

Abstract
Purpose:
To determine the tolerability, pharmacokinetics and mechanisms of temozolomide resistance in children with relapsed or refractory leukemia.
Patients and Methods:
Cohorts of 3–6 patients received 200 mg/m2/day or 260 mg/m2/day of temozolomide by mouth daily for 5 days every 28 days. Toxicities, clinical response, and pharmacokinetics (PK) were evaluated. Pretreatment leukemia cell O6-methylguanine-DNA- methyltransferase (MGMT) activity, tumor and plasma MGMT promoter methylation, and microsatellite instability (MSI) were examined in 14/16 study patients and in tissue bank samples from children with ALL not treated with temozolomide (MGMT:n=67; MSI:n=65).
Results:
Sixteen patients (9 female, 7 male) (ALL=8, AML=8), median age 11 years (range 1–19 years), received either 200 mg/m2/day (9 enrolled, 3 evaluable for toxicity) or 260 mg/m2/day (7 enrolled, 3 evaluable for toxicity) of temozolomide. No dose limiting toxicities occurred. The mean clearance of temozolomide was 107 mL/min/m2 with a volume of distribution of 20 L/m2; and half-life of 109 min. MGMT activity in leukemia cells was quite variable and was highest in patients with relapsed ALL. Only one patient had MSI. Two patients had a PR; neither had detectable MGMT activity with methylated MGMT promoters and both were MSI-stable.
Conclusion:
Temozolomide was well tolerated at doses as high as 260 mg/m2/day for five days in children with relapsed/refractory leukemia. Increased MGMT activity may account for the temozolomide resistance in children with relapsed leukemia. Leukemia cell MGMT activity was higher in pediatric ALL than AML (p<.0001).
Keywords: methyl guanine methyltransferase, MGMT, alkyl guanine transferase, AGT, promoter methylation, microsatellite instability, acute lymphoblastic leukemia, acute myelogenous leukemia
INTRODUCTION
Although cure rates for children with newly diagnosed leukemia approach 85–90%, the prognosis for children with relapsed leukemia remains poor and novel treatment approaches are needed. Temozolomide, an imidazole tetrazinone approved for the treatment of high-grade glioma,1,2 inhibits cell growth in leukemia cell lines and leukemia xenografts.3,4 In a phase 1 study of temozolomide in adults with leukemia, two patients achieved a complete response (CR) and two achieved a CR without platelet recovery (CRp) with durable remissions (8–14 months). The dose-limiting toxicity (DLT) was prolonged bone marrow aplasia and the maximum tolerated dose (MTD) was 200 mg/m2/day for 7 days with courses repeated every 28 days.5
Temozolomide resistance correlates with activity of the DNA repair enzyme MGMT.6–8 Increased MGMT activity is associated with inferior outcomes in adult and pediatric malignant gliomas.9,10 Several in vitro studies have shown that increased MGMT activity can contribute to temozolomide resistance in leukemia;11,12 however, correlations between patient leukemia cell MGMT activity and temozolomide resistance have not been directly examined.
Hypermethylation of CpG islands near gene promoter regions results in transcriptional inactivation of many genes.13 MGMT promoter methylation is associated with both decreased MGMT activity14 and increased response to temozolomide in both CNS tumors15,16 and diffuse large-B cell lymphomas.17 However some patients with leukemia have evidence of MGMT promoter hypermethylation;18,19 and it is not known whether MGMT methylation correlates with temozolomide response.
Although increased MGMT activity is associated with temozolomide resistance in AML patients and leukemia cell lines, 3,20 some leukemia cells remain resistant to temozolomide despite MGMT inhibition.3 Temozolomide resistance in these tumor cells results from mismatch repair mutations,21–24 which are frequently associated with microsatellite instability (MSI).24 MSI is uncommon in adults with de novo AML;25 however, the frequency of MSI and its contribution to temozolomide resistance in pediatric leukemia has not previously been examined.
In this study we examined the efficacy, toxicities and pharmacokinetics of oral temozolomide given daily for 5 days at two dose levels, 200 mg/m2/day and 260 mg/m2/day. In addition, we evaluated MGMT enzymatic activity, MGMT promoter methylation, and MSI in leukemic blasts from patients enrolled on this trial and banked leukemia samples.
PATIENTS AND METHODS
Patient Eligibility:
Patients between the ages of 1 and 21 years (inclusive) with refractory acute lymphoblastic leukemia (ALL) or acute myelogenous leukemia (AML) were eligible for this trial (> 25% blasts on bone marrow aspirate). Other eligibility criteria included: 1) a Karnofsky or Lanksy performance score ≥ 50, 2) recovery from the acute toxic effects of prior chemotherapy, radiotherapy or immunotherapy with a minimum elapsed period of at least 7 days since the last dose of corticosteroids or hematopoietic growth factors, 3) at least 3 months since the last stem cell transplant, and 4) at least 3 months since prior craniospinal radiation, pelvic radiation or TBI, at least 2 weeks since local palliative radiation, and at least 6 weeks since other substantial radiation. Patients could have no evidence of active graft-versus-host disease. Patients were required to have adequate renal function (serum creatinine below the upper limits of normal for age OR a GFR ≥ 70 ml/min/1.73m2); adequate liver function (serum bilirubin ≤ 1.5 mg/dL, ALT ≤5 x the institutional upper limit of normal for age, and albumin ≤ 2 g/dL); a platelet count ≤ 20,000/μL, and a hemoglobin ≤ 8g/dL. Transfusion support was allowed if necessary to meet hematologic parameters. Study exclusion criteria included: CNS leukemia, pregnancy or lactation in women of child-bearing age, uncontrolled infection, hyperleukocytosis (WBC > 100,000 cells/μL), receipt of concomitant enzyme-inducing anticonvulsants, or concomitant use of other experimental agents. Hydroxyurea was permitted up to 24 hours before the start of therapy if needed for cytoreduction. Institutional Review Board approval and informed consent from the patient or their parent(s) and assent, as appropriate, were obtained in accordance with federal and institutional policies.
Dosage and drug administration:
Temozolomide capsules (Schering-Plough Research Institute, Kenilworth, NJ) were administered orally one hour (h) before or 2 h after meals. Capsules could be opened and the contents resuspended in apple juice or applesauce for children unable to swallow capsules.
Trial design:
The starting dose of temozolomide was 200 mg/m2/dose for 5 days, 70% of the adult MTD.5 Subsequent planned dose escalations were in increments of 30%. Courses were repeated every 28 days if there was no evidence of progressive disease, the platelet count was ≤ 20,000/μL, the hemoglobin ≤ 8 g/dL (with transfusion support) and any other treatment-related adverse events were ≤ grade 1. Doses were not held or omitted for hematologic toxicity. A minimum of three patients assessable for DLT were entered at each dose level. Dose levels were expanded to six patients evaluable for toxicity if one patient experienced a DLT during the first course of therapy.
Toxicity assessment:
Adverse events were graded according to the NCI Common Terminology Criteria for Adverse Events (CTCAE v. 3.0). Non-hematologic DLT was defined as any grade 3 or 4 adverse event attributable to temozolomide with the specific exclusion of grade 3 nausea or vomiting, grade 3 hepatic transaminase (AST or/or ALT) elevation returning to ≤ grade 1 before the next treatment course, and grade 3 fever or infection.
Response assessment:
Complete response (CR) was defined as the attainment of an M1 bone marrow (< 5% blasts and adequate marrow cellularity) with no evidence of circulating blasts or extramedullary disease. Partial response (PR) was defined as the complete disappearance of circulating blasts and achievement of M2 marrow status (≥ 5% to < 25% blast cells with adequate cellularity). Progressive disease (PD) was defined as an increase of at least 25%, or an absolute increase of at least 5,000 cells/mm3 (whichever was greater) in the number of circulating leukemia cells, development of extramedullary disease, or other laboratory or clinical evidence of PD. Stable disease (SD) was defined as failing to fulfill the criteria for a CR, PR or PD.
Pharmacokinetic studies:
Blood samples (2 mL) were collected in heparinized tubes prior to and at 0.5, 1, 2, 3, 4, 6, and 8 h after the first temozolomide dose. Plasma was separated and frozen at −80°C until analysis. Plasma temozolomide concentrations were measured using a modification of a previously described high-performance liquid chromatography assay.27 In brief, plasma treated with phosphoric acid (20 μg/ml) underwent solid phase extraction using 30 mg/mL StrataX reverse phase columns (Phenomenex, Torrance, CA). Plasma was eluted with acetonitrile and evaporated to dryness under nitrogen at 37°C. Samples were reconstituted in 1% phosphoric acid and injected onto a Luna (2) C18, 3 μ, 4.6 mm x 250 mm column with a C18, 3 μ, 2 mm X 3 mm guard column (Phenomenex, Torrance, CA) and eluted with a mobile phase of 0.5 % acetic acid/methanol (88/12, v/v). Peaks were monitored on a Waters 996 photodiode array detector at 330 nm (Waters Associates, Milford, MA). Recovery of temozolomide was 79 ± 2% and the intra- and inter-assay precisions were 1.7% and 3.9%. The lower limit of quantitation for temozolomide was 0.012 μg/mL.
Temozolomide pharmacokinetic analyses were performed using both compartmental and noncompartmental methods. Time to maximum concentration (Tmax), peak plasma concentration (Cmax), and under the concentration-time curve (AUC), apparent volume of distribution (Vd-ss/F) were calculated using non-compartmental methods.28 One- and two-compartment models were fit to the concentration-time data using ADAPT II (Biomedical Simulations Resources, University of Southern California, Los Angeles, CA) with maximum likelihood estimation.29 Akaike’s Information Criterion (AIC) was used to select the best model fit.30 Individual patient clearance (Cl/F), the absorption half-life, and the elimination- half lives for each phase were estimated from the model.28
Tumor cell isolation:
Pretreatment blood and bone marrow leukemia cells were collected, shipped, and processed as previously described.26 After plasma removal, samples with <90% tumor cells were sorted to >90% purity using a DakoMation cell sorter26. Viability of each patient sample was ≤ 90% as determined by trypan blue exclusion. Plasma, tumor cells, and normal lymphocytes (when available) were frozen in RPMI 1640 medium supplemented with 20% bovine growth serum and 10% DMSO.26 An additional 67 previously banked leukemia specimens were obtained from the Texas Children’s Cancer Center tissue bank using an IRB approved protocol.
MGMT activity:
MGMT enzyme activity in PBMC or BMA tumor cell lysates was determined by the removal of O6-[3H]methylguanine from a [3H] methylated DNA substrate and quantitated as fmol O6-[3H]methylguanine / total protein lysate.31 All samples contained >90% leukemia cells except patient #5 (44% blasts), #12 (75% blasts) and #15 (50% blasts). Elevated MGMT activity and low MGMT activity were defined as > 2 standard deviation away from the mean MGMT activity from 4 normal volunteers.
MGMT promoter methylation:
MGMT promoter DNA methylation patterns were determined using either tumor cell DNA or free DNA isolated from patient plasma using methylated-specific PCR as described previously.16
Microsatellite instability assay:
Genomic DNA from pretreatment blasts was extracted and amplified using three MSI multiplex reaction mixtures containing NCI panel markers (BAT-25, BAT-26, D2S123, D17S250, D5S346),32 quasimonomorphic mononucleotide markers (NR-21, NR-22, NR-24, BAT-25, BAT-26),33 and an alternative panel of markers (D18S35, TP53-DI, TP53-PENTA, D1S2883, and FGA) .34 Tumor cell MSI was compared to peripheral blood or non-malignant lymphocytes obtained from the same patient. Each sample was scored according to number of markers positive or negative for MSI. Tumors were categorized into one of three categories; MSI-high (>40% of markers unstable), MSI-low (less than 30% of markers unstable), or MSI-stable using NCI standards.35
Statistics:
The Wilcoxon rank-sum test was used to compare MGMT activity by leukemia subtype (ALL vs. AML) and patient disease status (newly diagnosed vs. relapsed). Three banked specimens had total protein concentrations below the level of quantitation (0.1 mg/mL) and these samples were removed from analysis. The impact of potential confounders , such as age, sex, race and WBC count, on MGMT levels was assessed in stratified analyses using the Wilcoxon rank-sum test. Statistical analyses were applied to patient data and banked samples separately.
RESULTS
From July 2004 until April 2006, 16 pediatric patients (8 with AML, 8 with ALL) were enrolled (Table 1). Ten patients were not fully assessable for toxicity because they had early progressive disease (PD) and either received non-protocol therapy before completing the first course of temozolomide (n=8) or died from PD during the first course of treatment (n=2). None of these patients experienced unusual or severe temozolomide-related toxicities. Three patients were fully assessable for toxicity at each dose level.
Table 1.
Patient Characteristics for Eligible Patients (n=16)
| Characteristic | Number |
|---|---|
| Age (years) | |
| Median (Range) | 11 (1–19) |
| Sex (Male:Female) | 7:9 |
| Race/ethnicity | |
| White | 7 |
| Black or African American | 4 |
| Asian | 3 |
| Hispanic | 1 |
| Other | 1 |
| Diagnosis | |
| ALL | 8 |
| AML | 8 |
| Prior Therapy | |
| Chemotherapy Regimens | |
| Median (Range) | 4 (1–7) |
| Prior Radiation | 8 |
| Prior Bone Marrow Transplant | 10 |
| Karnofsky/Lansky | |
| Median (Range) | 90 (60–100) |
Toxicity:
Temozolomide was well tolerated in children with recurrent or refractory leukemia. There were no DLTs at the two dose levels evaluated. Non-DLTs related to temozolomide included 1 case each of grade 3 nausea, vomiting, fever with neutropenia, pneumonia, and elevated serum transaminases levels.
Response:
One five year-old male with AML (M1 subtype) treated with 200 mg/m2 temozolomide had a partial response (PR) with a decrease in bone marrow blasts from 60% to 10%, received four courses before developing PD. A 14-year-old female with AML/MDS (M2 subtype) also had a PR, with a decrease in bone marrow blasts from 56% to 6% after one course of temozolomide. Her parents refused further treatment and she developed PD six weeks after cessation of therapy. Four patients had PD after the completion of course one and the remaining ten patients had early PD.
Pharmacokinetics:
Pharmacokinetic samples were obtained from 10 patients (Table 2). Temozolomide was both rapidly absorbed and eliminated from plasma. Based on the non-compartmental analysis, the time to maximal concentration was 74 ± 50 minutes and the terminal half-life was 109 ± 24 minutes. The Vd was 20 ± 5.2 L/m2, and the apparent oral systemic Cl was 107 ± 31 mL/min/m2. Both AUC and Cmax increased in proportion with dose. Compartmental analysis resulted in a mean elimination t1/2 of 111 ± 37 minutes. Compartmental and non-compartmental t1/2, Cl and Vd were similar.
Table 2:
Summary of Day 1 pharmacokinetic parameters from 10 pediatric patients treated with temozolomide.
| Parametera | 200 mg/m2
(n=6)b |
260 mg/m2 (n=4)b |
|---|---|---|
| Tmax (min) | 82 ± 57 | 61± 36 |
| Cmax (μg/mL) | 9.3 ± 3.0 | 13 ± 2.8 |
| AUC0–24h (μg/m•h) | 28 ± 7.3 | 46 ± 6.4 |
| AUC0−∞ (μg/ml•h) | 30 ± 7.8 | 51 ± 8.7 |
| Vd-ss/F (L/m2) | 21 ± 5.7 | 17 ± 3.1 |
| Cl/F (mL/min/m2) | 120 ± 38 | 90 ± 21 |
| t1/2 abs (min) | 19 ± 18 | 8.7 ± 3.4 |
| t1/2 el (min) | 100± 24 | 129 ± 45 |
Noncompartmental analyses were used to determine Tmax, Cmax, AUC and Vd;compartmental analyses were used to determine Cl and t1/2.
Mean ± standard deviation
Assessment of pre-treatment MGMT enzyme activity:
We assessed MGMT enzyme activity in 12 patients (Table 3). Patients had a wide range of MGMT activity (<100 to >2500 fmol/mg protein) compared to normal volunteers, who had an average MGMT activity of 771 ± 170 fmol/mg protein (Figure 1). MGMT activity appeared to correlate with leukemia subtype; 3/7 patients with AML had MGMT activity below the level of detection (<5%) while no ALL patients had undetectable MGMT activity. Although 4/5 patients with ALL had elevated MGMT enzyme activity (>1100 fmol/mg protein), no patients with AML had elevated MGMT activity (Table 3). There was no correlation between patient MGMT activity and age, sex or race. The two patients with a PR to temozolomide had undetectable MGMT activity.
Table 3: Mechanisms of temozolomide resistance:
MGMT activity and microsatellite instability in peripheral blood or bone marrow aspirate samples (n=14)
| Pt | Dx | Response | MGMT activity (fmol/mg protein) |
MGMT promoter methylation (plasma)2 |
MGMT promoter methylation (cells) 2 |
MSI3 |
|---|---|---|---|---|---|---|
| 1 | Infant ALL | PD | 356 | U | U | Stable |
| 3 | ALL | PD | ND | U | ND | Stable |
| 5 | ALL | PD | 1607 | U | U | Stable |
| 6 | ALL | SD | 1756 | U | U | Stable |
| 11 | ALL | PD | >2500 | U | ND | Stable |
| 13 | ALL | PD | 2393 | U | U | Stable |
| 15 | AML | PD | 390 | M | M# | Stable |
| 2 | AML | PD | Undetectable | M# | M# | Stable |
| 7 | AML | PD | 430 | U | ND | Stable |
| 8 | AML | PD | 837 | U | U | Stable |
| 9 | AML | PR | Undetectable | M# | M# | Stable |
| 12 | AML | PD | 835 | U | U | Stable |
| 14 | AML | PR | Undetectable | M | ND | Stable |
| 16 | AML | PD | ND | U | ND | MSI-high |
U=unmethylated, M= methylated, M#= both methylated and unmethylated PCR products present
Microsatellite instability
Figure 1: Summary of MGMT enzyme activity in an independent set of leukemia samples:
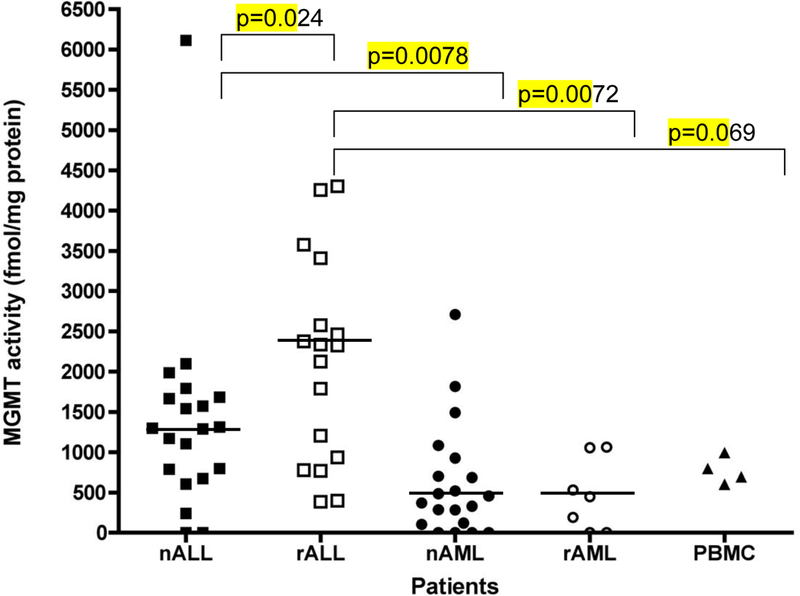
MGMT activity was determined in 67 pediatric patients with newly-diagnosed ALL (nALL)(n=21), relapsed ALL (rALL) (n=19), newly-diagnosed AML (nAML) (n=20), relapsed AML (rAML)(n=7), and PBMC from healthy volunteers (n=4). Median values for each group noted as solid line. Differences that are statistically different are noted (Wilcoxon rank-sum test).
We further explored MGMT activity in an independent set of 67 banked ALL and AML samples obtained from the Texas Children’s Hospital tissue tumor bank using an IRB-approved protocol. Three banked specimens had total protein concentrations below the level of quantitation (0.1 mg/mL) and were excluded from analysis . As shown in Figure 1, MGMT activity correlated with leukemia subtype. ALL lymphoblasts had significantly higher MGMT activity than AML myeloblasts (p<0.0001). The highest MGMT activity was found in specimens from patients with newly-diagnosed ALL. In general, specimens from patients with relapsed ALL had higher MGMT activity than specimens from either newly-diagnosed ALL (p=0.02), or relapsed AML (p<0.01). MGMT activity did not correlate with sex but it appeared to correlate with race (whites: 1316 ± 1227 fmol/mg protein vs. non-whites: 623 ± 761 fmol/mg protein; p=0.048). Differences in MGMT activity between leukemia subtypes, however, remained significant after controlling for this potential confounder.
Assessment of MGMT promoter methylation:
MGMT promoter methylation correlated with MGMT activity in 12 patients. (Table 3, Figure 2). Patients with MGMT promoter methylation had little or no MGMT activity, whereas patients with an unmethylated MGMT promoter had MGMT activity present. Although the two patients with a PR to temozolomide had methylated MGMT promoters and no MGMT expression, patients with no objective responses had both unmethylated (n=10) and methylated (n=2) MGMT promoters, implying that factors other than MGMT activity can result in temozolomide resistance. MGMT promoter methylation status was concordant in plasma and leukemia blasts in the 9 patients where both were evaluated.
Figure 2: MGMT promoter methylation patterns in patients treated with temozolomide.
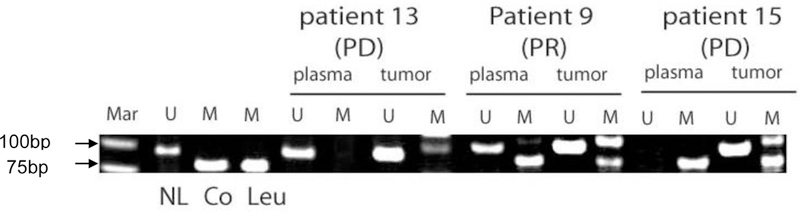
Genomic DNA was extracted prior to temozolomide treatment and MGMT promoter methylation determined by methylation-specific PCR. U= unmethylated, M=methylated, Mar= 50bp marker, NL= normal lymphocytes, Co- SW-48 colon carcinoma, Leu = U937 AML cells. PD = progressive disease, PR = partial response. PCR product at 110 bp in methylated PCR reaction is non-specific.
Microsatellite instability:
Thirteen of the 14 patients tested were MSI-stable, whereas only one patient was MSI-high (Table 3). This patient had MSI at multiple MSI loci (Figure 3A). MSI was also examined in 65 banked leukemia samples. As shown in Figure 3B, samples from patients with newly diagnosed ALL or AML were MSI-stable or MSI-low. In contrast, samples from 7/18 patients with relapsed ALL were MSI-high and 2/18 patients were MSI-low. Several patients with relapsed AML (3/7) were also MSI-low. This suggests that MSI is common in pediatric patients with relapsed leukemia.
Figure 3: Microsatellite instability in pediatric leukemia:
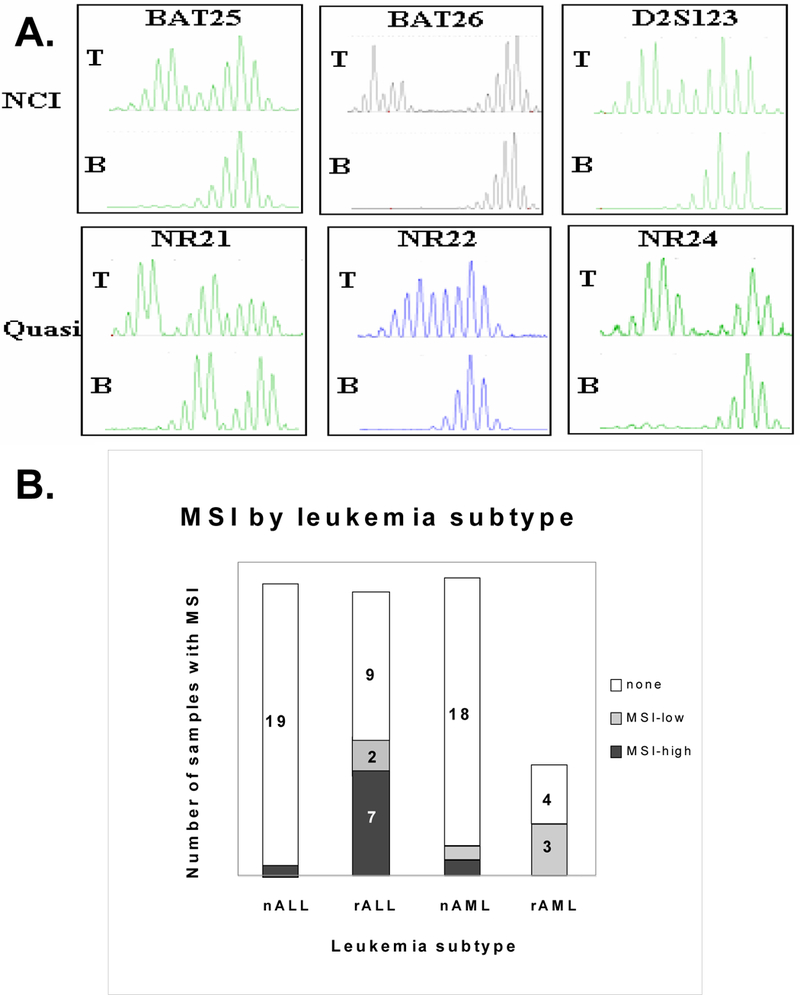
(A) Representative fluorescent-based microsatellite alterations from a patient demonstrating microsatellite instability in the leukemia cells (T), but not the normal lymphocytes (B) for the NCI MSI markers BAT-25, BAT-26, and D2S123 32 and the quasimonomorphic MSI markers NR-21, NR-22 and NR-2433. (B) Microsatellite instability was assessed in 65 pediatric leukemia samples categorized by subtype and relapse status. Microsatellite instability was determined at 13 alleles. Samples were classified as stable, having instability at a single site (MSI-low), or having instability at multiple sites (MSI-high), consistent with a mutator phenotype (RER+).
DISCUSSION
In this study we examined the toxicities, response and pharmacokinetics of temozolomide given as a single agent to pediatric patients with relapsed or refractory leukemia. Temozolomide drug disposition in children with leukemia was similar to that observed in pediatric patients with solid tumors36 as well as in adults.28,37–40 Due to the high number of patients with PD during the first treatment course, the MTD of temozolomide in pediatric patients with relapsed leukemia was not determined. However, three evaluable patients tolerated 260 mg/m2 without evidence of DLTs. Of the sixteen patients enrolled, 2 had objective responses.
Another objective of this trial was to examine the mechanisms of temozolomide resistance. Increased MGMT enzymatic activity has been correlated with temozolomide resistance in both glioma and leukemia cell lines.1,3,41 Patients in this study with elevated MGMT activity were also resistant to temozolomide. MGMT expression is decreased by MGMT promoter methylation, which is common in many tumor types, and MGMT methylation may predict temozolomide sensitivity.42 However, others have found only a moderate association between MGMT promoter methylation and MGMT enzyme activity43–45. In our study, 12 samples showed concordance between MGMT activity and MGMT promoter methylation.
We also noted a concordance between MGMT methylation in tumor cells and plasma. Since only tumor DNA is abnormally methylated (normal tissue remains unmethylated), plasma MGMT methylation could be used to monitor disease response and detect early relapse in patients whose tumors have methylated MGMT promoters.46 Balana et al. reported a correlation between plasma MGMT promoter methylation and chemotherapy response in patients with glioblastoma multiforme,16 suggesting that further research in this area is warranted.
Some tumors remain resistant to temozolomide despite low MGMT activity due to mismatch repair pathway defects,47 which may arise from mutations or gene silencing of mismatch repair enzymes. 23 Mismatch repair defects and the resulting MSI, which are common in leukemia cell lines,22,24,48,49 are less common in primary pediatric leukemias (approx. 10% prevalence) 48,50–52 and in adults with newly diagnosed leukemia.53,54 MSI, however, is more prevalent in relapsed leukemia,55–57 treatment-related AML,25,58,59 and adult T-cell ALL.60 In our study, only one patient was MSI-high. Since several patients with PD had stable MSI (Table 3), it did not appear that MSI was a major determinant of temozolomide resistance. However, MSI stability may be necessary for temozolomide sensitivity since previous work has shown that leukemia cells with mismatch repair defects are resistant to temozolomide despite low MGMT activity.
In summary, we determined that in children with relapsed and refractory leukemia, temozolomide could be tolerated at doses of 260 mg/m2/day for five days. In this heavily pre-treated population, however, we did not observe significant clinical activity. As patients with relapsed AML in general had lower MGMT activity than patients with relapsed ALL, further study of temozolomide in this leukemia subtype should be considered.
Acknowledgements:
Thanks to Anu Gannavarapu, Gaye Jenkins, John Hyatt and Alexander Aleksic for technical assistance.
Supported and funded by: NCI grants UO1-CA97452 (COG), U01CA63187 (University of Chicago Cancer Research Center), K12CA90433 (TMH), The Lady Tata Memorial Fund (TMH), the Scott Carter NCCF Research Fellowship (TMH).
Prior presentation of work: The patient data from this study was presented in abstract form at the American Society of Hematology meeting, December, 2005.
Footnotes
Disclaimers: none.
References
- 1.Friedman HS, Kerby T, and Calvert H Temozolomide and treatment of malignant glioma. Clin.Cancer Res, 6: 2585–2597, 2000. [PubMed] [Google Scholar]
- 2.Stupp R, Mason WP, van den Bent MJ, Weller M, Fisher B, Taphoorn MJ, Belanger K, Brandes AA, Marosi C, Bogdahn U, Curschmann J, Janzer RC, Ludwin SK, Gorlia T, Allgeier A, Lacombe D, Cairncross JG, Eisenhauer E, and Mirimanoff RO Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N.Engl.J Med, 352: 987–996, 2005. [DOI] [PubMed] [Google Scholar]
- 3.Tentori L, Graziani G, Gilberti S, Lacal PM, Bonmassar E, and D’Atri S Triazene compounds induce apoptosis in O6-alkylguanine-DNA alkyltransferase deficient leukemia cell lines. Leukemia, 9: 1888–1895, 1995. [PubMed] [Google Scholar]
- 4.Messinger Y, Reaman GH, Ek O, and Uckun FM Evaluation of temozolomide in a SCID mouse model of human B-cell precursor leukemia. Leuk Lymphoma, 33: 289–293, 1999. [DOI] [PubMed] [Google Scholar]
- 5.Seiter K, Liu D, Loughran T, Siddiqui A, Baskind P, and Ahmed T Phase I study of temozolomide in relapsed/refractory acute leukemia. J Clin Oncol, 20: 3249–3253, 2002. [DOI] [PubMed] [Google Scholar]
- 6.Baer JC, Freeman AA, Newlands ES, Watson AJ, Rafferty JA, and Margison GP Depletion of O6-alkylguanine-DNA alkyltransferase correlates with potentiation of temozolomide and CCNU toxicity in human tumour cells. Br.J Cancer, 67: 1299–1302, 1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Friedman HS, McLendon RE, Kerby T, Dugan M, Bigner SH, Henry AJ, Ashley DM, Krischer J, Lovell S, Rasheed K, Marchev F, Seman AJ, Cokgor I, Rich J, Stewart E, Colvin OM, Provenzale JM, Bigner DD, Haglund MM, Friedman AH, and Modrich PL DNA mismatch repair and O6-alkylguanine-DNA alkyltransferase analysis and response to Temodal in newly diagnosed malignant glioma. J Clin.Oncol., 16: 3851–3857, 1998. [DOI] [PubMed] [Google Scholar]
- 8.Gerson SL Clinical relevance of MGMT in the treatment of cancer. J Clin.Oncol, 20: 2388–2399, 2002. [DOI] [PubMed] [Google Scholar]
- 9.Friedman HS, Dolan ME, Pegg AE, Marcelli S, Keir S, Catino JJ, Bigner DD, and Schold SC Jr. Activity of temozolomide in the treatment of central nervous system tumor xenografts. Cancer Res, 55: 2853–2857, 1995. [PubMed] [Google Scholar]
- 10.Pollack IF, Hamilton RL, Sobol RW, Burnham J, Yates AJ, Holmes EJ, Zhou T, and Finlay JL O6-methylguanine-DNA methyltransferase expression strongly correlates with outcome in childhood malignant gliomas: results from the CCG-945 Cohort. J Clin.Oncol, 24: 3431–3437, 2006. [DOI] [PubMed] [Google Scholar]
- 11.Taverna P, Catapano CV, Citti L, Bonfanti M, and D’Incalci M Influence of O6-methylguanine on DNA damage and cytotoxicity of temozolomide in L1210 mouse leukemia sensitive and resistant to chloroethylnitrosoureas. Anticancer Drugs, 3: 401–405, 1992. [DOI] [PubMed] [Google Scholar]
- 12.Tentori L, Orlando L, Lacal PM, Benincasa E, Faraoni I, Bonmassar E, D’Atri S, and Graziani G Inhibition of O6-alkylguanine DNA-alkyltransferase or poly(ADP-ribose) polymerase increases susceptibility of leukemic cells to apoptosis induced by temozolomide. Mol.Pharmacol, 52: 249–258, 1997. [DOI] [PubMed] [Google Scholar]
- 13.Esteller M, Hamilton SR, Burger PC, Baylin SB, and Herman JG Inactivation of the DNA repair gene O6-methylguanine-DNA methyltransferase by promoter hypermethylation is a common event in primary human neoplasia. Cancer Res, 59: 793–797, 1999. [PubMed] [Google Scholar]
- 14.Costello JF, Futscher BW, Tano K, Graunke DM, and Pieper RO Graded methylation in the promoter and body of the O6-methylguanine DNA methyltransferase (MGMT) gene correlates with MGMT expression in human glioma cells. J Biol.Chem, 269: 17228–17237, 1994. [PubMed] [Google Scholar]
- 15.Esteller M, Garcia-Foncillas J, Andion E, Goodman SN, Hidalgo OF, Vanaclocha V, Baylin SB, and Herman JG Inactivation of the DNA-repair gene MGMT and the clinical response of gliomas to alkylating agents. N.Engl.J Med, 343: 1350–1354, 2000. [DOI] [PubMed] [Google Scholar]
- 16.Balana C, Ramirez JL, Taron M, Roussos Y, Ariza A, Ballester R, Sarries C, Mendez P, Sanchez JJ, and Rosell RO (6)-methyl-guanine-DNA methyltransferase Methylation in Serum and Tumor DNA Predicts Response to 1,3-Bis(2-Chloroethyl)-1-Nitrosourea but not to Temozolamide Plus Cisplatin in Glioblastoma Multiforme. Clin Cancer Res, 9: 1461–1468, 2003. [PubMed] [Google Scholar]
- 17.Esteller M, Gaidano G, Goodman SN, Zagonel V, Capello D, Botto B, Rossi D, Gloghini A, Vitolo U, Carbone A, Baylin SB, and Herman JG Hypermethylation of the DNA repair gene O(6)-methylguanine DNA methyltransferase and survival of patients with diffuse large B-cell lymphoma. J Natl.Cancer Inst, 94: 26–32, 2002. [DOI] [PubMed] [Google Scholar]
- 18.Rossi D, Capello D, Gloghini A, Franceschetti S, Paulli M, Bhatia K, Saglio G, Vitolo U, Pileri SA, Esteller M, and Gaidano G Aberrant promoter methylation of multiple genes throughout the clinico-pathologic spectrum of B-cell neoplasia. Haematologica, 89: 154–164, 2004. [PubMed] [Google Scholar]
- 19.Matsushita C, Yang Y, Takeuchi S, Matsushita M, Van Dongen JJ, Szczepanski T, Bartram CR, Seo H, Koeffler HP, and Taguchi H Aberrant methylation in promoter-associated CpG islands of multiple genes in relapsed childhood acute lymphoblastic leukemia. Oncol.Rep, 12: 97–99, 2004. [PubMed] [Google Scholar]
- 20.D’Atri S, Piccioni D, Castellano A, Tuorto V, Franchi A, Lu K, Christiansen N, Frankel S, Rustum YM, Papa G, and . Chemosensitivity to triazene compounds and O6-alkylguanine-DNA alkyltransferase levels: studies with blasts of leukaemic patients. Ann.Oncol, 6: 389–393, 1995. [DOI] [PubMed] [Google Scholar]
- 21.Liu L, Markowitz S, and Gerson SL Mismatch repair mutations override alkyltransferase in conferring resistance to temozolomide but not to 1,3-bis(2-chloroethyl)nitrosourea. Cancer Res, 56: 5375–5379, 1996. [PubMed] [Google Scholar]
- 22.Hangaishi A, Ogawa S, Mitani K, Hosoya N, Chiba S, Yazaki Y, and Hirai H Mutations and loss of expression of a mismatch repair gene, hMLH1, in leukemia and lymphoma cell lines. Blood, 89: 1740–1747, 1997. [PubMed] [Google Scholar]
- 23.Taverna P, Liu L, Hanson AJ, Monks A, and Gerson SL Characterization of MLH1 and MSH2 DNA mismatch repair proteins in cell lines of the NCI anticancer drug screen. Cancer Chemother.Pharmacol, 46: 507–516, 2000. [DOI] [PubMed] [Google Scholar]
- 24.Gu L, Cline-Brown B, Zhang F, Qiu L, and Li GM Mismatch repair deficiency in hematological malignancies with microsatellite instability. Oncogene, 21: 5758–5764, 2002. [DOI] [PubMed] [Google Scholar]
- 25.Casorelli I, Offman J, Mele L, Pagano L, Sica S, D’Errico M, Giannini G, Leone G, Bignami M, and Karran P Drug treatment in the development of mismatch repair defective acute leukemia and myelodysplastic syndrome. DNA Repair (Amst), 2: 547–559, 2003. [DOI] [PubMed] [Google Scholar]
- 26.Horton TM, Pati D, Plon SE, Thompson PA, Bomgaars LR, Adamson PC, Ingle AM, Wright JJ, Brockman AH, Paton M, and Blaney S A Phase 1 Study of the Proteasome Inhibitor Bortezomib in Pediatric Patients with Refractory Leukemia: a Children’s Oncology Group Study. Clin Cancer Res . 2007. In press. [DOI] [PubMed] [Google Scholar]
- 27.Shen F, Decosterd LA, Gander M, Leyvraz S, Biollax J, and Lejeune F Determination of temozolomide in human plasma and urine by high-performance liquid chromatography after solid-phase extraction. J Chromatogr.B Biomed.Appl, 667: 291–300, 1995. [DOI] [PubMed] [Google Scholar]
- 28.Rudek MA, Donehower RC, Statkevich P, Batra VK, Cutler DL, and Baker SD Temozolomide in patients with advanced cancer: phase I and pharmacokinetic study. Pharmacotherapy, 24: 16–25, 2004. [DOI] [PubMed] [Google Scholar]
- 29.DiArgenio DZ and Schumitsky A ADAPT II Users Guide: pharmacokinetic/pharmacodynamic systems analysis software. (4). 1997. Los Angeles, CA, Biomedical systems resource. [Google Scholar]
- 30.Yamaoka K, Nakagawa T, and Uno T Application of Akaike’s information criterion (AIC) in the evaluation of linear pharmacokinetic equations. J Pharmacokinet.Biopharm., 6: 165–175, 1978. [DOI] [PubMed] [Google Scholar]
- 31.Dolan ME, Roy SK, Fasanmade AA, Paras PR, Schilsky RL, and Ratain MJ O6-benzylguanine in humans: metabolic, pharmacokinetic, and pharmacodynamic findings. J Clin.Oncol, 16: 1803–1810, 1998. [DOI] [PubMed] [Google Scholar]
- 32.Boland CR, Thibodeau SN, Hamilton SR, Sidransky D, Eshleman JR, Burt RW, Meltzer SJ, Rodriguez-Bigas MA, Fodde R, Ranzani GN, and Srivastava S A National Cancer Institute Workshop on Microsatellite Instability for cancer detection and familial predisposition: development of international criteria for the determination of microsatellite instability in colorectal cancer. Cancer Res, 58: 5248–5257, 1998. [PubMed] [Google Scholar]
- 33.Suraweera N, Duval A, Reperant M, Vaury C, Furlan D, Leroy K, Seruca R, Iacopetta B, and Hamelin R Evaluation of tumor microsatellite instability using five quasimonomorphic mononucleotide repeats and pentaplex PCR. Gastroenterology, 123: 1804–1811, 2002. [DOI] [PubMed] [Google Scholar]
- 34.Frazier ML, Sinicrope FA, Amos CI, Cleary KR, Lynch PM, Levin B, and Luthra R Loci for efficient detection of microsatellite instability in hereditary non-polyposis colorectal cancer. Oncol.Rep, 6: 497–505, 1999. [DOI] [PubMed] [Google Scholar]
- 35.Goel A, Arnold CN, Niedzwiecki D, Chang DK, Ricciardiello L, Carethers JM, Dowell JM, Wasserman L, Compton C, Mayer RJ, Bertagnolli MM, and Boland CR Characterization of sporadic colon cancer by patterns of genomic instability. Cancer Res, 63: 1608–1614, 2003. [PubMed] [Google Scholar]
- 36.Estlin EJ, Lashford L, Ablett S, Price L, Gowing R, Gholkar A, Kohler J, Lewis IJ, Morland B, Pinkerton CR, Stevens MC, Mott M, Stevens R, Newell DR, Walker D, Dicks-Mireaux C, McDowell H, Reidenberg P, Statkevich P, Marco A, Batra V, Dugan M, and Pearson AD Phase I study of temozolomide in paediatric patients with advanced cancer. United Kingdom Children’s Cancer Study Group. British Journal of Cancer, 78: 652–661, 1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Dhodapkar M, Rubin J, Reid JM, Burch PA, Pitot HC, Buckner JC, Ames MM, and Suman VJ Phase I trial of temozolomide (NSC 362856) in patients with advanced cancer. Clin.Cancer Res, 3: 1093–1100, 1997. [PubMed] [Google Scholar]
- 38.Hammond LA, Eckardt JR, Baker SD, Eckhardt SG, Dugan M, Forral K, Reidenberg P, Statkevich P, Weiss GR, Rinaldi DA, Von Hoff DD, and Rowinsky EK Phase I and pharmacokinetic study of temozolomide on a daily-for-5-days schedule in patients with advanced solid malignancies. J Clin.Oncol., 17: 2604–2613, 1999. [DOI] [PubMed] [Google Scholar]
- 39.Brada M, Judson I, Beale P, Moore S, Reidenberg P, Statkevich P, Dugan M, Batra V, and Cutler D Phase I dose-escalation and pharmacokinetic study of temozolomide (SCH 52365) for refractory or relapsing malignancies. Br.J Cancer, 81: 1022–1030, 1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Reid JM, Stevens DC, Rubin J, and Ames MM Pharmacokinetics of 3-methyl-(triazen-1-yl)imidazole-4-carboximide following administration of temozolomide to patients with advanced cancer. Clin.Cancer Res, 3: 2393–2398, 1997. [PubMed] [Google Scholar]
- 41.Tisdale MJ Antitumor imidazotetrazines--XV. Role of guanine O6 alkylation in the mechanism of cytotoxicity of imidazotetrazinones. Biochem.Pharmacol, 36: 457–462, 1987. [DOI] [PubMed] [Google Scholar]
- 42.Gerson SL MGMT: its role in cancer aetiology and cancer therapeutics. Nat.Rev.Cancer, 4: 296–307, 2004. [DOI] [PubMed] [Google Scholar]
- 43.Yeager ND, Dolan ME, Gastier JM, Gross TG, Delaney S, Frick J, Ruymann FB, and Ewesuedo R O6-methylguanine-DNA methyltransferase activity and promoter methylation status in pediatric rhabdomyosarcoma. J Pediatr Hematol.Oncol, 25: 941–947, 2003. [DOI] [PubMed] [Google Scholar]
- 44.Nakayama K, Inokuchi K, and Dan K Hypermethylation of the putative tumor-suppressor genes DCC, p51/63 and O6-methylguanine-DNA methyltransferase (MGMT) and loss of their expressions in cell lines of hematological malignancies. J Nippon Med.Sch, 72: 270–277, 2005. [DOI] [PubMed] [Google Scholar]
- 45.Pieper RO, Patel S, Ting SA, Futscher BW, and Costello JF Methylation of CpG island transcription factor binding sites is unnecessary for aberrant silencing of the human MGMT gene. J Biol.Chem, 271: 13916–13924, 1996. [DOI] [PubMed] [Google Scholar]
- 46.Soejima H, Zhao W, and Mukai T Epigenetic silencing of the MGMT gene in cancer. Biochem.Cell Biol, 83: 429–437, 2005. [DOI] [PubMed] [Google Scholar]
- 47.Levati L, Marra G, Lettieri T, D’Atri S, Vernole P, Tentori L, Lacal PM, Pagani E, Bonmassar E, Jiricny J, and Graziani G Mutation of the mismatch repair gene hMSH2 and hMSH6 in a human T-cell leukemia line tolerant to methylating agents. Genes Chromosomes.Cancer, 23: 159–166, 1998. [PubMed] [Google Scholar]
- 48.Ribeiro EM, Rodriguez JM, Coser VM, Sotero MG, Fonseca Neto JM, Pasquini R, and Cavalli IJ Microsatellite instability and cytogenetic survey in myeloid leukemias. Braz.J Med Biol Res, 35: 153–159, 2002. [DOI] [PubMed] [Google Scholar]
- 49.Kodera T, Kohno T, Takakura S, Morishita K, Hamaguchi H, Hayashi Y, Sasaki T, and Yokota J Microsatellite instability in lymphoid leukemia and lymphoma cell lines but not in myeloid leukemia cell lines. Genes Chromosomes.Cancer, 26: 267–269, 1999. [DOI] [PubMed] [Google Scholar]
- 50.Inokuchi K, Ikejima M, Watanabe A, Nakajima E, Orimo H, Nomura T, and Shimada T Loss of expression of the human MSH3 gene in hematological malignancies. Biochem.Biophys.Res.Commun, 214: 171–179, 1995. [DOI] [PubMed] [Google Scholar]
- 51.Takeuchi S, Seriu T, Tasaka T, Koike M, Cho SK, Park S, Slater J, Mufti I, Hatta Y, Miyoshi I, Bartram CR, and Koeffler HP Microsatellite instability and other molecular abnormalities in childhood acute lymphoblastic leukaemia. Br.J Haematol, 98: 134–139, 1997. [DOI] [PubMed] [Google Scholar]
- 52.Reato G, Basso G, Putti MC, Cignetti A, Guarini A, and Foa R Microsatellite analysis in childhood acute lymphoblastic leukemia. Haematologica, 83: 403–407, 1998. [PubMed] [Google Scholar]
- 53.Krskova-Honzatkova L, Cermak J, Sajdova J, Stary J, Sedlacek P, and Sieglova Z Microsatellite instability in hematological malignancies. Leuk.Lymphoma, 43: 1979–1986, 2002. [DOI] [PubMed] [Google Scholar]
- 54.Ohyashiki JH, Ohyashiki K, Aizawa S, Kawakubo K, Shimamoto T, Iwama H, Hayashi S, and Toyama K Replication errors in hematolgoical neoplasias: genomic instability in progresion of disease is different among different types of leukemia. Clin Cancer Res, 2: 1583–1589, 1996. [PubMed] [Google Scholar]
- 55.Tasaka T, Lee S, Spira S, Takeuchi S, Nagai M, Takahara J, and Koeffler HP Microsatellite instability during the progression of acute myelocytic leukaemia. Br.J Haematol, 98: 219–221, 1997. [DOI] [PubMed] [Google Scholar]
- 56.Uehara E, Takeuchi S, Tasaka T, Matsuhashi Y, Yang Y, Fujita M, Tamura T, Nagai M, and Koeffler HP Aberrant methylation in promoter-associated CpG islands of multiple genes in therapy-related leukemia. Int.J Oncol, 23: 693–696, 2003. [PubMed] [Google Scholar]
- 57.Das-Gupta EP, Seedhouse CH, and Russell NH Microsatellite instability occurs in defined subsets of patients with acute myeloblastic leukaemia. Br.J Haematol, 114: 307–312, 2001. [DOI] [PubMed] [Google Scholar]
- 58.Sheikhha MH, Tobal K, and Liu Yin JA High level of microsatellite instability but not hypermethylation of mismatch repair genes in therapy-related and secondary acute myeloid leukaemia and myelodysplastic syndrome. Br.J Haematol, 117: 359–365, 2002. [DOI] [PubMed] [Google Scholar]
- 59.Olipitz W, Hopfinger G, Aguiar RC, Gunsilius E, Girschikofsky M, Bodner C, Hiden K, Linkesch W, Hoefler G, and Sill H Defective DNA-mismatch repair: a potential mediator of leukemogenic susceptibility in therapy-related myelodysplasia and leukemia. Genes Chromosomes.Cancer, 34: 243–248, 2002. [DOI] [PubMed] [Google Scholar]
- 60.Hatta Y, Yamada Y, Tomonaga M, Miyoshi I, Said JW, and Koeffler HP Microsatellite instability in adult T-cell leukaemia. Br.J Haematol, 101: 341–344, 1998. [DOI] [PubMed] [Google Scholar]