

Summary
Many tumors produce platelet-derived growth factor (PDGF)-DD, which promotes cellular proliferation, epithelial-mesenchymal transition, stromal reaction, and angiogenesis through autocrine and paracrine PDGFRβ signaling. By screening a secretome library, we found that the human immunoreceptor NKp44 encoded by NCR2 and expressed on natural killer (NK) cells and innate lymphoid cells recognizes PDGF-DD. PDGF-DD engagement of NKp44 triggered NK cell secretion of IFN-γ and TNF-α that induced tumor cell growth arrest. A distinctive transcriptional signature of PDGF-DD-induced cytokines and the downregulation of tumor cell cycle genes correlated with NCR2 expression and greater survival in glioblastoma. NKp44 expression in mouse NK cells controlled the dissemination of tumors expressing PDGF-DD more effectively than control mice, an effect enhanced by blockade of the inhibitory receptor CD96 or CpG-oligonucleotide treatment. Thus, whilst cancer cell production of PDGF-DD supports tumor growth and stromal reaction, it concomitantly activates innate immune responses to tumor expansion.
Keywords: NK cell, innate lymphoid cells, cancer, PDGF-D, NKp44, cytokines, cell cycle
Graphical Abstract
eTOC
The growth factor PDGF-DD, expressed by multiple types of tumor, is a stimulatory ligand for human NK cell receptor NKp44.
Introduction
Natural killer (NK) and innate lymphoid cells (ILCs) lack highly variable antigen-specific receptors yet express multiple germline-encoded receptors that recognize ligands associated with pathogens (Arase et al., 2002; Li et al., 2013) and cellular stress (Raulet et al., 2013). Such receptors transmit activating intracellular signals by recruiting the protein tyrosine kinases Syk or ZAP70, either directly or through the adaptors DAP12 and FcRγ. These immunoreceptor tyrosine-based activation motif (ITAM) signaling pathways are similar to those induced by antigen-specific receptors (Bezbradica and Medzhitov, 2012). The human NKp44 receptor (encoded by NCR2) is expressed by activated NK cells (Vitale et al., 1998), innate lymphoid cells of group 1 (ILC1) (Fuchs et al., 2013) and group 3 (ILC3) (Cella et al., 2009), and plasmacytoid DCs (Fuchs et al., 2005), signals through DAP12 and has been implicated in the recognition of transformed cells (Cantoni et al., 1999; Carrega et al., 2015; Sivori et al., 2000; Vitale et al., 1998). NKp44 has no mouse orthologue. NKp44 ligands have previously been reported (Baychelier et al., 2013; Ho et al., 2008; Rosental et al., 2011; Vieillard et al., 2005) but their physiological relevance in vivo thus far remains elusive.
The platelet-derived growth factor (PDGF) family encompasses four polypeptides that assemble into five dimeric isoforms (PDGF-AA, PDGF-BB, PDGF-AB, PDGF-CC, and PDGF-DD) and engage two receptor tyrosine kinases, PDGFRα and PDGFRβ, which are mainly expressed on cells of mesenchymal origin (Andrae et al., 2008). PDGFR signaling contributes to the embryonal development of multiple organs and blood vessels, and promotes wound healing in the adult. Cancer cells frequently produce PDGFs, which trigger autocrine and paracrine PDGFR signaling and promote tumor growth, proliferation, stromal recruitment, angiogenesis, epithelial-mesenchymal transition, and metastasis. A homologue of cellular PDGF encoded by the simian sarcoma virus v-sis oncogene also promotes neoplastic transformation through PDGFR (Deuel et al., 1983).
By screening a human secretome protein library with an NKp44-GFP reporter cell line, here we found that NKp44 recognizes PDGF-DD, which has been shown to stimulate neoplastic transformation and tumor growth through PDGFRβ (LaRochelle et al., 2002; Li and Eriksson, 2003; Reigstad et al., 2005; Wu et al., 2013; Xu et al., 2005). Engagement of NKp44 by PDGF-DD triggered NK cell secretion of IFN-γ, TNF-α, and other proinflammatory cytokines and chemokines, which in turn induced the downregulation of tumor cell cycle genes and tumor growth arrest. Meta-analysis of a publicly available database of gene expression profiles for different human cancers revealed a remarkable positive correlation between NCR2 expression, upregulation of PDGF-DD-induced NK cell cytokine genes, and downregulation of tumor cell cycle genes, along with greater survival in glioblastoma (GBM). In vivo analysis of NCR2-transgenic mice demonstrated that NKp44 limits the dissemination of PDGF-DD-expressing tumor cells, which was enhanced by checkpoint blockade with anti-CD96 or by CpG-oligonculeotide (ODN) treatment. We conclude that PDGF-DD not only supports tumor growth and stromal expansion through PDGFRβ but also facilitates recognition of tumor cells by innate immune cells through NKp44.
Results
PDGF-DD is a ligand for NKp44
Physiological ligands of NKp44 have remained elusive for almost 20 years. To address this outstanding question, we performed an unbiased screen of a secretome protein library containing approximately 4000 purified human and mouse secreted proteins or ectodomains of single-pass transmembrane proteins for NKp44 ligands (Gonzalez et al., 2010). To perform the screen, we transfected a chimeric receptor consisting of the human NKp44 extracellular and transmembrane domains fused to the CD3ζ intracellular signaling domain into a reporter cell line that expresses GFP under the control of NFAT responsive elements. Ligand engagement of the NKp44-CD3ζ fusion protein in the reporter cells leads to Ca2+ mobilization and consequently NFAT transactivation of GFP expression (Arase et al., 2002). NKp44-GFP reporter cells were seeded into wells coated with individual secretome library proteins and GFP expression detected by flow cytometry (Figure 1A). The top 30 secreted protein hits from the screen were individually re-tested and amongst these PDGF-D was validated as a putative NKp44 ligand (Figure 1B and Table S1). PDGF-D is a member of the PDGF family that signals through PDGFRβ (Andrae et al., 2008; Reigstad et al., 2005; Li and Eriksson, 2003). PDGF-D differs from PDGF-A and PDGF-B in having an unusual N-terminal domain known as complement subcomponent C1r/C1s, Uegf, and Bmp1 (CUB) domain (Figure 1C) (Bergsten et al., 2001). PDGF-D is secreted as a dimer and must undergo extracellular proteolytic processing of the CUB domain by proteases, such as urokinase plasminogen activator and matriptase (Bergsten et al., 2001; Huang and Kim, 2015; Ustach and Kim, 2005; Ustach et al., 2010), to liberate active homodimeric PDGF-DD that binds to and activates PDGFRβ signaling (Figure 1C).
Figure 1. PDGF-DD is a ligand for NKp44.

(A) A secretome library screen with NKp44-GFP reporter cells identified 30 proteins (red) as putative NKp44 ligands including PDGF-D; y-axis, screen 1; x-axis, screen 2 (normalized fluorescence signal, NFS).
(B) GFP expression from NKp44-GFP reporter cells incubated with dilution series of the top 30 secretome library hits (PDGF-D, red; protein hits 1-29, black). Plate immobilized anti-NKp44 (blue) and anti-NKp30 (green) mAbs were used as positive and negative controls.
(C) Schematic of PDGFD, PDGF-D, and PDGF-DD isoforms: signal peptide (SP, white); CUB domain (blue); growth factor domain (GFD, red); black triangle, protease cleavage site; black lines, disulfide bonds.
(D, E) Recombinant PDGF-D and PDGF-DD activate NKp44-GFP reporter cells that is blocked by anti-PDGF-D.
(F) NKp44-Fc binds to recombinant PDGF-DD but not PDGF-D in solid phase. BSA, recombinant Chikungunya virus (CHIK) E2 protein and IL-17A were used as negative controls. Recombinant CUB-TEV-PDGFD contains a TEV cleavage site between CUB and GFD.
(G) Dose-dependent GFP expression from NKp44-GFP reporter cells stimulated with PDGF-DD.
(H) PDGF-DD has 3.3 ± 0.4 μM affinity for NKp44 as determined by surface plasmon resonance (SPR).
(I) PDGF-DD but not PDGF-D binds NKp44 by SPR. PDGF-D or PDGF-DD binding were investigated in the solid phase and NKp44 in the mobile phase (two-fold dilution series from 36 μM to 36 nM). A subset of the concentrations tested is shown. Data are represented as mean ± SEM (****, P ≤ 0.0001).
NKp44-GFP reporter cells expressed GFP when cultured in serum-containing medium with either recombinant PDGF-D (Figure 1D) or the processed PDGF-DD form (Figure 1E). Corroborating the specificity of these interactions, a mAb against PDGF-D blocked NKp44-GFP reporter activation by PDGF-D and PDGF-DD (Figure 1D, E). However, a chimeric protein consisting of the NKp44 ectodomain fused to the Fc domain of human IgG (NKp44-Fc) specifically bound to PDGF-DD but not to PDGF-D by solid phase binding assay, demonstrating that NKp44 effectively binds to active PDGF-DD (Figure 1F). PDGF-D most likely activates NKp44-GFP reporter cells because it is processed by serum proteases contained in the culture medium. PDGF-DD-induced activation of NKp44-GFP reporter cells was dose-dependent and saturated at high PDGF-DD concentrations (Figure 1G). Biacore analysis determined that PDGF-DD has a 3.3 ± 0.4 μM affinity for the Ig superfamily (IgSF) domain of NKp44 (Figure 1H), within the known range of the PDGF/PDGFR interactions (Shim et al., 2010), and confirmed that NKp44 does not bind to unprocessed PDGF-D (Figure 1I). NKp44 ligands (NKp44L) have been proposed, such as viral hemagglutinins (Ho et al., 2008), and the nuclear proteins Proliferating Cell Nuclear Antigen (PCNA) (Rosental et al., 2011) and Mixed-lineage leukemia protein 5 (MLL5) (Baychelier et al., 2013; Vieillard et al., 2005). We did not detect NKp44 interaction with either EL4 cells over-expressing PCNA (Figure S1A–C), or CHO cells transfected with three different influenza virus hemagglutinins (Figure S1D, E) using NKp44-Fc binding and/or activation of NKp44-GFP reporter cells. Similarly, no interaction of NKp44 was detected with human CD4+ T cells stimulated with an HIV gp41-derived peptide, which purportedly induces cell-surface expression of MLL5 (Figure S1F, G), or with Jurkat T cells, which should express MLL5 on cell surface (Figure S1H) (Baychelier et al., 2013). Potential low affinity interactions of NKp44 with these putative ligands and their physiological relevance remain unclear. Altogether, these results show that the proteolytically processed ‘active’ PDGF-DD growth factor is a bona fide ligand for NKp44.
PDGF-DD triggers NK cell activation and cytokine secretion via NKp44
We first investigated whether PDGF-DD evokes ITAM signaling in human NK cells. The human NK cell line NK92 expresses NKp44. PDGF-DD induced Ca2+ mobilization in NK92, which was blocked by anti-NKp44 (Figure 2A). Furthermore, PDGF-DD induced phosphorylation of AKT, ERK and its downstream target FOXO3A in primary human NK cells (Figure 2B). Since activated human NK cells express the NKp44/DAP12 receptor complex, but not PDGFRα or PDGFRβ (Figure S2A), we conclude that PDGF-DD binding to NKp44 triggers ITAM signaling in human NK cells.
Figure 2. PDGF-DD triggers cytokine secretion pathways in NK cells via NKp44 and DAP12.
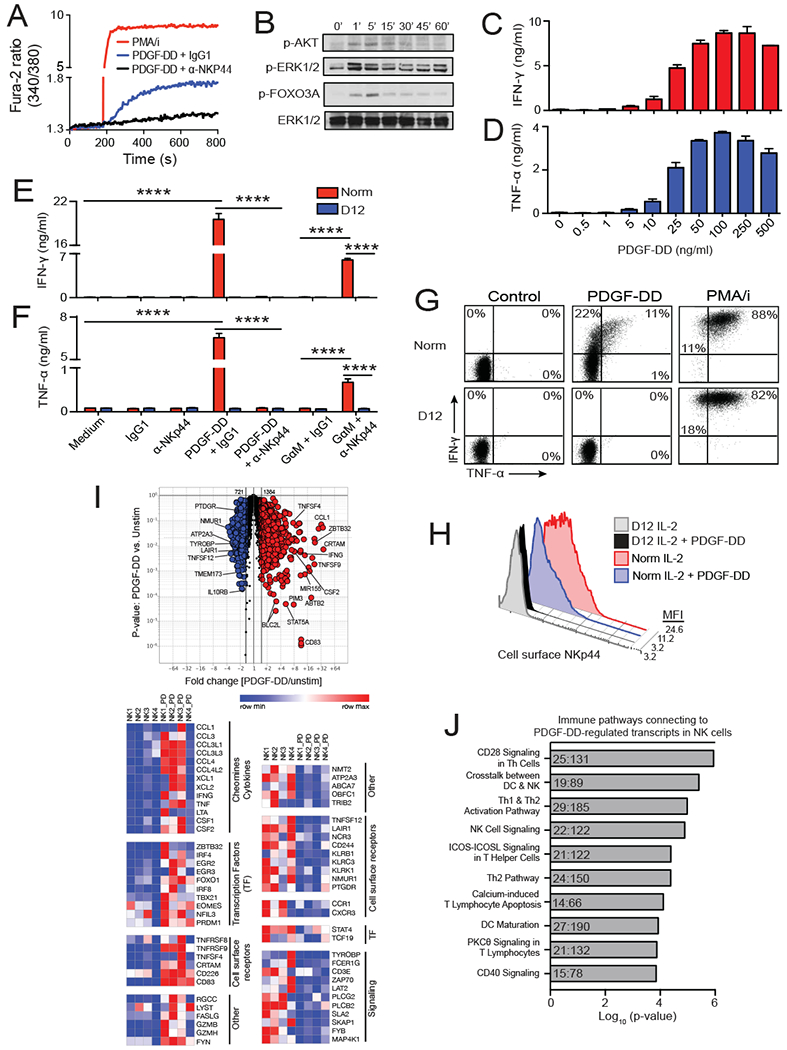
(A) PDGF-DD induces calcium signaling in NK92 cells, which is blocked by anti-NKp44 mAb.
(B) IL-2 cultured NK cells were stimulated with PDGF-DD for the indicated time points and phosphorylated AKT, ERK1/2 and FOXO3A and total ERK1/2 determined by immunoblotting.
(C, D) PDGF-DD stimulates dose-dependent NK cell secretion of IFN-γ and TNF-α. NK cells were kept in IL-2 medium throughout the assay.
(E, F) IFN-γ and TNF-α secretion by PDGF-DD-stimulated NK cells derived from either normal donor (Norm) or DAP12-deficient (D12) patient. Induction of IFN-γ and TNF-α is blocked by anti-NKp44. In the absence of PDGF-DD, IFN-γ and TNF-α can be induced by cross-linking NKp44 with GαM+α-NKp44 as surrogate NKp44 ligand.
(G) Representative dotplots of intracellular IFN-γ and TNF-α staining of PDGF-DD-stimulated IL-2-cultured NK cells from normal (Norm) or DAP12-deficient (D12) donors (percentage expression indicated in each quadrant). PMA/i was used as positive control.
(H) Representative histograms of NKp44 surface expression on IL-2-cultured NK cells from normal (Norm) or DAP12-deficient (D12) donors before and after PDGF-DD stimulation.
(I) Volcano plot and heatmap analysis of RNA-seq transcriptional profile induced by PDGF-DD in 4 NK cell donors. Transcripts in red were upregulated at least 1.5-fold by PDGF-DD (NK;1-4_PD) versus unstimulated NK cells (NK;1-4), those in blue were similarly downregulated.
(J) Top 10 highest scoring IPA Cellular Immune Response pathways generated from transcripts differentially expressed (±1.5-fold) in PDGF-DD-stimulated NK cells, as in (I). Ratios in columns indicate total PDGF-DD-regulated genes to the total number of genes in each pathway. Data represented as mean ± SEM (****, P ≤ 0.0001).
We next determined the impact of PDGF-DD-NKp44 interactions on NK cell functions. Incubation of IL-2-activated NKp44+ NK cells from normal donors with increasing concentrations of PDGF-DD induced dose-dependent cell surface expression of CD107a (Figure S2B), which is indicative of cytokine secretion and/or the release of lytic granules. Induction of CD107a expression was blocked by anti-NKp44 (Figure S2C), corroborating that PDGF-DD acted via NKp44 and not through other receptors. Similar induction of CD107a was observed when NK cells were stimulated with an NKp44 surrogate ligand, i.e. anti-NKp44 antibody crosslinked with a secondary antibody (Figure S2C). Although PDGF-DD clearly induced cell surface expression of CD107a, forced expression of PDGF-DD in target cell lines normally resistant to NK cell killing was insufficient to elicit NK cell cytotoxicity and only modestly enhanced antibody-dependent cellular cytotoxicity, which relies on activation of FcγRIII (CD16) on NK cells by an antibody bound to target cells (data not shown). These results are consistent with previous studies showing that NKp44 promotes target cell lysis only in combination with other activating receptors (Sivori et al., 2000; Vitale et al., 1998).
In contrast, PDGF-DD alone was sufficient to induce robust and dose-dependent IFN-γ and TNF-α secretion by IL-2-activated NKp44+ NK cells (Figure 2C, D), which was blocked by anti-NKp44 (Figure 2E, F), further supporting specific PDGF-DD/NKp44 interaction. Induction of IFN-γ and TNF-α was also obtained by stimulating NK cells using antibody-mediated cross-linking of NKp44 as surrogate ligand (Figure 2E, F). PDGF-DD stimulated the secretion of IFN-γ and TNF-α from NK cells derived from normal donors but not from an individual carrying a homozygous deletion of the TYROBP gene that encodes for DAP12 (Figure 2E, F). These DAP12-deficient NK cells lack cell surface expression of NKp44 as well as all DAP12-associated receptors (Fuchs et al., 2005). Intracellular staining for IFN-γ and TNF-α revealed that PDGF-DD stimulation of normal NK cells, but not DAP12-deficient NK cells, induced IFN-γ-producing cells as well as bifunctional cells secreting both IFN-γ and TNF-α (Figure 2G). Cell surface staining for NKp44 of PDGF-DD-stimulated NK cells showed downregulation of NKp44 (Figure 2H), probably reflecting ligand-induced internalization of this receptor. To provide further evidence that active PDGF-DD induces NK cell cytokine secretion, we engineered a construct encoding a full-length PDGF-D protein with a tobacco etch virus (TEV) protease cleavage site (CUB-TEV-PDGFD) in front of the growth factor domain. Incubation of CUB-TEV-PDGFD with TEV protease released active PDGF-DD (Figure S2D), which effectively induced NK cell secretion of IFN-γ and TNF-α (Figure S2E, F). CUB-TEV-PDGFD was in part active, probably because CUB-TEV-PDGFD is cleaved into PDGF-DD by serum proteases during the time required to perform the assay.
We asked how PDGF-DD impacts global gene expression in NK cells by RNA sequencing (RNA-seq) of unstimulated and PDGF-DD stimulated NK cells from four different donors. Collectively, induction of 1384 unique transcripts and downregulation of 721 transcripts was evident in the transcriptional profiles of PDGF-DD-activated NK cells compared to unstimulated NK cells (Figure 2I). Induced genes encoded IFN-γ, TNF-α and other proinflammatory cytokines including LTα and GM-CSF, as well as chemokines, such as CCL3, CCL4, CCL4L2, XCL1, and XCL2 (Figure 2I). Cell surface markers of activation, such as TNFSF4 and TNFRSF9 (CD134L and CD137), and transcription factors involved in ITAM signaling, cellular activation and proliferation, such as IRF4, EGRs, and EOMES, were also upregulated (Figure 2I). Downregulated genes encoded components of the ITAM signaling pathways, suggesting a feedback mechanism in which the functional activation of ITAM signaling mediators is paralleled by their reduced transcription. Ingenuity Pathway Analysis (IPA) of PDGF-DD-induced transcriptional changes corroborated the induction of pathways involved in NK- and T-cell activation, proliferation and survival as well as IFN-γ signaling (Figure 2J). Thus, PDGF-DD binding and signaling through the NKp44/DAP12 receptor complex leads to pronounced NK cell activation characterized by secretion of proinflammatory cytokines and chemokines.
PDGF-DD triggers cytokine secretion in ILC3 and ILC1
We sought to test whether PDGF-DD also activated human ILC3 and intraepithelial ILC1, since these cells were initially revealed through their distinctive expression of NKp44 in mucosal tissues (Cella et al., 2009; Fuchs et al., 2013), and have been implicated in cancer immunosurveillance (Carrega et al., 2015; Dadi et al., 2016). Although NKp44+ ILC3 isolated from tonsil can produce IL-22, GM-CSF and TNF-α, PDGF-DD selectively induced TNF-α (Figure 3A, B and Figure S3). IL-23 induced IL-22, as reported (Cella et al., 2009), while concurrent PDGF-DD and IL-23 stimulation did not further increase TNF-α or IL-22 production (Figure 3A, B and Figure S3). This result corroborates the observation that stimulation of NKp44 in ILC3 with a specific mAb as a surrogate ligand selectively induces TNF-α (Glatzer et al., 2013). We also determined the impact of PDGF-DD on ILC3 cultured in vitro with IL-1β and IL-2, a condition that converts ILC3 into IFN-γ-producing ILC1-like cells (Cella et al., 2010). PDGF-DD promoted secretion of both IFN-γ (Figure 3C) and TNF-α (Figure 3D) in a dose dependent fashion that was blocked by anti-NKp44, but not a control antibody. We finally tested the impact of PDGF-DD on the transcriptome of tonsilar intraepithelial ILC1 expanded in vitro with IL-2. In comparison to unstimulated ILC1, PDGF-DD induced the expression of 127 genes and downregulation of 102 genes (Figure 3E). PDGF-DD-induced genes were similar to those induced in NK cells, including IFNG, CCL1, CCL3, CCL4 and IRF4 (Figure 3E). IPA of PDGF-DD-induced changes in ILC1 gene expression profiles confirmed the involvement of lymphocyte activation and IFN-γ signaling pathways (Figure 3F). We conclude that PDGF-DD induces an NKp44-dependent proinflammatory gene program in ILC3, converted ILC3 and intraepithelial ILC1.
Figure 3. PDGF-DD/NKp44 interaction induces cytokine secretion by ILC1 and ILC3.

(A) Representative histograms of intracellular TNF-α content of freshly isolated tonsil ILC3s stimulated with PDGF-DD and/or IL-23 (percentage cytokine-positive cells is indicated in each histogram).
(B) Percentage of TNF-α secreting cells in PDGF-DD-stimulated tonsil ILC3s from 6 different donors (***, P ≤ 0.001).
(C, D) ILC1 derived in vitro by IL-2-induced conversion of tonsil ILC3 secrete IFN-γ and TNF-α following stimulation with PDGF-DD. Anti-NKp44 but not control anti-NKp46 mAb blocks cytokine secretion. Data represented as mean (n = 3) ± SEM (****, P ≤ 0.0001).
(E) Volcano plot and heatmap analysis of transcripts differentially expressed between PDGF-DD-stimulated (ILC1_1/2PD) versus unstimulated tonsilar ILC1 (ILC1_1/2) isolated from two different donors. Transcripts represented in red were upregulated at least 1.5-fold in PDGF-DD-stimulated versus unstimulated ILC1, and those in blue were similarly downregulated.
(F) Top 5 highest scoring IPA Cellular Immune Response pathways generated from transcripts differentially expressed (±1.5-fold) in PDGF-DD-stimulated tonsil ILC1. Ratios in columns indicate total PDGF-DD-regulated genes to the total number of genes in each pathway.
PDGF-DD-activated NK cells induce tumor cell growth arrest
We next investigated the impact of the PDGF-DD/NKp44 interactions on tumor cells. It has been shown that CD4+ Th1 cells can induce tumor cell growth arrest through the secretion of IFN-γ and TNF-α (Braumüller et al., 2013). Since PDGF-DD/NKp44 interactions induced IFN-γ and TNF-α secretion by NK cells, ILC3 and ILC1, we tested the hypothesis that this interaction might promote cell cycle arrest in tumor cell lines. Melanoma cell lines treated with supernatants from NK cells stimulated with PDGF-DD plus control IgG (NKDD+IgG) proliferated less and had more cells arrested in G1 than did cells treated with supernatants from NK cells stimulated with PDGF-DD plus a blocker of NKp44 (NKDD+αNKp44) (Figure 4A). Tumor cell growth arrest even persisted through one or two additional cell passages after replacing NKDD+IgG supernatants with complete growth medium lacking cytokines (Figure 4B, C). The growth inhibitory effect of NKDD+IgG supernatants was neutralized by the addition of mAbs to IFN-γ and TNF-α (Figure 4D). Thus, cytokine-containing NKDD+IgG supernatants instigate a transient suppression of tumor cell growth.
Figure 4. PDGF-DD binding to NKp44 induces NK cell secretion of cytokines that mediate tumor growth arrest.

(A) Meljuso melanoma cells were cultured with supernatants from NK cells stimulated with either PDGF-DD plus IgG1 (NKDD+IgG sup) or PDGF-DD plus anti-NKp44 (NKDD+α-NKp44 sup) or recombinant IFN-γ and TNF-α. Representative dotplots (left panel) show BrdU incorporation (y-axis) and 7-AAD staining for total DNA content (x-axis). The percentages of cells in different stages of the cell cycle (G1, S and G2/M) are indicated in each gate. Cells in the different stages of the cell cycle are quantified (right panel).
(B, C) Colo38 or Meljuso melanoma cells were cultured in either NKDD+IgG sup, NKDD+α-NKp44 sup or complete medium, washed, then re-plated in complete medium and the percentage (%) growth relative to control medium recorded for 3 passages.
(D) Colo38 cells were cultured in either complete medium or NKDD+IgG sup with (+) or without anti-IFN-γ and anti-TNF-α (Abs) before washing and passaging in complete medium as described above.
(E-G) Colo38 or Meljuso cells were pre-cultured with NKDD+IgG sup, NKDD+α-NKp44 sup or complete medium. IL-8 or IL-6 secretion in culture medium was measured at the end of the first passage.
(H) Representative dotplots of CADM1 (assessed by CRTAM-Fc), CD112, CD155 and CD95 (Fas) expression from Colo38 cells exposed to NKDD+IgG sup or NKDD+α-NKp44 sups (24h).
(I) RNA-seq profiles of human melanoma (Colo38 and Meljuso), breast cancer (MCF7) and ovarian cancer (OVCA) cell lines exposed to either NKDD+IgG sup or NKDD+α-NKp44 sup (48h). Transcripts in red were upregulated at least 1.5-fold and those in blue were similarly downregulated. Data are represented as mean ± SEM (***, P ≤ 0.001; ****, P ≤ 0.0001; ns, not significant).
Although arrested in G1, tumor cells treated with NKDD+IgG supernatants remained metabolically active. They secreted more IL-8 (Figure 4E, F) and IL-6 (Figure 4G), which may induce the recruitment of inflammatory cells. Tumor cells treated with NKDD+IgG supernatants up-regulated CADM1, CD112 and CD155, which can either activate or inhibit NK cell-mediated lysis through engagement of the NK cell receptors CRTAM (which binds CADM1) and CD226/CD96/TIGIT (all of which bind CD112 and/or CD155) (Figure 4H) (Blake et al., 2016). Additionally, tumor cells upregulated CD95 (Fas) (Figure 4H) that can cause apoptosis upon encountering NK cells expressing CD95L (FasL).
Finally, we examined global gene expression in tumor cell-lines treated with either NKDD+IgG or NKDD+αNKp44 supernatants by RNA-seq. NKDD+IgG supernatants induced the expression of numerous unique transcripts (738), many of which reflected the impact of IFN-γ and TNF-α, such as MHC-I, MHC-II, various IFN-γ-inducible components of the antigen-processing pathway (Figure 4I). IL8 and CD155 were also upregulated. Transcripts that were downregulated in comparison to unstimulated cells (1192) included many cell cycle genes (Figure 4I). I PA analysis confirmed that treatment of cancer cells with PDGF-DD-activated NK cell supernatants induced pathways involving IFN-γ signaling, antigen presentation, and cell cycle control of chromosomal replication (Figure S4). Altogether, these data show that tumor cells exposed to PDGF-DD-induced NK cell secreted cytokines undergo cell cycle arrest but also express inflammatory cytokines, chemokines and cell-surface ligands that can broadly influence the immune response to tumors.
Expression of NKp44 correlates with distinct cytokine and cell cycle gene expression signatures in GBM
We sought to determine the potential impact of the PDGF-DD/NKp44 interaction in clinical settings. Multiple human tumors, including lung, ovarian, renal, brain, prostate, breast and pancreatic cancer, have been shown to express PDGFD mRNA (LaRochelle et al., 2002; Lokker et al., 2002), whilst a marked increase of PDGF-D protein was reported in lung and ovarian cancer (LaRochelle et al., 2002). We extended PDGF-D protein analysis to normal and tumor tissues. In normal tissues, PDGF-D was mainly produced in some epithelial cells and, to a lesser extent, in vascular endothelial cells and a few other cells of the vascular tunica media (Figure S5A–I and data not shown). Strong PDGF-D expression was detected in tumor cells of carcinomas of head, neck, bladder, lung, colon, breast, melanomas and ovarian cancer (Figure S5J–T). PDGF-D was expressed in GBM (Figure 5A) and was highly associated with vascular proliferation (Figure 5B). Thus, PDGF-D is expressed in cancer cells and/or stroma of many human tumors with aggressive features.
Figure 5. Core signatures of PDGF-DD-activated NK cells and growth arrested tumor cells correlate with NCR2 expression in TCGA GBM cohort.

(A, B) PDGF-D expression in tissue sections of GBM, as determined by immunohistochemistry (representative of 5 cases). Arrowheads indicate PDGF-D+ hyperplastic blood vessels.
(C) Expression of signature core cytokine genes by NK cells from different donors kept in medium alone (Unstim), or stimulated with PDGF-DD or PMA/i.
(D) Expression of signature core cell cycle genes in different tumor cells exposed to medium alone, NKDD+α-NKp44 supernatant, or NKDD+IgG supernatant.
(E) Correlation data for individual cytokine genes (y-axis) in the core signatures of PDGF-DD-activated NK cells versus NCR2 expression (x-axis) extracted for TCGA GBM. Each dot represents a tumor case (n = 539 patients). The statistical significance of the correlation was determined using the Pearson’s correlation coefficient. A red linear regression line is shown in each plot.
(F) Infiltration of NKp44+ (red) and CD3ε+ (green) lymphocytes in tissue sections of a GBM case (representative of 8) including a case of gliosarcoma, as assessed by immunofluorescence.
(G) Correlation data for NCR2 (y-axis) versus NCR3 (x-axis) expression in the TCGA GBM cohort.
(H) Correlation data for individual cell cycle genes (y-axis) in core signatures of growth arrested tumor cells versus NCR2 expression (x-axis) extracted for TCGA GBM.
(I, J) Kaplan-Meier survival curves for the first (Q1) and fourth quartile (Q4) of the canonical cytokine and canonical cell cycle variate of NCR2 expression, respectively.
We next defined a core expression signature for the 9 most highly induced cytokine genes in PDGF-DD-activated NK cells from different donors (Figure 5C) and a core signature of 34 most significantly downregulated cell cycle genes in human ovarian carcinoma, breast carcinoma and melanoma cell lines exposed to NKp44DD+IgG supernatants (Figure 5D). We interrogated The Cancer Genome Genome Atlas (TCGA, https://cancergenome.nih.gov/), a comprehensive map of the transcriptional changes in multiple types of cancer, for correlations between NCR2 expression and the cytokine gene signature in various human cancers. Remarkable correlations were found in the TCGA GBM cohort (539 patients); NCR2 expression positively correlated with 6 of 7 available core cytokines extracted from the GBM TCGA cohort (Figure 5E). The canonical correlation of NCR2 with the cytokine variate was 0.60 (Table S2). The correlation between NCR2 expression and the NK cell cytokine gene signature in GBM suggested that NKp44+ NK cells might infiltrate GBM. Indeed, we observed numerous NKp44+ cells along with relatively few T cells infiltrating GBM in tissue sections obtained from several patients (Figure 5F); however, a GBM variant known as gliosarcoma was infiltrated by more T cells and fewer NKp44+ cells, which suggests that lymphocytic infiltrates of GBM vary somewhat. Further supporting NK cell infiltration in GBM, NCR2 expression in the GBM TCGA cohort was positively correlated with expression of the NK cell-specific gene NCR3, which encodes the activating receptor NKp30 (Figure 5G). These results indicate that infiltration of NKp44+ NK cells is present in many, but not all, GBM and positively correlates with the expression of proinflammatory NK cell cytokine genes, such as IFNG and TNF.
We next determined the correlation between NCR2 and the expression of the tumor cell cycle gene signature (see Figure 5D) for GBM. Strikingly, NCR2 expression negatively correlated with the expression of 25 of 27 available core cell cycle genes in the GBM TCGA cohort (Figure 5H). The canonical correlation of NCR2 with the cell cycle variate was 0.68 (Table S3). We next examined if NCR2 expression was correlated with greater overall survival in the GBM cohort. GBM patients in the upper quartile (Q4) expression of the canonical cytokine (Figure 5I) and cell cycle (Figure 5J) variates of NCR2 had greater overall survival compared to GBM patients in the lower quartile (Q1) (p = 0.0266 and p 0.0093, respectively). We also observed a trend towards increased survival from the 1st to the 4th quartile of NCR2 expression, but it was not statistically significant, probably because the dynamic range of NCR2 expression in TCGA GBM mRNA was limited (data not shown). Thus, meta-analysis of TCGA database indicates that overall survival correlates not only on the expression of NCR2 but also on the downstream effects of NKp44 engagement by PDGF-DD. While other types of tumors also showed associations of NCR2 expression with NK cell core cytokine signature, and/or downregulation of tumor cell cycle genes, correlations with overall survival were not significant (data not shown), suggesting that the contribution of PDGF-DD/NKp44 interaction to control of tumor progression may be context-dependent.
PDGF-DD binding to NKp44 limits tumor growth in vivo
Since NKp44 is encoded only in humans, we developed bacterial artificial chromosome (BAC) NCR2 transgenic (tg) mice to determine the impact of PDGF-DD/NKp44 interactions on cancer in vivo. Two NCR2-tg lines were generated; in both, expression of the NKp44 transgene mirrored NKp44 expression in the human immune system, although NKp44 was expressed at slightly higher levels in one line. In steady state, NKp44 was found on a small subset of splenic and lymph node NK cells (Figure 6A and Figure S6A, B). NKp44 was also expressed on a larger subset of ILC3 and few NK/ILC1 cells in the small intestine (Figure S6C, D). Upon in vitro activation with IL-2 and IL-15, NK cells showed a marked upregulation of NKp44, which was increased by further stimulation with inflammatory cytokines (Figure 6B). To determine the impact of PDGF-DD/NKp44 interaction on anti-tumor NK cell activity in vivo, we chose the B16 metastatic tumor model, which has been shown to be sensitive to NK cells, particularly to their IFN-γ secretion capacity (Takeda et al., 2011). We generated B16F10 melanoma cells stably expressing either a bicistronic gene encoding human PDGFD and GFP (B16-PDGFD) or the backbone vector (B16-pMX) as control cells (Figure 6C). Incubation of tissue culture supernatants from B16-PDGFD cells with human NK cells promoted the secretion of IFN-γ and TNF-α, which was dependent on NKp44 and PDGF-DD (Figure 6D, E), whereas control supernatants from B16-pMX cells did not activate NK cells unless recombinant PDGF-DD was added to the cultures (Figure S6E, F). Two weeks after intravenous injection of B16-PDGFD cells, NCR2-tg mice had fewer surface metastases (Figure 6F and Figure S6G), diminished mean tumor area (Figure 6G and Figure S6H), as well as augmented expression of Ifng (Figure 6H), but not Tnf (Figure 6I) in lungs compared to non-tg littermates. B16-PDGFD cells formed more lung metastases in NCR2-tg mice treated with blocking anti-NKp44 compared to a control mAb (Figure 6J), corroborating that higher rejection of B16-PDGFD cells in NCR2-tg mice depends on NKp44. Moreover, lung surface metastases formed by B16-pMX control cells did not differ between NCR2-tg and non-transgenic littermate mice indicating that NCR2-tg mice had no inherent advantage over non-tg mice in restricting B16F10 cells (Figure 6K). We conclude that PDGF-DD/NKp44 interactions facilitate NK cell control of tumor expansion in vivo.
Figure 6. NKp44 restricts tumor cells expressing PDGFD in vivo.

(A) Representative dotplots of NKp44 expression in CD3−NK1.1+ NK cells (NK), CD3+NK1.1+ NKT cells (NKT), and CD3+NK1.1− T lymphocytes (T) isolated from spleen and mesenteric lymph nodes (mLN) of NCR2-tg (founder #1) and non-tg mice. The percentages of cells are indicated in each gate.
(B) NKp44 expression on NK cells from spleens of NCR2-tg and non-tg mice activated in vitro with IL-2 and IL-15 ± IL-1β and TNF-α.
(C) GFP expression of B16F10 melanoma cells stably transduced with pMX-IRES-eGFP retroviral vector (B16-pMX) or pMX-IRES-eGFP encoding PDGFD (B16-PDGFD).
(D, E) Tissue culture supernatants from B16-PDGFD cells elicit IFN-γ and TNF-α secretion from human NK cells, which is blocked by anti-PDGF-D or anti-NKp44.
(F) Quantification (left panel) and representative photos (right panel) of surface lung metastases formed in NCR2-tg or non-tg littermate mice injected with B16-PDGFD cells.
(G) Mean tumor area (μM2) ± SEM and representative histochemical images of lung metastases from NCR2-tg (n = 6) or non-tg (n = 7) mice injected with B16-PDGFD cells.
(H, I) Day 17 quantitative RT-PCR of Ifng and Tnf transcripts in lungs of NCR2-tg and non-tg mice injected with B16-PDGFD cells.
(J) Day 17 surface lung metastases in NCR2-tg injected with B16-PDGFD cells and treated with either control or anti-NKp44 antibodies.
(K) Day 17 surface lung metastases in NCR2-tg and non-tg mice injected with B16-pMX control cells (ns, not significant). Data are represented as mean ± SEM (*, P ≤ 0.05; **, P ≤ 0.01; ****, P ≤ 0.0001; ns, not significant).
NKp44-PDGF-DD interaction enhances checkpoint blockade
We sought to determine whether NKp44-PDGF-DD interaction would synergize with checkpoint blockade in tumor rejection. We first examined NK cell expression of NKp44 along with the inhibitory checkpoint receptors PD-1 (Kline and Gajewski, 2010), TIGIT (Blake et al., 2016), and CD96 (Blake et al., 2016) in the lungs and lymph nodes of NCR2-tg mice. In tumor free NCR2-tg mice, NK cells did not express NKp44, CD96, PD1 or TIGIT (Figure 7A and data not shown). In contrast, NKp44 and CD96, but not PD-1 or TIGIT, were expressed on NK cells in the lungs and lymph nodes of NCR2-tg mice that had been intravenously injected with B16-PDGFD cells (Figure 7B, C). Moreover, the CD96 ligand, CD155, was induced on B16F10 cells by IFN-γ and TNF-α (Figure 7D). Thus, we hypothesized that blockade of the CD96-CD155 interaction might augment NK cell activation and limit B16-PDGFD cell metastases (Blake et al., 2016). Indeed, anti-CD96 treatment lowered the number of B16-PDGFD surface lung metastases formed in NCR2-tg and non-tg littermates. Importantly, B16-PDGFD surface lung metastases were fewer in NCR2-tg mice compared to non-tg mice receiving anti-CD96 immunotherapy (Figure 7E). Thus, PDGF-DD/NKp44 interaction and anti-CD96 immunotherapy cooperate in NK cell control of B16-PDGFD cell metastases.
Figure 7. PDGF-DD binding to NKp44 augments immunotherapies.

(A) NK cell expression of NKp44 and CD96 in the lungs of NCR2-tg and non-tg mice.
(B) Representative plots of NK cells from day 17 lungs and mLN of NCR2-tg mice injected with B16-PDGFD cells, showing expression of NKp44, CD96, PD-1, or TIGIT.
(C) Percentages of NKp44+ NK cells expressing CD96, PD-1, or TIGIT in day 17 lungs and mLN of NCR2-tg mice injected with B16-PDGFD cells.
(D) The CD96 ligand CD155, and CD95 are upregulated on B16F10 cells stimulated with IFN-γ and TNF-α.
(E) Quantification of day 17 surface lung metastases from NCR2-tg and non-tg mice injected with B16-PDGFD cells and treated with anti-CD96 mAb or control antibodies.
(F) Mean tumor volumes ± SEM from NCR2-tg or non-tg mice injected subcutaneously with B16-PDGFD cells and treated with CpG-ODN or left untreated (arrows indicate intratumoral CpG-ODN injections).
(G, H) Representative dotplots and percentage of NKp44+ cells in NK cells isolated from B16-PDGFD tumors of NCR2-tg or non-tg mice treated with CpG-ODN or untreated.
NKp44 engagement boosts CpG-induced control of subcutaneous melanoma
In contrast to the lung B16 metastatic model, NK cells have little impact on subcutaneous B16 melanoma, probably due a poorly immunogenic environment that prevents NK cell activation. However, it has been shown that CpG-ODN immunotherapy can stimulate NK cell recruitment, activation and NK cell-dependent regression of subcutaneous B16 melanoma (Liu et al., 2008). We examined whether the NKp44-PDGF-DD axis can impact the ability of CpG-ODN to restrict the growth of subcutaneous melanoma. In the absence of CpG treatment, tumor growth of subcutaneous B16-PDGF-D melanoma was equally unrestricted in NCR2-tg or non-tg littermates (Figure 7F). Conversely, in mice receiving CpG-ODN, growth of B16-PDGFD tumors was restricted sooner in NCR2-tg compared to non-tg littermates (Figure 7F). The earlier control of B16-PDGFD growth in NCR2-tg following CpG-ODN was paralleled by a marked induction of NKp44 expression in intratumoral NK cells (Figure 7G, H). These results show that CpG-ODN treatment can restrict outgrowth of a solid tumor more effectively when infiltrating NK cells are activated by tumor-expressed PDGF-D.
Discussion
We have demonstrated that NKp44 recognizes PDGF-DD, enabling NK cells, as well as ILC3 and ILC1, to detect cells secreting PDGF-DD. The identification of PDGF-DD as a ligand for NKp44 establishes a new paradigm in NK cell biology. Activating NK cell receptors have been shown to recognize cell surface ligands encoded by pathogens (Arase et al., 2002; Li et al., 2013) or induced by cellular stress during viral infections (Raulet et al., 2013). NKp44 is the first activating NK cell receptor found to recognize a cellular growth factor. In tumor settings, cancer cell production of PDGF-DD is known to stimulate tumor growth and angiogenesis through PDGFRβ. However, our data indicate that in tumors infiltrated by activated NK cells, PDGF-DD also exposes tumor cells to innate immune recognition through NKp44, inducing a proinflammatory reaction that restricts cancer cell growth in vitro and in vivo. Since the simian sarcoma virus contains a v-sis oncogene that encodes a functional homologue of PDGF (Deuel et al., 1983), NKp44-mediated recognition of PDGF-DD may reflect an evolutionary strategy for NK surveillance of transforming viruses. While studies have mostly focused on the impact of NK cells on Epstein-Barr virus-driven lymphomas (Dolcetti et al., 2013), future investigations will establish whether NK cells control other oncogenic viruses, what is the impact of NKp44 on their immunosurveillance and whether other NK cell receptors recognize viral growth factors and/or their cellular counterparts.
Meta-analysis of TCGA datasets suggests that PDGF-DD/NKp44 interaction may have clinical impact in the control of GBM. Higher expression of the canonical cytokine and cell cycle variates of NCR2 correlated with greater overall survival of GBM patients. Moreover, NKp44+ NK cells infiltrated GBM, although not in all cases, and PDGF-DD protein or PDGFD mRNA were expressed in glioma cell lines and primary glioblastoma tumor tissues (Lokker et al., 2002). While PDGF-DD was shown to promote mitogenic pathways in glioblastoma cells through PDGFRβ (Lokker et al., 2002; Nazarenko et al., 2012), it may also alert tumor-infiltrating innate immune cells through engagement of NKp44. Appropriate clinical studies beyond these correlative data will be important to test this hypothesis. We envision that therapies aimed at blocking PDGFRβ may favor PDGF-DD-mediated stimulation of NKp44 and control of GBM growth. Additionally, we have shown that NKp44-PDGF-DD interactions synergized with blockade of the inhibitory receptor CD96 in controlling lung metastatic melanoma and enhanced the control of subcutaneous melanoma by CpG-ODN treatment. Thus, combination of NKp44 activation therapies with checkpoint blockade or adjuvant therapy may be a valid strategy for control of GBM and other tumors infiltrated by NK cells.
Although many tumors express PDGF-D and NCR2, correlative studies of TCGA cohorts did not support a significant impact of PDGF-DD/NKp44 interactions with tumor progression beyond GBM. NK cells or ILCs may not infiltrate these tumors, the tumor environment may prevent NK cell activation or expression of NKp44. The continuing stimulation of NKp44 by PDGF-DD could result in exhaustion/anergy of innate immune cells and paradoxically promote tumor growth, as observed with chronic activation of other NK cell receptors (Groh et al., 2002). Sustained production of IFN-γ may also be co-opted in the tumor environment (Nirschl et al., 2017). Finally, it is possible that the PDGF-DD/NKp44 contribution is obscured by other concurrent innate and/or adaptive immune responses (Gajewski et al., 2013). Overall, the impact of PDGF-DD/NKp44 interactions may be more complex than just the control of cell growth observed in our experimental settings and may have diverse effects on tumor editing and progression in different contexts.
PDGF-DD contributes to angiogenesis, embryonal development of various organs, formation of the placenta during pregnancy, and supports wound healing (Li and Eriksson, 2003; Reigstad et al., 2005). In diseases, PDGF-DD has been implicated in vascular diseases (Chen et al., 2005), mesangioproliferative glomerulonephritis, and fibrosis (Reigstad et al., 2005). Polymorphisms in PDGFD have even been associated with coronary artery disease (Nikpay et al., 2015) and serum levels of IFN-γ (Ahola-Olli et al., 2017). Thus, PDGF-DD/NKp44 interactions play wider biological roles beyond cancer.
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Dr. Marco Colonna (mcolonna@wustl.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Mice
Mice were of mixed sexes. Mice within experiments were age and sex matched. All mice were between 8-12 weeks of age at the time off use. Mice used in this study were NCR2-tg and non-tg littermate mice. Mice were housed under specific pathogen free conditions. Mice did not undergo any procedures prior to their stated use. Sex matched littermate mice were randomly assigned to experimental groups for anti-CD96 and CpG 2216 DNA treatments. All studies performed on mice were done in accordance with the Institutional Animal Care and Use Committee at Washington University in St. Louis and approved all protocols used in this study.
Human tissues and primary cells
Tonsils were obtained from children undergoing elective tonsillectomy (Children’s Hospital, WUSM). Peripheral blood products were obtained from the pheresis unit of the WUSM Center of Advanced Medicine. Informed consent was obtained from all patients for the use of human tissue and cells. Human tissues (normal and cancer) for histology were obtained under the approval of the institutional review boards of Washington University in St Louis and the University of Brescia, respectively. Tonsilar lymphocyte suspensions and peripheral blood mononuclear cells (PBMC) were prepared as previously described (Cella et al., 2009, 2010). Briefly, CD56+ cells were enriched from PBMC and tonsil lymphocytes by magnetic cell sorting using CD56 MicroBeads (Miltenyi Biotec). NK cells and ILC subsets were sorted from CD56+ cells, as previously reported (Cella et al., 2010). Human CD4+ T cells were enriched from PBMC by magnetic cell sorting using CD4 MicroBeads (Miltenyi Biotec). Mouse LPLs were prepared as previously described (Lefrançois and Lycke, 2001). Formalin-fixed paraffin-embedded (FFPE) human tissues used for this study were retrieved from the archive of the Department of Pathology (ASST Spedali Civili di Brescia, University of Brescia, Brescia Italy) and included reactive lymphoid organs (tonsils and lymph nodes) and normal non-lymphoid tissue (colon, stomach, thyroid, salivary gland, breast, lung, liver, pancreas, skin, kidney, bladder, testis, prostate). PDGF-D analysis was also screened on a set of seventy-five human neoplasms (FFPRE blocks) including primary (13 cases) and metastatic (22 cases) melanoma, glioblastomas (5 cases), gastrointestinal stromal tumor (4 cases) and a set of carcinomas from different sites including lung (5 cases), ovary (2 cases), breast (2 cases), bladder (2 cases), colon (5 cases), prostate (6 cases), stomach (2 cases), kidney (5 cases), head and neck (2 cases). NKp44 staining was performed on frozen material from 10 cases of glioblastoma (6 cases, Brescia; four, Wash U), which included one case of gliosarcoma GBM variant (Wash U).
Cell lines
NKp30 and NKp44 reporter cells were maintained in 10% FBS in RPMI 1640 supplemented with sodium pyruvate, GlutaMAX, and penicillin/streptomycin. NKp44 and NKp30 reporter cells were based on the 2B4 NFAT-GFP cells developed by Arase et al (Arase et al., 2002). The sex of the mouse from which the 2B4 T cell hybridoma was derived has not been reported (Arase et al., 2002; Hedrick et al., 1982). Colo38 is a human female malignant melanoma cell line (Chang et al., 2005). Meljuso is a human female cutaneous melanoma cell line (Deschodt-Lanckman et al., 1990). OVCA is a human female ovarian serous adenocarcinoma cell line (Bast et al., 1981). MCF7 is a human female breast carcinoma cell line (Soule et al., 1973). NK92 is a human male NK cell lymphoblastic leukemia/lymphoma cell line (Gong et al., 1994). Jurkat is a human male T lymphoblastic leukemic line (Schneider et al., 1977). B16F10 is a male mouse melanoma cell-line (Nakamura et al., 2002). J558L are a mouse plasma cell myeloma cell line. The sex of the mouse from which the J558L myeloma cell line was derived has not been reported (Oi et al., 1983). EL4 are a mouse thymic lymphoma cell-line (Gorer, 1950). The sex of the mouse from which the EL4 cell line was derived has not been reported. The Phoenix amphotropic packaging cell-line and HEK293F are derivatives of the human female embryonic kidney (HEK) 293 cell-line (Lin et al., 2014). CHO are a female Chinese hamster ovary cell-line.
METHOD DETAILS
Preparation of NCR2 transgenic mice
The RP11-380E17 BAC (~300 kb) was chosen because it encoded the complete human NCR2 gene on chromosome 6p21.1 in addition to sufficient upstream (~142kb) and downstream (~142kb) sequence to include all the necessary elements required for correct expression of NKp44. The RP11-380E17 BAC clone was purchased from Invitrogen, isolated and purified using the Qiagen Maxi-prep kit. PCR using the following primers located in genomic regions covering NCR2: 5kb upstream (Forward, 5’-GTGGCAAGAGCTGAGCATCG-3’; Reverse, 5’-GCCCCAAGTTAGGGGGTGAC-3’), the ATG (Forward, 5’-GCTTGTGTGAGTGAGTGGCG-3’; Reverse, 5’-CTCTGCTGGACTGGCGATCT-3’), mid-gene (Forward, 5’-TGCTCGTCTGGTGGTGAGTG-3’; Reverse, 5’-CTCTGGCCGATTCCCCTCTG-3’), Stop codon (Forward, 5’-GGCGAGCAAGGCTTGGAAAC-3’; Reverse, 5’-CCCGCCACCTATGGACTCAC-3’), and 5 kb downstream (Forward, 5’-GGCTAAGAGGGGCCATCACG-3’; Reverse, 5’-AGGATACAGGGGCAGGTGTTG-3’), served to screen for positive BAC clones containing the complete NCR2 gene ready for pronuclear injection. Purified BAC DNA was then used to generate BAC transgenic mice on a CBA/C57BL/6J background in the Washington University School of Medicine Mouse Transgenic and Gene Targeting Core Facility using standard transgenic methods. PCR on tail-snip DNA using human-specific primers corresponding to an internal site on the transgene (Forward, 5’-ATCATTCTCCAGGCTCTCAGGCACAATCCA-3’; Reverse, 5’-CATGGTGACAGTGAAGAAGCCAGCATCAGG-3’) confirmed that the BAC had fully integrated. We used 2 transgenic founders to establish 2 independent transgenic lines for further characterization. We then crossed each of these lines further onto the C57BL/6J background to obtain mice homozygous for Klrb1 (encoding the NK1.1 antigen) at the C57BL/6J Natural Killer Receptor (NKR) locus. Mice were maintained on a standard 12 hour light/dark cycle with food and water available ad libitum. All procedures were conducted in strict accordance with the NIH Guide for the Care and Use of Laboratory Animals and all studies performed on mice were approved by the Washington University School of Medicine Institutional Animal Care and Use Committee.
Screening of the GNF secretomics library
A cell line with GFP reporter readout driven by NFAT-responsive promoter elements that stably expresses an integrated construct encoding a fusion protein of the NKp44 extracellular and transmembrane domains with the intracellular cytoplasmic tail of CD3ζ (NKp44 reporter cells) was generated for screening a library of secreted proteins (Gonzalez et al., 2010) for NKp44 ligands. Engagement of the chimeric NKp44-CD3ζ receptor results in GFP expression. GFP reporter cells expressing a fusion protein consisting of the NKp30 extracellular and transmembrane domains with the intracellular cytoplasmic tail of CD3ζ (NKp30 reporter cells) were also generated. Library proteins were coated onto a 384-well TC treated plate in duplicate at 5-fold dilution from a stock in a final volume of 25μl/well. Stock solutions varied in concentration ranging from ~0.1 to ~50 μM. The proteins were allowed to adsorb overnight at 4°C. The following day, protein coated plates were washed with 80ul of PBS and NKp44 or NKp30 reporter cells, previously cultured to 70% confluence in T175 flasks, were seeded into protein coated plates at a density of 35,000 cells per well in 50 μl volume of culture medium. After 24 hours incubation at 37°C, 5 μl/well of a 2 μg/ml DAPI dilution was added to each well for detection of dead cells, bringing the final volume/well to 55 μl. Flow cytometry was used to acquire GFP and DAPI signal from each well. Candidate proteins were retested in the same 384 well format using serial dilutions from library stocks as indicated. The GNF Secretomics library (Gonzalez et al., 2010) screened contains purified versions of known and predicted mouse and human secreted proteins as well as extracellular domains of single pass transmembrane proteins. Anti-NKp44 and anti-NKp30 were immobilized to plates using goat anti-mouse Ig and used as positive and negative controls, respectively. Results of initial screen are expressed as normalized fluorescence signal (NFS).
Immobilization of antibodies, proteins and peptides
2% BSA, or 5 μg/ml of proteins or antibodies were immobilized in 100μl PBS onto tissue culture (TC) or ELISA plates, as previously described (Barrow et al., 2011). Briefly, all wells were washed 3 times in Tris buffered saline (10 mM Tris-HCl pH 7.5, 150 mM NaCl) + 0.05% Tween-20 (TBS-T) to remove excess proteins before blocking in 5% BSA in TBS-T for 1 hour at 37°C prior to performing binding assays.
Solid-phase binding assay
Solid-phase binding assays were performed as previously described (Barrow et al., 2015). After blocking, NKp44-Fc fusion protein was diluted in 100μl of TBS-T + 0.1% BSA and incubated for 1 hour at room temperature (RT) in ELISA plate wells coated with the different proteins. Wells were then washed five times in TBS-T before incubating with 100μl 1:5000 HRP-conjugated goat anti-human Ig (Southern Biotech) in 100μl of TBS-T for 1 hour at RT. Wells were then washed a further five times in TBS-T before developing with O-Phenylenediamine dihydrochloride and the absorbance at 492 nM recorded using a plate reader. BSA, recombinant Chikungunya virus (CHIK) E2 protein and IL-17A were used as negative controls. Recombinant CUB-TEV-PDGFD contains a TEV cleavage site between CUB and GFD.
Calcium imaging using flexstation III
NK92 cells were loaded with 1 μM Fura-2 AM in Ringer’s buffer (135 mM NaCl, 5 mM KCl, 1 mM CaCl2, 1 mM MgCl2, 5.6 mM Glucose, and 10 mM Hepes, pH 7.4) for 40 min in the dark, washed, and then plated in 96-well plates at a density of 105 cells/well with primary antibodies. Fura-2 excitation ratios were measured by alternatively exciting the dye at 340 and 380 nm, at a frequency of ~1 image pair every 4 s and collecting emission at 510 nm using Flexstation III (Molecular Devices) equipped with SoftMax Pro 5 software (Molecular Devices). Recombinant PDGF-DD was injected after 180 secs to a final concentration of 1 μg/ml.
Immunoblotting
IL-2 cultured NK cells were serum-starved in RPMI 1640 medium for two hours at 37Ό prior to stimulation with 250 ng/ml PDGF-DD for the indicated time points. Following PDGF-DD stimulation, NK cells were placed on ice, washed once in ice cold PBS before resuspending in ice cold lysis buffer (50 mM Tris pH 8.0, 150 mM NaCl, 1% Triton X-100) containing 1 mM PMSF, 1 mM Na3VO4, leupeptin and apoprotinin (1 μg/ml each), and 1× phosphatase inhibitor cocktail 3 (Sigma Aldrich Cat. Number P0044) on ice for 10 mins. After centrifugation at 13,000 rpm for 15 mins at 4°C to pellet cellular debris, sample supernatants were removed and 4× SDS-PAGE loading buffer (200 mM Tris-HCl pH 6.8, 8% sodium dodecyl sulfate, 0.4% bromophenol blue, 40% glycerol and 400 mM dithiothreitol) added. Lysates were heated to 95°C for 10 minutes and run on a 4-12% bis-tris gel (Nupage). Proteins were transferred to nitrocellulose and blocked for 1 hour at RT in 5% BSA in TBS-T. Membranes were incubated in primary antibody overnight at 4°C. Membranes were subsequently washed and incubated in Goat anti-rabbit HRP (Southern Biotech) for 1 hour at RT, washed, and developed using SuperSignal West Pico Chemiluminescent Substrate.
Plasmids
Human PDGFD (residues E184-R370) and NKP44 (residues G18-S130 including a C-terminal His6 tag) were cloned into the Ndel and Notl restriction sites of the pET21 bacterial expression vector. Full-length PDGFD (residues 1-370), with or without an internal Tobacco Etch Virus (TEV) protease recognition sequence (ENLYFQS) preceding the cytokine domain (R254), a C-terminal TEV protease site and His6 tag, was cloned downstream of the CMV promoter in the IRES-RFP mammalian expression vector pFM-1.2. The NKp44 ectodomain and transmembrane region was cloned in frame with human CD3ζ into the pEF6/V5-His mammalian expression vector. The NKp44 extracellular domain was cloned into the pCD4 vector to make the resulting NKp44-Fc vector containing the exons for the hinge, CH2 and CH3 regions of human IgG1, the guanosine phosphotransferase gene conferring resistance to mycophenolic acid, and the κ promoter for expression in mouse myeloma. Cloning of the CRTAM-Fc fusion protein to detect expression of Necl-2, has been described before (Boles et al., 2005). Full-length human PDGFD or PCNA were cloned into the EcoRI and Notl sites of the pMX-IRES-eGFP retroviral expression vector, respectively.
Generation of stable cell-lines
GFP reporter cells were generated by electroporation of the NKp44-CD3ζ fusion construct or NKp30-CD3ζ fusion constructs, respectively, and selection of stable clones with Blasticidin. J558L myeloma cells were electroporated with pCD4 NKp44-Fc vector and secreting transfectants selected in mycophenolic acid xanthine (Sigma-Aldrich). For the generation of stable B16F10 and EL4 cells, the Phoenix amphotropic retroviral packaging cell-line was transfected with either pMX-IRES-eGFP or pMX-IRES-eGFP (Kitamura et al., 2003) encoding either PDGFD or PCNA, respectively. Cells were infected with the resulting amphotropic retroviruses and single-cell clones stably expressing similar levels of GFP selected in the presence of puromycin.
Recombinant Proteins
Human PDGFD (E184-R370) and NKp44 (G18-S130) were expressed in BL21 (DE3) codon plus E. coli cells by autoinduction (Studier, 2005). The proteins were refolded from inclusion bodies denatured in 7 M guanidinium hydrochloride, 30 mM β-mercaptoethanol centrifuged for 10 min at 4°C and diluted to 2 M guanidinium hydrochloride in 50 mM sodium acetate pH 5.2. The supernatant was filtered and refolded by rapid, serial dilution (1 ml injections, hourly) in 1 liter of refolding buffer (400 mM L-arginine, 100 mM Tris-base pH 8.3, 2 mM EDTA, 0.5 mM oxidized glutathione, 5 mM reduced glutathione, and 0.2 mM phenylmethanesulfonyl fluoride) at 4Ό. The recombinant proteins were concentrated using a stirred cell concentrator with YM10 membrane (Millipore), centrifuged to remove aggregates and purified on an S-75 size exclusion chromatography column equilibrated in 20 mM HEPES pH 7.4 and 150 mM NaCl, 0.01% sodium azide. Full-length PDGFD and TEV cleavable PDGFD proteins were transiently expressed in serum-free HEK-293F suspension cells and purified by Ni-NTA agarose affinity (Qiagen) and Superdex 200 gel filtration chromatography, cleaved where appropriate by the addition of TEV protease, and samples at 0 and 24 hours were analyzed by SDS-PAGE. Recombinant PDGF-DD used for in vitro cell stimulations was purchased from R&D systems. NKp44-Fc fusion protein was purified from J558L TC supernatants, as previously described.
Surface plasmon resonance
Affinity analysis was performed using SPR (BIAcore T100, GE Company) at 25G in 10 mM HEPES, 150 mM NaCl, 3 mM EDTA and 0.0 5% v/v surfactant P20. PDGFD, PDGF-DD or a control protein (IL17A or Chikungunya E2 glycoprotein) was immobilized (500-1000 Response Units) onto a CM5 chip (GE healthcare) using amine-coupling chemistry. Bacterial expressed NKP44 (residues 18-130 with a C-terminal His6 tag) was injected at a flow rate of 20 μl/min at concentrations ranging from 0.036 μM to 36.8 μM for 5 minutes to saturate binding and then allowed to dissociate for 20 minutes. Under these conditions, regeneration of the chip surface was not necessary. The observed binding curves were double referenced to a control protein (human IL-17A or Chikungunya E2 glycoprotein) as well as buffer in the absence of NKp44. Curves were analyzed by a steady-state fit for a 1:1 interaction, and a nonlinear least squares fit was used to evaluate the fit of the curve to the observed data.
Cell culture
Unless indicated otherwise stated, all cells were cultured in a 5% CO2 incubator at 37°C. All primary cells and cell-lines were cultured in RPMI 1640 (Sigma-Aldrich) supplemented with kanamycin sulfate, sodium pyruvate, GlutaMAX, non-essential amino-acids and 10% serum (complete medium), and either 10% fetal bovine serum (FBS) for primary cells or 10% bovine calf serum (BCS) for tumor cell-lines with the exception of J558L cells stably expressing NKp44-Fc that were cultured in complete RPMI 1640 medium containing 1% BCS. NK92 and primary mouse and human NK cells were cultured with IL-2 (10 ng/ml). CD3−CD19− NKp44+CCR6+ tonsilar cells were cultured with human IL-2 (10 ng/ml) and IL-1β (10 ng/ml) for 21 days prior to stimulation. For microarray analysis, CD3−CD19− NKp44+CD103+ ILC1 were expanded in IL-2 (10 ng/ml, Peprotech) for 10 days prior to stimulation.
Reporter cell assays
NKp44 GFP reporter cells (105) were mixed 1:1 with target cells or directly stimulated with proteins in 96-well plates for 16h before immunostaining with APC-conjugated anti-NKp44 antibodies and the expression of GFP subsequently analyzed by flow cytometry.
Generation of cytokine-conditioned TC supernatants
IL-2 cultured human NK cells were incubated with 250 ng/ml PDGF-DD (R&D Systems) in 24-well plates for 16h in the presence of 1 μg/ml IgG1 (NKDD+IgG sup) or blocking anti-NKp44 3.43.14 mAb (NKDD+α-NKp44 sup). After 16h, supernatants were pooled, centrifuged and filtered to remove cellular debris, and then stored at −80°C until further use. B16-pMX and B16-PDGFD stable cell-lines were cultured in 6-well plates for 72h. After 72h, supernatants were centrifuge, filtered and then stored at +4°C until further use.
Tumor cell cycle analysis
For analysis of the cell cycle by flow cytometry, Meljuso, cells were cultured in 1:2 NK cell-conditioned TC supernatants (NKDD+IgG sup or NKDD+α-NKp44 sup) or complete RPMI 1640 medium with 10 ng/ml each of human IFN-γ and TNF-α (Peprotech) for 48h. Meljuso cells were pulsed with 1 mM BrdU prior to removal from TC plates using trypsin/1 mM EDTA. Single cell suspensions were then washed twice to remove excess BrdU prior to staining with 7-aminoactinomycin D (7-AAD) and APC-conjugated antibodies to BrdU using the Becton Dickinson APC BrdU Flow kit, according to the manufacturer’s instructions and then analyzed by flow cytometry. Electronic gates were set according to each stage of the cell cycle and the percentage of events in each gate is indicated.
Tumor growth inhibition assays
Meljuso and Colo38 cancer cell-lines were cultured in complete medium or 1:2 NK cell-conditioned supernatants (NKDD+IgG sup or NKDD+α-NKp44 sup) for 48h and 72h, respectively, with or without 2 μg/ml of blocking antibodies to IFN-γ and TNF-α, respectively in 12-well plates. Cells were removed using a trypsin/EDTA solution to generate single-cell suspensions and washed twice in complete medium before enumerating the number of live cells by trypan blue exclusion using a hemacytometer (Thermofisher Scientific). A total of 4 × 104 cells per well were then cultured in 24-well plates in 1ml complete RPMI 1640 medium in the absence of cytokines. At the end of each passage, TC supernatants were removed for the detection of inflammatory cytokines and chemokines, and the total number of cells per well enumerated by trypan blue exclusion before replating at 4 × 104 cells per well in 24-well for the next passage. The number of cells at the end of each passage (as determined by confluence/cell density in control wells) was recorded for a total of 3 passages and the percentage growth relative to control medium determined.
Sorting of ILC subsets
Enriched tonsilar CD56+ cells were stained with PerCP-Cy5.5-conjugated antibodies to CD3 and CD19, PE-conjugated anti-NKp44, and biotin-conjugated antibody to CCR6 followed by APC-conjugated streptavidin, and FITC-conjugated anti-CD103 and sorted on a FACS Aria II (BD Biosciences) into CD3− CD19−NKp44+CCR6+ cells (NKp44+ ILC3) and CD3−CD19−NKp44+CD103+ cells (NKp44+CD103+ ILC1). Purities of sorted populations were typically above 99%.
NK cell treatments
Unless indicated otherwise, NK cells were stimulated with 250 ng/ml recombinant PDGF-DD (R&D Systems) or 1 μg/ml CUB-TEV-PDGFD or neat B16F10 TC supernatants with or without addition of soluble blocking antibodies (5 μg/ml) for 16h in all assays. For CD107 degranulation assays, CD3−CD56+ NK cells (500,000 cells) were stimulated in 96-well round bottom plates. FITC-conjugated anti-CD107a antibody was added at the start of the culture. Cells were cultured for 5 hours with 2 μM monensin (Sigma Aldrich) added after the first hour of culture. NK cells were then stained with APC-conjugated mAb to CD56 prior to flow cytometry. For intracellular cytokine staining, NK cells were stimulated in 96-well flat bottom plates. Cells were cultured for 8h with 2 μM monensin added after two hours of culture, before surface staining with PE-conjugated anti-NKp44 and PE-Cy7-conjugated anti-CD56 followed by staining for intracellular cytokines with APC-conjugated antibodies to IFN-γ and FITC-conjugated antibodies to TNF-α. For analysis of the upregulation of cell surface NKp44 on mouse NK cells, splenocytes were isolated from NCR2-tg and non-tg mice and, following red blood cell lysis, cultured in 10 ng/ml each of human IL-2 and IL-15 (both Peprotech) for a total of four days with some splenocyte cultures additionally stimulated with 4 ng/ml mouse IL-1β and 10 ng/ml mouse TNF-α (both Peprotech) after 2 days before immunostaining staining with APC-conjugated antibody to NK1.1, FITC-conjugated antibody to mouse CD3 and PE-Cy7-conjugated antibody to NKp44 and DAPI and cell surface NKp44 expression on live (DAPI-) CD3−NK1.1+ NK cells determined by flow cytometry.
ILC1 and ILC3 treatments
Unless indicated otherwise, ILCs were stimulated with 250 ng/ml PDGF-DD with or without soluble blocking antibodies (5 μg/ml) for 16 h for all assays. For detection of intracellular cytokines in ILC3, enriched CD56+ tonsilar cells were stimulated with 250 ng/ml PDGF-DD for 6h with 2 μM monensin added after the two hours of culture, before either surface staining with APC-conjugated anti-NKp44 and the expression of intracellular cytokines detected with PE-conjugated antibodies to IL-22 and FITC-conjugated antibodies to TNF-α (as in Figure 3A) or surface staining with PerCP-Cy5.5-conjugated antibodies to CD3 and CD19, APC-conjugated anti-NKp44, PE-Cy7-conjugated anti-CD56, BV421-conjugated anti-CCR6 and BV605-conjugated anti-CD103 and the expression of intracellular cytokine detected with FITC-conjugated antibodies to TNF-α (as in Figure 3B).
Detection of cancer cell-surface ligands for NK cell receptors
Colo38 and Meljuso cells were cultured with 1:2 NK cell-conditioned supernatants (NKDD+IgG sup or NKDD+α-NKp44 sup) in 12-well TC plates for 48h before cells were removed using PBS/1 mM EDTA and stained with FITC-conjugated antibodies to CD95/Fas and PE-conjugated antibodies to CD112 and either biotin-conjugated antibodies to CD155/PVR or CRTAM-Fc followed by biotin-conjugated anti-human IgG. Biotin-conjugates were detected with APC-conjugated streptavidin. B16F10 cells were cultured with 10 ng/ml of either murine TNF-α, IFN-γ or TNF-α and IFN-γ (both Peprotech) for 48h prior to staining with FITC-conjugated antibodies to CD95/Fas and biotin-conjugated antibodies to CD155 followed by APC-conjugated streptavidin and propidium iodide. Cells were washed and cell surface expression of the different antigens on live (PI−) cells determined by flow cytometry. Percentage expression is indicated in each gate or quadrant.
HIV gp41 peptide stimulation of human CD4+ T cells
Human CD4+ T cells were enriched from peripheral blood by magnetic cell sorting using CD4 MicroBeads (Miltenyi Biotec) and 105 human CD4+ T cells were stimulated with or without 5 μg/ml of HIV gp41-derived peptide (NH2-PWNASWSNKSLDDIW-CO2H) in 200 μl complete RPMI 1640 (Vieillard et al., 2005). After 16h incubation, CD4+ T cells were immunostained with 10 μg/ml NKp44-Fc followed biotin-conjugated anti-human IgG and APC-conjugated streptavidin and FITC-conjugated anti-CD4 and then analyzed by flow cytometry. The percentage expression is indicated in each gate.
Analysis of cellular cytokine secretion
IL-2 cultured NK cells or converted tonsil ILC3 (IL-2 and IL-1β) were stimulated in 96-well plates (105 cells/well) with or without blocking antibodies and the secretion of IFN-γ and TNF-α in the TC supernatants determined using the Th1/Th2/Th17 cytometric bead array (BD Biosciences). In some experiments, IFN-γ and TNF-α were induced by cross-linking NKp44 with GαM + α-NKp44 as a surrogate NKp44 ligand. NK cells and ILCs were kept in cytokine-containing medium throughout the assay. PMA/ionomycin was included in some experiments as positive control for cytokine secretion. In tumor cell growth inhibition assays, the secretion of IL-6 and IL-8 by tumor cell-lines at the end of each passage was determined using the Human Inflammatory cytokine CBA (BD Biosciences). The concentration of IL-6 or IL-8 present was normalized relative to the cell number following each passage.
Microarray and data analysis
Tonsilar ILC1 cells were sorted from two donors as previously described (Cella et al., 2010; Fuchs et al., 2013) (Cella et al., 2010; Fuchs et al., 2013). Briefly, expanded tonsil ILC1 were stimulated with or without 250 ng/ml PDGF-DD for 16h before RNA isolation using the RNeasy Plus Micro Kit (QIAGEN), amplified, and hybridized to the Affymetrix Human Gene (v.1.0) ST arrays. RNA yields from each subset were comparable. Array data were analyzed as previously described (Robinette et al., 2015). Pathway analysis of microarray data was performed using IPA software (Qiagen).
RNA-seq and data analysis
Confluent Colo38 and Meljuso (both melanoma), OVCA (ovarian) and MCF7 (breast) cancer cell-lines were cultured with 1:2 NK cell-conditioned supernatants (NKDD+IgG sup or NKDD+α-NKp44 sup), or complete medium in 12-well TC plates (~0.5-1.0 x 106 cells) for 48h before RNA isolation. IL-2 cultured NK cells (1 x 106) from four different donors were stimulated with 250 ng/ml PDGF-DD or PMA/ionomycin or left unstimulated in 12-well plates for 8 h before RNA isolation. Total RNA was isolated from NK cells and cancer cell-lines using Trizol followed by Qiagen RNAeasy columns. 500 ng total RNA was used to make lllumina libraries using the TruSeq Stranded Total RNA library prep kit. Libraries were sequenced on an Illumina HiSeq 1000, single-end 50 bp run, to a sequencing depth of between 28 million and 60 million mapped reads per sample. Reads were aligned to a transcriptome file with BWA v0.5.9 (Li and Durbin, 2009), and the number of reads aligning to each transcript counted with SAMtools (Li et al 2009). These counts were converted to Reads per Million (RPM) by dividing each count by the total number of mapped reads for that sample, and multiplying by 106. Gene expression signatures were derived from RNA-seq data using the RPM values of the transcripts for each NK cell donor (Fig 2I). The up- or down-regulation of a transcript in response to PDGF-DD stimulation was determined according to the following criteria. First, a background level of gene expression was established in unstimulated NK cells from the four different donors cultured in IL-2. Transcripts in PDGF-DD-stimulated NK cells with at least eight RPM and at least two-fold expression over unstimulated NK cells were declared to be upregulated in that sample. Similarly, transcripts from PDGF-DD-stimulated NK cells with background expression of at least eight RPM and at most half the expression in unstimulated NK cells were declared to be downregulated. Finally, gene expression was declared to be upregulated upon stimulation with PDGF-DD, if upregulated in at least three of the donor samples, and downregulated, if downregulated in at least two donors. Using gene expression data for each cancer cell-line (Fig 4I; MCF, Meljuso, OVCA, or Colo38), the upregulation or downregulation of a gene in response to treatment with supernatants from PDGF-DD-stimulated NK cells (NKDD+IgG sup) was determined according to criteria analogous to that used for the stimulation of NK cells with PDGF-DD or PMA/i described above, where the background expression of a transcript was established by taking the geometric mean of its RPM in media or upon stimulation with control supernatant from NK cells stimulated with PDGF-DD but with the addition of anti-NKp44 blocking mAbs (NKDD+αNKp44 sup). Pathway analysis of RNA-seq data was performed using IPA software (Qiagen).
Analysis of TCGA datasets
TCGA GBM mRNA gene expression data obtained using the Affymetrix HT Human Genome U133a microarray platform (n = 539 patients) was downloaded through the UCSC data portal (https://xenabrowser.net) and matched to the gene expression data (Fig. 4C, D). 7 of 9 cytokine genes (Fig 4C) and 27 of 34 cell cycle genes (Fig. 4D) were matched with the GBM cohort. Scatter plots were generated plotting a cytokine or cell cycle gene with NCR2. Simple linear regression lines were fitted with correlation between two genes gauged by the Pearson correlation coefficient. The canonical correlation analysis (CCA), a dimension reduction statistical technique, was performed to investigate the overall correlation between a set of genes with NCR2. For CCA, each of the cytokine and cell cycle gene were at centered to have zero mean. A canonical cytokine variate was constructed as the linear combination of the set of centered cytokine genes and a canonical cell cycle variate as the linear combination of the centered cell cycle genes such that the resulting canonical cytokine and cell cycle variate each showed maximized positive correlation with NCR2. The canonical correlation between the constructed canonical cytokine variate with NCR2 and the canonical cell cycle variate with NCR2 were finally reported as a measure of potential absolute magnitude of the overall correlation between NCR2 with the set of cytokine genes and cell cycle genes. The canonical variates are always derived from CCA as being positively correlated with NCR2. The direction of each individual gene’s associated linear coefficient, however, indicate the positive or negative correlation direction with the canonical covariate and thus with NCR2.
Tissue immunostaining
For PDGF-D immunostaining of normal and cancer tissues, 4 μM thick sections were deparaffinized with xylene and rehydrated with decreasing concentrations of ethanol. Tissue sections were incubated with rabbit anti-PDGF-D antibody (dilution 1:250) after appropriate antigen retrieval (microwave oven; 2 x 5 minutes at max Watt and 3 x 5 minutes at 750 Watt in EDTA buffer pH 8.0) and revealed by Labeled Polymer-HRP followed by DAB. Immunostained sections were photographed using the DP-70 Olympus digital camera mounted on the Olympus BX60 microscope. For NKp44 immunostaining of GBM, 7 μM frozen sections were dried overnight before fixing in ice cold acetone for 10 mins followed by rehydration in PBS. Slides were blocked in 10% goat serum in PBS before incubating with anti-NKp44 and anti-CD3ε antibodies followed by Alexa 488-conjugated anti-mouse IgG1 and Alexa 568-conjugated anti-mouse IgG2a secondary antibodies supplemented with DAPI. Immunostained sections were imaged on a Zeiss LSM880 Laser Scanning Confocal Microscope. Images were then processed with Zen 2.1 (Zeiss).
Tumor cell injections
Adherent B16-pMX or B16-PDGFD stable cells were removed from TC plates using a Trypsin/1 mM EDTA solution and washed twice in PBS before twice passing through a 100 μM cell filter and resuspended in PBS, and 200 μl containing a total of 2 x 105 cells injected into the tail vein of either NCR2-tg or non-tg littermate mice, respectively. After 17 days, total surface lung metastases were counted. To establish subcutaneous tumors in mice, 2 x 106 cells in 200 μl of PBS were implanted into the rear hind flanks of mice. Tumor growth was monitored by measurement of tumor size with a caliper every other day. Tumor volume was determined by the formula: length x width2/2. At the experimental end point, mice were sacrificed, and tumors were removed and processed for flow cytometric analysis.
Measurement of lung tumor area
Formalin-fixed and paraffin embedded mouse lung tissues were sectioned at 3-4 μM and the slides stained with hematoxylin and eosin (H & E). H & E Slides were digitalized using an Aperio ScanScope CS Slide Scanner (Aperio Technologies) at 40X magnification. Individual tumors were manually selected using Aperio ImageScope (Leica Biosystems Imaging); a software that automatically provides the value of every selected tumor area in μM2 as output.
Quantitative RT-PCR
Total RNA was isolated with TRIzol Reagent (Invitrogen) and single-strand Cdna synthesized using Superscript III reverse transcriptase kit (Invitrogen). Real-time PCR was performed using SYBR Green real-time PCR master mix (Thermo-Fisher) using a StepOnePlus detection system (Applied Biosystems). Primers were to mouse interferon-γ and TNF-α and cyclophilin was used as housekeeping gene.
GENERAL STATISTICAL METHODS
Data in figures are presented as mean ± SEM. Statistical significance was determined using GraphPad Prism 7.0. Statistical differences were determined by two-tailed Student’s t-test (between two groups) and a one-way ANOVA among multiple groups and a P-value of < 0.05 was considered significant.
DATA AND SOFTWARE AVAILABILITY
Microarray data has been deposited at GEO:GSE107043.
RNA-seq data has been deposited at GEO:GSE107047.
Supplementary Material
Figure S1. NKp44 does not interact with PCNA, influenza virus hemagglutinins (HA) or the putative NKp44 ligand upregulated by an HIV gp41-derived peptide. Related to Figure 1.
(A) Generation of EL4 cell-line clones stably over-expressing PCNA. Transfection of constructs encoding nuclear PCNA into tumor cell-lines was proposed to induce cell-surface PCNA expression (Rosental et al., 2011). EL4 cells were transduced with retrovirus-containing supernatants generated from the transfection of phoenix amphotropic packaging cells with a pMX-PCNA-IRES-eGFP retroviral vector and five single-cell clones that stably over-express high levels of GFP were selected (EL4-PCNA cells). The mean fluorescent intensity (MFI) of GFP expression for each clone is displayed.
(B) PCNA is not expressed on the cell-surface of parental EL4 cells or EL4-PCNA cell clones as determined by staining with anti-PCNA mAb. The Percentage of PCNA+ cells are indicated. Anti-Thy1.2 is used as positive control.
(C) EL4-PCNA cell clones do not activate NKp44-GFP reporter cells. 105 NKp44-GFP reporter cells were mixed 1:1 with either EL4 or EL4-PCNA cell clones and incubated overnight before determining GFP expression on NKp44+ cells by flow cytometry. PDGF-DD was used to stimulate NKp44-GFP reporter cells as a positive control (+ PDGF-DD). Percentage of GFP+ cells are indicated.
(D) Influenza virus HA does not interact with NKp44. CHO cells were transiently transfected with expression plasmids encoding either the Hong Kong/97 H5 (HK/97), Vietnam/04 H5 (Viet/04) or WSN/33 H1 influenza virus type A HAs (kindly provided by Adolfo Garcia-Sastre). Binding of anti-HA antibodies (left histograms) and NKp44-Fc (right histograms) to transfected CHO cells was determined by flow cytometry. As negative controls, CHO cells were either mock transfected with pcDNA3.1 or left untransfected. MFI of HA expression for each transfection is displayed.
(E) CHO cells transfected with influenza HAs do not activate NKp44-GFP reporter cells. 105 NKp44-GFP reporter cells were mixed 1:1 with CHO cells transfected with plasmids encoding the different influenza HAs in 96-well plates, incubated for 16h and GFP expression from NKp44-GFP reporter cells determined by flow cytometry. As negative controls, CHO cells were either mock transfected or left untransfected. PDGF-DD was used to stimulate NKp44-GFP reporter cells as a positive control. Percentages of GFP+ cells are indicated.
(F, G) The HIV gp41 peptide described by Vieillard et al. does not upregulate an NKp44 ligand (NKp44L) on human CD4+ T cells. Human CD4+ T cells from two different donors (d1, d2) were incubated overnight with (+) or without (−) the HIV gp41-derived peptide (HIV gp41) that was reported to upregulate an NKp44L (Vieillard et al. 2005), later identified as the nuclear antigen MLL5 (Baychelier et al., 2013). NKp44-Fc binding to CD4+ T cells (dotplots) (F) or GFP expression from NKp44-GFP reporter cells mixed 1:1 overnight with CD4+ T cells (G) either unpulsed or pulsed with HIV gp41 peptide. PDGF-DD was used to stimulate NKp44-GFP reporter cells as a positive control.
(H) Jurkat T cells do not activate NKp44-GFP reporter cells. NKp44-GFP reporter cells were mixed 1:1 overnight with or without Jurkat T cells, which were reported to express MLL5 on the cell surface (Baychelier et al., 2013). GFP expression was measured by flow cytometry. PDGF-DD was used to stimulate NKp44-GFP reporter cells as a positive control.
Figure S2. Expression of NKp44 and PDGFR-α/β in human NK cells. Related to Figure 2.
(A) Polyclonal NK cells cultured in IL-2 medium were stained either with isotype control mAbs or mAbs to CD3, CD56 in combination with mAbs to NKp44, PDGFRα or PDGFRβ. CD3−CD56+ human NK cells express NKp44 but not PDGFRα or PDGFRβ.
(B) PDGF-DD stimulates dose-dependent NK cell surface CD107a expression.
(C) Cell surface CD107a expression by PDGF-DD-stimulated NK cells. Upper panels, representative dotplots and percentage expression in each gate; lower panel, quantification. Cell-surface CD107a is blocked by soluble anti-NKp44. In the absence of PDGF-DD, CD107a expression was induced by mAb-mediated cross-linking of NKp44 (α-NKp44 + goat anti-mouse - GαM). Induction of CD107a by PMA/i was used as positive control.
(D) Generation of a TEV cleavable PDGF-D construct (CUB-TEV-PDGFD). After expression and purification from 293F cells, CUB-TEV-PDGFD proteins were incubated with (+TEV) or without (−TEV) TEV protease at 30°C for 0 or 24 hours (h). Samples were then boiled in SDS-PAGE loading buffer with (+) or without (−) DTT before resolving on 4-10% SDS-PAGE gels (Molecular mass, kD).
(E) IFN-γ and TNF-α secretion by NK cells is enhanced by TEV cleavage of CUB-TEV-PDGFD. Recombinant PDGF-DD was added to some cultures as positive control. Cleavage of CUB-TEV-PDGFD by proteases in serum containing medium likely explains cytokine production by NK cells in wells stimulated with CUB-TEV-PDGFD without TEV protease.
(F) The enhanced secretion of IFN-γ and TNF-α by NK cells stimulated with CUB-TEV-PDGFD +/−TEV wells is blocked by anti-NKp44 and anti-PDGF-D mAbs. Data are represented as mean (n = 3) ± SEM. (****, p ≤ 0.0001).
Figure S3. PDGF-DD binding to NKp44 induces TNF-α secretion by ILC3. Related to Figure 3.
Representative dotplots of intracellular TNF-α and IL-22 content and cell surface NKp44 from freshly isolated tonsil ILC3 stimulated with PDGF-DD and/or IL-23. The percentage of cytokine-positive cells is indicated in each quadrant.
Figure S4. Ingenuity Pathway Analysis of growth arrested cancer cell-lines exposed to supernatants from PDGF-DD activated NK cells. Related to Figure 4.
Top 10 highest scoring IPA canonical pathways generated from transcripts differentially expressed (±1.5-fold) in cancer cells cultured in NKDD+IgGsup versus control NKDD+α-NKp44 sup. Ratios in columns indicate total NKDD+IgG sup-regulated genes to the total number of genes in each pathway.
Figure S5. Expression of PDGF-D in normal and cancerous human tissues. Related to Figure 5.
(A-I) Sections from normal tissues are derived from: (A) stomach, (B) skin, (C) colon, (D) bladder, (E) lung, (F) kidney, (G) prostate, (H) pancreas, and (I) testis. Reactivity is evident in tissue epithelial compartments (brown staining). Scale bar = 100 μM.
(J-X) Sections from human carcinomas are from different primary sites including: (J) head and neck, (K, L) lung, (M) bladder, (N, O) colon, (P) breast, (Q) ovarian cancer, (R, S) primary melanoma, (T-X) melanoma metastasis to the (T, U) skin, (V) lung, (W) lymph nodes and (X) brain. All sections were stained with anti-PDGF-D antibody. Scale bar = 100 μM.
Figure S6. Reduced dissemination of tumor cells expressing PDGFD in vivo in a second NCR2-tg line. Related to Figure 6.
(A, B) Representative dotplots and quantification of NKp44 expression in CD3− NK1.1+ (NK) cells isolated from the spleen and mesenteric lymph nodes (mLN) of a second NCR2-tg founder line (founder #2). The percentages of cells are indicated in each gate.
(C, D) Representative dotplots and quantification of NKp44 expression in ILC1/NK cells, ILC2, CCR6+ ILC3, NKp46+ ILC3, and NKp46−CCR6− (DN) ILC3 isolated from the small intestine of NCR2-tg mice. The percentages of cells are indicated in each gate. Lamina propria mononuclear cells were stained for T-bet, Roryt, GATA3, NKp46, NK1.1, CCR6, lineage markers (CD3, CD19, CD5) and analyzed by flow cytometry. Cells were gated on Lineage− cells. ILC1/NK: T-bet+NK1.1+GATA3−Rorγt−; ILC2: GATA3+T-berRoryt−; ILC3: Roryt+GATA3int.
(E, F) Tissue culture supernatants from B16-pMX control cells do not stimulate IFN-γ or TNF-α unless 250 ng/ml recombinant PDGF-DD (+ PDGF-DD) is added to B16-pMX control supernatants (sup) as a positive control for NK cell activation.
(G) Quantification (upper panel) and representative photos (lower panel) of surface metastases from the lungs of non-tg and NCR2-tg (founder #2) at Day 17 post-injection with 2 × 105 B16F10 cells stably expressing PDGFD (B16-PDGFD) (*, p ≤ 0.05).
(H) Mean tumor area (μM2) ± SEM and representative histochemical images of lung metastases from non-tg (n = 7) and NCR2-tg (founder #2; n = 10) mice injected with B16-PDGFD cells.
Highlights.
PDGF-DD is a ligand for the human NK/ILC receptor NKp44 encoded by NCR2
PDGF-DD-NKp44 interaction stimulates NK cell secretion of IFN-γ, TNF and chemokines
PDGF-DD-induced NK cell cytokines trigger tumor cell cycle arrest
NCR2 transgenic mice control the growth of PDGF-DD expressing cancer cells in vivo
Acknowledgments
We would like to thank Susan Gilfillan, Bob Schreiber, Gavin Dunn, Sheila Stewart, Fabio Luna, Whitney Barnes, Todd Fehniger and Maximilian Rosario for helpful experimental advice and comments. A.D.B. was awarded a Marie Curie International Outgoing Fellowship. M.B. is supported by Fondazione Beretta (Brescia, Italy). W.V. is supported by AIRC (IG 2014/15378) and Ministero della Salute (Ricerca Finalizzata RF-2010-2315888). M.Cella is supported by NIH grant RO1 CA176695.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Supplementary Information
Supplementary Information includes 6 supplementary figures and 3 tables.
References
- Ahola-Olli AV, Würtz P, Havulinna AS, Aalto K, Pitkänen N, Lehtimäki T, Kähönen M, Lyytikäinen L-P, Raitoharju E, Seppälä I, et al. (2017). Genome-wide Association Study Identifies 27 Loci Influencing Concentrations of Circulating Cytokines and Growth Factors. Am. J. Hum. Genet 100, 40–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrae J, Gallini R, and Betsholtz C (2008). Role of platelet-derived growth factors in physiology and medicine. Genes Dev. 22, 1276–1312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arase H, Mocarski ES, Campbell AE, Hill AB, and Lanier LL (2002). Direct recognition of cytomegalovirus by activating and inhibitory NK cell receptors. Science 296, 1323–1326. [DOI] [PubMed] [Google Scholar]
- Barrow AD, Raynal N, Andersen TL, Slatter DA, Bihan D, Pugh N, Cella M, Kim T, Rho J, Negishi-Koga T, et al. (2011). OSCAR is a collagen receptor that costimulates osteoclastogenesis in DAP12-deficient humans and mice. J. Clin. Invest 121, 3505–3516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barrow AD, Palarasah Y, Bugatti M, Holehouse AS, Byers DE, Holtzman MJ, Vermi W, Skjødt K, Crouch E, and Colonna M (2015). OSCAR Is a Receptor for Surfactant Protein D That Activates TNF-α Release from Human CCR2+ Inflammatory Monocytes. J. Immunol. Baltim. Md 1950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bast RC, Feeney M, Lazarus H, Nadler LM, Colvin RB, and Knapp RC (1981). Reactivity of a monoclonal antibody with human ovarian carcinoma. J. Clin. Invest 68, 1331–1337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baychelier F, Sennepin A, Ermonval M, Dorgham K, Debré P, and Vieillard V (2013). Identification of a cellular ligand for the natural cytotoxicity receptor NKp44. Blood 122, 2935–2942. [DOI] [PubMed] [Google Scholar]
- Bergsten E, Uutela M, Li X, Pietras K, Ostman A, Heldin CH, Alitalo K, and Eriksson U (2001). PDGF-D is a specific, protease-activated ligand for the PDGF beta-receptor. Nat. Cell Biol. 3, 512–516. [DOI] [PubMed] [Google Scholar]
- Bezbradica JS, and Medzhitov R (2012). Role of ITAM signaling module in signal integration. Curr. Opin. Immunol 24, 58–66. [DOI] [PubMed] [Google Scholar]
- Blake SJ, Dougall WC, Miles JJ, Teng MWL, and Smyth MJ (2016). Molecular Pathways: Targeting CD96 and TIGIT for Cancer Immunotherapy. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 22, 5183–5188. [DOI] [PubMed] [Google Scholar]
- Boles KS, Barchet W, Diacovo T, Cella M, and Colonna M (2005). The tumor suppressor TSLC1/NECL-2 triggers NK-cell and CD8+ T-cell responses through the cell-surface receptor CRTAM. Blood 106, 779–786. [DOI] [PubMed] [Google Scholar]
- Braumüller H, Wieder T, Brenner E, Aßmann S, Hahn M, Alkhaled M, Schilbach K, Essmann F, Kneilling M, Griessinger C, et al. (2013). T-helper-1-cell cytokines drive cancer into senescence. Nature 494, 361–365. [DOI] [PubMed] [Google Scholar]
- Cantoni C, Bottino C, Vitale M, Pessino A, Augugliaro R, Malaspina A, Parolini S, Moretta L, Moretta A, and Biassoni R (1999). NKp44, a triggering receptor involved in tumor cell lysis by activated human natural killer cells, is a novel member of the immunoglobulin superfamily. J. Exp. Med 189, 787–796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carrega P, Loiacono F, Di Carlo E, Scaramuccia A, Mora M, Conte R, Benelli R, Spaggiari GM, Cantoni C, Campana S, et al. (2015). NCR(+)ILC3 concentrate in human lung cancer and associate with intratumoral lymphoid structures. Nat. Commun 6, 8280. [DOI] [PubMed] [Google Scholar]
- Cella M, Fuchs A, Vermi W, Facchetti F, Otero K, Lennerz JKM, Doherty JM, Mills JC, and Colonna M (2009). A human natural killer cell subset provides an innate source of IL-22 for mucosal immunity. Nature 457, 722–725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cella M, Otero K, and Colonna M (2010). Expansion of human NK-22 cells with IL-7, IL-2, and IL-1beta reveals intrinsic functional plasticity. Proc. Natl. Acad. Sci. U. S. A 107, 10961–10966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang C-C, Campoli M, Restifo NP, Wang X, and Ferrone S (2005). Immune selection of hot-spot beta 2-microglobulin gene mutations, HLA-A2 allospecificity loss, and antigen-processing machinery component downregulation in melanoma cells derived from recurrent metastases following immunotherapy. J. Immunol. Baltim. Md 1950 174, 1462–1471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J, Han Y, Lin C, Zhen Y, Song X, Teng S, Chen C, Chen Y, Zhang Y, and Hui R (2005). PDGF-D contributes to neointimal hyperplasia in rat model of vessel injury. Biochem. Biophys. Res. Commun 329, 976–983. [DOI] [PubMed] [Google Scholar]
- Dadi S, Chhangawala S, Whitlock BM, Franklin RA, Luo CT, Oh SA, Toure A, Pritykin Y, Huse M, Leslie CS, et al. (2016). Cancer Immunosurveillance by Tissue-Resident Innate Lymphoid Cells and Innate-like T Cells. Cell 164, 365–377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deschodt-Lanckman M, Vanneste Y, Loir B, Michel A, Libert A, Ghanem G, and Lejeune F (1990). Degradation of alpha-melanocyte stimulating hormone (alpha-MSH) by CALLA/endopeptidase 24.11 expressed by human melanoma cells in culture. Int. J. Cancer 46, 1124–1130. [DOI] [PubMed] [Google Scholar]
- Deuel TF, Huang JS, Huang SS, Stroobant P, and Waterfield MD (1983). Expression of a platelet-derived growth factor-like protein in simian sarcoma virus transformed cells. Science 221, 1348–1350. [DOI] [PubMed] [Google Scholar]
- Dolcetti R, Dal Col J, Martorelli D, Carbone A, and Klein E (2013). Interplay among viral antigens, cellular pathways and tumor microenvironment in the pathogenesis of EBV-driven lymphomas. Semin. Cancer Biol. 23, 441–456. [DOI] [PubMed] [Google Scholar]
- Fuchs A, Cella M, Kondo T, and Colonna M (2005). Paradoxic inhibition of human natural interferon-producing cells by the activating receptor NKp44. Blood 106, 2076–2082. [DOI] [PubMed] [Google Scholar]
- Fuchs A, Vermi W, Lee JS, Lonardi S, Gilfillan S, Newberry RD, Cella M, and Colonna M (2013). Intraepithelial type 1 innate lymphoid cells are a unique subset of IL-12- and IL-15-responsive IFN-γ-producing cells. Immunity 38, 769–781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gajewski TF, Schreiber H, and Fu Y-X (2013). Innate and adaptive immune cells in the tumor microenvironment. Nat. Immunol 14, 1014–1022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glatzer T, Killig M, Meisig J, Ommert I, Luetke-Eversloh M, Babic M, Paclik D, Blüthgen N, Seidl R, Seifarth C, et al. (2013). RORγt+ innate lymphoid cells acquire a proinflammatory program upon engagement of the activating receptor NKp44. Immunity 38, 1223–1235. [DOI] [PubMed] [Google Scholar]
- Gong JH, Maki G, and Klingemann HG (1994). Characterization of a human cell line (NK-92) with phenotypical and functional characteristics of activated natural killer cells. Leukemia 8, 652–658. [PubMed] [Google Scholar]
- Gonzalez R, Jennings LL, Knuth M, Orth AP, Klock HE, Ou W, Feuerhelm J, Hull MV, Koesema E, Wang Y, et al. (2010). Screening the mammalian extracellular proteome for regulators of embryonic human stem cell pluripotency. Proc. Natl. Acad. Sci. U. S. A 107, 3552–3557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorer PA (1950). Studies in antibody response of mice to tumour inoculation. Br. J. Cancer 4, 372–379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Groh V, Wu J, Yee C, and Spies T (2002). Tumour-derived soluble MIC ligands impair expression of NKG2D and T-cell activation. Nature 419, 734–738. [DOI] [PubMed] [Google Scholar]
- Ho JW, Hershkovitz O, Peiris M, Zilka A, Bar-Ilan A, Nal B, Chu K, Kudelko M, Kam YW, Achdout H, et al. (2008). H5-type influenza virus hemagglutinin is functionally recognized by the natural killer-activating receptor NKp44. J. Virol 82, 2028–2032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang W, and Kim H-RC (2015). Dynamic regulation of platelet-derived growth factor D (PDGF-D) activity and extracellular spatial distribution by matriptase-mediated proteolysis. J. Biol. Chem 290, 9162–9170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitamura T, Koshino Y, Shibata F, Oki T, Nakajima H, Nosaka T, and Kumagai H (2003). Retrovirus-mediated gene transfer and expression cloning: powerful tools in functional genomics. Exp. Hematol 31, 1007–1014. [PubMed] [Google Scholar]
- Kline J, and Gajewski TF (2010). Clinical development of mAbs to block the PD1 pathway as an immunotherapy for cancer. Curr. Opin. Investig. Drugs Lond. Engl 2000 11, 1354–1359. [PubMed] [Google Scholar]
- LaRochelle WJ, Jeffers M, Corvalan JRF, Jia X-C, Feng X, Vanegas S, Vickroy JD, Yang X-D, Chen F, Gazit G, et al. (2002). Platelet-derived growth factor D: tumorigenicity in mice and dysregulated expression in human cancer. Cancer Res. 62, 2468–2473. [PubMed] [Google Scholar]
- Lefrançois L, and Lycke N (2001). Isolation of mouse small intestinal intraepithelial lymphocytes, Peyer’s patch, and lamina propria cells. Curr. Protoc. Immunol Chapter 3, Unit 3.19. [DOI] [PubMed] [Google Scholar]
- Li H, and Durbin R (2009). Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinforma. Oxf. Engl 25, 1754–1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X, and Eriksson U (2003). Novel PDGF family members: PDGF-C and PDGF-D. Cytokine Growth Factor Rev. 14, 91–98. [DOI] [PubMed] [Google Scholar]
- Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R, and 1000 Genome Project Data Processing Subgroup (2009). The Sequence Alignment/Map format and SAMtools. Bioinforma. Oxf. Engl 25, 2078–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li SS, Kyei SK, Timm-McCann M, Ogbomo H, Jones GJ, Shi M, Xiang RF, Oykhman P, Huston SM, Islam A, et al. (2013). The NK receptor NKp30 mediates direct fungal recognition and killing and is diminished in NK cells from HIV-infected patients. Cell Host Microbe 14, 387–397. [DOI] [PubMed] [Google Scholar]
- Lin Y-C, Boone M, Meuris L, Lemmens I, Van Roy N, Soete A, Reumers J, Moisse M, Plaisance S, Drmanac R, et al. (2014). Genome dynamics of the human embryonic kidney 293 lineage in response to cell biology manipulations. Nat. Commun 5, 4767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu C, Lou Y, Lizee G, Qin H, Liu S, Rabinovich B, Kim GJ, Wang YH, Ye Y, Sikora AG, et al. (2008). Plasmacytoid dendritic cells induce NK celldependent, tumor antigen-specific T cell cross-priming and tumor regression in mice. J. Clin. Invest 118, 1165–1175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lokker NA, Sullivan CM, Hollenbach SJ, Israel MA, and Giese NA (2002). Platelet-derived growth factor (PDGF) autocrine signaling regulates survival and mitogenic pathways in glioblastoma cells: evidence that the novel PDGF-C and PDGF-D ligands may play a role in the development of brain tumors. Cancer Res. 62, 3729–3735. [PubMed] [Google Scholar]
- Nakamura K, Yoshikawa N, Yamaguchi Y, Kagota S, Shinozuka K, and Kunitomo M (2002). Characterization of mouse melanoma cell lines by their mortal malignancy using an experimental metastatic model. Life Sci. 70, 791–798. [DOI] [PubMed] [Google Scholar]
- Nazarenko I, Hede S-M, He X, Hedrén A, Thompson J, Lindström MS, and Nistér M (2012). PDGF and PDGF receptors in glioma. Ups. J. Med. Sci 117, 99–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nikpay M, Goel A, Won H-H, Hall LM, Willenborg C, Kanoni S, Saleheen D, Kyriakou T, Nelson CP, Hopewell JC, et al. (2015). A comprehensive 1,000 Genomes-based genome-wide association meta-analysis of coronary artery disease. Nat. Genet 47, 1121–1130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nirschl CJ, Suárez-Fariñas M, Izar B, Prakadan S, Dannenfelser R, Tirosh I, Liu Y, Zhu Q, Devi KSP, Carroll SL, et al. (2017). IFN-Dependent Tissue-Immune Homeostasis Is Co-opted in the Tumor Microenvironment. Cell 170, 127–141.e15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oi VT, Morrison SL, Herzenberg LA, and Berg P (1983). Immunoglobulin gene expression in transformed lymphoid cells. Proc. Natl. Acad. Sci. U. S. A 80, 825–829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raulet DH, Gasser S, Gowen BG, Deng W, and Jung H (2013). Regulation of ligands for the NKG2D activating receptor. Annu. Rev. Immunol 31, 413–441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reigstad LJ, Varhaug JE, and Lillehaug JR (2005). Structural and functional specificities of PDGF-C and PDGF-D, the novel members of the platelet-derived growth factors family. FEBS J. 272, 5723–5741. [DOI] [PubMed] [Google Scholar]
- Robinette ML, Fuchs A, Cortez VS, Lee JS, Wang Y, Durum SK, Gilfillan S, Colonna M, and Immunological Genome Consortium (2015). Transcriptional programs define molecular characteristics of innate lymphoid cell classes and subsets. Nat. Immunol 16, 306–317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosental B, Brusilovsky M, Hadad U, Oz D, Appel MY, Afergan F, Yossef R, Rosenberg LA, Aharoni A, Cerwenka A, et al. (2011). Proliferating cell nuclear antigen is a novel inhibitory ligand for the natural cytotoxicity receptor NKp44. J. Immunol. Baltim. Md 1950 187, 5693–5702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider U, Schwenk HU, and Bornkamm G (1977). Characterization of EBV-genome negative “null” and “T” cell lines derived from children with acute lymphoblastic leukemia and leukemic transformed non-Hodgkin lymphoma. Int. J. Cancer 19, 621–626. [DOI] [PubMed] [Google Scholar]
- Shim AH-R, Liu H, Focia PJ, Chen X, Lin PC, and He X (2010). Structures of a platelet-derived growth factor/propeptide complex and a platelet-derived growth factor/receptor complex. Proc. Natl. Acad. Sci. U. S. A 107, 11307–11312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sivori S, Parolini S, Marcenaro E, Castriconi R, Pende D, Millo R, and Moretta A (2000). Involvement of natural cytotoxicity receptors in human natural killer cell-mediated lysis of neuroblastoma and glioblastoma cell lines. J. Neuroimmunol 107, 220–225. [DOI] [PubMed] [Google Scholar]
- Soule HD, Vazguez J, Long A, Albert S, and Brennan M (1973). A human cell line from a pleural effusion derived from a breast carcinoma. J. Natl. Cancer Inst. 51, 1409–1416. [DOI] [PubMed] [Google Scholar]
- Studier FW (2005). Protein production by auto-induction in high density shaking cultures. Protein Expr. Purif. 41, 207–234. [DOI] [PubMed] [Google Scholar]
- Takeda K, Nakayama M, Sakaki M, Hayakawa Y, Imawari M, Ogasawara K, Okumura K, and Smyth MJ (2011). IFN-γ production by lung NK cells is critical for the natural resistance to pulmonary metastasis of B16 melanoma in mice. J. Leukoc. Biol 90, 777–785. [DOI] [PubMed] [Google Scholar]
- Ustach CV, and Kim H-RC (2005). Platelet-derived growth factor D is activated by urokinase plasminogen activator in prostate carcinoma cells. Mol. Cell. Biol 25, 6279–6288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ustach CV, Huang W, Conley-LaComb MK, Lin C-Y, Che M, Abrams J, and Kim H-RC (2010). A novel signaling axis of matriptase/PDGF-D/ß-PDGFR in human prostate cancer. Cancer Res. 70, 9631–9640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vieillard V, Strominger JL, and Debré P (2005). NK cytotoxicity against CD4+ T cells during HIV-1 infection: a gp41 peptide induces the expression of an NKp44 ligand. Proc. Natl. Acad. Sci. U. S. A 102, 10981–10986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vitale M, Bottino C, Sivori S, Sanseverino L, Castriconi R, Marcenaro E, Augugliaro R, Moretta L, and Moretta A (1998). NKp44, a novel triggering surface molecule specifically expressed by activated natural killer cells, is involved in non-major histocompatibility complex-restricted tumor cell lysis. J. Exp. Med 187, 2065–2072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Q, Hou X, Xia J, Qian X, Miele L, Sarkar FH, and Wang Z (2013). Emerging roles of PDGF-D in EMT progression during tumorigenesis. Cancer Treat. Rev. 39, 640–646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu L, Tong R, Cochran DM, and Jain RK (2005). Blocking platelet-derived growth factor-D/platelet-derived growth factor receptor beta signaling inhibits human renal cell carcinoma progression in an orthotopic mouse model. Cancer Res. 65, 5711–5719. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. NKp44 does not interact with PCNA, influenza virus hemagglutinins (HA) or the putative NKp44 ligand upregulated by an HIV gp41-derived peptide. Related to Figure 1.
(A) Generation of EL4 cell-line clones stably over-expressing PCNA. Transfection of constructs encoding nuclear PCNA into tumor cell-lines was proposed to induce cell-surface PCNA expression (Rosental et al., 2011). EL4 cells were transduced with retrovirus-containing supernatants generated from the transfection of phoenix amphotropic packaging cells with a pMX-PCNA-IRES-eGFP retroviral vector and five single-cell clones that stably over-express high levels of GFP were selected (EL4-PCNA cells). The mean fluorescent intensity (MFI) of GFP expression for each clone is displayed.
(B) PCNA is not expressed on the cell-surface of parental EL4 cells or EL4-PCNA cell clones as determined by staining with anti-PCNA mAb. The Percentage of PCNA+ cells are indicated. Anti-Thy1.2 is used as positive control.
(C) EL4-PCNA cell clones do not activate NKp44-GFP reporter cells. 105 NKp44-GFP reporter cells were mixed 1:1 with either EL4 or EL4-PCNA cell clones and incubated overnight before determining GFP expression on NKp44+ cells by flow cytometry. PDGF-DD was used to stimulate NKp44-GFP reporter cells as a positive control (+ PDGF-DD). Percentage of GFP+ cells are indicated.
(D) Influenza virus HA does not interact with NKp44. CHO cells were transiently transfected with expression plasmids encoding either the Hong Kong/97 H5 (HK/97), Vietnam/04 H5 (Viet/04) or WSN/33 H1 influenza virus type A HAs (kindly provided by Adolfo Garcia-Sastre). Binding of anti-HA antibodies (left histograms) and NKp44-Fc (right histograms) to transfected CHO cells was determined by flow cytometry. As negative controls, CHO cells were either mock transfected with pcDNA3.1 or left untransfected. MFI of HA expression for each transfection is displayed.
(E) CHO cells transfected with influenza HAs do not activate NKp44-GFP reporter cells. 105 NKp44-GFP reporter cells were mixed 1:1 with CHO cells transfected with plasmids encoding the different influenza HAs in 96-well plates, incubated for 16h and GFP expression from NKp44-GFP reporter cells determined by flow cytometry. As negative controls, CHO cells were either mock transfected or left untransfected. PDGF-DD was used to stimulate NKp44-GFP reporter cells as a positive control. Percentages of GFP+ cells are indicated.
(F, G) The HIV gp41 peptide described by Vieillard et al. does not upregulate an NKp44 ligand (NKp44L) on human CD4+ T cells. Human CD4+ T cells from two different donors (d1, d2) were incubated overnight with (+) or without (−) the HIV gp41-derived peptide (HIV gp41) that was reported to upregulate an NKp44L (Vieillard et al. 2005), later identified as the nuclear antigen MLL5 (Baychelier et al., 2013). NKp44-Fc binding to CD4+ T cells (dotplots) (F) or GFP expression from NKp44-GFP reporter cells mixed 1:1 overnight with CD4+ T cells (G) either unpulsed or pulsed with HIV gp41 peptide. PDGF-DD was used to stimulate NKp44-GFP reporter cells as a positive control.
(H) Jurkat T cells do not activate NKp44-GFP reporter cells. NKp44-GFP reporter cells were mixed 1:1 overnight with or without Jurkat T cells, which were reported to express MLL5 on the cell surface (Baychelier et al., 2013). GFP expression was measured by flow cytometry. PDGF-DD was used to stimulate NKp44-GFP reporter cells as a positive control.
Figure S2. Expression of NKp44 and PDGFR-α/β in human NK cells. Related to Figure 2.
(A) Polyclonal NK cells cultured in IL-2 medium were stained either with isotype control mAbs or mAbs to CD3, CD56 in combination with mAbs to NKp44, PDGFRα or PDGFRβ. CD3−CD56+ human NK cells express NKp44 but not PDGFRα or PDGFRβ.
(B) PDGF-DD stimulates dose-dependent NK cell surface CD107a expression.
(C) Cell surface CD107a expression by PDGF-DD-stimulated NK cells. Upper panels, representative dotplots and percentage expression in each gate; lower panel, quantification. Cell-surface CD107a is blocked by soluble anti-NKp44. In the absence of PDGF-DD, CD107a expression was induced by mAb-mediated cross-linking of NKp44 (α-NKp44 + goat anti-mouse - GαM). Induction of CD107a by PMA/i was used as positive control.
(D) Generation of a TEV cleavable PDGF-D construct (CUB-TEV-PDGFD). After expression and purification from 293F cells, CUB-TEV-PDGFD proteins were incubated with (+TEV) or without (−TEV) TEV protease at 30°C for 0 or 24 hours (h). Samples were then boiled in SDS-PAGE loading buffer with (+) or without (−) DTT before resolving on 4-10% SDS-PAGE gels (Molecular mass, kD).
(E) IFN-γ and TNF-α secretion by NK cells is enhanced by TEV cleavage of CUB-TEV-PDGFD. Recombinant PDGF-DD was added to some cultures as positive control. Cleavage of CUB-TEV-PDGFD by proteases in serum containing medium likely explains cytokine production by NK cells in wells stimulated with CUB-TEV-PDGFD without TEV protease.
(F) The enhanced secretion of IFN-γ and TNF-α by NK cells stimulated with CUB-TEV-PDGFD +/−TEV wells is blocked by anti-NKp44 and anti-PDGF-D mAbs. Data are represented as mean (n = 3) ± SEM. (****, p ≤ 0.0001).
Figure S3. PDGF-DD binding to NKp44 induces TNF-α secretion by ILC3. Related to Figure 3.
Representative dotplots of intracellular TNF-α and IL-22 content and cell surface NKp44 from freshly isolated tonsil ILC3 stimulated with PDGF-DD and/or IL-23. The percentage of cytokine-positive cells is indicated in each quadrant.
Figure S4. Ingenuity Pathway Analysis of growth arrested cancer cell-lines exposed to supernatants from PDGF-DD activated NK cells. Related to Figure 4.
Top 10 highest scoring IPA canonical pathways generated from transcripts differentially expressed (±1.5-fold) in cancer cells cultured in NKDD+IgGsup versus control NKDD+α-NKp44 sup. Ratios in columns indicate total NKDD+IgG sup-regulated genes to the total number of genes in each pathway.
Figure S5. Expression of PDGF-D in normal and cancerous human tissues. Related to Figure 5.
(A-I) Sections from normal tissues are derived from: (A) stomach, (B) skin, (C) colon, (D) bladder, (E) lung, (F) kidney, (G) prostate, (H) pancreas, and (I) testis. Reactivity is evident in tissue epithelial compartments (brown staining). Scale bar = 100 μM.
(J-X) Sections from human carcinomas are from different primary sites including: (J) head and neck, (K, L) lung, (M) bladder, (N, O) colon, (P) breast, (Q) ovarian cancer, (R, S) primary melanoma, (T-X) melanoma metastasis to the (T, U) skin, (V) lung, (W) lymph nodes and (X) brain. All sections were stained with anti-PDGF-D antibody. Scale bar = 100 μM.
Figure S6. Reduced dissemination of tumor cells expressing PDGFD in vivo in a second NCR2-tg line. Related to Figure 6.
(A, B) Representative dotplots and quantification of NKp44 expression in CD3− NK1.1+ (NK) cells isolated from the spleen and mesenteric lymph nodes (mLN) of a second NCR2-tg founder line (founder #2). The percentages of cells are indicated in each gate.
(C, D) Representative dotplots and quantification of NKp44 expression in ILC1/NK cells, ILC2, CCR6+ ILC3, NKp46+ ILC3, and NKp46−CCR6− (DN) ILC3 isolated from the small intestine of NCR2-tg mice. The percentages of cells are indicated in each gate. Lamina propria mononuclear cells were stained for T-bet, Roryt, GATA3, NKp46, NK1.1, CCR6, lineage markers (CD3, CD19, CD5) and analyzed by flow cytometry. Cells were gated on Lineage− cells. ILC1/NK: T-bet+NK1.1+GATA3−Rorγt−; ILC2: GATA3+T-berRoryt−; ILC3: Roryt+GATA3int.
(E, F) Tissue culture supernatants from B16-pMX control cells do not stimulate IFN-γ or TNF-α unless 250 ng/ml recombinant PDGF-DD (+ PDGF-DD) is added to B16-pMX control supernatants (sup) as a positive control for NK cell activation.
(G) Quantification (upper panel) and representative photos (lower panel) of surface metastases from the lungs of non-tg and NCR2-tg (founder #2) at Day 17 post-injection with 2 × 105 B16F10 cells stably expressing PDGFD (B16-PDGFD) (*, p ≤ 0.05).
(H) Mean tumor area (μM2) ± SEM and representative histochemical images of lung metastases from non-tg (n = 7) and NCR2-tg (founder #2; n = 10) mice injected with B16-PDGFD cells.
Data Availability Statement
Microarray data has been deposited at GEO:GSE107043.
RNA-seq data has been deposited at GEO:GSE107047.