titer in spleen 3 d p.i. Additional groups of mice infected with the same set of viruses were depleted of NK cells 24 h before virus infection. Deletion of MATp1, but not MATp2, resulted in a >10-fold decrease in viral load compared with WT MCMV (Fig. 5 A). This phenotype was lost in mice depleted of NK cells. Furthermore, we analyzed the impact of MATp1 on mice lacking T and B lymphocytes (BALB/c SCID) and still found attenuation of scMATp1 MCMV virus compared with WT MCMV (Fig. 5 B), confirming the importance of NK cells for the control of scMATp1 MCMV. However, virus lacking MAT was not attenuated in TAP1-deficient mice (BALB/c TAP1−/−; Fig. 5 C), thereby confirming that MATp1 exerts its function by modulating MHC I. Moreover, the absence of MATp1 also resulted in the attenuation of scMATp1 MCMV in mice of H-2k haplotype (CBA/J and BALB.K), but not in congenic mice of H-2b haplotype BALB.B (Fig. 5, D–F). In BALB.B mice, the MHC I locus has been exchanged with that of C57BL/6, while the rest of the genome is from BALB/c. Therefore, these mice possess Ly49A, which cannot bind H-2Dd MHC I (Babić et al., 2010), which further emphasizes that the Ly49A–MHC I interaction is responsible for MATp1-mediated viral missing self evasion.
Figure 5.

Virus lacking MATp1 is attenuated in vivo in an NK- and MHC I–dependent manner. (A–F) Viral titers in spleen at day 3 p.i. in various mouse strains of different haplotypes (indicated in parentheses). Mice were infected i.v. with 2 × 105 PFU of WT or indicated MAT mutant MCMVs. Viral titers were assessed by plaque assay. Mice that were depleted of NK cells received 20 µl of α-AGM1 i.p. 24 h before virus infection. The data are representative of at least two or three independent experiments with 4–5 mice per group. Unpaired two-tailed Mann–Whitney test was used for statistical analysis. (G) Frequency and total number of splenic NK cells. Splenocytes from mock-infected, WT, or scMATp1 MCMV–infected BALB/c mice were isolated on indicated days p.i. and absolute number (#) and frequency (%) of CD3−CD19−NKp46+ populations (NK cells) were determined. Mice were infected as described in A. (H) Proliferation of NK cells. BALB/c mice were infected as described in A. 2 h before sacrifice, mice were injected with BrdU i.p. BrdU incorporation by total NK cells in mice was assessed by staining with α-BrdU antibody. (I) Expression of KLRG1 in NK cells from BALB/c mice, infected as described in A and stained for KLRG1 at indicated time points. (J) Frequency of CD107a expression of CD3−CD19−NKp46+ population 3 d p.i. was assessed. Representative plots are shown. In G–J, results were obtained by flow cytometry and represent at least three independent experiments with five mice per group. Unpaired two-tailed Student’s test was used. Graphs show mean with SEM as error bars. ****, P ≤ 0.0001; ***, P ≤ 0.001; **, P ≤ 0.01; *, P ≤ 0.05.
To further elaborate on these findings, we examined whether better control of scMATp1 MCMV resulted in any alternations in NK cell numbers, phenotype, and function compared with WT MCMV–infected mice. Both the frequency and number of NK cells were higher following scMATp1 MCMV infection in comparison to WT MCMV–infected mice on days 3 and 7 p.i. (Fig. 5 G). The observed phenotype was a consequence of higher NK cell proliferation in mice infected with scMATp1 MCMV (Fig. 5 H). Better viral control of scMATp1 MCMV resulted in more activated NK cells through increased expression of KLRG1 (Fig. 5 I). No major changes in maturation status of NK cells or any differences in cytokine secretion between the two groups of infected mice were observed (Fig. S4, A–D). However, NK cells from scMATp1 MCMV–infected mice degranulated more on day 3 p.i. (Fig. 5 J), which is in agreement with lower viral titers in spleen and better control of virus by NK cells in Fig. 5 A. All in all, our data show that MATp1 impairs NK cell proliferation and activation through the Ly49A–H-2Dd axis, which leads to inhibition of NK cells and inadequate control of WT MCMV.
MATp1 helps infected cells to avoid missing self–dependent killing by NK cells
The results presented so far suggest that MATp1 should prevent lysis of infected cells via missing self recognition. To that aim, we tested the ability of NK cells to lyse targets infected with MCMV that expresses MATp1 or with mutant lacking this protein. B12 fibroblasts (H-2d) were infected and, after 16 h p.i., cocultured with splenic cells from naive BALB/c mice for 4 h. It is well described that uninfected B12 cells are sensitive to NK cell lysis and that WT MCMV reduces their sensitivity to NK cell killing as a consequence of viral down-regulation of NKG2D ligands (Krmpotić et al., 2002; Babić et al., 2010; Fig. 6 A). Importantly, scMATp1 MCMV–infected cells were more susceptible to NK cell killing than WT MCMV–infected targets (Fig. 6 A). Similarly, NK cells that were cocultured with scMATp1 MCMV–infected targets expressed higher levels of surface CD107a in the NK cell degranulation assay (Fig. 6 B). All of this further demonstrates that MATp1 prevents missing self recognition and NK cell activation and killing.
Figure 6.

MATp1 helps infected cells to avoid missing self–dependent killing by NK cells. (A) NK cell in vitro killer assay. Immortalized B12 cells (SV40-transformed BALB/c fibroblasts, targets) were infected with WT or scMATp1 MCMV or left uninfected, labeled with CPD eF670, and after 16 h p.i., cocultured with splenocytes from naive BALB/c mice for another 4 h. Specific lysis of target cells was assessed by staining with PI and measured by flow cytometry. Data are representative of two independent experiments. Data points on the graph represent the mean of a technical triplicate for each sample. (B) Degranulation of NK cells after coincubation with target cells infected with indicated viruses. IL-15–expanded NK cells were cocultured with CPD-labeled and infected targets: B12 cells. Infection and labeling of B12 cells were performed as described in A. Antibody against CD107a was added together with NK cells, and Brefeldin and Monensin after 1 h, followed by another 4 h of incubation. CD107a expression on NK cell population was assessed by flow cytometry. Data are displayed as frequency (%) and mean fluorescence intensity of CD107+ population, and a representative experiment is shown (of three). In each experiment, technical triplicates were analyzed. (C) Conjugate formation between NK and target cells. Target B12 cells were infected and CPD labeled as described in A. IL-15–expanded NK cells were CFSE-labeled and cocultured with CPD+ targets in an E:T ratio of 1:1. After coincubation for indicated times, the reaction was stopped with 1% PFA in PBS, and CDP+CFSE+ doublets were analyzed on a flow cytometer. Graph shows the percentage of infected target cells that are in complex with NK cells. (D) Conjugate formation at 10 min after incubation shown on graph C is dependent on MHC I, as incubation of B12 target cells with α-H-2Dd antibody (34-5-8S) reduced the amount of conjugates by nearly half. (E) Conjugate formation between Ly49A-RCs and target cells. The experiment was performed as described in C, but instead of NK cells, Ly49A-RCs were used as effectors. (F) Conjugate formation between Ly49A-RCs and B12 targets can be prevented by the addition of α-H-2Dd antibody as in D. In C–F, the data are representative of two or three independent experiments where statistical significance of indicated groups compared with WT MCMV–infected samples is shown. One-way ANOVA was used in B, D, and F and two-way ANOVA with post hoc test in A, C, and E. Graphs show mean with SEM as error bars. ****, P ≤ 0.0001; ***, P ≤ 0.001; **, P ≤ 0.01; *, P ≤ 0.05.
To further elaborate the role of MATp1 on interactions between NK cell receptors and infected targets, specifically Ly49A–H-2Dd, we investigated whether NK cells form tight conjugates with infected cells in the presence of MATp1 (WT MCMV). Ly49A receptors contribute to conjugate formation and cell adhesion between NK cell and its targets (Back et al., 2007). We thus hypothesized that NK cells would form less-stable conjugates with scMATp1 MCMV–infected targets, since there is less altered-self H-2Dd on the target’s surface, which would lead to weaker Ly49A–H-2Dd interactions. To test this, target B12 cells were infected and labeled with cell proliferation dye eFluor 670 (CPD eF670). After 20 h of infection, targets were cocultured with CFSE-labeled, IL-15–expanded NK cells from BALB/c mice for the indicated time and analyzed by flow cytometry. IL-15–expanded NK cells showed activated phenotype with no alternations in Ly49 expression compared with naive NK cells, as expected (Fig. S4, E and F). We gated on the CPD eF670+CFSE+ population, which represents NK cells and targets that have formed tight conjugates in a given time. Interestingly, mock-infected cells started to form conjugates with NK cells after 5 min, surpassing the conjugate formation of NKs and infected cells, probably due to the high level of MHC I molecules as well as NKG2D ligands (Fig. 6 C). Nevertheless, WT MCMV–infected targets formed a higher percentage of conjugates with NK cells than scMATp1 MCMV–infected cells, especially after 10 min of incubation (Fig. 6 C). This phenomenon could be reverted by blocking Ly49A–MHC I interaction with α-H-2Dd antibody but not by the addition of irrelevant antibody (Fig. 6 D). In the end, to analyze the observed phenotype on a level of a single Ly49 receptor, we used Ly49A-RCs, since they only express Ly49A receptors. As with NK cells, we labeled Ly49A-RCs with CFSE and incubated them with CPD eF670–labeled and 20-h-infected B12 targets for the indicated times. While there was no difference in conjugate formation between groups in the first 5 min of incubation, WT MCMV–infected targets engaged Ly49A-RCs better than scMATp1 MCMV–infected ones at later time points (Fig. 6 E). Moreover, the percentage of conjugates between WT MCMV-infected targets and Ly49A-RCs stayed stable for ≥20 min, while the percentage of conjugates with scMATp1 MCMV–infected targets declined. Interestingly, conjugate formation of Ly49A-RCs and uninfected targets followed conjugate formation of scMATp1 MCMV–infected targets. This indicates that Ly49A engages its ligands on WT MCMV-infected cells with stronger stability and affinity than on uninfected cells. As in Fig. 6 D, conjugate formation could be prevented by blocking of Ly49A–MHC I interaction by the addition of α-H-2Dd antibody but not irrelevant antibody (Fig. 6 F). Addition of α-H-2Dd antibody had no effect on conjugate formation when target cells were infected with scMATp1 MCMV, which is in accordance with our previous results showing that there are hardly any MHC I molecules present on the surface of scMATp1 MCMV–infected cells (Fig. 2, A and B; and Fig. 3 A).
MATp1 is necessary for infected cell recognition by activating Ly49 receptors, which led to its high variability among field MCMV isolates and loss of recognition
Several activating Ly49 receptors recognize the m04/MHC I complex. However, similar to the inhibitory receptors, this also requires another, so far unknown viral component (Kielczewska et al., 2009). We thus asked whether MATp1 is the missing component required for recognition of MCMV-infected cells by the activating Ly49 receptors D2PWK/Pas, LBALB, and PMA/My. To test this hypothesis, we employed RCs for these three activating receptors and cocultured them with MEFs infected with WT MCMV or our MAT mutant viruses. MCMV mutant lacking m04 was used as a control. Removal of either MATp1 or m04 from MCMV resulted in complete failure of all three activating Ly49 receptors to recognize infected cells (Fig. 7, A–C). We conclude that MATp1 is the missing viral component that, together with MHC I and m04, is needed for the specific recognition of infected cells by activating Ly49L, Ly49P, and Ly49D2 receptors in an MHC I haplotype–restricted manner.
Figure 7.

MATp1 is necessary for infected cell recognition by activating Ly49 receptors, which led to its high variability among field MCMV isolates and loss of recognition. (A–C) Reporter cell assay with activating Ly49L-, Ly49P-, and Ly49D2-RCs coincubated with BALB/c (H-2d; A and B) and BALB.K (H-2k; C) MEFs. (A) Representative histograms showing activation of Ly49L-RCs after coincubation with BALB/c MEFs infected with indicated viruses at 1.5 PFU/cell. Activation of Ly49-RCs was measured after 24 h of cocultivation by measuring the frequency of GFP expressing Ly49-RCs. Both m04 and MATp1 were necessary for the proper activation Ly49L-, Ly49P-, and Ly49D2-RCs. All the experiments were performed at least three times, except two times with Ly49D2-RCs. One-way ANOVA with post hoc test was used for statistical analysis. Graphs show mean with SEM as error bars. ****, P ≤ 0.0001; ***, P ≤ 0.001; **, P ≤ 0.01; *, P ≤ 0.05. (D) Ly49L receptor specifically recognizes altered-self H-2Dd in an MHC I– and m04-dependent manner. Primary BALB/c MEFs were infected with indicated viruses and then incubated with 10 µg of indicated antibody for 1 h at 4°C before the addition of RCs. Ly49L-RCs were cocultivated with targets for 24 h, as described previously. Addition of α-H-2Dd antibody (34-5-8S) completely abrogated recognition of either infected or mock-infected cells by Ly49A-RCs, demonstrating that they indeed recognize H-2Dd. Incubation with α-m04.16 antibody partially blocked Ly49L-RC activation, reducing it to the level of activation seen when Ly49-RCs are incubated with mock-infected MEFs. α-MATp1 antibody was unable to block interaction between MCMV-infected cells and Ly49L-RCs. Perpendicular dashed lines denote MFI of Ly49A-RCs after incubation with WT MCMV–infected MEFs. Representative histograms are shown; experiment was performed three times with equal results. (E) MATp1 ORF is highly variable among different field isolates. Whole-genome sequences of MCMV strains Smith (gB acc. no. NC_004065.1), G4 (gB acc. no. EU579859), K181 (gB acc. no. AM886412), C4C (gB acc. no. HE610453), C4D (gB acc. no. HE610456), and WP15B (gB acc. no. EU579860) were aligned in software Geneious, with Smith MCMV sequence used as a reference. MATp1 ORF is the only variable part of the MAT transcript, and sequence similarity is <30% throughout the whole MATp1 ORF, while the rest of the MAT transcript is conserved (Corbett et al., 2007; Smith et al., 2008). (F and G) Reporter cell assay with Ly49-RCs coincubated with BALB/c MEFs infected with MCMV field isolates as described in A. Bar charts of Ly49L-, Ly49P-, and Ly49A-RCs incubated with BALB/c MEFs infected with indicated viruses that represent three or more independent experiments with indicated RCs. Representative plots are shown in Fig. S5 C. One-way ANOVA with post hoc test was used for statistical analysis. Graphs show mean with SEM as error bars. ****, P ≤ 0.0001; ***, P ≤ 0.001; **, P ≤ 0.01; *, P ≤ 0.05. norm., normalized.
Having demonstrated that MATp1- and m04-modified MHC I molecules are recognized by both activating and inhibitory receptors in the context of MCMV infection, we wondered whether these two viral MHC I modulators can do the same outside the context of viral infection. To that aim, we used previously generated and well-characterized NIH-3T3 cells (H-2q haplotype) stably transfected with either just H-2Dk or with both H-2Dk and m04, transiently transfected them with vector bearing MATp1 ORF, and then performed RC assays using inhibitory Ly49A- and activating Ly49L-RCs (Fig. S5, A and B). A greater proportion of Ly49A-RCs were activated and to a greater extent when coincubated with transfectants coexpressing m04 and MATp1. However, Ly49L-RCs did not react to either transfectant. It is important to note that m04 is an abundant protein, while MAT is the most abundant transcript. We have not been able to achieve such high levels of expression in transfectants, which might explain the absence of Ly49L-RC activation. Another possible explanation could be that other viral-derived or virus-induced cellular factors are needed for activating Ly49 recognition.
It has already been demonstrated that Ly49P-RCs recognize MCMV-infected cells in an MHC I–dependent manner (Desrosiers et al., 2005). To demonstrate that the same is true for Ly49L, we performed Ly49L-RC assays after target cells had been incubated with irrelevant antibody, α-H-2Dd, α-m04.16, or α-MATp1 antibody, as described previously for Ly49A-RCs. Addition of either α-H-2Dd or α-m04 antibody completely abrogated recognition of WT MCMV–infected cells by Ly49L-RCs (Fig. 7 D). As before, α-MATp1 antibody was not able to block activation of Ly49L-RCs.
Interestingly, we observed a striking degree of variability strictly localized within the MATp1 ORF (Fig. 7 E). This was very similar to that observed for m04 (Smith et al., 2008). Therefore, we asked whether these differences would affect recognition of infected cells by activating Ly49 receptors. As shown in Fig. S5 C and Fig. 7 F, only one field isolate (G4), whose MATp1 is highly similar to the Smith laboratory strain, managed to activate Ly49L- and Ly49P-RCs, whereas all others failed. On the other hand, inhibitory Ly49A receptor recognized all cells infected with field isolates; however, the level of Ly49A engagement differed among them (Fig. 7 G). Specifically, Ly49A-RCs engage cells infected with G4 and WP15B at levels similar to WT MCMV. We conclude that variability of m04, MATp1, and MHC I haplotypes determine the outcome of the interaction between NK cells and the infected cells. Altogether, these results strongly support an important role of m04 and MATp1 in the evolution of MHC I–restricted, MCMV-specific Ly49 receptors.
Discussion
Here we unravel a novel molecular mechanism by which MCMV evades missing self recognition. We and others previously reported that certain inhibitory Ly49 receptors can engage MHC I in a complex with the MCMV m04 protein, which counteracts the action of the other MCMV-encoded MHC I down-regulators m06 and m152 (Babić et al., 2010). Thereby, a small number of altered MHC I molecules are escorted to the cell surface, efficiently engage inhibitory Ly49 receptors, and prevent NK cell activation via missing self recognition. However, m04 on its own cannot bring MHC I to the cell surface and consequently fails to prevent missing self–dependent virus control, which suggested the role of an additional viral component (Lu et al., 2006). We have now identified that viral component crucial for successful evasion of missing self and have evidence that MCMV has devised an elaborate mechanism of ensuring surface expression of altered-self MHC I molecules with higher affinities for inhibitory Ly49A receptors. It uses two proteins, m04 and MATp1, to selectively target only those MHC I that are ligands for inhibitory Ly49 receptors and important for regulation of NK cell behavior, education, and licensing. MATp1 ensures proper formation and egress of the m04–MHC I complex despite the low affinity of m04 for H-2Dd and H-2Dk molecules (Berry et al., 2014). Importantly, this explains previous observations that in MCMV infection, licensed NK cells are inhibited and do not contribute significantly to virus control (Orr et al., 2010). This is further supported by strong NK-dependent attenuation of the virus lacking MATp1 in vivo.
Remarkably, we show that specific recognition of infected cells by the activating Ly49 receptors P, L, and D2 is absolutely dependent on MATp1/m04-modified altered-self MHC I, since the absence of any of these three abrogated recognition. It is established that specific recognition by activating Ly49P receptor in MA/My mice confers protection against MCMV infection in the context of the protective H-2k locus (Desrosiers et al., 2005; Dighe et al., 2005). Our results on Ly49P-RCs demonstrate that MATp1 is essential for this recognition. On the other hand, the BALB/c (H-2d haplotype) strain that bears the activating Ly49L receptor remains MCMV sensitive. Moreover, Pyzik et al. (2011) showed that Ly49L binds to the H-2k locus of MCMV-infected BALB.K mice better than to H-2d in BALB/c, and yet BALB.K mice are still not protected against the virus in the early times of infection. The Ly49L+ NK population has an important contribution to virus control in mice of H-2k haplotype (BALB.K). However, this appears at later time points, when they help CD8 T cells take control over the infection. There are several potential mechanisms to explain these phenomena. First, both activating Ly49L and inhibitory Ly49 receptors (e.g., Ly49A, C, or G) expressed on the same NK cell compete for the same ligands (H-2D) on target cells. Second, MHC I molecules on target cells are rapidly down-regulated during infection to evade the CD8 T cell response. By doing so, MCMV limits the availability of ligands for Ly49 receptors, which further contributes to the competitive environment between different Ly49 receptors to bind MHC I molecules. Low levels of MHC I ligands in BALB strains are consequently mostly recognized and bound by inhibitory Ly49 receptors, which contributes to inhibitory signaling, as confirmed by our results on BALB.B mice. MCMV modulates and down-regulates almost all ligands for major activating receptors: NCR1, NKG2D, DNAM1, and NK1.1 (Lisnić et al., 2015). The virus cannot allow this to happen with Ly49L ligands due to the role of MHC I molecules in NK-cell missing self–mediated killing. In general, inhibitory signals in NK cells overrule activating signals (Long et al., 2013), probably to prevent excessive inflammation and autoimmunity. This is even more important when inhibitory and activating receptors share the same ligand. Thus, this represents a fundamental difference between recognition of infected cells in C57BL/6 mice (mediated exclusively by Ly49H) and in BALB strains (mediated by MATp1/m04 altered-self MHC I). Furthermore, unlike MHC I–like m157, MCMV altered-self MHC I molecules could impact CD8+ T cell responses. Whether this impact is in favor of the host or the virus needs to be addressed and is currently being investigated in our laboratory.
Our work provides further examples of how CMV can be a strong driving force in the evolution of Ly49 NK cell receptors. It has already been suggested that CMVs and CMV-like viruses could be an important driving force in the evolution of immune receptors, especially receptors that recognize MHC I or MHC I–like molecules (Abi-Rached and Parham, 2005; Carrillo-Bustamante et al., 2013, 2014, 2015a,b). A well-established example is MCMV-encoded MHC I–like m157, which can be recognized by inhibitory Ly49I129/J and MCMV-specific Ly49HC57BL/6 activating receptor that, when present in a mouse strain, grants this strain MCMV resistance (Arase and Lanier, 2004; Carlyle et al., 2008; Zeleznjak et al., 2017). We are the first, however, to demonstrate that the evolution of Ly49 receptors could be driven by MCMV-modified altered-self MHC I molecules. To avoid lysis by NK cells via missing self, MCMV evolved a novel strategy to strengthen inhibition of NK cells by inhibitory receptors in spite of MHC I down-regulation. As a countermeasure, the host evolved activating Ly49 receptors to recognize these altered-self MHC I molecules engaged in complex with viral proteins. In some humans, activating KIR+ NK cells also expand (Béziat et al., 2013; Della Chiesa et al., 2014; Liu et al., 2016). Like their homologues Ly49 receptors, KIRs also recognize MHC I molecules. It is likely that activating KIR receptors recognize virus altered-self MHC I similar to those we describe here. To date, no other natural pathogen has demonstrated such propensity to drive NK or other innate immune cells to generate virus-specific innate cell receptors to counter its action. CMV’s impact on NK cell populations can have significant influence on human health and vaccine responses. Better experimental models are required to predict these outcomes (Goodier et al., 2018). Our findings have the potential to serve as a mouse model for further studies of the role of CMV in the evolution of NK cell receptors, NK cell responses during coinfection, and CMV–mediated expansion of NK cells.
Materials and methods
Mice
All mice used in experiments were housed and bred under specific pathogen–free conditions at the Central Animal Facility of the Medical Faculty, University of Rijeka. 8–12-wk-old mice were used in all experiments and were age- and sex-matched within experiments. All experiments were approved by the Animal Welfare Committee and Responsible Expert of the University of Rijeka, Faculty of Medicine, Rijeka, Croatia, as well as by the Veterinary Department of the Ministry of Agriculture, and have been performed in accordance with the Croatian Animal Protection Act, which has been matched with existing European Union legislation. Animals were randomly assigned to groups and housed in cages in groups, maximum six animals/cage. Animals belonging to the same experimental group were housed together and were not housed with animals from other experimental groups.
BALB/c NCR1gfp/gfp mice were a kind gift from O. Mandelboim (Hebrew University, Jerusalem, Israel; Gazit et al., 2006). BALB/c NCR1gfp/+ mice were generated by crossing BALB/c and BALB/c NCR1gfp/gfp mice.
Primary cells and cell culture
Primary BALB/c (H-2d), BALB.K (H-2k), BALB.B (H-2b), and C57BL/6 (H-2b) MEFs were prepared as described in (Brizić et al., 2018). SV40-transformed BALB/c fibroblasts (B12, H-2d) were generated as described (Del Val et al., 1991). Both primary cells and immortalized cell lines were cultivated in DMEM supplemented with either 3% or 10% FCS, respectively. All Ly49-RCs were a kind gift from S. Vidal (McGill University, Montreal, Canada). Ly49-RCs (2B4-NFAT-GFP) were generated by retroviral transduction as described in Arase et al. (2002) and cocultured in RPMI 1640 media (Roswell Park Memorial Institute) supplemented with 10% FCS. Primary mouse NK cells derived from BALB/c or BALB/c NCR1gfp/+ mice were cocultured in RPMI 1640 with 10% FCS and expanded with IL-15 (10 ng/ml; PeproTech) for 6 d before enrichment and stimulation (Assmann et al., 2017). Low levels of IL-2 (100 U/ml; PeproTech) were added to NK cells during experiments that required longer periods of incubation. All cells were tested for Mycoplasma using MycoAlert Micoplasma Detection Kit (Lonza) and were found to be negative.
Viruses and infection conditions
All viruses were propagated on primary BALB/c MEFs and titrated by standard plaque assay as described in Brizić et al. (2018). For all in vitro analyses, primary MEFs were infected with 1.5 PFU/cell, while B12 was infected with 3 PFU/cell. Adherent cells were infected in two ways: either by incubating adherent cell layer for 30 min in a small volume of virus suspension at 37°C and in atmosphere with 5% CO2, followed by 30 min centrifugation at 800 g (centrifugal enhancement), or by incubating cells in suspension at concentration of 107 cells/ml with the virus for 30 min with occasional agitation 37°C and in atmosphere with 5% CO2. The two infection methods yield comparable infection levels (unpublished data). Animals were infected i.v. with 2 × 105 PFU/mouse of virus. For NK depletion experiments, mice were injected i.p. with 20 µl of rabbit α-asialo GM1 (Wako Chemicals) 24 h before infection according to the manufacturer’s instructions. Viral titers were assessed 3 d p.i. by plaque assay (Brizić et al., 2018).
WT MCMV refers to the bacterial artificial chromosome (BAC)-derived MCMV strain pSM3fr, previously shown to be biologically equivalent to the MCMV strain Smith (VR-1399; Wagner et al., 2013). Construction of Δm04 (Wagner et al., 2002), ΔMAT (Δm169-m170; Marcinowski et al., 2012), ΔMATp2 (Δm169; Wagner and Koszinowski, 2004), and miR-27 mut MCMV (m169 mut; Marcinowski et al., 2012) were described previously using the full-length MCMV BAC pSM3fr. WT MCMV isolates K181 (GenBank accession no. AM886412), G4 (GenBank accession no. EU579859), c4C (GenBank accession no. HE610453), c4D (GenBank accession no. HE610456), and WP15B (GenBank accession no. EU579860; Smith et al., 2008, 2013) were a kind gift from A. Redwood (University of Western Australia, Perth, Western Australia, Australia). MATp1 is located at 229164–228844 nt on Smith MCMV sequence, GenBank accession no. GU305914.1.
Generation of ΔMATp1 and scMATp1 MCMV viruses
ΔMATp1 and scMATp1 MCMV were constructed according to the published literature (Tischer et al., 2010). Briefly, to generate ΔMATp1 MCMV virus, a disruption cassette containing kanamycin resistance gene (KanR) and I-SceI restriction site from plasmid pEP-SaphA was PCR amplified using primers (deluORF forward, 5′-TCGGTGGTCTCTCTCTGCCTGTCTGTGGTGAGCTGTGCTCCGGCGGTTCCCGCGTCCGTCCCAGTATACACTCCGCTAGC-3′; Del-uORF reverse, 5′-GGACCGCGTTGCTCATCGCGAGTCCGGCGTCGCGGGGAGCGAAAAGCTGAGACGGACGCGGGAACCGCCGGAGCACAGCTCACCACAGACTAATGCTCTGCCAGTGTTAC-3′; pEP-Kan forward, 5′-CCAGTATACACTCCGCTAGC-3′; and pEP-Kan reverse, 5′-TAATGCTCTGCCAGTGTTAC-3′). Escherichia coli containing the entire WT MCMV genome (BAC-MCMV) within the bacterial chromosome were transformed with the amplified disruption cassette, resulting in the replacement of MATp1 ORF following homologous recombination. The kanamycin-carrying cassette was then removed from kanamycin-resisting clones by inducible endonuclease I-SceI, which induces double-strand break in the target sequence present in the integrated disruption cassette. Recombination resulted in removal of the kanamycin gene and formation of the precise MATp1 deletion in BAC-MCMV. The replacement of MATp1 ORF with scMATp1 sequence was analogous to the previously described procedure of MATp1 deletion in ΔMATp1 MCMV virus. The only difference is that the disruption cassette, besides the selection marker KanR, I-SceI, and side homology sites, also contained the sequence encoding scMATp1 ORF, which was organized as a direct repeat. In short, the same disrupting cassette from plasmid pEP-SaphA was amplified using sets of primers that contained restriction sites for endonuclease PstI (Rep-suORF forward, 5′-GGAAGGCTGCAGGAAAATTGCTTCGAAGCCGAAGATAACGCTGTCAGTGGAATGACCATGCACCAGTATACACTCCGCTAGC-3′; Rep-suORF-KanR, 5′-AAGGAACTGCAGTAATGCTCTGCCAGTGTTAC-3′). The cassette was then inserted into plasmid pUC57-suORF, which included the scMATp1 sequence and a single restriction site for PstI, resulting in generation of the pUC57-suORF-Kan vector. The replacement cassette was then PCR amplified using pUC57-suORF-Kan vector as a template (scr-uORF forward, 5′-CTCTCTGCCTGTCTGTGGTGAGCTGTGCTCCGGCGGTTCCCGCGTCCGTCATGATCACCAACATCTGTGATACG-3′; scr-uORF reverse, 5′-GGACCGCGTTGCTCATCGCGAGTCCGGCGTCGCGGGGAGCGAAAAGCTGATTACCTGGACCTCGAACAGTC-3′). This was followed by homologous recombination of the cassette with the BAC-MCMV, removal of the kanamycin gene from the cassette by I-SceI, and replacement of MATp1 with scMATp1.
Generation of α-m04 and α-MATp1 antibodies
DNA sequence encoding for the extracellular domain of protein m04 without the signal sequence (amino acids at positions 24–219) was cloned into the pFUSE-hIgG1-Fc2 vector (InvivoGen) using primers 5′-CAAGGCCATGGACCGTAATGACAATGAATGTGAA-3′ (forward) and 5′-AGAACCCATGGACGTGTTTGGTGACTCATTC-3′ (reverse). The m04-pFuse-hIgG1-Fc2 construct obtained was sequenced and then used to transfect the HEK293 cell line. Secreted m04-Fc protein was purified from the supernatant by affinity chromatography using protein G columns (ÄKTA; GE Healthcare) and used to immunize BALB/c mice and produce monoclonal antibodies, as described previously (Yokoyama et al., 2013). Two clones were then selected for affinity purification on protein G columns and further analyses: m04.16 (IgG2a) and m04.17 (IgG1). Both clones recognize MCMV Smith encoded m04 in flow cytometry and immunoprecipitation.
For detection in FACS, an aliquot of α-m04.17 and α-m04.16 was biotin-labeled with Pierce EZ-LINK Sulfo-NHS-SS-biotin labeling kit (Thermo Fisher Scientific) according to the manufacturer’s instructions. Briefly, affinity-purified antibodies were labeled with 20× molar excess of biotin. After labeling, excess unbound biotin was removed by dialysis using Slide-A-Lyzer Dialysis tubes (Thermo Fisher Scientific) against PBS. The efficacy of biotin labeling was checked by staining WT and Δm04 MCMV–infected cells with biotinylated m04 antibodies and streptavidin and comparing the signal with cells stained with either biotin-labeled α-m04 or original, unlabeled α-m04 antibody and detected with α-mouse Ig antibody.
For MATp1 antibody generation, the DNA sequence encoding putative MATp1 was cloned into pQE30 vector (Qiagen) using primers 5′-TTGGATCCGTGGTCTCTCTCTGCCTGTCT-3′ and 5′-TTAAGCTTTTAGATGGTGTTGATACACGT-3′. The plasmid obtained was sequenced and then transformed into bacteria E. coli Bl21 (DE3) strain (Qiagen). Production of His-tagged MATp1 protein was induced with the addition of isopropyl-β-d-thiogalactoside according to the manufacturer’s instructions (QIAExpressionist; Qiagen). The protein was purified from bacterial lysates by affinity chromatography on Ni-Sepharose columns. Purified MATp1 protein was then used to immunize BALB/c mice in the same manner as described for the generation of m04 antibodies. All antibodies generated and/or produced in the Center for Proteomics (α-m04, α-MATp1, 34-5-8S, 34–2-12, irrelevant isotype-matched controls) were affinity purified on protein G columns.
Ribosome profiling
6 × 105 NIH-3T3 fibroblasts were infected with WT MCMV Smith strain for 0 and 48 h at an multiplicity of infection of 10 using centrifugal enhancement (800 g for 30 min). Ribo-seq was performed as described (Rutkowski et al., 2015). All libraries were quality checked by Agilent Bioanalyzer and sequenced on a HiSeq2000 with 50-nt single-end reads at the Beijing Genomics Institute.
RC assay
The RC assay was performed as described in Babić et al. (2010) with some minor modifications. Briefly, primary MEFs were infected with indicated viruses for 16 h before addition of RCs. Ly49-RCs were added at a ratio of 1:3 and were cocultured for another 24 h in 12-well plates followed by analysis on a flow cytometer. Dead cells were excluded by propidium iodide (PI; Sigma-Aldrich). For mAb blockade experiments, 10 µg of 34-5-8S, α-m04.16, or α-MATp1 antibodies were added to each well 16 h p.i. and 1 h before the addition of RCs. The RC assay was then performed as in Babić et al. (2010).
Metabolic labeling and immunoprecipitation
All immunoprecipitations used adherent primary MEFs, which were previously infected with MCMV or left uninfected. Unless otherwise indicated, cells were infected for 24 h with 1.5 PFU/cell of virus. In case of IFN-γ pretreatment, mock-infected cells were treated with 0.1 µg/ml recombinant IFN-γ (PeproTech) for 24 h. Immunoprecipitations depicted in Fig. 2 were performed as previously described (Halenius et al., 2011). Cells cultured in six-well plates were washed with PBS, incubated for 30 min in methionine- and cysteine-free medium, and then metabolically labeled (Easytag Express [35S] Met-Cys protein labeling mix; PerkinElmer) with 100 µCi/ml for 2 h before lysis of cells. For pulse-chase experiments, cells were washed with chase medium (DMEM supplemented with 3% FCS, unlabeled methionine, and cysteine) at the end of the labeling period, after which they were cultured in chase medium for the period indicated in the figures. Approximately 1 × 106 cells were lysed in 1 ml of digitonin lysis buffer (140 mM NaCl, 20 mM Tris, pH 7.6, 5 mM MgCl2, and 1% digitonin [Calbiochem]). Protease inhibitor cocktail (Sigma-Aldrich) was added to the lysis buffer shortly before use. Lysates were cleared from membrane debris at 13,000 rpm for 30 min at 4°C. Lysates were incubated with antibodies for 1 h at 4°C in an overhead tumbler before immune complexes were retrieved by protein A–Sepharose (GE Healthcare). Sepharose pellets were washed four times with increasing NaCl concentrations (0.15 to 0.5 M in lysis buffer containing 0.2% detergent). Immune complexes were dissociated at 95°C for 5 min in sample buffer containing 40 mM dithiothreitol, samples were cooled on ice, and then iodoacetamide (80 mM) was added to the samples before gel loading. Proteins were separated by SDS-PAGE using 10–12.5% gradient gel.
Western blot analysis
Unless otherwise specified, cell lysates were prepared with radioimmunoprecipitation assay buffer (25 mM Tris, 150 mM NaCl, 1% Na-deoxycholate, and 0.1% SDS) with the addition of Complete Protease Inhibitor Cocktail (Roche). The concentration of proteins was determined with bicinchoninic acid assay (Pierce BCA Protein Assay Kit; Thermo Fisher Scientific) according to the manufacturer’s instructions. Proteins were separated on 12% SDS-PAGE gels in Laemmli buffer on constant voltage. Following separation, proteins were transferred to Amersham Hybond PVDF membranes with 0.2-µm pores (GE Healthcare) using Trans-blot semidry transfer system (Bio-Rad) in Bjerrum Schafer-Nielsen transfer buffer (48 mM Tris, 39 mM glycine, and 20% methanol, pH 9.2). Membranes were blocked with 3% BSA in PBS or 5% nonfat milk in Tris-buffered saline and Tween 20 for ≥30 min at room temperature, followed by primary antibody in the same blocking overnight at 4°C with constant gentle agitation. Secondary antibodies labeled with peroxidase were always added in 5% nonfat milk in Tris-buffered saline and Tween 20. Blots were visualized on ImageQuant LAS 4000 mini-instrument (GE Healthcare). Antibodies generated in house (Center for Proteomics) were as follows: α-m04.10 (HR-MCMV-01), α-MATp1 (described above), α-MATp2 (m169.03), α-m06 (clone CROMA 229; HR-MCMV-02), and α-m152.05 (HR-MCMV-11). For loading control, the following antibodies against housekeeping genes were used: α-actin (clone C4; Merck) and α-cofilin (clone D3F9; Cell Signaling Technology). All antibodies generated by us in the Center for Proteomics are available from the Center for Proteomics website at https://products.capri.com.hr/.
Immunoprecipitation using Dynabeads without metabolic labeling
Epoxy Dynabeads (Coimmunoprecipitation kit; Thermo Fisher Scientific) were coupled with α-m04.16 or 34-5-8S (H-2Dd) antibodies according to the manufacturer’s instructions. The efficiency of coupling was verified by staining aliquots of antibody-coupled and uncoupled beads with goat α-mouse IgG-PE (BD PharMingen) and measuring fluorescence by flow cytometry. For all immunoprecipitations, primary BALB/c MEFs were used, infected with the indicated viruses at 1.5 PFU/cell for 24 h. The cells were collected by scraping, washing in PBS, and then lysing in lysis buffer (20 mM Tris-HCl, 150 mM NaCl, 1 mM EGTA, 1% Triton-X, and Protease Complete Mini EDTA-free protease inhibitors [Roche]) for 30 min with mild, constant agitation in a cold room. Cellular debris was removed by centrifugation at 2,600 g for 30 min at 4°C, and the lysates were then incubated with antibody-coupled Dynabeads (Thermo Fisher Scientific) for 30 min at room temperature with constant agitation. After incubation, coprecipitates were extensively washed according to the manufacturer’s instructions and eluted with elution buffer. Eluates were mixed with denaturing sample buffer, denatured at 80°C for 5 min, loaded on 12% SDS-PAGE, and then transferred and detected as described above for Western blot analysis.
To verify that MHC I was coimmunoprecipitated, a small aliquot of coimmunoprecipitation mixture was taken before elution of coprecipitated proteins and stained with 32-2-12 mAb (recognizes α3 domain of H-2Dd MHC I) or isotype control and then checked by flow cytometry. m04 coprecipitation with H-2Dd was also followed by staining an aliquot of coprecipitation mixture with biotinylated α-m04.17 or isotype control and streptavidin-PE (Thermo Fisher Scientific) and measured in flow cytometry using a BD FACSAriaIIu flow cytometer and DiVa software.
PLA
PLA was performed using a Duolink In Situ Green Fluorescent kit (Sigma-Aldrich) according to the manufacturer’s instructions with minor modifications. Primary BALB/c MEFs were infected with WT, Δm04, or scMATp1 MCMV in solution as described previously and then seeded on glass slides in 12-well plates. 24 h p.i., the cells were fixed with 4% paraformaldehyde (PFA), permeabilized with 0.1% Triton X-100 in PBS, and labeled with antibodies. Because α-m04.16, α-H-2Dd, and α-MATp1 antibodies are all from mice, α-m04.16 and α-H-2Dd (34-5-8S) antibodies were directly labeled with oligonucleotides using Duolink ProbeMaker kits (Sigma-Aldrich), according to the manufacturer’s instructions, and used in the following combinations: for detection of MHC I and m04 interactions, α-H-2Dd directly labeled with PLUS oligonucleotides and α-m04.16 unlabeled; for m04 and MATp1 interactions, directly labeled α-m04.16-MINUS and unlabeled α-MATp1; and for MATp1 and MHC I interactions, directly labeled α-H-2Dd PLUS and unlabeled α-MATp1. The cells were first stained with unlabeled antibodies, followed by secondary α-mouse oligonucleotide-labeled antibodies and then, after washing, with primary oligonucleotide-labeled antibodies. Following oligonucleotide ligation, DNA amplification was left overnight. Samples were then mounted using Mowiol mounting medium and analyzed at room temperature with an Olympus FV300 confocal laser scanning microscope using a PlanApo 60× NA1.4 oil objective (Olympus) and FluoView acquisition software.
NK cytotoxicity assay
Target B12 (H-2d) cells were infected with 3 PFU/cell of WT, Δm04, or scMATp1 MCMV or left uninfected followed by labeling with CPD eF670 (Thermo Fisher Scientific) according to the manufacturer’s instructions. After 16 h of infection, cells were cocultured with splenocytes from naive BALB/c mice for 4 h at effector-to-target (E:T) ratios adjusted to the number of NK cells: 4:1, 1:1, or 0.5:1. Specific lysis of target cells was assessed by staining each sample with 1 µg/ml PI (Sigma-Aldrich) and measured by flow cytometer. As a control, samples containing target cells only were stained with PI to measure spontaneous death of cells. NK cell cytotoxicity was calculated by following formula: [(% CPD+PI+ cell specific lysis − % CPD+PI+ cell spontaneous lysis)/(100 − % CPD+ PI+ cell spontaneous lysis)] × 100, as described in Babić et al. (2010). All tested groups, including different E:T ratios, were done in triplicate.
NK cell degranulation assay
IL-15–expanded primary NK cells from naive BALB/c orBALB/c NCR1+/gfp mice were used as effectors in this assay. Target B12 cells were infected as described above. After 16 h of infection, NK cells were cocultured with targets at E:T ratio 1:1, mixed, and centrifuged at 150 g for 3 min. Antibody against lysosome-associated membrane protein-1 (CD107a; Thermo Fisher Scientific) was added together with NK cells, IL-2 (100 U/ml; PeproTech), and IL-12 (100 pg/ml; PeproTech). After 1 h of incubation at 37°C, Brefeldin (Thermo Fisher Scientific) and Monensin (Thermo Fisher Scientific) were added to the cells, which were again mixed, centrifuged at 150 g for 3 min, and left for another 4 h of incubation in the total volume of 200 µl. CD107a expression on NK cells was assessed by flow cytometer. All assays were done in triplicate.
Effector–target conjugate formation
NK conjugate formation was performed as described in Back et al. (2007). In short, B12 target cells were infected for 20 h and labeled with CPD eF670 as above. A few hours before cocultivation with targets, pure NK cells or Ly49-RCs were labeled with CFSE (Molecular Probes) and resuspended in 10% RPMI 1640. Effectors and targets were mixed in a 1:1 ratio. Tubes were centrifuged at 150 g for 2 min at 4°C followed by incubation at 37°C. After the indicated time points (0, 5, 10, and 20 min), cells were briefly mixed and fixed with 1% PFA in PBS. For mAb blockade experiments, 10 µg of α-MHC I (34-5-8S) or isotype-matched antibody was incubated with targets 1 h before the addition of effector cells. Conjugate formation then proceeded in the same way as described above. Samples were run on a flow cytometer where the double-positive population of effectors and targets (CFSE+CPD+) was assessed. All assays were done in duplicate or triplicate.
Flow cytometry and intracellular cytokine staining
Single-cell suspensions of spleen were prepared according to standard protocols. Flow cytometric analysis were performed by using α-mouse CD3e (145-2C11), CD19 (1D3), NKp46 (29A1.4), CD69 (H1.2F3), KLRG1 (2F1), CD27 (LG.7F9), CD11b (M1/70), CD107 (1D4B), IFN-γ (XMG1.2), and Granzyme B (NGZB) purchased from Thermo Fisher Scientific, preceded by blocking of Fc receptors using 2.4G2 antibody (generated in-house). Directly conjugated Ly49 antibodies were as follows: Ly49A/L FITC (YE1/48; BioLegend), Ly49A/D PE (12A8; Thermo Fisher Scientific), and Ly49G biotin (AT-8; Thermo Fisher Scientific). Unlabeled Ly49A (JR9), Ly49A/L (YE1/48), and Ly49C/I (5E6) were a kind gift from W. Held (Ludwig Institute for Cancer Research, Epalinges, Switzerland). Intracellular staining, permeabilization, and fixation of cells were done with the Fixation/Permeabilization kit (Thermo Fisher Scientific). PMA (Sigma-Aldrich) and ionomycin (Sigma-Aldrich) were used to stimulate NK cells as positive controls. Stimulation of NK cells was done in the presence of Brefeldin A (1,000×; Thermo Fisher Scientific) and, if CD107a was assessed, with Monensin (1,000×; Thermo Fisher Scientific). Splenic NK cells were enriched using NK Cell Isolation Kit II, mouse (Miltenyi Biotec) according to the manufacturer’s instructions. The cell proliferation assay was performed by giving infected mice 2 mg/ml of BrdU (BD PharMingen) i.p. 2 h before sacrifice. To detect incorporated BrdU, cells were stained according to the manufacturer's protocol (BrdU flow kit; BD PharMingen). Fixable Viability Dye (1,000×, Thermo Fisher Scientific) or PI was used to stain dead cells.
For surface MHC I and m04 expression analysis, primary MEFs were stained for m04 (α-m04.17 or α-m04.16, generated in-house) followed by staining with isotype-specific secondary antibody: goat α-mouse Ig-PE (BD PharMingen) or rat α-mouse IgG1 PercP-eFluor710 (Thermo Fisher Scientific). Cells were then stained for MHC I using directly conjugated antibodies: α-H-2Dd PE (34-5-8S; recognizes α1-α2 subunit loaded with peptide; Thermo Fisher Scientific), α-H-2Dd PE (34-2-12S, recognizes α3 subunit independently of peptide presence; BD PharMingen), α-H-2Kd PE (SF1-1.1.1; Thermo Fisher Scientific), α-H-2Ld PE (30-5-7S, binds peptide loaded MHC I; Thermo Fisher Scientific), α-H-2Db FITC (28-14-8; Thermo Fisher Scientific), or α-H-2Kb APC (AF6-88.5; Thermo Fisher Scientific). Irrelevant isotype-matched control or secondary antibody only was used as a negative control. For detection of m04 in immunoprecipitation assays, α-m04.16 and α-m04.17 were used. Streptavidin PE (Thermo Fisher Scientific) was used to stain α-m04-biotin antibody. All data were acquired using FACSAria or FACSVerse (BD Biosciences) and analyzed using FlowJo software (TreeStar).
Generation of transient MATp1 transfectants
To generate m04-MATp1 transfectants, we took advantage of already-existing NIH-3T3-Dk cells (Kielczewska et al., 2009) stably transfected with m04 and transiently transfected them with plasmid containing MATp1 ORF using X-tremeGENE 9 transfection reagent (Sigma-Aldrich) according to the manufacturers’ instructions. MATp1 ORF was amplified from the CMV genome and cloned using the following primers: forward, 5′-AATCCATGGatgaggtccaggtcgtgtga-3′; reverse, 5′-AATGCTAGCttagatggtgttgatacacg-3′ (capital letters denote nucleotides added to 5′ ends of the primers that encode recognition sequences for restriction endonucleases NcoI and NheI, respectively). MATp1 ORF was cloned into pINFUSE-mIgG2b-Fc1 plasmid (InvivoGen) following digestion with NcoI and NheI restriction endonucleases. The plasmid in question contains a murine IgG Fc fragment with introns, which is excised from the plasmid after digestion with NcoI and NheI. Therefore, the newly constructed pMATp1 plasmid contains MATp1 under strong EF1-HTLV promoter with no protein tags that could compromise its function.
Statistical analyses
Statistical significance was calculated by two-tailed unpaired Student’s t test, one- and two-way ANOVA with appropriate post hoc tests (Dunnett’s and Sidak’s multiple comparison tests for one-way ANOVA and Tukey’s multiple comparisons test for two-way ANOVA), or unpaired two-tailed Mann–Whitney U test using GraphPad Prism 8 software. Unless otherwise noted, data are presented as mean ± SEM. A value of P > 0.05 was deemed not statistically significant and was not marked on the figures.
Data and software availability
All sequencing data have been deposited in the Gene Expression Omnibus (GEO) database and are available through reviewer’s account (accession no. GSE102375; reviewer token cnobuwsupbefboz).
Online supplemental material
Figs. S1 and S2 summarize characterization of the MAT locus and show characteristics of scMATp1 MCMV virus together with Western blot analysis of MCMV’s proteins m06 and m152 in MEFs infected with WT MCMV and other mutant MCMV viruses (Fig. S2, G and H). Fig. S3 includes validation of newly generated α-m04 antibodies, surface levels of H-2Dd in MHC I–deficient cells, surface m04 expression in infected BALB.K and BALB.B MEFs, and the scheme of the coimmunoprecipitation protocol of MHC I and m04 using epoxy Dynabeads. Fig. S4 depicts maturation stages, cytokine production, and degranulation of NK cells from WT MCMV and scMATp1 MCMV–infected BALB/c mice. In addition, it shows that IL-15–expanded NK cells have identical surface levels of all tested Ly49 receptors compared with NK cells from naive BALB/c mice. Fig. S5 shows how transfection of pMATp1 into NIH-Dk-m04 cells results in stronger activation of inhibitory Ly49A-RCs. Moreover, it includes representative plots of Ly49L-RC assay on primary BALB/c MEFs infected with various MCMV field isolates where, of all field isolates tested, only one (G4) could activate Ly49L-RCs.
Supplementary Material
Acknowledgments
We thank D. Rumora, E. Ražić, S. Slavić Stupac, K. Mikić, S. Malić, D. Rebić, A. Miše, E. Marinović, and M. Samsa for the excellent technical support. We also thank A. Gerbin, J. Živković, I. Nenadić, A. Čosić, and A. Šarlija for excellent administrative support.
This work has been supported by the Croatian Science Foundation under project 7132 (A. Krmpotić), University of Rijeka under projects 13.06.1.1.01 (S. Jonjić) and 13.06.1.1.02 (A. Krmpotić), European Regional and Development Fund (K.01.1.1.01.0006) awarded to the Scientific Centre of Excellence for Virus Immunology and Vaccines and cofinanced by the European Regional Development Fund (S. Jonjić), Medical Research Council Fellowship grant G1002523 and National Health Service Blood and Transplant grant WP11-05 (L. Dölken), and Deutsche Forschungsgemeinschaft–funded Research Unit: Advanced Concepts in Cellular Immune Control of Cytomegalovirus (FOR 2830); Project: “Solving the m04 paradox: Evasion of missing self recognition and CD8 T cell killing by MAT uORF” (JO 1634/1-1; S. Jonjić and V. Juranić Lisnić).
The authors declare no competing interests.
Author contributions: Conceptualization: V. Juranić Lisnić, B. Lisnić, L. Dölken, A. Krmpotić, and S. Jonjić. Investigation, validation, and formal analysis: J. Železnjak, V. Juranić Lisnić, B. Popović, B. Lisnić, M. Babić, A. Halenius, A. L’Hernault, F. Erhard, and T.L. Roviš. Methodology: J. Železnjak, V. Juranić Lisnić, B. Lisnić, L. Dölken, F. Erhard, A. Halenius, H. Hengel, S. Jonjić, and A. Krmpotić. Visualization: J. Železnjak, B. Lisnić, and V. Juranić Lisnić. Resources: S.M. Vidal, H. Hengel, and A.J. Redwood. Writing: J. Železnjak, V. Juranić Lisnić, A. Krmpotić, and S. Jonjić. Data curation: A.J. Redwood, F. Erhard, and L. Dölken. Supervision: V. Juranić Lisnić, A. Krmpotić, H. Hengel, L. Dölken, and S. Jonjić. Funding acquisition and project administration: S. Jonjić, A. Krmpotić, V. Juranić Lisnić, B. Lisnić, and L. Dölken.
References
- Abi-Rached L., and Parham P.. 2005. Natural selection drives recurrent formation of activating killer cell immunoglobulin-like receptor and Ly49 from inhibitory homologues. J. Exp. Med. 201:1319–1332. 10.1084/jem.20042558 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aguilar O.A., Mesci A., Ma J., Chen P., Kirkham C.L., Hundrieser J., Voigt S., Allan D.S., and Carlyle J.R.. 2015. Modulation of Clr Ligand Expression and NKR-P1 Receptor Function during Murine Cytomegalovirus Infection. J. Innate Immun. 7:584–600. 10.1159/000382032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aguilar O.A., Berry R., Rahim M.M.A., Reichel J.J., Popović B., Tanaka M., Fu Z., Balaji G.R., Lau T.N.H., Tu M.M., et al. . 2017. A Viral Immunoevasin Controls Innate Immunity by Targeting the Prototypical Natural Killer Cell Receptor Family. Cell. 169:58–71.e14. 10.1016/j.cell.2017.03.002 [DOI] [PubMed] [Google Scholar]
- Arase H., and Lanier L.L.. 2002. Virus-driven evolution of natural killer cell receptors. Microbes Infect. 4:1505–1512. 10.1016/S1286-4579(02)00033-3 [DOI] [PubMed] [Google Scholar]
- Arase H., and Lanier L.L.. 2004. Specific recognition of virus-infected cells by paired NK receptors. Rev. Med. Virol. 14:83–93. 10.1002/rmv.422 [DOI] [PubMed] [Google Scholar]
- Arase H., Mocarski E.S., Campbell A.E., Hill A.B., and Lanier L.L.. 2002. Direct recognition of cytomegalovirus by activating and inhibitory NK cell receptors. Science. 296:1323–1326. 10.1126/science.1070884 [DOI] [PubMed] [Google Scholar]
- Assmann N., O’Brien K.L., Donnelly R.P., Dyck L., Zaiatz-Bittencourt V., Loftus R.M., Heinrich P., Oefner P.J., Lynch L., Gardiner C.M., et al. . 2017. Srebp-controlled glucose metabolism is essential for NK cell functional responses. Nat. Immunol. 18:1197–1206. 10.1038/ni.3838 [DOI] [PubMed] [Google Scholar]
- Babić M., Pyzik M., Zafirova B., Mitrović M., Butorac V., Lanier L.L., Krmpotić A., Vidal S.M., and Jonjić S.. 2010. Cytomegalovirus immunoevasin reveals the physiological role of “missing self” recognition in natural killer cell dependent virus control in vivo. J. Exp. Med. 207:2663–2673. 10.1084/jem.20100921 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Back J., Chalifour A., Scarpellino L., and Held W.. 2007. Stable masking by H-2Dd cis ligand limits Ly49A relocalization to the site of NK cell/target cell contact. Proc. Natl. Acad. Sci. USA. 104:3978–3983. 10.1073/pnas.0607418104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bagchi S., Fredriksson R., and Wallén-Mackenzie Å.. 2015. In Situ Proximity Ligation Assay (PLA). Methods Mol. Biol. 1318:149–159. 10.1007/978-1-4939-2742-5_15 [DOI] [PubMed] [Google Scholar]
- Berry R., Vivian J.P., Deuss F.A., Balaji G.R., Saunders P.M., Lin J., Littler D.R., Brooks A.G., and Rossjohn J.. 2014. The structure of the cytomegalovirus-encoded m04 glycoprotein, a prototypical member of the m02 family of immunoevasins. J. Biol. Chem. 289:23753–23763. 10.1074/jbc.M114.584128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Béziat V., Liu L.L., Malmberg J.A., Ivarsson M.A., Sohlberg E., Björklund A.T., Retière C., Sverremark-Ekström E., Traherne J., Ljungman P., et al. . 2013. NK cell responses to cytomegalovirus infection lead to stable imprints in the human KIR repertoire and involve activating KIRs. Blood. 121:2678–2688. 10.1182/blood-2012-10-459545 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brizić I., Lisnić B., Brune W., Hengel H., and Jonjić S.. 2018. Cytomegalovirus Infection: Mouse Model. Curr. Protoc. Immunol . e51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brodin P., Lakshmikanth T., Johansson S., Kärre K., and Höglund P.. 2009. The strength of inhibitory input during education quantitatively tunes the functional responsiveness of individual natural killer cells. Blood. 113:2434–2441. 10.1182/blood-2008-05-156836 [DOI] [PubMed] [Google Scholar]
- Brune W. 2013. Molecular basis of cytomegalovirus host species specificity. In Reddehase MJ, ed. Cytomegaloviruses: From Molecular Pathogenesis to Intervention. Caister Academic Press, Poole, UK: 322–329. [Google Scholar]
- Carlyle J.R., Mesci A., Fine J.H., Chen P., Bélanger S., Tai L.H., and Makrigiannis A.P.. 2008. Evolution of the Ly49 and Nkrp1 recognition systems. Semin. Immunol. 20:321–330. 10.1016/j.smim.2008.05.004 [DOI] [PubMed] [Google Scholar]
- Carrillo-Bustamante P., Keşmir C., and de Boer R.J.. 2013. Virus encoded MHC-like decoys diversify the inhibitory KIR repertoire. PLOS Comput. Biol. 9:e1003264 10.1371/journal.pcbi.1003264 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carrillo-Bustamante P., Keşmir C., and de Boer R.J.. 2014. Quantifying the Protection of Activating and Inhibiting NK Cell Receptors during Infection with a CMV-Like Virus. Front. Immunol. 5:20 10.3389/fimmu.2014.00020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carrillo-Bustamante P., Kesmir C., and de Boer R.J.. 2015a Can Selective MHC Downregulation Explain the Specificity and Genetic Diversity of NK Cell Receptors? Front. Immunol. 6:311 10.3389/fimmu.2015.00311 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carrillo-Bustamante P., Keşmir C., and de Boer R.J.. 2015b A Coevolutionary Arms Race between Hosts and Viruses Drives Polymorphism and Polygenicity of NK Cell Receptors. Mol. Biol. Evol. 32:2149–2160. 10.1093/molbev/msv096 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chalifour A., Scarpellino L., Back J., Brodin P., Devèvre E., Gros F., Lévy F., Leclercq G., Höglund P., Beermann F., and Held W.. 2009. A Role for cis Interaction between the Inhibitory Ly49A receptor and MHC class I for natural killer cell education. Immunity. 30:337–347. 10.1016/j.immuni.2008.12.019 [DOI] [PubMed] [Google Scholar]
- Corbett A.J., Forbes C.A., Moro D., and Scalzo A.A.. 2007. Extensive sequence variation exists among isolates of murine cytomegalovirus within members of the m02 family of genes. J. Gen. Virol. 88:758–769. 10.1099/vir.0.82623-0 [DOI] [PubMed] [Google Scholar]
- Corbett A.J., Coudert J.D., Forbes C.A., and Scalzo A.A.. 2011. Functional consequences of natural sequence variation of murine cytomegalovirus m157 for Ly49 receptor specificity and NK cell activation. J. Immunol. 186:1713–1722. 10.4049/jimmunol.1003308 [DOI] [PubMed] [Google Scholar]
- Della Chiesa M., Falco M., Bertaina A., Muccio L., Alicata C., Frassoni F., Locatelli F., Moretta L., and Moretta A.. 2014. Human cytomegalovirus infection promotes rapid maturation of NK cells expressing activating killer Ig-like receptor in patients transplanted with NKG2C-/- umbilical cord blood. J. Immunol. 192:1471–1479. 10.4049/jimmunol.1302053 [DOI] [PubMed] [Google Scholar]
- Del Val M., Schlicht H.J., Ruppert T., Reddehase M.J., and Koszinowski U.H.. 1991. Efficient processing of an antigenic sequence for presentation by MHC class I molecules depends on its neighboring residues in the protein. Cell. 66:1145–1153. 10.1016/0092-8674(91)90037-Y [DOI] [PubMed] [Google Scholar]
- Desrosiers M.P., Kielczewska A., Loredo-Osti J.C., Adam S.G., Makrigiannis A.P., Lemieux S., Pham T., Lodoen M.B., Morgan K., Lanier L.L., and Vidal S.M.. 2005. Epistasis between mouse Klra and major histocompatibility complex class I loci is associated with a new mechanism of natural killer cell-mediated innate resistance to cytomegalovirus infection. Nat. Genet. 37:593–599. 10.1038/ng1564 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dighe A., Rodriguez M., Sabastian P., Xie X., McVoy M., and Brown M.G.. 2005. Requisite H2k role in NK cell-mediated resistance in acute murine cytomegalovirus-infected MA/My mice. J. Immunol. 175:6820–6828. 10.4049/jimmunol.175.10.6820 [DOI] [PubMed] [Google Scholar]
- Fernandez N.C., Treiner E., Vance R.E., Jamieson A.M., Lemieux S., and Raulet D.H.. 2005. A subset of natural killer cells achieves self-tolerance without expressing inhibitory receptors specific for self-MHC molecules. Blood. 105:4416–4423. 10.1182/blood-2004-08-3156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fredriksson S., Gullberg M., Jarvius J., Olsson C., Pietras K., Gústafsdóttir S.M., Ostman A., and Landegren U.. 2002. Protein detection using proximity-dependent DNA ligation assays. Nat. Biotechnol. 20:473–477. 10.1038/nbt0502-473 [DOI] [PubMed] [Google Scholar]
- Gazit R., Gruda R., Elboim M., Arnon T.I., Katz G., Achdout H., Hanna J., Qimron U., Landau G., Greenbaum E., et al. . 2006. Lethal influenza infection in the absence of the natural killer cell receptor gene Ncr1. Nat. Immunol. 7:517–523. 10.1038/ni1322 [DOI] [PubMed] [Google Scholar]
- Goodier M.R., Jonjić S., Riley E.M., and Juranić Lisnić V.. 2018. CMV and natural killer cells: shaping the response to vaccination. Eur. J. Immunol. 48:50–65. 10.1002/eji.201646762 [DOI] [PubMed] [Google Scholar]
- Halenius A., Hauka S., Dölken L., Stindt J., Reinhard H., Wiek C., Hanenberg H., Koszinowski U.H., Momburg F., and Hengel H.. 2011. Human cytomegalovirus disrupts the major histocompatibility complex class I peptide-loading complex and inhibits tapasin gene transcription. J. Virol. 85:3473–3485. 10.1128/JVI.01923-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hengel H., Reusch U., Gutermann A., Ziegler H., Jonjic S., Lucin P., and Koszinowski U.H.. 1999. Cytomegaloviral control of MHC class I function in the mouse. Immunol. Rev. 168:167–176. 10.1111/j.1600-065X.1999.tb01291.x [DOI] [PubMed] [Google Scholar]
- Janßen L., Ramnarayan V.R., Aboelmagd M., Iliopoulou M., Hein Z., Majoul I., Fritzsche S., Halenius A., and Springer S.. 2016. The murine cytomegalovirus immunoevasin gp40 binds MHC class I molecules to retain them in the early secretory pathway. J. Cell Sci. 129:219–227. 10.1242/jcs.175620 [DOI] [PubMed] [Google Scholar]
- Juranic Lisnic V., Babic Cac M., Lisnic B., Trsan T., Mefferd A., Das Mukhopadhyay C., Cook C.H., Jonjic S., and Trgovcich J.. 2013. Dual analysis of the murine cytomegalovirus and host cell transcriptomes reveal new aspects of the virus-host cell interface. PLoS Pathog. 9:e1003611 10.1371/journal.ppat.1003611 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kärre K., Ljunggren H.G., Piontek G., and Kiessling R.. 1986. Selective rejection of H-2-deficient lymphoma variants suggests alternative immune defence strategy. Nature. 319:675–678. 10.1038/319675a0 [DOI] [PubMed] [Google Scholar]
- Kavanagh D.G., Gold M.C., Wagner M., Koszinowski U.H., and Hill A.B.. 2001a The multiple immune-evasion genes of murine cytomegalovirus are not redundant: m4 and m152 inhibit antigen presentation in a complementary and cooperative fashion. J. Exp. Med. 194:967–978. 10.1084/jem.194.7.967 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kavanagh D.G., Koszinowski U.H., and Hill A.B.. 2001b The murine cytomegalovirus immune evasion protein m4/gp34 forms biochemically distinct complexes with class I MHC at the cell surface and in a pre-Golgi compartment. J. Immunol. 167:3894–3902. 10.4049/jimmunol.167.7.3894 [DOI] [PubMed] [Google Scholar]
- Kielczewska A., Pyzik M., Sun T., Krmpotic A., Lodoen M.B., Munks M.W., Babic M., Hill A.B., Koszinowski U.H., Jonjic S., et al. . 2009. Ly49P recognition of cytomegalovirus-infected cells expressing H2-Dk and CMV-encoded m04 correlates with the NK cell antiviral response. J. Exp. Med. 206:515–523. 10.1084/jem.20080954 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim S., Poursine-Laurent J., Truscott S.M., Lybarger L., Song Y.J., Yang L., French A.R., Sunwoo J.B., Lemieux S., Hansen T.H., and Yokoyama W.M.. 2005. Licensing of natural killer cells by host major histocompatibility complex class I molecules. Nature. 436:709–713. 10.1038/nature03847 [DOI] [PubMed] [Google Scholar]
- Kleijnen M.F., Huppa J.B., Lucin P., Mukherjee S., Farrell H., Campbell A.E., Koszinowski U.H., Hill A.B., and Ploegh H.L.. 1997. A mouse cytomegalovirus glycoprotein, gp34, forms a complex with folded class I MHC molecules in the ER which is not retained but is transported to the cell surface. EMBO J. 16:685–694. 10.1093/emboj/16.4.685 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krmpotić A., Busch D.H., Bubić I., Gebhardt F., Hengel H., Hasan M., Scalzo A.A., Koszinowski U.H., and Jonjić S.. 2002. MCMV glycoprotein gp40 confers virus resistance to CD8+ T cells and NK cells in vivo. Nat. Immunol. 3:529–535. 10.1038/ni799 [DOI] [PubMed] [Google Scholar]
- Lisnić B., Lisnić V.J., and Jonjić S.. 2015. NK cell interplay with cytomegaloviruses. Curr. Opin. Virol. 15:9–18. 10.1016/j.coviro.2015.07.001 [DOI] [PubMed] [Google Scholar]
- Liu L.L., Landskron J., Ask E.H., Enqvist M., Sohlberg E., Traherne J.A., Hammer Q., Goodridge J.P., Larsson S., Jayaraman J., et al. . 2016. Critical Role of CD2 Co-stimulation in Adaptive Natural Killer Cell Responses Revealed in NKG2C-Deficient Humans. Cell Reports. 15:1088–1099. 10.1016/j.celrep.2016.04.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Long E.O., Kim H.S., Liu D., Peterson M.E., and Rajagopalan S.. 2013. Controlling natural killer cell responses: integration of signals for activation and inhibition. Annu. Rev. Immunol. 31:227–258. 10.1146/annurev-immunol-020711-075005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu X., Kavanagh D.G., and Hill A.B.. 2006. Cellular and molecular requirements for association of the murine cytomegalovirus protein m4/gp34 with major histocompatibility complex class I molecules. J. Virol. 80:6048–6055. 10.1128/JVI.00534-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marcinowski L., Tanguy M., Krmpotic A., Rädle B., Lisnić V.J., Tuddenham L., Chane-Woon-Ming B., Ruzsics Z., Erhard F., Benkartek C., et al. . 2012. Degradation of cellular mir-27 by a novel, highly abundant viral transcript is important for efficient virus replication in vivo. PLoS Pathog. 8:e1002510 10.1371/journal.ppat.1002510 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orr M.T., Murphy W.J., and Lanier L.L.. 2010. ‘Unlicensed’ natural killer cells dominate the response to cytomegalovirus infection. Nat. Immunol. 11:321–327. 10.1038/ni.1849 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pyzik M., Charbonneau B., Gendron-Pontbriand E.M., Babić M., Krmpotić A., Jonjić S., and Vidal S.M.. 2011. Distinct MHC class I-dependent NK cell-activating receptors control cytomegalovirus infection in different mouse strains. J. Exp. Med. 208:1105–1117. 10.1084/jem.20101831 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pyzik M., Dumaine A., Charbonneau B., Fodil-Cornu N., Jonjic S., and Vidal S.M.. 2014. Viral MHC class I-like molecule allows evasion of NK cell effector responses in vivo. J. Immunol. 193:6061–6069. 10.4049/jimmunol.1401386 [DOI] [PubMed] [Google Scholar]
- Rahim M.M., and Makrigiannis A.P.. 2015. Ly49 receptors: evolution, genetic diversity, and impact on immunity. Immunol. Rev. 267:137–147. 10.1111/imr.12318 [DOI] [PubMed] [Google Scholar]
- Rahim M.M., Wight A., Mahmoud A.B., Aguilar O.A., Lee S.H., Vidal S.M., Carlyle J.R., and Makrigiannis A.P.. 2016. Expansion and Protection by a Virus-Specific NK Cell Subset Lacking Expression of the Inhibitory NKR-P1B Receptor during Murine Cytomegalovirus Infection. J. Immunol. 197:2325–2337. 10.4049/jimmunol.1600776 [DOI] [PubMed] [Google Scholar]
- Reusch U., Muranyi W., Lucin P., Burgert H.G., Hengel H., and Koszinowski U.H.. 1999. A cytomegalovirus glycoprotein re-routes MHC class I complexes to lysosomes for degradation. EMBO J. 18:1081–1091. 10.1093/emboj/18.4.1081 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rutkowski A.J., Erhard F., L’Hernault A., Bonfert T., Schilhabel M., Crump C., Rosenstiel P., Efstathiou S., Zimmer R., Friedel C.C., and Dölken L.. 2015. Widespread disruption of host transcription termination in HSV-1 infection. Nat. Commun. 6:7126 10.1038/ncomms8126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith L.M., McWhorter A.R., Masters L.L., Shellam G.R., and Redwood A.J.. 2008. Laboratory strains of murine cytomegalovirus are genetically similar to but phenotypically distinct from wild strains of virus. J. Virol. 82:6689–6696. 10.1128/JVI.00160-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith L.M., McWhorter A.R., Shellam G.R., and Redwood A.J.. 2013. The genome of murine cytomegalovirus is shaped by purifying selection and extensive recombination. Virology. 435:258–268. 10.1016/j.virol.2012.08.041 [DOI] [PubMed] [Google Scholar]
- Tischer B.K., Smith G.A., and Osterrieder N.. 2010. En passant mutagenesis: a two step markerless red recombination system. Methods Mol. Biol. 634:421–430. 10.1007/978-1-60761-652-8_30 [DOI] [PubMed] [Google Scholar]
- Wagner M., and Koszinowski U.H.. 2004. Mutagenesis of viral BACs with linear PCR fragments (ET recombination). Methods Mol. Biol. 256:257–268. [DOI] [PubMed] [Google Scholar]
- Wagner F.M., Brizic I., Prager A., Trsan T., Arapovic M., Lemmermann N.A., Podlech J., Reddehase M.J., Lemnitzer F., Bosse J.B., et al. . 2013. The viral chemokine MCK-2 of murine cytomegalovirus promotes infection as part of a gH/gL/MCK-2 complex. PLoS Pathog. 9:e1003493 10.1371/journal.ppat.1003493 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagner M., Gutermann A., Podlech J., Reddehase M.J., and Koszinowski U.H.. 2002. Major histocompatibility complex class I allele-specific cooperative and competitive interactions between immune evasion proteins of cytomegalovirus. J. Exp. Med. 196:805–816. 10.1084/jem.20020811 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yokoyama W.M., Christensen M., Dos Santos G., Miller D., Ho J., Wu T., Dziegelewski M., and Neethling F.A.. 2013. Production of monoclonal antibodies. Curr. Protoc. Immunol . 102:Unit 2.5. [DOI] [PubMed] [Google Scholar]
- Zeleznjak J., Popovic B., Krmpotic A., Jonjic S., and Lisnic V.J.. 2017. Mouse cytomegalovirus encoded immunoevasins and evolution of Ly49 receptors - Sidekicks or enemies? Immunol. Lett. 189:40–47. 10.1016/j.imlet.2017.04.007 [DOI] [PubMed] [Google Scholar]
- Ziegler H., Thale R., Lucin P., Muranyi W., Flohr T., Hengel H., Farrell H., Rawlinson W., and Koszinowski U.H.. 1997. A mouse cytomegalovirus glycoprotein retains MHC class I complexes in the ERGIC/cis-Golgi compartments. Immunity. 6:57–66. 10.1016/S1074-7613(00)80242-3 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All sequencing data have been deposited in the Gene Expression Omnibus (GEO) database and are available through reviewer’s account (accession no. GSE102375; reviewer token cnobuwsupbefboz).