

Abstract
Bioassay-guided fractionation of a colon cancer growth inhibitory extract of the fungus Paraconiothyrium sp. led to the isolation of eight new versiol derivatives (1, 3-8, 10) along with two known compounds. The structures were elucidated by interpretation of combined MS and 2D NMR spectroscopic data. Compounds 8, 9, and 10 showed cell growth inhibition against COLO205 and KM12 cells, and both 8 and 9 displayed selectivity in their inhibition of melanoma cell lines in the NCI 60 one dose test. In addition, compound 8 and the crude Paraconiothyrium sp. extract showed potent dose dependent inhibitory effects in the five dose NCI 60 cell line assay.
Graphical Abstract
Colon cancer is the second most prevalent cancer in the United States.1 Like most cancers, colon cancers have the ability to invade tissues and metastasize to other parts of the body.1,2 New colon cancer drugs such as the targeted monoclonal antibodies cetuximab, panitumumab, and bevacizumab have recently been introduced, which inhibit the epidermal growth factor receptor or vascular endothelial growth factor A.3,4 In spite of some improvements in results with these new biologic agents, about 50% of colon cancer patients ultimately die from metastatic disease.2,5 Therefore, it is important to discover new agents from natural products that will improve the outcomes for patients who would otherwise succumb to the disease. During a search for anti-proliferative agents against various cancer types, we found that the crude extract of the fungus Paraconiothyrium sp. (Montagnulaceae) exhibited significant growth inhibition against COLO205 and KM12 colon cancer cell lines. Isoprenoid decalin and diterpene lactone derivatives, some with cytotoxic activity, have been previously reported from this fungus.6,7 Versiol represents a metabolite that has been previously isolated from several fungi.8,9 The skeleton is composed of an isoprenoid decalin ring system and shows an unusual substitution and oxidation pattern. However, there have been no reports of versiol derivatives being found in the genus Paraconiothyrium, nor have anti-cancer activities been noted. We performed bioassay-guided fractionation and isolation based on the anti-proliferative activity against two colon cancer cell lines. This yielded eight new compounds and two known compounds, and three of the metabolites were further tested in the NCI 60 screen to assess their selectivity profiles.
RESULTS AND DISCUSSION
The Paraconiothyrium sp. fungal extract was provided by the Natural Product Discovery Institute (NPDI) and the extract dissolved in CH2Cl2−MeOH, 1:1, v/v was initially fractionated by two separate diol DIO Spe-ed SPE cartridges, and each cartridge was sequentially eluted with hexane, CH2Cl2, EtOAc, and MeOH to give four fractions (A−D). The colon cancer cytotoxicity of the EtOAc fraction [IC50 = 5.9 μg/mL (COLO205), IC50 = 5.5 μg/mL (KM12)] was more potent than the other fractions (Supporting Figure S51), thus, it was chromatographed on an ODS silica gel column and six fractions (A−F) were collected, respectively. These fractions were subjected to C18 HPLC to yield eight new compounds, craterellone F (1), isoversiol A (3), isoversiol B (4), isoversiol C (5), isoversiol D (6), isoversiol E (7), calbistrin F (8), and conionaphthalenediol (10), along with two known compounds, craterellone E (2) and calbistrin A (9).
Compound 1 was obtained as a brown amorphous powder. Its molecular formula was determined to be C16H24O3 based on HRESIMS analysis and 13C NMR data (Table 1). In the 13C NMR data, resonances for one quaternary, two methyl, four methylene, four methine, four olefinic, and one carbonyl carbon were observed. Two methyl signals (H3-14 and H3-16), and four olefinic protons (H-6, H-7, H-15a, and H-15b) were observed in the 1H-NMR data. The COSY spectrum indicated two spin systems, one a sequence including H-10 and H-1 through H-7, and another as H-12–H-13. Interpretation of the COSY and HMBC data revealed a bicyclic trans-decalin ring system with a side chain (Figure 1). HMBC correlations from H-1 (δH 3.60) to C-3 (δC 25.7) and C-5 (δC 30.3), from H3-14 (δH 0.84) to C-3 (δC 25.7), C-2 (δC 44.0), and C-4 (δC 42.1), and from H-3 (δH 1.96) to C-1 (δC 66.5) and C-5 (δC 30.3) determined the position of the oxygenated methine (C-1, δC 66.5) and the methyl carbon (C-14, δC 22.5) of the A ring. HMBC correlations from H2-15 (δH 4.80 and 4.30) to C-7 (δC 127.3), C-8 (δC 149.6), and C-9 (δC 56.6), and from H-7 (δH 6.04) to C-15 (δC 111.8), suggested that the exocyclic double bond extended from C-8. In addition, the signals correlating the carbonyl carbon at C-11 (δC 211.9) with H2-12 (δH 2.72, 2.49), H-13 (δH 3.64), H3-16 (δH 1.20), and H-10 (δH 1.64), and HMBC correlations from H3-16 to C-8 (δC 149.6), C-9 (δC 56.6) and, C-10 (δC 47.4) indicated that compound 1 was a bicyclic polyketide derivative similar to compound 2, craterellone E, which was isolated from a Chinese fungus, Craterellus odoratus,10 and also isolated in this study. Using ROESY correlations, vicinal couplings could be observed from H-10 to a deshielded methine at H-1, as well as from H-1 to methylene protons at H-2a/2b, and H-2a/2b to H3-14. In addition, 1,3 diaxial ROESY correlations from H-3 to H-5 and H-5 to H3-16 supported the relative configuration of the hexahydronaphthalene skeleton bearing a side chain with a carbonyl group such as were seen in decumbenone A, decumbenone B, and craterellone C.10,11 In addition, the carbon chemical shifts for both epimers were theoretically calculated using DFT to compare with the experimental data (Supporting Figure S63), supporting the relative configuration shown at C-1. Therefore, the structure of 1 was determined as shown and named craterellone F, since it differs from craterellone E only in the position of the hydroxy group at C-1 versus C-4.
Table 1.
1H (600 MHz) and 13C (150 MHz) NMR Data for Compounds 1, 3, 4, 5, 6, 7, 8, and 10 in DMSO-d6
| position | 1 | 3 | 4 | 5 | ||||
|---|---|---|---|---|---|---|---|---|
| δc | δH (J in Hz) | δc | δH (J in Hz) | δc | δH (J in Hz) | δc | δH (J in Hz) | |
| 1 | 66.5 | 3.60, m | 65.6 | 3.91, m | 64.8 | 4.05, m | 69.8 | 3.85, m |
| 2 | 44.0 | 1.60, 1.04, m | 44.2 | 1.57,1.01, m | 39.5 | 1.71, 1.10, m | 76.1 | 3.60, brt (6.7) |
| 3 | 25.7 | 1.96, m | 26.3 | 1.86, m | 25.6 | 2.45, m | 33.4 | 2.31, m |
| 4 | 42.1 | 1.87, 0.74, m | 42.1 | 1.7, 0.64, m | 133.4 | 5.50, s | 131.9 | 5.37, s |
| 5 | 30.3 | 2.46, m | 31.2 | 2.30, t (11) | 132.3 | 131.6 | ||
| 6 | 134.9 | 5.63, d (9.8) | 129.9 | 5.28, d (9,8) | 127.8 | 5.84, d (9.8) | 127.1 | 5.84, d (9.5) |
| 7 | 127.3 | 6.04, dd (9.8, 2.5) | 134.6 | 5.24 dd (9.8, 2.2) | 134.4 | 5.30, d (9.8) | 134.7 | 5.32, d (9.5) |
| 8 | 149.6 | 73.4 | 73.8 | 73.5 | ||||
| 9 | 56.6 | 56.7 | 57.2 | 57.1 | ||||
| 10 | 47.4 | 1.64, d (13) | 46.2 | 1.65, d (12.3) | 42.0 | 2.7, m | 42.2 | 2.80, m |
| 11 | 211.9 | 214.1 | 213.7 | 213.7 | ||||
| 12 | 41.5 | 2,72, dt (17, 6.6, 6.6), 2.49, m | 45.1 | 2,93, dt (18, 6.4, 6.4), 2.65, m | 44.7 | 2,90, dt (18, 7.1, 7.1), 2.60, m | 44.7 | 2,95, dt (18, 7.2, 7.2), 2.70, m |
| 13 | 57.2 | 3.64, m | 56.8 | 3.58, m | 57.3 | 3.60, m | 57.0 | 3.60, m |
| 14 | 22.5 | 0.84, d (6.6) | 22.6 | 0.81, d (7.1) | 21.6 | 0.94, dd (16, 6.9) | 18.3 | 1.02, d (7.0) |
| 15 | 111.8 | 4.80, 4.30, brs | 28.1 | 0.93, s | 26.8 | 0.99, brs | 26.8 | 0.98, m |
| 16 | 20.2 | 1.2, brs | 13.8 | 1.37, s | 14.5 | 1.3, brs | 14.7 | 1.33, s |
| 1-OH | 4.13, s | |||||||
| position | 6 | 7 | 8 | 10 | ||||
|---|---|---|---|---|---|---|---|---|
| δc | δH (J in Hz) | δc | δH (J in Hz) | δc | δH (J in Hz) | δc | δH (J in Hz) | |
| 1 | 69.7 | 4.06, m | 208.6 | 69.0 | 5.85, m | 67.9 | 5.95, brs | |
| 2 | 42.5 | 1.78, 1.29, m | 50.2 | 2.14,2.10, m | 35.2 | 2.03, 1.30, m | 30.9 | 1.96, 1.91, m |
| 3 | 23.7 | 1.80, m | 39.9 | 1.90, 1.28, m | 26.7 | 2.41, m | 29.0 | 2.03, m |
| 4 | 47.9 | 1.70, 1.10, m | 34.3 | 1.85, m | 135.6 | 5.70, s | 69.7 | 4.30, m |
| 5 | 70.0 | 38.4 | 2.13, m | 130.5 | 139.5 | |||
| 6 | 128.2 | 5.37, d (10) | 127.7 | 5.40, d (9,8) | 128.6 | 5.98, d (9.7) | 128.2 | 7.06, d (7.7) |
| 7 | 136.1 | 5.42, d (10) | 136 | 5.35, d (9.8) | 131.2 | 5.58, d (9.7) | 130.3 | 7.14, d (7.7) |
| 8 | 72.6 | 72.7 | 77.1 | 136.4 | ||||
| 9 | 56.0 | 53.9 | 53.4 | 136.1 | ||||
| 10 | 40.8 | 2.19, m | 54.6 | 2.87, m | 39.5 | 2.80, m | 131.2 | |
| 11 | 213.6 | 212.4 | 209.5 | 17.8 | 0.98, d (6.6) | |||
| 12 | 44.0 | 2.92, dt (19, 6.6), 2.69, m | 44.2 | 2.92, dt (19, 6.6), 2.69, m | 47.5 | 2.70, dd (14, 5.5), 2.32, m | 20.7 | 2.21, s |
| 13 | 56.6 | 3.60, m | 56.8 | 3.56, t (7.1) | 91.3 | 5.10, d (5.5) | 14.7 | 2.04, s |
| 14 | 21.1 | 0.93, m | 22.3 | 0.98, d (7.0) | 21.2 | 0.98, d (7.0) | ||
| 15 | 26.7 | 0.92, s | 27.9 | 0.94, s | 23.2 | 1.14, s | ||
| 16 | 14.0 | 1.38, s | 13.1 | 1.28, s | 17.6 | 1.22, s | ||
| 1’ | 174.8 | 174.8 | ||||||
| 2’ | 44.3 | 2.30, m | 44.3 | 2.38, m | ||||
| 3’ | 79.4 | 3.79, d (9.8) | 79.3 | 3.91, d (9.9) | ||||
| 4’ | 136.2 | 136.2 | ||||||
| 5’ | 126.9 | 5.19, t (7.3) | 127.8 | 5.88, d (10.8) | ||||
| 6’ | 21.9 | 2.14, dd (14, 7.3) | 124.3 | 6.20, dd (15, 7.2) |
||||
| 7’ | 42.8 | 2.45, t (7.3) | 139.7 | 5.64, dd (15, 7.2) |
||||
| 8’ | 208.6 | 33.7 | 2.60, m | |||||
| 9’ | 30.2 | 2.05, s | 42.4 | 2.18, 2.08, m | ||||
| 10’ | 14.6 | 0.73, d (7.1) | 174.3 | |||||
| 11’ | 10.6 | 1.46, s | 14.5 | 0.70, d (6.9) | ||||
| 12’ | 11.2 | 1.58, brs | ||||||
| 13’ | 20.4 | 0.98, d (6.6) | ||||||
in DMSO-d6
Figure 1.

1H-1H COSY, key HMBC, and ROESY correlations of compound 1.
Compound 3 was isolated as a brown amorphous powder. It was assigned the molecular formula C16H24O3 from HRESIMS. The 13C NMR data revealed signals for one quaternary carbon, three methyl, four methylene, four methine, two olefinic, and one carbonyl group. The two olefinic protons were coupled to one another appearing as a doublet (δH 5.28, J6,7 = 9.8 Hz) and doublet of doublets (δH 5.22, J = 9.8 Hz and 2.2 Hz). Analysis of the COSY data led to the establishment of the structure fragments from C-1 to C-7, C-5 to C-10, and C-12 to C-13 (Figure 2). Two olefinic protons at H-6 and H-7 (δH 5.28, 5.24) and two methyl signals at H3-15 and H3-16 (δH 0.93, 1.37) were linked to an oxygenated carbon at C-8 (δC 73.4) by the HMBC correlations of H-6, H-7, H3-15, and H3-16 with C-8. The olefinic proton at H-7 (δH 5.24), two methyl signals at H3-15 (δH 0.93) and H3-16 (δH 1.37), one oxygenated methine proton at H-1 (δH 3.91), and two methine protons at H-5 (δH 2.30) and H-10 (δH 1.65) were also correlated to the quaternary carbon at C-9 (δC 56.7), which led to the establishment of the structure of a decalin ring. This was also supported by the HMBC correlations from H-6 (δH 5.28) to C-4 (δC 42.1) and H-10 (δH 1.65) to C-2 (δC 44.2) and C-4 (δC 42.1). The carbonyl carbon was determined to be located at C-11 (δC, 214.1) by the HMBC correlations of C-11 from H-10 (δH 1.65), H3-16 (δH 1.37), H2-12 (δH 2.93, 2.65) and H2-13 (δH 3.58). Further HMBC correlations of H-1 (δH 3.91) with C-3 (δC 26.3) and C-5 (δC 31.2) and H3-14 (δH 0.81) with C-2 (δC 44.2), C-3 (δC 26.3), and C-4 (δC 42.1) established the remainder of a decalin ring, closely related to versiol, a fungal metabolite isolated from Penicillium decumbens.11 The relative configuration of 3 was assigned by a ROESY experiment. ROESY correlations from H3-15 to H-10, H-10 to H-1, H-1 to H2-2, and H2-2 to H3-14 as well as H-3 to H-5 and H-5 to H3-16 were observed. The relative configuration of 3 was consistent with that of 1 in terms of polyketide biosynthesis as co-metabolites (Supporting Figure S49). Owing to the similarity between the structure of 3 and versiol, compound 3 was named isoversiol A.
Figure 2.

1H-1H COSY, key HMBC, and ROESY correlations of compound 3.
Compound 4 was purified as a white amorphous powder, and its molecular formula was determined to be C16H22O3 by positive HRESIMS. Its 1H and 13C NMR data were similar to those of 3, except for additional olefinic signals at C-4 and C-5 and the missing methane and methylene carbons at C-4 and C-5. Comparison of the NMR spectra of 4 with that of 3 revealed that the methylene signal at C-4 was replaced by a methine signal (δC 133.4, δH 5.5) in 4, suggesting the same planar structure as versiol. According to the ROESY data, compound 4 possessed a different relative configuration from that of versiol. The ROESY correlation from H3-15 to H-10, H-10 to H-1, H-1 to H2-2, and H2-2 to H3-14 indicated that 4 had similar relative configuration as that of 3. The absolute configuration of C-1 in 4 was also deduced as S- based on chemical shift differences from the corresponding Mosher’s esters (Supporting Figure S50). Consequently, the structure of compound 4 (isoversiol B) was established as shown.
Compound 5 was a white amorphous powder. Its molecular formula was determined to be C16H22O4 by positive HRESIMS,.16 units more than 4, indicating the likely addition of an oxygen atom. Its 1H and 13C NMR spectra were also similar to those of 4, except for the deshielded shift at C-2 due to an hydroxy group. Comparison of the NMR and MS spectra of 5 and 4 revealed that the methylene signal of 4 was replaced by an oxygenated methine (δC 76.1, δH 3.06) signal. Through HMBC correlations of H-2 (δH 3.06) with C-10 (δC 42.2), C-3 (δC 33.4), and C3-14 (δC 18.3) and H-4 (δH 5.37) with C-2 (δC 76.1) the new hydroxy group was determined to be attached at C-2. The relative configuration was also confirmed by ROESY correlations. The correlation from axial H-10 to H-1 established H-1 as equatorial, in common with other hexahydronaphthalene moieties from this Paraconiothyrium sp. fungus. Secondly, 1,3 diaxial ROESY correlations from H-10 to H3-15, and from the axial H-2 to axial H-10 were observed. Consequently, the structure of compound 5 was established as isoversiol C.
Compound 6 was obtained as a white amorphous powder and its molecular formula was established as C16H24O4 by positive HRESIMS, 16 units more than 3. Its 1H and 13C NMR data were similar to those of 3, except for an additional hydroxy group at C-5. Comparison of the NMR spectra of 6 with that of 3 revealed that the methine carbon was replaced by an oxygenated carbon, which was in agreement with the deshielded signal for C-5 (δC 70.0) and the HMBC correlation from H-1 (δH 4.06), H-3 (δH 1.80), H-6 (δH 5.37), H-7 (δH 5.42), and H-10 (δH 2.19) to C-5 (δC 70.0). ROESY correlations from H-1 to H-10, H-10 to H3-15 as well as H-1 to methylene protons at H-2a/2b, and H-2a/2b to H3-14 were observed. Therefore, the chemical structure of 6 was assigned as isoversiol D.
Compound 7 was isolated as a white amorphous powder and its molecular formula was established as C16H22O3 by positive HRESIMS. Comparison of the NMR data of 7 with that of 6 indicated that an oxygenated methine group was replaced by a carbonyl group at C-1 (δC 208.6). HMBC correlations between the carbonyl carbon (δC 208.6) and H-5 (δH 2.13), H-10 (δH 2.87), H2-2 (δH 2.14, 2.10), H-3b (δH, 1.28) indicated that the position of the carbonyl was at C-1. The methyl group (C-14, δC 22.3) attached at C-4 was located by the HMBC correlations between H3-14 (δH 0.98) and C-3 (δC 39.9), C-4 (δC 34.3), and C-5 (δC 38.4). In the ROESY spectrum, the cross peaks from H-5 to H3-16 and H-10 to H3-15 were easily detected. In addition, another 1, 3-diaxial correlation was observed between H-10 and H3-14 was also detected, supporting that the relative configuration of 7 (isoversiol E) as drawn.
Compound 8 was isolated as a brown amorphous powder, for which the molecular formula was determined as C27H38O7 by positive HRESIMS. The presence of two ketones and one ester carbonyl were indicated by the 13C NMR data. The 1H, 13C, and HSQC NMR data also supported the existence of six methyl, four methylene, six olefinic and four other oxygenated carbons. The COSY spectrum showed spin systems of H-1 through H-4, H-6–H-7, and H-12–H-13 as well as H-10’–H-2’–H-3’ and H-5’ through H-7’. HMBC correlations from H-1, H-3, H-4, H-6, H-7, and H-10 to C-5 and H-1, H-2, H-4, and H-6 to C-10, and H-6, H-7, and H-10 to C-8 suggested that fragments C-1–C-4 and C-6–C-9 were connected through two carbons, C-5 and C-10 (Figure 3). An isolated methyl (δH 0.98, δC 21.2, C-14) was linked to C-3 supported by the HMBC correlations from H3-14 to C-2, C-3, and C-4. The HMBC cross peaks from H-15 to C-7, C-8, and C-9 and H-16 to C-8, C-9, and C-10 suggested that two methyl protons at H3-15 (δH 1.14) and H3-16 (δH 1.22) were linked by two carbons at C-8 (δC 77.1) and C-9 (δC 53.4). Thus, the carbon skeleton of compound of the ring system of 8 was similar to that of 3 and 4. HMBC correlations between the proton of the oxygen-bearing methine (H-13, δH 5.10), methylene (H2-12, δH 2.70, 2.32), and methyl (H3-16, δH 1.22) with the carbonyl carbon (C-11, δC 209.5) established the hexahydronaphthalene system similar to compound 9 (calbistrin A), which was previously isolated from Penicillium restrictum12 and was also isolated in our study. Further HMBC correlation between the proton of the oxygen-bearing methine (H-1, δH 5.85) and the carbonyl carbon C-1’ (δC 174.8) implied the presence of an ester substitution at C-1. HMBC correlations of H-2’, H-3’, H-5’, H-6’ to C-4’ and H-3’, H-6’, H-7’ to C-5’ suggested that fragments C-2’–C-3’ and C-5’–C-7’ were connected through a carbon at C-4. The locations of two methyl carbons were determined to be at C-2’ and C-4’, respectively, through the cross peaks from H3-10’ to C-1’, C-2’, C-3’ and H3-11’ to C-3’, C-4’, C-5’ in the HMBC spectrum. The signals correlating the methyl signal (H3-9’, δH 2.05) of the methyl ketone group with the methylene carbon (C-7’, δC 42.8) indicated that the methyl ketone group was connected to C-7’. The relative configuration of 8 was determined by ROESY data. Strong correlations of H-10’/H-3’/ H-5’/ H-6a’ as well as H-2’/H3-11’ supported an E-configuration of the double bond C-4’–C-5’ of the side chain. Correlations of H-15/H-10/H-1/H-2b/H3-14 indicated that these protons of the hexahydronaphthalene ring are on the same face of the molecule, as in calbistrin A (9). The ROESY correlation between H-2a and H3-16 also supported that H3-16 is co-facial with H-2b. The configuration of C-1 was tentatively proposed on biosynthetic grounds of other hexahydronaphthalene moieties bearing a side chain with a carbonyl group.12,13 Taking into account all of the above described spectroscopic data, and since 8 was isolated in this study as a co-metabolite with the known fungal metabolite 9 (calbistrin A), compound 8 was named calbistrin F.
Figure 3.

1H-1H COSY, key HMBC, and ROESY correlations of compound 8.
Compound 10 was isolated as a white amorphous powder and its molecular formula was established as C26H36O6 by negative HRESIMS. The 1H and 13C NMR and HSQC data displayed signals for six methyls, three oxygenated methines, five olefinic methines, and two carboxyls. The connectivity of H-1 through H-4 and H-11, H-6–H-7, H-11’–H-3’, H-5’ through H-9’, and H-13’–H-8’ was confirmed through COSY analysis which led to the establishment of the structure fragments C-1-C-4, C-6–C-7, C-11’–C-3, and C-5’–C-9’. In the HMBC spectrum, the locations of two oxygenated methines at C-1 (δC 67.9) and C-4 (δC 69.7) were determined, respectively, by the cross peaks from H-2 and H-3 to C-1, H-2, H-3 to C-4, and H-1, H-4 to C-2 (Figure 4). The correlated signals from H3-11 to C-2, C-3, and C-4 suggested that a methyl group (δH 0.98, δC 17.8, C-11) was attached to C-3 (δC 29.0). Two methyl signals at H3-12 (δH 2.21) and H3-13 (δH 2.04), and two olefinic protons at H-6 (δH 7.06) and H-7 (δH 7.14) had cross peaks with C-7 (δC 130.3), C-8 (δC 136.4), and C-9 (δC 136.1) suggesting that two methyl protons were located at C-8 and C-9 in the benzene ring. The HMBC cross peaks from H-1, H-2, and H3-12 to C-10 and H-1, H-3, H-4, H-6, and H-7 to C-5 suggested that fragments C-1–C-4 and C-6–C-9 were connected through two quaternary carbons, C-5 (δC 139.5) and C-10 (δC 131.2), forming the hydroxy naphthalenol ring. The HMBC correlation between the oxygenated methine proton at H-1 (δH 5.95) and the carbonyl group at C-1’ (δC 174.8) implied the presence of an ester at C-1 (δC 67.9). COSY experiments showed that the side chain contained two spin systems, one from the singlet methyl (δH 0.70, H3-11’) to an oxygenated methine proton (δH 3.91, H-3’) and another from an olefinic proton (δH 5.88, H-5’) to a methylene proton (δH 2.18, 2.08, H2-9’). HMBC correlations from H-2’, H-3’, H-5’, and H-6’ to C-4’ and H-3’, H-6’, and H-7’ to C-5’ suggested that fragments C-2’–C-3’ and C-5’–C-9’ were connected through a quaternary carbon at C-4 (δC 69.7). In the HMBC spectrum, the locations of three methyl carbons at C-11’ (δc 14.5), C-12’ (δc 11.2), and C-13’ (δc 20.4) were determined to be attached at C-2’ (δc 44.3), C-4’ (δc 136.2), and C-8’ (δc 33.7), respectively, through the cross peaks from H3-11’ to C-1’, C-2’, and C-3’ and H3-12’ to C-3’, C-4’, and C-5’, and H3-13’ to C-7’, C-8’, C-9’, and C-10’. The signals correlating the carbonyl carbon (δc 174.3, C-10’) of the carboxyl group with the methylene proton (δH 2.6, H2-8’), methine proton (δH 2.18, 2.08, H2-9’), and methyl proton (δH 0.98, H3-13’) indicated that the carboxyl group was connected at C-9’ (δC 42.4) at the terminal of the side chain. ROESY correlation peaks of H-2’/H-12’/H-6’/H-8’ and H-11’/H-3’/H-5’/H-7’/H-13’ led to E-configurations at the double bonds C-4’–C-5’ and C-6’–C-7’ and the large 1H-1H coupling constants (15.2 Hz) between H-6’ and H-7’ also supported that compound 10 contains an all trans side chain. ROESY correlations of H-1/H-2b/H-3/H-4 indicated that the protons of the hydroxy naphthalenol ring are on the same face. Taking into account all of the above described spectroscopic data, compound 10 was assigned the relative structure as illustrated and named conionaphthalenediol.
Figure 4.

1H-1H COSY, key HMBC, and ROESY correlations of compound 10.
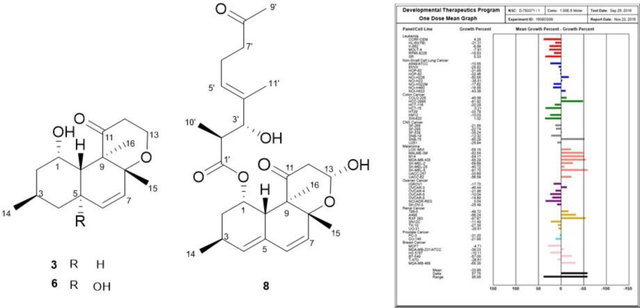
We investigated the inhibitory activity of these compounds with a two-day drug exposure assay against colon cancer cells. In our study, compound 8 (IC50 << 1.25 μM in both cell lines), 9 (IC50 << 1.25 μM in both cell lines), and 10 (IC50 = 12.5 μM (COLO205), IC50=6.5 μM (KM12)) showed potent cell growth inhibition in a dose-dependent manner (Supporting Figure S52). To further assess the biologic effect of these compounds on the viability of tumor cells, we used the NCI-60 screen, a platform containing 60 different cancer cell lines14,15. Calbistrin F (8) and E (9) with −34 and −3 mean percent cell growth inhibition at 10 μM, respectively, had superior activity against the NCI 60 cell lines to conionaphtalenediol (10), which inhibited cancer cells with an average growth compared to control of only 89 percent despite the structural similarity (Supporting Figures S53–S58). The slight structural differences in the hexahydronaphthalene moiety with a cyclic hemiacetal greatly affected inhibitory activity. Compounds 8 and 9 also showed potent selective inhibition of melanoma cancer cell lines including MALME-3M, M14, SK-MEL-2, SK-MEL-5, and UACC-62. These results showed a sufficient growth-inhibitory activity comparable to those of adriamycin, positive control, when tested at a single concentration to warrant full dose-response testing against all 60 cell lines.16–18 In addition, callbstrin F (8) showed an enhanced antiproliferative effect against the entire panel of nine kinds of cancer cell lines including colon cell lines at the mean GI50, mean TGI (total growth inhibition), and mean LC50 (50% cell death) levels in the five dose NCI 60 cell tests (Supporting Figure S59, 60). The results of testing the crude Paraconiothyrium sp. extract also showed a similar pattern to those of calbstrin F (8), validating the bioassay-guided isolation (Supporting Figure S61, 62). These results suggest that the major active compound, calbstrin F, could lead to a potential therapeutic antitumor agent.
EXPERIMENTAL SECTION
General Experimental Procedures.
Optical rotations ([α]25D) were measured on a PerkinElmer 241 polarimeter in a 100 × 2 mm cell (units 10−1 deg cm2 g−1). UV absorption spectra were obtained using a Varian Cary 50 Bio UV–visible spectrophotometer. NMR spectra were acquired on a Bruker Avance III spectrometer operating at 600 MHz for 1H and 150 MHz for 13C and equipped with a 3 mm cryogenically cooled probe. 1H and 13C spectra were referenced to the residual deuterated solvent peaks. HRESIMS data were acquired on an Agilent 6520 Accurate Mass Q-TOF instrument with internal reference masses calibrated at 121.05087 and 922.00979, both within 5 ppm. Diol SPE fractionation of the extract was performed on DIO Spe-ed SPE cartridges, and subsequent fractions were separated on a RP-C18 column attached to a model UA-6 UV detector and Foxy 200 fraction collector (Teledyne Isco). Final purifications were performed using a Varian ProStar 218 solvent delivery module HPLC equipped with a Varian ProStar 325 UV–vis detector, operating under Star 6.41 chromatography workstation software. All solvents and chemicals used were of analytical grade.
Fungal material and Extraction.
Fungal strain NPDI-J3940 was isolated as Mycelia sterilia from desert soil collected in Arizona. Fermentations were carried out by the FERMEX method.19 Briefly, 1-liter roller bottle fermentation vessels were inoculated from seed (corn steep liquor, tomato paste, oat flour, glucose and trace elements solution) onto vermiculite impregnated with a medium containing (per liter distilled water) 75 g glycerol, 10 grams glucose and 5 grams each Bacto Yeastolate, soybean meal and Hunt’s tomato paste. After 20 days incubation at 22°C, the contents of sixteen roller bottles were thoroughly extracted with 2.5 liters of methylethylketone; solvent was evaporated under vacuum to afford ca. 3.3 grams of gummy, brown residue.
To determine the taxonomic position of the strain, extracted DNA was amplified using PCR with primers for ITS4. PCR products were sequenced by Genomics BioScience and Technology Co., Ltd. The conserved regions of the 18S rRNA genes of the strain were compared with other sequences in the public database using the BLAST program show the highest match to Paraconiothyrium sp (Montagulaceae) (99.0). The resulting sequences were submitted to GenBank (BankIt2007192, KY948085).
Isolation.
The extract was loaded onto two separate diol DIO Spe-ed SPE cartridges (112 mg each), and each cartridge was sequentially eluted with 7.0 mL each of n-hexane, CH2Cl2, EtOAc, and MeOH to give four fractions (A−D). The colon cancer cytotoxicity of fraction C (IC50 = 5.9 μg/mL (COLO205), IC50= 5.5 μg/mL (KM12)) was more potent than the other fractions (Supporting Figure S51), thus, it was chromatographed on an ODS column (MeOH−H2O; gradient from 1:9 to 9:1) with 180 drop fractions collected. On the basis of UV absorption at 220 nm, six fractions (A−F) were collected by combining tubes 5−10, 11−19, 20, 21–24, 25, and 26–30, respectively. Compounds 3 (0.6 mg) and 4 (1.4mg) were separately obtained from fractions C and E by MPLC on a ODS gel column (MeOH−H2O; gradient from 1:9 to 9:1). Fraction A was further purified by HPLC using a 250 ×10 mm Phenomenex Luna 5 μ C18 (2) 100 Å column. The detector wavelength was set to 210 nm, and the solvent flow rate was 4.0 mL/min. Elution began with 10% MeOH−H2O, followed by a linear gradient for 30 min of 10% to 60% MeOH−H2O to yield compounds 1 (1.0 mg), 2 (0.9 mg) and 7 (0.4 mg). Fraction B was also purified using the same C18 HPLC column and identical conditions to those used for fraction A. Of the four fractions (B1−B4) that were collected, fraction B2 was found to be solely composed of compound 5 (1.1 mg), while fraction B4 appeared to be a mixture of two closely related compounds. B4 was then subjected to further HPLC purification on the same C18 column using an isocratic elution with 50% MeOH−H2O to yield compounds 6 (1.0 mg) and 10 (0.7 mg). Compounds 8 (2.3 mg) and 9 (1.8 mg) were obtained from fraction F by C-18 HPLC column with a gradient for 30 min of 30% to 80% MeOH−H2O.
Craterellone F (1):
brown amorphous powder; [α]25D +10 (c 0.3, MeOH); UV (MeOH) λmax (logε) 230 nm; 1H NMR (600 MHz, DMSO-d6) and 13C NMR (150 MHz, DMSO-d6) data, see Table 1; HRESIMS found m/z 287.1602 [M+Na]+ (calcd for C16H24O3Na, 287.1623).
Isoversiol A (3):
brown amorphous powder; [α]25D +51 (c 0.3, MeOH); UV (MeOH) λmax (logε) 223 nm; 1H NMR (600 MHz, DMSO-d6) and 13C NMR (150 MHz, DMSO-d6) data, see Table 1; HRESIMS found m/z 265.1792 [M+H]+ (calcd for C16H25O3 265.1804).
Isoversiol B (4):
white amorphous powder; [α]25D +43 (c 0.3, MeOH); UV (MeOH) λmax (logε) 225 nm; 1H NMR (600 MHz, DMSO-d6) and 13C NMR (150 MHz, DMSO-d6) data, see Table 1; HRESIMS found m/z 263.1609 [M+H]+ (calcd for C16H23O3 263.1647).
Isoversiol C (5):
white amorphous powder; [α]25D +34 (c 0.3, MeOH); UV (MeOH) λmax (logε) 224 nm; 1H NMR (600 MHz, DMSO-d6) and 13C NMR (150 MHz, DMSO-d6) data, see Table 1; HRESIMS found m/z 279.1589 [M+H]+ (calcd for C16H23O4 279.1596).
Isoversiol D (6):
white amorphous powder; [α]25D +47 (c 0.3, MeOH); UV (MeOH) λmax (logε) 225 nm; 1H NMR (600 MHz, DMSO-d6) and 13C NMR (150 MHz, DMSO-d6) data, see Table 1; HRESIMS found m/z 303.1575 [M+Na]+ (calcd for C16H24O4Na, 303.1572).
Isoversiol E (7):
white amorphous powder; [α]25D +76 (c 0.3, MeOH); UV (MeOH) λmax (logε) 231 nm; 1H NMR (600 MHz, DMSO-d6) and 13C NMR (150 MHz, DMSO-d6) data, see Table 1; HRESIMS found m/z 263.1628 [M+H]+ (calcd for C16H23O3, 263.1647).
Calbistrin F (8):
brown amorphous powder; [α]25D +36 (c 0.3, MeOH); UV (MeOH) λmax (logε) 225 nm; 1H NMR (600 MHz, DMSO-d6) and 13C NMR (150 MHz, DMSO-d6) data, see Table 1; HRESIMS found m/z 497.2512 [M+Na]+ (calcd for C27H38O7Na, 497.2515).
Conionaphthalenediol (10):
white amorphous powder; [α]25D −37 (c 0.3, MeOH); UV (MeOH) λmax (logε) 229 nm; 1H NMR (600 MHz, DMSO-d6DMSO-d6) and 13C NMR (150 MHz, DMSO-d6) data, see Table 1; HRESIMS found m/z 443.2456 [M-H]+ (calcd for C26H35O6, 443.2434).
Antiproliferative Bioassay.
The assay used in this study was an in vitro antitumor assay using the XTT endpoint developed by the Assay Development and Screening section of the MTL (Molecular Targets Laboratory). The colon cancer cell lines, COLO205 and KM12, were used for bioassay-guided isolation. In brief, cells were harvested and plated (45 μL) at a seeding density of 3.0 × 105 cells per well for the COLO205 cell line and 2.5 × 105 cells per well for the KM12 cell line into a 384-well “assay plate” and then incubated for 24 h. DMSO solutions of the test materials (8 μL) were diluted 1:25 with medium (192 μL) and then subjected to five 2:1 serial dilutions (100 μL each) on a 96-well plate. Duplicate 40 μL aliquots of each sample concentration were then transferred to a 384-well dilution plate, which could accommodate the duplicate samples from two 96-well plates. A 5 μL portion of each solution on the dilution plate was transferred to the cell cultures on the 384-well assay plate to give a final volume of 50 μL and a DMSO concentration of 0.4%. Cells were incubated for 48 h at 37 °C in the presence of the test samples and then treated with the tetrazolium salt XTT (2,3-bis[2-methoxy-4-nitro-5-sulfophenyl]-2H-tetrazolium-5-carboxanilide). Viable cells reduced the XTT to a colored formazan product, and after an additional 4 h incubation period the amount of formazan produced was quantified by absorption at 450 nm, using a 650 nm reference. Pure compounds were tested in the assay described above and in the NCI 60 cell assay.15
Computational Details.
Molecular mechanics were performed using Macromodel interfaced to the Maestro program (Version 2015.3, Schrödinger). All conformational searches used the OPLS_2005 force field. Conformers having internal relative energies within 3 kcal/mol were subjected to geometry optimization on Gaussian 09 at the DFT level with the B3LYP functional and the 6–31G(d) basis set. Single point calculations in DMSO with the B3LYP functional and the 6–311+G(d,p) basis set were then employed to provide the shielding constants of carbon and proton nuclei. Meanwhile, the same procedure was applied on TMS (tetramethylsilane). Final 1H and 13C chemical shifts were obtained as the results of the Boltzmann weighted average. The theoretical chemical shifts were calculated according to an equation
Where is the calculated shift for nucleus x (in ppm); σx is the shielding constant for nucleus x; σTMS is the shielding constant and chemical shift of TMS computed at the same level of theory.
The mean absolute error (MAE) was defined as after removing systematic errors during the chemical shift calculations. The correlation coefficient (R2) were determined from a plot of σcalc. against σexp. for each particular compound. DP4 parameters were calculated by using the online applet at http://www-jmg.ch.cam.ac.uk/tools/nmr/.
Supplementary Material
Scheme 1.

Structures of compounds reported.
ACKNOWLEDGMENTS
This research was supported by the Intramural Research Program of the NIH, National Cancer Institute, Center for Cancer Research (1ZIA-BC01147005), and by the Developmental Therapeutics Program in the Division of Cancer Treatment and Diagnosis of the National Cancer Institute. Additionally, this work was supported by a grant from the KRIBB Research Initiative Program (Korean Biomedical Scientist Fellowship Program), Korea Research Institute of Bioscience and Biotechnology, Republic of Korea. We thank Heidi Bokesch for HRESIMS data collection, and Sarah Long and Dongdong Wang for helpful comments. This work utilized the computational resources of the NIH HPC Biowulf cluster.
Footnotes
Supporting Information. The NMR spectra of isolated compounds and their cell growth inhibitory activities in the NCI-60 cell assay.
REFERENCES
- (1).Siegel R; Naishadham D; Jemal A CA Cancer J. Clin 2013, 63, 11–30. [DOI] [PubMed] [Google Scholar]
- (2).de Gramont A; de Gramont A; Chibaudel B; Larsen AK; Tournigand C; André, T. Clin. Colorectal Cancer 2011, 10, 218–226. [DOI] [PubMed] [Google Scholar]
- (3).Sinha VR; Honey. Crit. Rev. Ther. Drug 2012, 29, 89–148. [DOI] [PubMed] [Google Scholar]
- (4).Stein A; Hiemer S; Schmoll HJ Drugs 2011, 71, 2257–2275. [DOI] [PubMed] [Google Scholar]
- (5).McKee TC; Rabe D; Bokesch HR; Grkovic T; Whitson EL; Diyabalanage T; VanWyk AW; Marcum SR; Gardella R, S.; Gustafson, K. R.; Linehan, W. M.; McMahon, J. B.; Bottaro, D. P. J. Nat. Prod 2012, 75, 1632–1636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Mohamed IE; Kehraus S; Krick A; König GM; Kelter G; Maier A; Fiebig HH; Kalesse M; Malek NP; Gross HJ Nat. Prod 2010, 73 2053–2056. [DOI] [PubMed] [Google Scholar]
- (7).Chen S; Zhang Y; Zhao C; Ren F; Liu X; Che Y Fitoterapia 2014, 99, 236–234. [DOI] [PubMed] [Google Scholar]
- (8).Stewart M, Capon RJ; Lacey E; Tennant S; Gill JH J. Nat. Prod 2005, 68, 581–584. [DOI] [PubMed] [Google Scholar]
- (9).McGahren WJ; Ellestad GA; Morton GO; Kunstmann MP J. Org. Chem 1976, 41, 66–71. [DOI] [PubMed] [Google Scholar]
- (10).Guo H; Feng T; Li ZH; Liu JK Nat. Prod. Bioprospect 2012, 2, 170–173. [Google Scholar]
- (11).Fujii Y; Asahara M; Ichinoe M; Nakajima H Phytochemistry 2002, 60, 703–738. [DOI] [PubMed] [Google Scholar]
- (12).Brill GM; Chen RH; Rasmussen RR; Whittern DN; McAlpine JB J. Antibiot 1993, 46, 39–47. [DOI] [PubMed] [Google Scholar]
- (13).Tatsuta K; ltoh M; Hirama R; Araki N; Kitagawa M Tetrahedron Lett. 1997, 38, 583–586. [Google Scholar]
- (14).Paull KD; Shoemaker RH; Hodes L; Monks A; Scudiero DA; Rubinstein L; Plowman J; Boyd MR J. Natl. Cancer Inst. 1989, 81, 1088–1092. [DOI] [PubMed] [Google Scholar]
- (15).Shoemaker RH Nat. Rev. Cancer 2006, 10, 813–823. [DOI] [PubMed] [Google Scholar]
- (16).da Cunha MG; Rosalen PL; Franchin M; de Alencar SM; Ikegaki M; Ransom T; Beutler JA Planta. Med 2016, 82, 190–194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Sunassee SN; Ransom T; Henrich CJ; Beutler JA; Covell DG; McMahon JB; Gustafson KR J. Nat. Prod 2014, 77, 2475–2480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Devkota KP; Covell D; Ransom T; McMahon JB; Beutler JA J. Nat. Prod 2013, 76, 710–714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Bills GF; Dombrowski AW; Goetz MA In: Methods in Molecular iology. Fungal Secondary Metabolism: Methods and Protocols; Keller NP; Turner G, Eds: Springer Verlag: Berlin, 2012; Chapter 5, pp 79–96. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.