

ABSTRACT
The group A streptococci are associated with a group of diseases affecting the heart, brain, and joints that are collectively referred to as acute rheumatic fever. The streptococcal immune-mediated sequelae, including acute rheumatic fever, are due to antibody and cellular immune responses that target antigens in the heart and brain as well as the group A streptococcal cross-reactive antigens as reviewed in this article. The pathogenesis of acute rheumatic fever, rheumatic heart disease, Sydenham chorea, and other autoimmune sequelae is related to autoantibodies that are characteristic of autoimmune diseases and result from the immune responses against group A streptococcal infection by the host. The sharing of host and streptococcal epitopes leads to molecular mimicry between the streptococcal and host antigens that are recognized by the autoantibodies during the host response. This article elaborates on the discoveries that led to a better understanding of the pathogenesis of disease and provides an overview of the history and the most current thought about the immune responses against the host and streptococcal cross-reactive antigens in group A streptococcal sequelae.
INTRODUCTION
Cross-reactive antigens are molecules on the group A streptococci that mimic host molecules and during infection or immunization induce an autoimmune response against host tissues, leading to the autoimmune group A streptococcal sequelae (1–25). “Molecular mimicry” is the term used to describe immunological cross-reactivity between the host and bacterial antigens. Immunological cross-reactions between streptococcal and host molecules have been identified involving antibodies or T cells that react with streptococcal components and tissue antigens (6, 7, 22, 24, 25). The advent of monoclonal antibodies (mAbs) and T cell clones/hybridomas has greatly advanced the identification of the host and streptococcal antigens responsible for immunological cross-reactions associated with immunization, infection, and autoimmune sequelae. The identification of cross-reactive antigens in group A streptococci and important in our understanding of the pathogenesis of autoimmune sequelae, such as rheumatic fever including carditis (8, 11) and Sydenham chorea (7), which may follow group A streptococcal infection (3).
Molecular mimicry between host and bacterial antigens was first defined as identical amino acid sequences shared between different molecules present in tissues and the bacterium (26–28). The investigation of molecular mimicry through the use of mAbs has identified other types of molecular mimicry. The second type of mimicry involves antibody recognition of similar structures such as alpha-helical coiled-coil molecules such as streptococcal M protein, and the host proteins myosin, keratin, tropomyosin, vimentin, and laminin that share regions containing 40% identity or less and cross-reactive sites are not completely identical (4, 29–43). A third type of molecular mimicry is revealed in immunological cross-reactions between molecules as diverse as DNA and proteins (36, 40, 44) or carbohydrates and peptides (45–47). The three types of molecular mimicry are summarized in Table 1. Studies of the cross-reactive antigens of the group A streptococci have contributed greatly to our knowledge about molecular mimicry, autoimmunity, and infection and the antibody molecules involved. The antibody molecules described as polyreactive or cross-reactive indicate recognition of multiple antigens. As described, the basis of the immunological cross-reactions may be identical or homologous amino acid sequences shared between two different proteins or may be epitopes shared between two entirely different chemical structures.
TABLE 1.
Types of immunological mimicry (cross-reactivity)
| Identical amino acid sequences in different proteins |
| Similar protein structures shared among different proteins |
| Diverse molecules such as DNA, carbohydrates, and proteins |
A theme in molecular mimicry suggests antibody recognition of an internal biomarker, often a structural protein, which cross-reacts with the streptococcal antigen and a cell surface antigen leading to cytotoxicity or to functional cell signaling (6, 7). Through these mechanisms, tissue is altered and T cells are recruited to the site of attack (48).
HISTORICAL PERSPECTIVE
Cross-reactive antigens and antibodies were first found to be associated with acute rheumatic fever (ARF) and group A streptococci when it was discovered that rheumatic fever sera or anti-group A streptococcal antisera reacted with human heart or skeletal muscle tissues (15, 17, 19). Antibodies against group A streptococci in the serum of rheumatic fever patients were shown to be absorbed from the sera with human heart extracts, and in contrast, the antiheart antibodies were shown to be absorbed with group A streptococci or streptococcal membranes (49, 50). Rabbit antisera produced against group A streptococcal cell walls reacted with human heart tissue, and antibodies produced against human heart tissue reacted with group A streptococcal antigens (49, 51, 52). In rheumatic fever, heart-reactive antibodies appeared to persist in patients with rheumatic recurrences, and there appeared to be a relationship between high titers of antiheart antibodies and recurrence of rheumatic fever (15, 19). Heart-reactive antibodies were reported to decline within 5 years of the initial rheumatic fever attack.
Subsequent research on this early work by Kaplan and Suchy (49) implicated the streptococcal cell wall containing the M protein as the cross-reactive antigen recognized by the antiheart antibodies, while studies by Zabriskie and Freimer (15, 17) implicated the streptococcal membrane as the site of the cross-reactive antigen. Beachey and Stollerman (53) and Widdowson and Maxted (54–56) reported an M-associated non-type-specific antigen and an M-associated protein, respectively, which were thought to be associated with immunological cross-reactions in ARF. In 1977, van de Rijn et al. demonstrated that highly purified peptides from group A streptococcal membranes reacted with the antiheart antibodies in rheumatic fever serum (57). Taken together, this evidence suggested that cross-reactive antigens were located in both the cell wall and membrane of the group A streptococci.
Evidence also supported the hypothesis that the group A polysaccharide was a cross-reactive antigen (58). Goldstein and colleagues showed that glycoproteins in heart valves contained the N-acetyl-glucosamine determinant that they proposed to be responsible for immune cross-reactivity of group A streptococci with heart tissues. Dudding and Ayoub demonstrated the persistence of anti-group A carbohydrate antibody in patients with rheumatic valvular disease (59). Lyampert in Russia also published studies suggesting that the group A streptococcal polysaccharide antigen induced responses against host tissues (60–62). McCarty suggested that the terminal O-linked N-acetyl-glucosamine might cross-react with antibodies against the group A carbohydrate and host tissues (63). Additional evidence suggested that the hyaluronic acid capsule of the group A streptococcus also might induce responses against joint tissues (64, 65). Administration of a peptidoglycan-polysaccharide complex prepared from group A streptococci to rats induced carditis and arthritis (66–71). The studies in the 1960s and 1970s left little doubt that group A streptococci induced autoantibodies against the heart and other host tissues and that streptococcal components could induce inflammatory lesions resembling arthritis and carditis in animal tissues.
CROSS-REACTIVE mAbs RECOGNIZE CARDIAC MYOSIN AND ALPHA-HELICAL COILED-COIL MOLECULES IN HEART AND BRAIN
mAbs cross-reactive with group A streptococci and human heart tissues were produced from mice immunized with streptococcal cell wall and membrane components (4, 33, 40, 72) and from rheumatic carditis patients (35). Figure 1 illustrates the cross-reactivity of an antistreptococcal mAb with myocardium. In 1985, Krisher and Cunningham identified myosin as a cross-reactive host tissue antigen that provided the link between streptococci and the heart (4). In 1987, it was demonstrated that cardiac myosin could induce myocarditis in genetically susceptible mice (73). Myosin is an alpha-helical coiled-coil molecule that was shown to be an important tissue target of the cross-reactive mAbs (72). Other host tissue antigens recognized by antistreptococcal mouse and human mAbs included the alpha-helical coiled-coil molecules tropomyosin (29, 40), keratin (29, 45, 74), vimentin (36), laminin (6, 30, 75), DNA (36), and N-acetyl-β-d-glucosamine (45–47), the immunodominant epitope of the group A polysaccharide. A summary of the cross-reactive antistreptococcal mAb host tissue targets is provided in Table 2. The tissue targets identified by the cross-reactive mAbs may be important in the manifestations of group A streptococcal rheumatic fever sequelae of arthritis, carditis, chorea, and erythema marginatum (76, 77). A summary of the cross-reactive human and mouse mAb specificities has been published in previous reviews (78–81).
FIGURE 1.

Reaction of mouse antistreptococcal mAb with a human tissue section of myocardium in a indirect immunofluorescence assay. Mouse IgM (20 μg/ml) was unreactive (not shown). mAbs were tested at 20 μg/ml. (Taken from reference 40 with permission from the Journal of Immunology.)
TABLE 2.
Cross-reactive monoclonal antibody host tissue targets
| Cardiac myosin |
| Skeletal myosin |
| Tropomyosin |
| Keratin |
| Vimentin |
| Laminin |
The cross-reactive antibodies were divided into three major subsets based on their cross-reactivity with (i) myosin and other alpha-helical molecules, (ii) DNA, or (iii) N-acetyl-glucosamine. Figure 2 illustrates the subsets of cross-reactive antistreptococcal/antimyosin mAbs. All three subsets were identified among mAbs from mice immunized with group A streptococcal components, but in humans, the predominant subset reacted with the N-acetyl-glucosamine epitope and myosin and related molecules. This result is not surprising, since rheumatic fever patients do not develop antinuclear antibodies during the course of their disease. Elevated levels of poly-reactive antimyosin antibodies found in ARF sera (35) and in animals immunized with streptococcal membranes or walls (17, 49, 50, 52) most likely account for the reactivity of these sera with myocardium and other tissues. Similar types of cross-reactive mouse and human antibodies have been investigated by Lange and colleagues (82) and by Young and colleagues (83), respectively.
FIGURE 2.
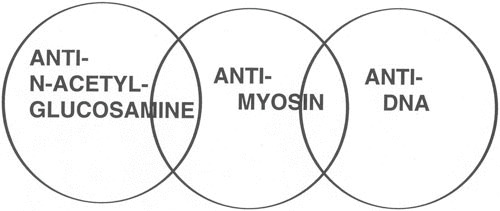
Human and murine antistreptococcal/antimyosin mAbs were divided into three subsets based on their reactivity with myosin and N-acetyl-glucosamine, the dominant group A carbohydrate epitope, with DNA and the cell nucleus, a property found among murine mAbs, and reactivity with myosin and a family of alpha-helical coiled-coil molecules. (Taken from reference 203 with permission from Indiana University School of Dentistry Press.)
Poly-specific or cross-reactive autoantibodies have emerged as a theme in autoimmunity and molecular mimicry (36, 84–86). The V-D-J region genes of the human and mouse cross-reactive mAbs have been sequenced (29, 30, 87), but there is no consensus sequence to explain the molecular basis of poly-specificity and cross-reactivity. It is worth noting that the three groups of reactivities in mice do not have specific antibody V gene families or VH and VL gene combinations that correlate with a specific reactivity. However, higher-avidity cross-reactive antimyosin mAb (87) was associated with reactivity with laminin and complement-mediated heart cell cytotoxicity (30, 75). The V genes of cross-reactive human mAbs from rheumatic fever were encoded by a heterogeneous group of VH3 family genes (VH-3, VH-8, VH-23, VH-30) and a VH4-59 gene segment (29). Wu et al. reported a similar group of V gene sequences for their human antistreptococcal/antimyosin antibodies produced from Epstein-Barr virus-transformed B cell lines (83). Many sequences were found to be either in germline configuration or were highly homologous with a previously sequenced germline V gene. It has been proposed that a germline antibody may be poly-reactive due to conformational rearrangement and configurational change, permitting binding of diverse molecules (88).
Cytotoxic mouse and human mAbs have identified the extracellular matrix protein laminin as a potential tissue target present in basement membrane surrounding myocardium and valve surface endothelium (6, 30, 75, 89, 90). Cross-reactive antibodies may become trapped in extracellular matrix that may act like a sieve to capture antibody and lead to inflammation in host tissues. The cross-reactive antibodies in rheumatic heart disease, even though they react with cardiac myosin, target valve surface endothelium and laminin (6) as shown in Fig. 3 and 4 and initiate the inflammation at the endocardium, where T cells then target the activated valve endothelium and infiltrate the valve (48) as shown in Fig. 5 and lead to eventual scarring and neovascularization of the normally avascular valve. Vascular cell adhesion molecule-1 (VCAM-1) expression has been found on the valve surface endothelium that attracts very late antigen-4 (VLA-4) on the activated T cell (48).
FIGURE 3.
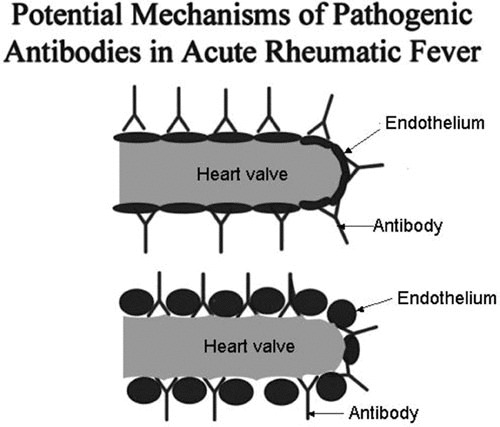
Diagram illustrating the potential mechanism of antibody in the pathogenesis of rheumatic heart disease. Cross-reactive antibody is shown binding directly to endothelium (top diagram) or binding to basement membrane of the valve (bottom diagram) exposed due to shear stress or damage by antibody and complement. (Reproduced with permission from ASM press).
FIGURE 4.

Reactivity of antistreptococcal/antimyosin mAb 3.B6 with normal human valve endothelium and myocardium. Formalin-fixed human mitral valve (A) and myocardium (B) were reacted with mAb 3.B6 at 10 μg/ml. mAb 3.B6 binding was detected using biotin-conjugated antihuman antibodies and alkaline phosphatase-labeled streptavidin followed by fast red substrate. Control sections did not react with human IgM at 10 μg/ml (C,D). (Reproduced with permission from Lippincott Williams and Wilkins).
FIGURE 5.
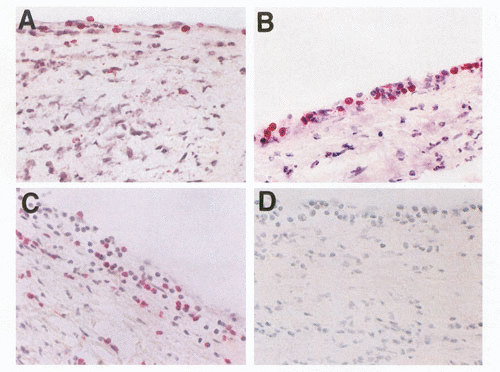
Adhesion and extravasation of T lymphocytes into an ARF valve in valvulitis. (A,B) Extravasation of CD4+ lymphocytes (stained red; original magnification, 200× and 400×, respectively). (C) Extravasation of CD8+ lymphocytes (stained red) into the valve through the valvular endothelium (magnification, 200×). An IgG1 isotype control mAb (IgG1) did not react with the same valve (not shown). (With permission from the Journal of Infectious Diseases, Chicago Press).
mAbs IDENTIFY STREPTOCOCCAL CROSS-REACTIVE ANTIGENS
Introduction
The discovery leading to the development of mAbs (91) was transformative for the investigation of autoantibodies and their specificities, including the cross-reactivities of the antibodies of the group A streptococci (4). Because a single mAb can represent a minor specificity and not the antibodies represented in the disease, the sera from patients with the disease should be investigated to prove that the mAbs reflect the disease and potentially its pathogenesis. Antimyosin mAbs identified cross-reactive antigens in streptococcal membranes (36, 92) and walls (36). The streptococcal M protein was shown to react with the heart or myosin cross-reactive mAbs (40, 92). In addition, a 60-kDa protein present in the cell membrane (93, 94) was cross-reactive with myosin in the heart, and a 67-kDa protein that was immunologically similar to class II major histocompatibility molecules was also identified (95). The data suggested that cross-reactive antigens were present in both the wall and membrane of the group A streptococci. The data support previous evidence from both Kaplan (49–52, 96) and Zabriskie (15, 17, 19). Although these data were once thought to be conflicting, it is clear that the cross-reactive antibodies recognize more than one antigen in the streptococcal cell. The data previously reported by Goldstein (58) are also supported by the evidence that a subset of cross-reactive mAbs recognized N-acetyl-β-d-glucosamine, the immunodominant epitope of the group A polysaccharide. N-acetyl-glucosamine is a major epitope of some of the cross-reactive mouse mAbs and virtually all of the human cross-reactive mAbs investigated (6, 45–47). The studies link together the cross-reactivity of the group A carbohydrate epitope, GlcNAc, human cardiac myosin, and streptococcal M protein. Antigenic redundancy due to cross-reactivity may be an important factor in triggering disease in a susceptible host. The cross-reactive antigens are now seen as separate entities recognized by mAbs that recognize more than one antigen molecule. Thus, the previous studies, which before seemed to be conflicting, were all correct. The use of mAbs has allowed dissection of the group A streptococcal cross-reactive antigens that would not have been possible using polyclonal sera. The following sections provide more evidence about the identification and analysis of the cross-reactive antigens of the group A streptococci. Table 2 summarizes the host tissue protein antigens targeted by antistreptococcal antibodies in disease and following immunization with group A streptococcal components. Table 3 summarizes the group A streptococcal cross-reactive antigens, of which the group A carbohydrate (6) and the M proteins (8, 97–101) are the most well investigated.
TABLE 3.
Cross-reactive antigens of group A streptococci
| M proteins |
| Hyaluronic acid capsule |
| N-acetyl-glucosamine/group A polysaccharide |
| 60-kDa wall-membrane antigen |
| 67-kDa antigen |
M Proteins and Rheumatic Fever
Investigation of the M proteins has provided important information about the sequence and primary structure of the molecule. The hypothesis that M proteins and myosin have immunological similarities was supported by the structural studies of Fischetti and colleagues (102–105) that demonstrated the seven-amino-acid-residue periodicity that is common among group A streptococcal M proteins and shared with proteins such as tropomyosin, myosin, desmin, vimentin, and keratin. Figure 6 illustrates the seven-residue periodicity and the homology characteristic of alpha-helical coiled-coil proteins such as tropomyosin and myosin. The cross-reactivity between group A streptococci and myosin was first identified by Krisher and Cunningham using mouse mAbs from mice immunized with group A streptococcal antigens (4). Subsequent studies by Dale, Beachey, Bronze, and colleagues using polyclonal sera and affinity purified antibodies further demonstrated immunological cross-reactivity between M proteins and myosin (37–39, 106). Studies using the cross-reactive mAbs also identified immunological cross-reactivity between streptococcal M proteins (both PepM [the N-terminal half of the molecule] and recombinant molecules) and cardiac and skeletal myosins (31, 36, 40).
FIGURE 6.
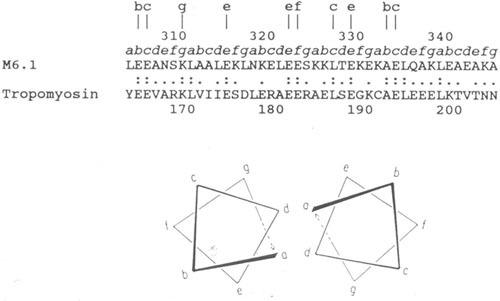
Sequence alignment of streptococcal M6 protein and human cardiac tropomyosin in a region exhibiting significant homology. Lowercase letters a to g directly above the sequence designate the position of these amino acids within the seven-residue periodicity in both segments. Lowercase letters at the top of the figure designate identities at external locations in the heptad repeat. Double dots indicate identities, and single dots indicate conservative substitutions. Within this segment of the streptococcal M6 molecule, 31% homology is observed with tropomyosin. Since both molecules are alpha-helical coiled-coil proteins, they contain the seven-residue repeat pattern where positions a and d are usually hydrophobic. Similar homologies are seen between M proteins and myosin heavy chains and any of the three laminin chains. (Reproduced from reference 40 with permission from the Journal of Immunology.)
Since streptococcal M proteins have been investigated for potential epitopes that were recognized by the cross-reactive mAbs, affinity purified antimyosin antibodies from ARF sera were reacted with peptides of streptococcal M5 protein. Affinity purified antimyosin antibodies from ARF reacted with an M5 amino acid sequence (residues 184 to 188) near the pepsin cleavage site (near the center of the molecule) in M5 and M6 proteins (40). The epitope, located in the B repeat region of M5 and M6 proteins, appeared to be a B cell epitope for antibody-mediated cross-reactivity with myosin (32, 40). An M5 peptide, containing the B repeat region epitope Gln-Lys-Ser-Lys-Gln (QKSKQ) was shown to inhibit antimyosin antibodies in ARF (40). Furthermore, an M5 peptide that contained the QKSKQ sequence induced antibodies in BALB/c mice against cardiac or skeletal myosins and the light meromyosin (LMM) fragment of myosin. This evidence further supported the previous findings indicating that the QKSKQ sequence is important in the antibody-mediated cross-reactions with myosin in ARF (40). M5 residues 164 to 197 were demonstrated to induce antibodies against sarcolemmal membrane of heart tissue (107), and M5 residues 84 to 116 induced heart-reactive antibody and reacted with antimyosin antibody purified from ARF sera (40, 107). Studies by Kraus and colleagues (42) demonstrated a vimentin cross-reactive epitope present in the M12 protein, and M protein peptides containing brain cross-reactive epitopes were localized to the M5 protein sequence 134 to 184 and the M19 sequence 1 to 24 (98, 99). A summary of the currently known myosin cross-reactive B cell epitopes in M5 protein is shown in Fig. 7, which illustrates the A, B, and C repeat regions of M protein.
FIGURE 7.
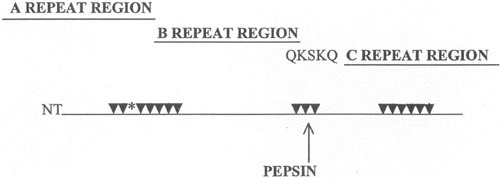
Antimyosin antibody cross-reactive sites in M5 protein A, B, and C repeat regions of the molecule. Arrows point to sites determined to induce human cardiac myosin cross-reactive antibody (32). The asterisk (*) marks the location of peptide NT4 containing several repeats of an epitope in cardiac myosins that causes myocarditis in MRL/++ and BALB/c mouse strains (32, 116). Site QKSKQ is the epitope determined to react with antimyosin antibody in ARF sera (40). (Reproduced from Effects of Microbes on the Immune System with permission from Lippincott Williams and Wilkins.)
Immunization of BALB/c mice with each of 23 overlapping synthetic peptides of M5 protein has revealed that the M5 peptides NT3-NT7, B2B3B, and C1A-C3 induce anti-human cardiac myosin antibodies as shown in Fig. 8. Similar overlapping synthetic peptides spanning the M5 protein molecule have been reported elsewhere (32). Titers of the anti-M5 peptide sera were 10 times greater against cardiac myosin than against skeletal myosin, tropomyosin, vimentin, and laminin (32). Certain M5 peptides appeared to induce a cardiac myosin-directed response in BALB/c mice. Furthermore, BALB/c mice developed mild myocarditis when immunized with M5 peptides NT4, NT5, NT6, B1A, and B3B (32). Peptides from the C-repeat region of M5 protein did not produce myocardial lesions when administered to BALB/c mice.
FIGURE 8.
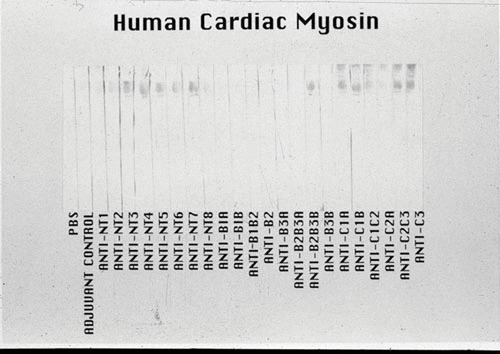
Reactivity of the antistreptococcal M5 peptide sera in a Western immunoblot of human cardiac myosin. The 200-kDa protein band of the purified human cardiac myosin, shown in the stained portion of the Western blot, reacted most strongly with antipeptide sera from mice immunized with the M5 peptides NT3-7, B2B3B, C1A, C1B, C2C3, and C3. Sera were tested at a 1:1000 dilution. A control antimyosin mAb CCM-52 (a gift from William Clark, Cardiovascular Research Institute, Michael Reese Hospital and Medical Center, Chicago, IL) reacted with the 200-kDa band present in our purified preparation of human cardiac myosin heavy chain. Purification of the human cardiac myosin heavy chain to homogeneity was previously described by Dell et al. (204). The Western blot confirms the data seen in the enzyme-linked immunosorbent assay with human cardiac myosin. (Reproduced from reference 32 with permission from ASM Press.)
The C-repeat region of class I M proteins contains the class I epitope, which is identified by reactivity with anti-M protein mAb 10B6 (108–111). Class I M protein serotypes were streptococcal strains associated with pharyngitis and rheumatic fever. Responses against the class I epitope were stronger in patients with rheumatic fever than in those with uncomplicated disease (112, 113). mAb 10B6, which recognizes the C-repeat epitope, also reacted with skeletal and cardiac myosins and the heavy meromyosin subfragment of myosin (112). The C-repeat class I epitope in M5 contains the amino acid sequence KGLRRDLDASREAK, which shares homology with RRDL, a conserved amino acid sequence found in cardiac and skeletal myosins. The myosin sequence RRDL is located in the heavy meromyosin subfragment of the cardiac and skeletal myosin heavy chain (112).
It was reported that reactivity of IgG from ARF sera was greater to the class I peptide sequence than IgG from uncomplicated pharyngitis (112). This was confirmed in a study by Mori and colleagues (114). However, the lower immune response to the class I epitope in uncomplicated pharyngitis most likely occurred because the patients with uncomplicated pharyngitis had been treated with penicillin (115). Antibodies against the class I epitope were affinity purified from ARF sera and shown to react with myosin (112).
Studies by Huber and Cunningham demonstrated that streptococcal M5 peptide NT4 containing the amino acid sequence GLKTENEGLKTENEGLKTE could produce myocarditis in MRL/++ mice, an autoimmune-prone strain (116). The studies demonstrated that the myocarditis was mediated by CD4+ T cells and class II major histocompatibility complex molecules. Antibodies against the IAk major histocompatibility molecule or antibody against CD4+ T cells abrogated the myocarditis (116). Amino acid sequence homology between the NT4 peptide of M5 and human cardiac myosin demonstrates 80% identity. Figure 9 illustrates the homology between the M5 peptide NT4 sequence and the cardiac myosin sequence. The mimicking sequence in NT4 is repeated four times in the M5 protein and is present in myosin only once. Repeated regions of the M protein that mimic cardiac myosin may be important in breaking tolerance in the susceptible host and producing autoimmune disease. The data support the hypothesis that epitopes in streptococcal M5 protein that mimic cardiac myosin may be important in breaking tolerance to this potent autoantigen. Table 4 summarizes the M protein amino acid sequences observed to induce inflammatory myocardial lesions in mice.
FIGURE 9.

Sequence homology between human cardiac myosin and peptide NT4 that causes myocarditis (32, 119). The homologous sequence repeats four times in the streptococcal M5 protein and in NT4 and once in cardiac myosin. Repeated sequences in M proteins that mimic cardiac myosin may be important in inducing inflammatory heart disease. (Adapted from reference 119 with permission from the Journal of Immunology.)
TABLE 4.
M5 protein sequences that produce myocarditis in micea
| Peptide | Amino acid sequence | Residues |
|---|---|---|
| A repeat region | ||
| NT4 | GLKTENEGLKTENEGLKTE | 40-58 |
| NT5 | KKEHEAENDKLKQQRDTL | 59-76 |
| NT6 | QRDTLSTQKETLEREVQN | 72-89 |
| B repeat region | ||
| B1A | TRQELANKQQESKENEKAL | 111-129 |
| B3B | GALKQELAKKDEANKISD | 202-219 |
Streptococcal M proteins not only mimic epitopes in cytoskeletal proteins, but they mimic epitopes in other strong bacterial and viral antigens, namely, heat shock protein (Hsp-65) (117) and coxsackie viral capsid proteins (31, 116, 118, 119). These immunological cross-reactions may be vital to the survival of the host and suggest that antibody molecules may recognize and neutralize more than a single infectious agent. Such antibodies may be an important first line of defense and would be highly advantageous for the host. Multiple pieces of evidence from experiments using synthetic peptides have shown that sites in the M protein mimic a site(s) in the VP1 capsid protein of coxsackie virus (31, 120). Further evidence demonstrated that some of the antistreptococcal/antimyosin mAbs neutralized enteroviruses and were cytotoxic for heart cells (31). This was extremely interesting because coxsackie viruses cause autoimmune myocarditis in susceptible hosts (121–123). The Hsp-65 antigen has been shown to play a role in the development of arthritis and diabetes (124, 125). Cross-reactive epitopes shared between streptococcal M proteins and Hsp-65 play a role in arthritis sequelae. Antibodies against heat shock protein 60 have been implicated in cytotoxicity against endothelium (126). Shared epitopes among pathogens may be important in molecular mimicry, may break tolerance to cryptic host molecules, and may influence the development of autoimmune diseases. Table 5 summarizes bacterial and viral proteins that have immunological similarities with M protein.
TABLE 5.
Bacterial and viral antigens with immunological similarities with group A streptococcal M proteins
| Heat shock protein 65 |
| Coxsackievirus VP1, 2, 3 capsid proteins |
| Group A streptococcal carbohydrate |
Identification of the Molecular Basis of Mimicry in the Streptococcal M Protein
Studies were undertaken to crystallize a portion of the streptococcal M1 protein (127) to evaluate its virulence properties when portions of the amino acid sequence were mutated, but also to investigate regions and structures responsible for eliciting or reacting with cross-reactive antibodies that participate in molecular mimicry. The structural study revealed irregularities and instabilities in the coiled coil of the M1 fragment crystal. Similar structural instabilities and irregularities occur in myosin and tropomyosin that had previously been demonstrated to cross-react with the streptococcal M proteins (37–40). Stabilization of the irregularities of the M protein structure, by creating mutants of the M protein gene to alter specific amino acids, enhanced the stability of the coiled coil and diminished the virulence properties of the M1 protein, including decreased cross-reactivity with autoantibodies that cross-reacted with heart tissues, cardiac myosin, and group A streptococcal M proteins (127). Loss of the heart or tissue cross-reactivity of the streptococcal M protein improves its potential for use as a vaccine antigen.
M Proteins and Cross-Reactive T Cells
M proteins have also been shown to stimulate human T lymphocyte responses (97, 128–135), including cytotoxic T lymphocytes from ARF patients (128, 136). PepM protein (pepsin-extracted M protein fragment) has been reported to be a superantigen that stimulated Vβ2, Vβ4, and Vβ8 bearing T cell subsets (133–135, 137). Other reports show that neither recombinant nor native forms of M proteins are superantigenic (138–140). A superantigenic site reported for the M5 protein has been localized to the amino acid sequence located in M5 residues 157 to 197 (KEQENKETIGTLKKILDETVKDKLAKE QKSKQNIGALKQEL) in the B3 repeat region (137). The site has a significant amount of sequence homology with other superantigens. The role of M protein or other superantigens in ARF may be to activate large numbers of T cells, including some that are cross-reactive and may lead to ARF.
Studies of T cell epitopes in rheumatic fever and in animal models focus on the M5 protein molecule because serotype M5 has often been associated with ARF outbreaks (141). T and B cell epitopes of the M5 protein were defined in earlier studies by Robinson and colleagues (131, 132), by Good and colleagues (129, 130), and in my laboratory (32).
Guilherme et al. isolated cross-reactive T cells from mitral valves, papillary muscle, and left atrium of rheumatic fever patients previously infected with M5 group A streptococci (97). T cell lines were responsive to several peptides of the streptococcal serotype M5 protein and proteins from heart tissue extracts. Sequences from the A and B repeat regions of M5 protein that stimulated the valvular T cells are shown in Table 6. In BALB/c mice using synthetic peptides, myosin-cross-reactive T cell epitopes from the A, B, and C repeat regions of the M5 protein were identified (32). Six dominant myosin cross-reactive T cell epitopes were located in the M5 molecule (32). Table 6 summarizes (i) the dominant myosin cross-reactive T cell epitopes of M5 protein in BALB/c mice (32), (ii) the M5 sequences recognized by T cell clones from rheumatic heart valves (97), and (iii) the M5 peptides reported by Good and colleagues to stimulate human T cells from normal controls and rheumatic fever patients that were responsive to myosin peptides (129, 130). An important correlation seen in Table 6 indicates that M5 peptides NT4/NT5 (GLKTENEGLKTENEGLKTE and KKEHEAENDKLKQQRDTL) and B1B2/B2 (VKDKIAKEQENKETIGTL and TIGTLKKILDETVKDKIA), which were dominant cross-reactive T cell epitopes in the BALB/c mouse, contain sequences similar to those reported to be recognized by T cells from rheumatic valves (97). Peptides NT4 and NT5 produced inflammatory infiltrates in the myocardium of animals immunized with those peptides (32). The collective evidence on cross-reactive T cells suggests that amino acid sequences in M5 protein that share homology with cardiac myosin may break tolerance and promote T cell-mediated inflammatory heart disease in animals and humans (32).
TABLE 6.
Summary of myosin or heart cross-reactive T cell epitopes of streptococcal M5 proteina
| Peptides | Sequencea | Origin of T cell clone or responsec |
|---|---|---|
| 1–25 | TVTRGTISDPQRAKEALDKYELENHb | ARF/valve (142) |
| 81–96 | DKLKQQRDTLSTQKETLEREVQNb | ARF/valve (142) |
| 163–177 | ETIGTLKKILDETVKb | ARF/valve (142) |
| 337–356 | LRRDLDASREAKKQVEKAL | Normal/PBL (130) |
| 347–366 | AKKQVEKALEEANSKLAALE | Mice (129)/normal PBL (130) |
| 397–416 | LKEQLAKQAEELAKLRAGKA | ARF/PBL (130) |
| NT4 40–58 | GLKTENEGLKTENEGLKTE | BALB/c/Lymph noded |
| NT5 59–76 | KKEHEAENDKLKQQRDTL | |
| B1B2 137–154 | VKDKIAKEQENKETIGTL | |
| B2 150–167 | TIGTLKKILDETVKDKIA | |
| C2A 254–271 | EASRKGLRRDLDASREAK | |
| C3 293–308 | KGLRRDLDASREAKKQ |
Taken from reference 32 with permission from ASM press.
The amino terminal TVTRGTIS sequence was taken from the M5 amino acid sequence published by Manjula et al. (201) and deviates from the M5 sequence published by Miller et al. (202) at positions 1 and 8. Other marked sequences ([81–96] and [163–177]) were taken from the PepM5 sequence as reported by Manjula (201). These two sequences are found in the sequence as 67–89 and 174–188, respectively, as reported by Miller et al. (202). All other sequences shown above are from the M5 gene sequence reported by Miller et al. (202).
PBL, peripheral blood lymphocytes, ARF, acute rheumatic fever.
BALB/c mice immunized with purified human cardiac myosin and the recovered lymph node lymphocytes were stimulated with each of the peptides in tritiated thymidine uptake assays.
T cells isolated and cloned from the peripheral blood or from human rheumatic mitral valves of human rheumatic heart disease patients recognized streptococcal M5 peptides and LMM peptides (21, 22, 25). The cross-reactive T cells taken from ARF and RHD rheumatic heart disease heart valves mainly recognize three regions (residues 1 to 25, 81 to 103, and 163 to 177) of the amino-terminal region of M5 protein, several valve-derived proteins, and peptides of the β-chain of the human cardiac LMM region, which is the smaller of two subunits produced by tryptic digestion of human cardiac myosin (21, 22, 25, 97). Mimicry between M proteins and cardiac myosin stimulates cross-reactive T cells in the peripheral blood of the host (25, 142, 143). The T cells then travel to the valve once the endothelium of the valve becomes activated and inflamed (48). This extravasation event at the valve endocardium allows the cross-reactive T cells to enter the valve, where they recognize and proliferate to valvular proteins such as vimentin (89), which may include cardiac myosin from papillary muscle. T cells that remain in the valve survive if they continue to be stimulated by host alpha helical proteins within the valve. The activated cross-reactive T cells when continually stimulated within the valve become pathogenic for the host and produce the TH1 mechanism of pathogenesis with scarring in the valve. Figure 10 illustrates the events in rheumatic carditis that result from immune responses against the cross-reactive antigens of the group A streptococci (144).
FIGURE 10.

Diagram representing the proposed immunopathogenesis of poststreptococcal rheumatic heart disease. Initially, B and T cells are activated by specific streptococcal antigens and superantigens, leading to strong immune responses against streptococcal and host antigens. The development of pathogenic clones of B and T lymphocytes is important in the development of the disease. Initially, antibodies develop against the group A carbohydrate, which are cross-reactive with the valve surface, and glycoproteins such as laminin and bind to the valve surface endothelium (endocardium), leading to inflammation and upregulation of cell adhesion molecules such as VCAM-1 on activated surface endothelium of the valve. M protein-reactive T cells enter the valve through the surface endothelium by binding to cell adhesion molecules such as VCAM-1 and extravasate into the valve (48). The formation of scar tissue in the valve followed by neovascularization allows the disease to continue in the valve. The specificity of the T cells in blood (25) and T cells entering the valve have been shown to react to M protein (22, 97, 144). T cell subsets include Th1 (IFNγ) (145) in the pathogenesis of proinflammatory responses and the development of the scarred and fibrotic valve. IL-17A has also been associated with rheumatic heart disease in humans and animal models, suggesting that Th17 cells are involved in disease (101, 146, 149).
The granulomatous Th1 cell response and the presence of interferon-γ (IFNγ) have been reported as dominant in rheumatic valves (145). Studies of rheumatic heart disease have suggested that Th17 cells in peripheral blood are elevated in rheumatic heart disease with concomitant downregulation of the Treg phenotype (146). In ARF and rheumatic heart disease, elevated numbers of Th17 cells, high interleukin-17A (IL-17A) levels, and decreased T regulatory cells were reported in peripheral blood (146). Although less is known about Th17 cell responses in rheumatic heart disease, Th17 cells are important in group A streptococcal infections and have been identified in nasopharyngeal and tonsillar lymphoid tissues in streptococcal infection animal models (147–149). Different routes of bacterial infection may induce long-lived Th1 memory cells and short-lived Th17 cell responses (148). IL-17 triggers effective immune responses against extracellular bacteria, such as promoting neutrophil responses (150–153).
The Lewis rat model of rheumatic heart disease also suggests the presence of IL-17A in the pathogenesis of experimental valvulitis induced by immunization with group A streptococcal antigens (13, 101). This finding does not preclude Th1, which is often found in tandem with Th17 responses in autoimmune disease models and in human autoimmune diseases (148). Although both Th1 and Th17 are proinflammatory, Th17 responses have been found to be more damaging in the chronic stages of disease leading to fibrosis, and IL-17A was dispensable in the earlier proinflammatory stages of myocarditis in mice (154) but promoted fibrosis in later stages and was associated with heart failure in humans (155).
A New Model of Valvular Heart Disease Induced by Group A or Group G Streptococcal M Proteins in the Lewis Rat
A new model of experimental autoimmune valvulitis and rheumatic heart disease was developed by Quinn et al. by immunization of the Lewis rat model with the recombinant M6 protein (23). The lesions that developed demonstrated characteristics of rheumatic heart disease in humans with verrucae (Fig. 11C) (23), Anitschkow cells, and edema with cellular infiltration into the endocardium of the valve (Fig. 11A and B) (8, 23). T cell lines from the model proliferated to both the rM6 protein and cardiac myosin. The model appeared to be very similar in appearance to the VCAM-1 activation (Fig. 11D) in humans and infiltration of CD4+ T cells into the VCAM-1 positive endothelium/endocardium (not shown) of the valve in rheumatic heart disease (48) (Fig. 12). More recent studies identified A and B region peptide epitopes of M5 protein that induced valvulitis in the Lewis rat (8). Repeat region peptides NT4, NT5/6, and NT7 (see Table 6) induced valvulitis similar to the intact pepsin fragment of M5 protein, PepM5. T cell lines from rats with valvulitis proliferated to M5 peptides NT5/6 and NT6. Passive transfer of an NT5/6-specific T cell line into naive rats produced valvulitis characterized by infiltration of CD4+ cells and upregulation of VCAM-1 in valvular endothelium, while another NT6-specific T cell line did not target the valve (8). Thus, the infiltrating T cell line appeared to be important in the induction of inflammation in the valve.
FIGURE 11.
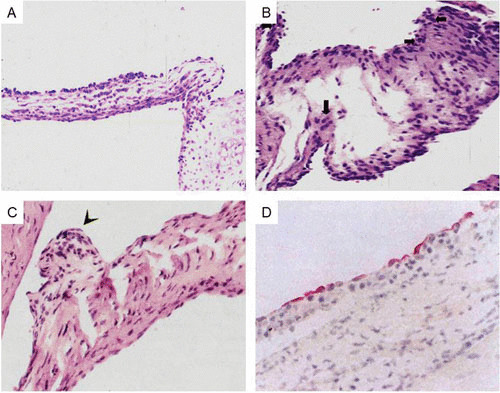
(A) Induction of valvulitis and cellular infiltration in hematoxylin- and eosin-stained heart valves from Lewis rats immunized with group A streptococcal M5 peptides NT1 to NT4/5 (AVTRGTINDPQRAKEALD amino acid [aa] residues 1 to 18; NT-2 KEALDKYELENHDLKTKN aa residues 14 to 31; NT-3 LKTKNEGLKTENEGLKTE aa residues 27 to 44; NT-4 GLKTENEGLKTENEGLKTE aa residues 40 to 58; NT-4/5 GLKTEKKEHEAENDKLK aa residues 54 to 70). (Taken from Kirvan et al. [8] with permission from the Journal of Translational Cardiology.) (B) Induction of valvulitis, edema, and cellular infiltration in hematoxylin- and eosin-stained heart valves from Lewis rats immunized with group A streptococcal M5 peptides NT1 to NT4/5 (AVTRGTINDPQRAKEALD aa residues 1 to 18; NT-2 KEALDKYELENHDLKTKN aa residues 14 to 31; NT-3 LKTKNEGLKTENEGLKTE aa residues 27 to 44; NT-4 GLKTENEGLKTENEGLKTE aa residues 40 to 58; NT-4/5 GLKTEKKEHEAENDKLK aa residues 54 to 70). (Taken from Kirvan et al. [8] with permission from the Journal of Translational Cardiology.) (C) Verrucous nodule observed on Lewis rat valve after immunization with group A streptococcal rM6 protein. (Taken from Quinn et al. [23] with permission from Infection and Immunity.) (D) VCAM-1 expressed on rheumatic valve. (Taken from Roberts et al. [48] with permission from the Journal of Infectious Diseases.)
FIGURE 12.

Extravasation of CD4+ lymphocytes into valve above Aschoff’s body in the subendocardium of the left atrial appendage. Original magnification, 200×. (A) Stained with anti-CD4 Mab; (B) stained with antibody isotype control. (Taken from Roberts et al. [48]).
Recent studies have used the Lewis rat model to demonstrate the induction of valvular heart disease by the C repeat region peptides of group A streptococcal M5 protein (12, 13). T cells from the model proliferated to M protein and cardiac myosin. Further studies by Ketheesan and colleagues demonstrated that exacerbation and increasing severity of valvulitis followed repeated group A streptococcal immunizations with group A streptococcal M protein (156). Lewis rats were immunized with group A streptococcal recombinant M5 protein and boosted once, twice, or three times. Anti-cardiac myosin antibodies and T cell responses increased with the booster immunizations. More severe lesions were observed in the repetitively immunized Lewis rats (156). Thus, the model demonstrates increased cardiac damage as seen in humans after repeated streptococcal infections. Immunization of the Lewis rat model with M protein from group G streptococci also led to valvular lesions (101). In the experimental autoimmune valvulitis model, both group A and group G M proteins and cardiac myosin induced proliferation of the T cells. IL-17A and IFNγ were found to be upregulated in the model (101). Thus, roles for IL17A in ARF and rheumatic heart disease are supported by the studies of patients with carditis.
Human Cardiac Myosin Peptide Epitopes in Rheumatic Heart Disease in Human Populations and Their Use in Monitoring Progression of Disease
Studies of humans with rheumatic fever and rheumatic heart disease have identified disease-specific epitopes of human cardiac myosin in the development of rheumatic carditis in humans (24). Immune responses to cardiac myosin were similar in rheumatic carditis among a small sample of worldwide populations, in which serum IgG targeted disease-specific human cardiac myosin epitopes in the S2 subfragment hinge region within S2 peptides containing amino acid residues 842 to 992 and 1164 to 1272 (Fig. 13) (24). Analysis of the anti-cardiac myosin autoantibodies in rheumatic carditis in a Pacific Islander family further confirmed the specific responses against cardiac myosin in rheumatic heart disease and confirmed the presence of potential rheumatogenic epitopes in the S2 region of human cardiac myosin (24). The study suggested that cardiac myosin epitopes targeted in rheumatic carditis were in the S2 hinge region of cardiac myosin and that the anti-cardiac myosin responses were similar among populations with rheumatic carditis worldwide regardless of the infecting group A streptococcal M protein serotype.
FIGURE 13.
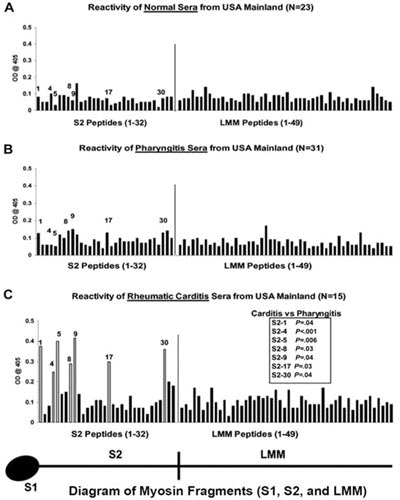
Reactivity of serum samples originating from the United States with human cardiac myosin peptides from the S2 and LMM regions in the enzyme-linked immunosorbent assay (1:100 serum dilution). (A) Mean reactivity of normal serum samples from control donors with no evidence of streptococcal infection or heart disease on the U.S. mainland against S2 and LMM peptides. These samples rarely reacted with any S2 or LMM peptide at an optical density of 0.2. (B) Mean reactivity of serum samples from patients with streptococcal pharyngitis in the United States against S2 and LMM peptides. These samples rarely reacted with any S2 or LMM peptide at an optical density of 0.2. (C) Serum immunoglobulin G from patients with rheumatic carditis in the United States. Serum samples from patients with rheumatic carditis from the United States reacted predominantly with peptides S2-1, S2-4, S2-5, S2-8, S2-9, S2-17, and S2-30, compared with the reactivity of serum samples from patients with pharyngitis in the United States against those same peptides (B). (C) Unadjusted Mann-Whitney P values for the comparison between carditis and pharyngitis on the United States mainland. The comparison for S2-4 is statistically significant on the basis of a two-sided alpha level adjusted to preserve the false-discovery rate at 5%. (Taken from Ellis et al. [24] with permission from the Journal of Infectious Diseases).
Further study of cardiac myosin peptide epitopes revealed that Australian patients with active rheumatic heart disease reacted significantly more to peptides from the S2 or LMM regions of human cardiac myosin than did healthy controls (11). Comparison of serum from active rheumatic heart disease patients to serum from patients treated for over a year with antibiotics revealed that the treatment led to a reduction in the anti-cardiac myosin peptide autoantibodies (11). Thus, identifying the immune response against the cardiac myosin peptides may monitor progression of rheumatic heart disease and may also determine effective treatment with a concomitant decline of the autoantibodies to cardiac myosin.
M Proteins and Poststreptococcal Acute Glomerulonephritis
Kefalides and colleagues showed that sera from patients with acute poststreptococcal glomerulonephritis contained antibodies against laminin, collagen, and other macromolecules found in the glomerular basement membrane (157). The epitope recognized in collagen was identified and shown to be located in the 7-S domain of type IV collagen (158). Streptococcal antigens, immunoglobulins, and complement were detected in the kidney glomeruli in acute poststreptococcal glomerulonephritis (159). Studies by Lange and Markowitz suggested that the glomerular basement membrane shared antigens with streptococcal M12 protein (160) and/or between the group A streptococcus and glomeruli (41, 161–163). Kraus and Beachey discovered a renal autoimmune epitope (Ile-Arg-Leu-Arg) in M protein (43). Evidence has suggested that certain M protein serotypes are associated with nephritis and that molecular mimicry or immunological cross-reactivity between glomeruli and M protein could be one mechanism by which antiglomerular antibodies are produced during infection. In animal models of nephritis induced by nephritogenic streptococci (M type 12), antiglomerular antibodies eluted from kidney glomeruli reacted with the type 12 streptococcal M protein (164). Furthermore, an antiglomerular mAb reacted with the M12 protein (163). These studies support immune-mediated mechanisms in poststreptococcal acute glomerulonephritis (AGN). Streptococcal and renal antigens may be an important source of mimicking antigen present in the kidney and play a role in binding immunoglobulin and complement with production of nephritis.
Novel Cross-Reactive Protein Antigens in Group A Streptococci
Antimyosin antibodies from ARF were used to identify group A streptococcal cross-reactive antigens other than M proteins. The cross-reactive antigens were detected in a Streptococcus pyogenes strain that had spontaneously lost its emm gene. The affinity purified antimyosin antibodies were used to screen gene libraries and the antigen preparations from this M protein-deficient streptococcus. A 60-kDa streptococcal actin-like protein (165), a 60-kDa wall-membrane protein (93, 94), and a 67-kDa protein from a cloned gene product (95) were identified. All of the antigens were unique and interesting. The 60-kDa actin-like molecule behaved biochemically like an actin and produced actin-like filaments observed by electron microscopy (165). The 67-kDa cloned gene product recognized by antimyosin antibody from ARF sera was novel in that it shared enough sequence homology with major histocompatibility molecules that it reacted with sera against mouse major histocompatibility complex class II molecules and exhibited a hydrophilicity plot almost identical to that of major histocompatibility complex class II molecules (95). The gene for the 67-kDa protein was present only in pathogenic streptococcal groups A, C, and G (95). This gene was not found in other streptococcal groups, Escherichia coli, or staphylococci when they were screened for hybridization with the gene probe. The role of the 67-kDa protein as a potential virulence factor in the pathogenesis of streptococcal infections or sequelae is not yet known.
IMMUNE RESPONSES TO N-ACETYL- β-d-GLUCOSAMINE, DOMINANT EPITOPE OF GROUP A POLYSACCHARIDE, IN THE PATHOGENESIS OF RHEUMATIC HEART DISEASE AND SYDENHAM’S CHOREA IN ARF
Immunological Cross-Reactivity Between Carbohydrates and Peptides
The structure of the group A polysaccharide is a polymer of rhamnose with N-acetyl-β-d-glucosamine linked to the rhamnose backbone. The N-acetyl-glucosamine is the immunodominant epitope of the group A carbohydrate that distinguishes S. pyogenes from other streptococcal species. The N-acetyl glucosamine and the group A polysaccharide have been identified in cross-reactions between streptococci and heart tissues (58, 61, 62). Recently, Shikhman, Adderson, and others reported that a subset of cross-reactive antimyosin/antistreptococcal mAbs from immunized mice and virtually all human mAbs from rheumatic carditis patients reacted strongly with an N-acetyl-glucosamine epitope of the group A streptococcal carbohydrate (29, 45–47). Furthermore, synthetic peptides from keratin (45, 47), coxsackie virus (46), and human cardiac myosin (29) reacted with mAbs against N-acetyl-glucosamine and myosin as previously described. In addition, a synthetic cytokeratin peptide, SFGSGFGGGY, mimicked N-acetyl-glucosamine in its reaction with antibodies to N-acetyl-glucosamine and lectins. Mimicry between the group A carbohydrate epitope and peptide molecules is a form of mimicry between chemically diverse molecules. Based on results with peptides altered at a single amino acid residue, it was deduced that aromatic and hydrophobic interactions were important in cross-reactive anticarbohydrate antibody binding to the peptide. Our data as well as that of others (166, 167) clearly link anticardiac myosin and anti-N-acetyl-glucosamine responses with antiheart cross-reactivity.
Rheumatic Heart Disease
Dudding and Ayoub described the elevation and persistence of anti-group A polysaccharide antibody in rheumatic valvulitis (59). Studies of the human antistreptococcal/antimyosin mAbs that reacted strongly with N-acetyl-glucosamine have revealed that one of the mAbs was cytotoxic for human endothelium and reacted with valvular endothelium in tissue sections of a human valve (6). The cytotoxic mAb recognized the extracellular matrix protein laminin that is part of the basement membrane underlying the valvular endothelium. These data suggest that human antibody cross-reactivity with valve tissues is established through antimyosin/anti-N-acetyl-glucosamine/antilaminin reactivity. Antibodies that react with valve endothelium may lead to inflammation at the valve surface and promote T cell infiltration of the valve in rheumatic heart disease. Based on our current knowledge as illustrated in Fig. 10 (143, 144), the proposed events in rheumatic valvulitis/carditis involve both antibodies and T cells. Cross-reactive antibodies target the valve and are believed to act against the group A carbohydrate and valve endothelium based on several lines of evidence described herein (6, 58, 59, 89, 96). T cells are cross-reactive with streptococcal M proteins and homologous alpha helical protein antigens such as myosin, laminin, tropomyosin, or vimentin and become activated and extravasate through activated endothelium into the valve, where they differentiate into CD4+ TH1 cells producing IFNγ and valvular scarring (21, 48, 97, 142–144, 168). VCAM-1 was upregulated on valve endothelium (endocardium) in rheumatic carditis, indicating that the endothelium of the valve in rheumatic carditis was activated (48). Evidence strongly supports the cross-reactive T cell component of the disease, where T cells are shown to penetrate the valve endothelium into an originally avascular valve (21, 32, 48, 97, 136, 142, 143, 168–174). T cells in peripheral blood of patients (24) and the valve (21, 22, 97, 175) have been shown to be cross-reactive with M protein and cardiac proteins, including cardiac myosin epitopes, and they are both CD4+ and CD8+ phenotypes, but the CD4+ phenotype and Th1 cells dominate. The development of scarring and fibrosis in the valve is part of the pathogenesis caused by IFNγ (3, 145, 175) production and IL-17A (101, 146), since the scarring promotes neovascularization and development of a blood supply into normally avascular valve tissue. T cells can subsequently enter the valve through blood vessels developed in the scar. The valve becomes predisposed to cellular infiltration through both the activated valve endocardial surface and the neovascularized scar tissue. Although antibodies develop against collagen in rheumatic heart disease (176, 177), they have not been found to be cross-reactive but may contribute to the pathogenesis of disease (178) once mimicry damages the valve and collagen is exposed to the immune system.
Recent reviews on rheumatic heart disease describe the collective studies and proposed pathogenesis of molecular mimicry in the disease (1–3, 179–184). Initial damage at the chordae tendineae would be sufficient to begin the process of damage to the endothelium of the valve (25) where T cells infiltrate and congregate as they recognize laminin and other valvular proteins that are part of the basement membrane (185). These mechanisms lead to the upregulation of inflammatory molecules such as VCAM-1 (48), which allows the valve to be infiltrated by the T cells, and then the continual damage of the valve with every repetitive streptococcal infection. Treatment with antibiotic prophylaxis as part of the guidelines from the American Heart Association for treatment of rheumatic fever is an important deterrent to the development of rheumatic fever and heart disease in children.
Sydenham Chorea and Pediatric Autoimmune Neuropsychiatric Disorder Associated with Streptococcal Infection (PANDAS)
Sydenham chorea is the major neurological manifestation and movement disorder of rheumatic fever. Human mAbs produced from disease were compared with sera from Sydenham chorea and used to identify the potential antibody targets as well as provide a better understanding of pathogenesis. Human Sydenham chorea-derived mAbs and serum IgG demonstrated specificity for mammalian lysoganglioside and the group A streptococcal carbohydrate epitope N-acetyl-β-d-glucosamine (7). Chorea-derived mAbs and acute serum antibodies, as well as cerebrospinal fluid from Sydenham chorea, reacted with lysoganglioside and targeted the surface of human neuronal cells. These antibodies also induced elevated calcium/calmodulin-dependent protein kinase II activity (CaM kinase II) (7). Although the mAbs were IgM, the serum antibodies that triggered the cell signaling in human neuronal cells were IgG. IgG can penetrate the blood-brain barrier when breached by bacterial components or other molecules such as epinephrine (186–188), and the IgG can reach brain tissues, where it can target specific cells in the brain tissue and produce disease.
Thus, the human chorea mAb 24.3.1 IgG was shown to penetrate the brain when the genetic information for the chorea antibody V genes were expressed in a transgenic mouse model (189). The antibody was expressed by B cells in the transgenic model and was produced by B cells and specific IgG accumulated in mouse serum. Once the blood-brain barrier was broken, the antibody that was tagged with a specific mouse allotype was observed in the basal ganglia, where the allotype-tagged chorea antibody IgG penetrated dopaminergic neurons in the ventral tegumental area or the substantia nigra as shown in Fig. 14 (189). The chorea antibodies were then shown in tritiated thymidine experiments to lead to production of excess dopamine by neuronal cells that could lead to the symptoms of chorea. Other animal models demonstrated that antibodies induced by streptococcal immunization led to deposition in the basal ganglia and the development of behaviors characteristic of Sydenham chorea, such as holding a food pellet or holding on to a beam or demonstrating obsessive compulsive grooming after water spray to fur (190, 191). Passive transfer of the autoantibodies to animals also led to behaviors characteristic of neuropsychiatric symptoms and obsessive compulsive disorders (192–194).
FIGURE 14.
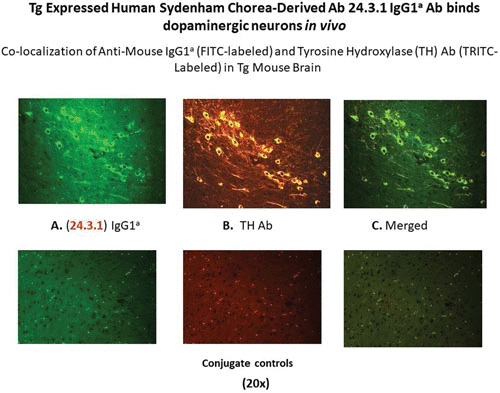
The human Sydenham chorea MAb 24.3.1 V gene expressed as the human V gene-mouse IgG1a constant region in transgenic (Tg) mice targets dopaminergic neurons. Chimeric Tg24.3.1 VH IgG1a antibody expressed in Tg mouse sera penetrated dopaminergic neurons in Tg mouse brain in vivo. Colocalization of Tg 24.3.1 IgG1a (anti-IgG1a Ab; left panel) and tyrosine hydroxylase antibody (anti-TH Ab; middle panel). TH is a marker for dopaminergic neurons. The left-hand panel shows IgG1a (labeled with fluorescein isothiocyanate [FITC]), the center panel shows TH Ab (labeled with tetramethylrhodamine isothiocyanate [TRITC]), and the right-hand panel shows a merged image (FITC-TRITC). Brain sections (basal ganglia) of VH24.3.1 Tg mouse (original magnification, 320×), showing (A) FITC-labeled anti-mouse IgG1a, (B) TRITC-labeled anti-TH Ab, and (C) the merged image. Controls treated with secondary antibody are negative. (Figure taken with permission from Cox et al. [189].)
Studies collectively demonstrated that the IgG from Sydenham chorea targeted a group of cross-reactive antigens in the brain (7–9, 195, 196). These include lysoganglioside, as mentioned above, and they also target tubulin, an intracellular protein abundant in the brain. More importantly, the cross-reactive antibodies have been shown to react with and signal the dopamine receptors D1 and D2 that have led to the characterization of Sydenham chorea as a dopamine receptor encephalitis where the autoantibodies signal the D2 receptor (189). The autoantibodies have been found in both serum and CSF of patients with Sydenham chorea, and the IgG autoantibodies as well as human mAbs signal human neuronal cells and activate CaMKII, which is an important signaling molecule in the brain. These cross-reactive antistreptococcal chorea-derived antibodies lead to the production and release of excess dopamine by neuronal cells in the basal ganglia that ultimately leads to chorea.
Similar antineuronal autoantibodies and autoantibody-mediated neuronal cell signaling are elevated in serum and CSF from PANDAS with the choreiform movements. In both Sydenham chorea and PANDAS, the presence of abnormally elevated antidopamine receptor autoantibodies suggests a dopamine receptor encephalitis (197, 198). Studies of mice infected intranasally with group A streptococci illustrated the importance of infection and its role in opening the blood-brain barrier that must be broken to allow IgG to penetrate the brain. Studies by Agalliu and Cleary and colleagues show that intranasal group A streptococcal infection leads to activated Th17 cells that traverse the olfactory bulb and open the blood-brain barrier to proteins such as IgG (199, 200). Upon opening the blood-brain barrier, IgG antineuronal autoantibodies can penetrate the brain and potentially lead to Sydenham chorea and PANDAS.
The studies suggest (i) that the antibodies against streptococci and brain in Sydenham chorea and related diseases produce central nervous system dysfunction through a neuronal signal transduction and subsequent excess dopamine release mechanism and (ii) that the molecular targets of the chorea antibodies include lysoganglioside and the dopamine receptors in neuronal cell membranes. The antineuronal autoantibodies also target the group A streptococcal carbohydrate epitope N-acetyl-β-d-glucosamine present on the rhamnose backbone of the carbohydrate and present in the cell membrane and wall of the group A streptococci as well as the intracellular brain protein tubulin. The diagram in Fig. 15 illustrates proposed events of how antineuronal autoantibodies against lysoganglioside and the dopamine receptors D1 and D2 may function in Sydenham chorea and PANDAS (3).
FIGURE 15.

Diagram of proposed events leading to the group A streptococcal sequelae Sydenham chorea and PANDAS. Autoantibodies against brain tissues in Sydenham chorea and PANDAS with piano-playing choreiform movements cross-react with the group A streptococcal carbohydrate, lysoganglioside, and dopamine receptors D1R and D2R. The antibodies in Sydenham chorea and PANDAS react with the surface of neuronal cells and trigger cell signaling events, leading to upregulation of calcium/calmodulindependent protein kinase II (CaMKII) and excess dopamine release that leads to the involuntary movements in Sydenham chorea or PANDAS with piano-playing choreiform movements. Both Sydenham chorea and PANDAS are likely to be a dopamine receptor encephalitis based on our data (189, 205) and those of Dale et al. (197, 198). (Taken from Nature Reviews Disease Primers with permission).
CONCLUSIONS
The theme in mimicry, as found in the above-described studies, suggests that cross-reactive antibodies target an intracellular biomarker such as myosin in the heart and tubulin in the brain, but for the antibodies to be pathogenic, they must target a cell surface antigen such as the extracellular matrix protein laminin in the valve endocardial basement membrane or dopamine receptors present at the surface of neuronal cells, leading to the initiation and manifestations of disease. In general, the autoantibodies present as a group of specificities in disease include the cross-reactive antigens recognized by the autoantibodies in disease sera. The cross-reactivity found in individual sera from the disease has been characterized using human mAbs that reflect the serum IgG in the disease.
Cross-reactive antibodies and cross-reactive T cells against group A streptococci are important in the pathogenesis of autoimmune sequelae that characterizes rheumatic fever following streptococcal infection. Cross-reactive antibodies have been defined as those that recognize host tissue alpha-helical coiled-coil antigens such as myosin, tropomyosin, keratin, vimentin, and laminin in the heart (79) and tubulin, lysoganglioside, and dopamine receptors in the brain (9). Cross-reactive antistreptococcal antibodies recognize peptide sequences in alpha-helical proteins such as the M protein virulence determinant of the group A streptococci as well as the N-acetyl-glucosamine molecule, the dominant epitope of the group A streptococcal carbohydrate (45–47). Glycosylated proteins are expected to be targets of the cross-reactive antistreptococcal antibodies. Historically, Goldstein et al. determined autoantibody reactivity with carbohydrates on heart valves (58), and Dudding and Ayoub demonstrated that the group A carbohydrate autoantibodies were an indicator of poor prognosis in valvular heart disease (59). This evidence supports our findings and confirms the importance of the autoantibodies against streptococcal antigens in the development of cross-reactivity that is the basis of the initial disease processes in rheumatic fever and rheumatic heart disease.
In rheumatic heart disease, laminin, an extracellular matrix molecule present in the basement membrane of the valve, may trap cross-reactive anticarbohydrate autoantibodies at the endocardial cell surface and lead to damage or inflammation of the endothelium. Activated endothelium leads to subsequent extravasation of streptococcal M protein/myosin-cross-reactive T cells into the valve (8, 48, 97, 142, 168). Cross-reactive group A streptococcal antigens with apparent roles in the pathogenesis of rheumatic heart disease include the M proteins and the group A polysaccharide. Amino acid sequences of streptococcal M5 protein that have been shown to be pathogenic in animals have also been reported to be recognized by T cells from rheumatic heart valves (21, 97, 142, 143, 168). The identification of human cardiac myosin cross-reactive B and T cell epitopes of M5 protein has been a step forward in understanding the cross-reactive epitopes that produce disease in animals and humans (32) and are hallmarks of molecular mimicry.
In Sydenham chorea, mimicry between the N-acetyl-glucosamine molecule of the group A carbohydrate and brain gangliosides potentially leads to antibodies that bind to the surface of neuronal cells and triggers induction of dopamine in the disease (7). In the pathogenesis of chorea, the development of IgG responses and higher-affinity cell surface reactive antibody that can bind strongly enough to alter host tissues may lead to disease. In carditis, T cells can enter the valve, but only those that are consistently stimulated with local antigens retain a strong immune response. The Th1 T cell subset and its proinflammatory IFNγ cytokine are dominant in valves with a potential role also for IL-17A and the Th17 subset of T cells in fibrosis and scarring.
Although the identity and pathogenic mechanisms of cross-reactive antigens and their cross-reactive antibodies and T cells is much clearer now than in the past, there will always be new lessons to be learned about the pathogenesis of the autoimmune sequelae of ARF and how cross-reactivity and mimicry of host tissues by the group A streptococci leads to autoimmune sequelae following streptococcal disease.
ACKNOWLEDGMENTS
My work was supported by grants HL35280 and HL56267 from the National Heart, Lung, and Blood Institute (NHLBI) and intramural funds from the National Institute of Mental Health (NIMH). M.W.C. is the recipient of an NHLBI merit award.
Deep appreciation is expressed to my students, postdoctoral fellows, and colleagues who contributed to the studies reviewed in this chapter.
REFERENCES
- 1.Cunningham MW. 2014. Rheumatic fever, autoimmunity, and molecular mimicry: the streptococcal connection. Int Rev Immunol 33:314–329 10.3109/08830185.2014.917411. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Cunningham MW. 2012. Streptococcus and rheumatic fever. Curr Opin Rheumatol 24:408–416 10.1097/BOR.0b013e32835461d3. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Carapetis JR, Beaton A, Cunningham MW, Guilherme L, Karthikeyan G, Mayosi BM, Sable C, Steer A, Wilson N, Wyber R, Zühlke L. 2016. Acute rheumatic fever and rheumatic heart disease. Nat Rev Dis Primers 2:15084 10.1038/nrdp.2015.84. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Krisher K, Cunningham MW. 1985. Myosin: a link between streptococci and heart. Science 227:413–415 10.1126/science.2578225. [PubMed] [DOI] [PubMed] [Google Scholar]
- 5.Galvin JE, Hemric ME, Kosanke SD, Factor SM, Quinn A, Cunningham MW. 2002. Induction of myocarditis and valvulitis in Lewis rats by different epitopes of cardiac myosin and its implications in rheumatic carditis. Am J Pathol 160:297–306 10.1016/S0002-9440(10)64373-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Galvin JE, Hemric ME, Ward K, Cunningham MW. 2000. Cytotoxic monoclonal antibody from rheumatic carditis reacts with human endothelium: implications in rheumatic heart disease. J Clin Invest 106:217–224 10.1172/JCI7132. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kirvan CA, Swedo SE, Heuser JS, Cunningham MW. 2003. Mimicry and autoantibody-mediated neuronal cell signaling in Sydenham chorea. Nat Med 9:914–920 10.1038/nm892. [PubMed] [DOI] [PubMed] [Google Scholar]
- 8.Kirvan CA, Galvin JE, Hilt S, Kosanke S, Cunningham MW. 2014. Identification of streptococcal m-protein cardiopathogenic epitopes in experimental autoimmune valvulitis. J Cardiovasc Transl Res 7:172–181 10.1007/s12265-013-9526-4. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kirvan CA, Cox CJ, Swedo SE, Cunningham MW. 2007. Tubulin is a neuronal target of autoantibodies in Sydenham’s chorea. J Immunol 178:7412–7421 10.4049/jimmunol.178.11.7412. [PubMed] [DOI] [PubMed] [Google Scholar]
- 10.Kirvan CA, Swedo SE, Snider LA, Cunningham MW. 2006. Antibody-mediated neuronal cell signaling in behavior and movement disorders. J Neuroimmunol 179:173–179 10.1016/j.jneuroim.2006.06.017. [PubMed] [DOI] [PubMed] [Google Scholar]
- 11.Gorton DE, Govan BL, Ketheesan N, Sive AA, Norton RE, Currie BJ, Towers RJ, Mascaro-Blanco AI, Cunningham MW. 2011. Cardiac myosin epitopes for monitoring progression of rheumatic fever. Pediatr Infect Dis J 30:1015–1016 10.1097/INF.0b013e31823058dd. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lymbury RS, Olive C, Powell KA, Good MF, Hirst RG, LaBrooy JT, Ketheesan N. 2003. Induction of autoimmune valvulitis in Lewis rats following immunization with peptides from the conserved region of group A streptococcal M protein. J Autoimmun 20:211–217 10.1016/S0896-8411(03)00026-X. [DOI] [PubMed] [Google Scholar]
- 13.Rush CM, Govan BL, Sikder S, Williams NL, Ketheesan N. 2014. Animal models to investigate the pathogenesis of rheumatic heart disease. Front Pediatr 2:116 10.3389/fped.2014.00116. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Husby G, van de Rijn I, Zabriskie JB, Abdin ZH, Williams RC Jr. 1976. Antibodies reacting with cytoplasm of subthalamic and caudate nuclei neurons in chorea and acute rheumatic fever. J Exp Med 144:1094–1110 10.1084/jem.144.4.1094. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zabriskie JB. 1967. Mimetic relationships between group A streptococci and mammalian tissues. Adv Immunol 7:147–188 10.1016/S0065-2776(08)60128-5. [DOI] [PubMed] [Google Scholar]
- 16.Zabriskie JB. 1985. Rheumatic fever: the interplay between host, genetics, and microbe. Lewis A. Conner memorial lecture. Circulation 71:1077–1086 10.1161/01.CIR.71.6.1077. [PubMed] [DOI] [PubMed] [Google Scholar]
- 17.Zabriskie JB, Freimer EH. 1966. An immunological relationship between the group. A streptococcus and mammalian muscle. J Exp Med 124:661–678 10.1084/jem.124.4.661. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zabriskie JB, Gibofsky A. 1986. Genetic control of the susceptibility to infection with pathogenic bacteria. Curr Top Microbiol Immunol 124:1–20 10.1007/978-3-642-70986-9_1. [PubMed] [DOI] [PubMed] [Google Scholar]
- 19.Zabriskie JB, Hsu KC, Seegal BC. 1970. Heart-reactive antibody associated with rheumatic fever: characterization and diagnostic significance. Clin Exp Immunol 7:147–159. [PMC free article] [PubMed] [Google Scholar]
- 20.Blank M, Krause I, Magrini L, Spina G, Kalil J, Jacobsen S, Thiesen HJ, Cunningham MW, Guilherme L, Shoenfeld Y. 2006. Overlapping humoral autoimmunity links rheumatic fever and the antiphospholipid syndrome. Rheumatology (Oxford) 45:833–841 10.1093/rheumatology/kel118. [PubMed] [DOI] [PubMed] [Google Scholar]
- 21.Faé K, Kalil J, Toubert A, Guilherme L. 2004. Heart infiltrating T cell clones from a rheumatic heart disease patient display a common TCR usage and a degenerate antigen recognition pattern. Mol Immunol 40:1129–1135 10.1016/j.molimm.2003.11.007. [PubMed] [DOI] [PubMed] [Google Scholar]
- 22.Faé KC, da Silva DD, Oshiro SE, Tanaka AC, Pomerantzeff PM, Douay C, Charron D, Toubert A, Cunningham MW, Kalil J, Guilherme L. 2006. Mimicry in recognition of cardiac myosin peptides by heart-intralesional T cell clones from rheumatic heart disease. J Immunol 176:5662–5670 10.4049/jimmunol.176.9.5662. [PubMed] [DOI] [PubMed] [Google Scholar]
- 23.Quinn A, Kosanke S, Fischetti VA, Factor SM, Cunningham MW. 2001. Induction of autoimmune valvular heart disease by recombinant streptococcal m protein. Infect Immun 69:4072–4078 10.1128/IAI.69.6.4072-4078.2001. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ellis NMJ, Kurahara DK, Vohra H, Mascaro-Blanco A, Erdem G, Adderson EE, Veasy LG, Stoner JA, Tam E, Hill HR, Yamaga K, Cunningham MW. 2010. Priming the immune system for heart disease: a perspective on group A streptococci. J Infect Dis 202:1059–1067 10.1086/656214. [PubMed] [DOI] [PubMed] [Google Scholar]
- 25.Ellis NMJ, Li Y, Hildebrand W, Fischetti VA, Cunningham MW. 2005. T cell mimicry and epitope specificity of cross-reactive T cell clones from rheumatic heart disease. J Immunol 175:5448–5456 10.4049/jimmunol.175.8.5448. [PubMed] [DOI] [PubMed] [Google Scholar]
- 26.Fujinami RS, Oldstone MBA. 1985. Amino acid homology between the encephalitogenic site of myelin basic protein and virus: mechanism for autoimmunity. Science 230:1043–1045 10.1126/science.2414848. [PubMed] [DOI] [PubMed] [Google Scholar]
- 27.Fujinami RS, Oldstone MBA, Wroblewska Z, Frankel ME, Koprowski H. 1983. Molecular mimicry in virus infection: crossreaction of measles virus phosphoprotein or of herpes simplex virus protein with human intermediate filaments. Proc Natl Acad Sci U S A 80:2346–2350 10.1073/pnas.80.8.2346. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Schwimmbeck PL, Oldstone MBA. 1989. Klebsiella pneumoniae and HLA B27-associated diseases of Reiter’s syndrome and ankylosing spondylitis. Curr Top Microbiol Immunol 145:45–56 10.1007/978-3-642-74594-2_4. [PubMed] [DOI] [PubMed] [Google Scholar]
- 29.Adderson EE, Shikhman AR, Ward KE, Cunningham MW. 1998. Molecular analysis of polyreactive monoclonal antibodies from rheumatic carditis: human anti-N-acetylglucosamine/anti-myosin antibody V region genes. J Immunol 161:2020–2031. [PubMed] [Google Scholar]
- 30.Antone SM, Adderson EE, Mertens NMJ, Cunningham MW. 1997. Molecular analysis of V gene sequences encoding cytotoxic anti-streptococcal/anti-myosin monoclonal antibody 36.2.2 that recognizes the heart cell surface protein laminin. J Immunol 159:5422–5430. [PubMed] [Google Scholar]
- 31.Cunningham MW, Antone SM, Gulizia JM, McManus BM, Fischetti VA, Gauntt CJ. 1992. Cytotoxic and viral neutralizing antibodies crossreact with streptococcal M protein, enteroviruses, and human cardiac myosin. Proc Natl Acad Sci U S A 89:1320–1324 10.1073/pnas.89.4.1320. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Cunningham MW, Antone SM, Smart M, Liu R, Kosanke S. 1997. Molecular analysis of human cardiac myosin-cross-reactive B- and T-cell epitopes of the group A streptococcal M5 protein. Infect Immun 65:3913–3923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Cunningham MW, Hall NK, Krisher KK, Spanier AM. 1986. A study of anti-group A streptococcal monoclonal antibodies cross-reactive with myosin. J Immunol 136:293–298. [PubMed] [Google Scholar]
- 34.Cunningham MW, Antone SM, Gulizia JM, McManus BA, Gauntt CJ. 1993. Alpha-helical coiled-coil molecules: a role in autoimmunity against the heart. Clin Immunol Immunopathol 68:118–123 10.1006/clin.1993.1106. [PubMed] [DOI] [PubMed] [Google Scholar]
- 35.Cunningham MW, McCormack JM, Talaber LR, Harley JB, Ayoub EM, Muneer RS, Chun LT, Reddy DV. 1988. Human monoclonal antibodies reactive with antigens of the group A Streptococcus and human heart. J Immunol 141:2760–2766. [PubMed] [Google Scholar]
- 36.Cunningham MW, Swerlick RA. 1986. Polyspecificity of antistreptococcal murine monoclonal antibodies and their implications in autoimmunity. J Exp Med 164:998–1012 10.1084/jem.164.4.998. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Dale JB, Beachey EH. 1986. Sequence of myosin-crossreactive epitopes of streptococcal M protein. J Exp Med 164:1785–1790 10.1084/jem.164.5.1785. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Dale JB, Beachey EH. 1985. Epitopes of streptococcal M proteins shared with cardiac myosin. J Exp Med 162:583–591 10.1084/jem.162.2.583. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Dale JB, Beachey EH. 1985. Multiple, heart-cross-reactive epitopes of streptococcal M proteins. J Exp Med 161:113–122 10.1084/jem.161.1.113. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Cunningham MW, McCormack JM, Fenderson PG, Ho MK, Beachey EH, Dale JB. 1989. Human and murine antibodies cross-reactive with streptococcal M protein and myosin recognize the sequence GLN-LYS-SER-LYS-GLN in M protein. J Immunol 143:2677–2683. [PubMed] [Google Scholar]
- 41.Kraus W, Dale JB, Beachey EH. 1990. Identification of an epitope of type 1 streptococcal M protein that is shared with a 43-kDa protein of human myocardium and renal glomeruli. J Immunol 145:4089–4093. [PubMed] [Google Scholar]
- 42.Kraus W, Seyer JM, Beachey EH. 1989. Vimentin-cross-reactive epitope of type 12 streptococcal M protein. Infect Immun 57:2457–2461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kraus W, Beachey EH. 1988. Renal autoimmune epitope of group A streptococci specified by M protein tetrapeptide Ile-Arg-Leu-Arg. Proc Natl Acad Sci U S A 85:4516–4520 10.1073/pnas.85.12.4516. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Putterman C, Diamond B. 1998. Immunization with a peptide surrogate for double-stranded DNA (dsDNA) induces autoantibody production and renal immunoglobulin deposition. J Exp Med 188:29–38 10.1084/jem.188.1.29. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Shikhman AR, Cunningham MW. 1994. Immunological mimicry between N-acetyl-beta-d-glucosamine and cytokeratin peptides. Evidence for a microbially driven anti-keratin antibody response. J Immunol 152:4375–4387. [PubMed] [Google Scholar]
- 46.Shikhman AR, Greenspan NS, Cunningham MW. 1993. A subset of mouse monoclonal antibodies cross-reactive with cytoskeletal proteins and group A streptococcal M proteins recognizes N-acetyl-beta-d-glucosamine. J Immunol 151:3902–3913. [PubMed] [Google Scholar]
- 47.Shikhman AR, Greenspan NS, Cunningham MW. 1994. Cytokeratin peptide SFGSGFGGGY mimics N-acetyl-beta-d-glucosamine in reaction with antibodies and lectins, and induces in vivo anti-carbohydrate antibody response. J Immunol 153:5593–5606. [PubMed] [Google Scholar]
- 48.Roberts S, Dunn T, Jankelow D, Duran C, Kosanke S, Cunningham MW. 2001. Immune mechanisms in rheumatic carditis: focus on valvular endothelium. J Infect Dis 183:507–511 10.1086/318076. [PubMed] [DOI] [PubMed] [Google Scholar]
- 49.Kaplan MH, Suchy ML. 1964. Immunologic relation of streptococcal and tissue antigens. II. Cross reactions of antisera to mammalian heart tissue with a cell wall constituent of certain strains of group A streptococci. J Exp Med 119:643–650 10.1084/jem.119.4.643. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kaplan MH, Svec KH. 1964. Immunologic relation of streptococcal and tissue antigens. III. Presence in human sera of stretpcoccal antibody cross reactive with heart tissue. Association with streptococcal infection, rheumatic fever, and glomerulonephritis. J Exp Med 119:651–666 10.1084/jem.119.4.651. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kaplan MH, Meyeserian M. 1962. An immunological cross-reaction between group-A streptococcal cells and human heart tissue. Lancet 1:706–710 10.1016/S0140-6736(62)91653-7. [DOI] [PubMed] [Google Scholar]
- 52.Kaplan MH. 1963. Immunologic relation of streptococcal and tissue antigens. I. Properties of an antigen in certain strains of group A streptococci exhibiting an immunologic cross reaction with human heart tissue. J Immunol 90:595–606. [PubMed] [Google Scholar]
- 53.Beachey EH, Stollerman GH. 1973. Mediation of cytotoxic effects of streptococcal M protein by nontype-specific antibody in human sera. J Clin Invest 52:2563–2570 10.1172/JCI107448. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Widdowson JP, Maxted WR, Grant DL, Pinney AM. 1971. The relationship between M-antigen and opacity factor in group A streptococci. J Gen Microbiol 65:69–80 10.1099/00221287-65-1-69. [PubMed] [DOI] [PubMed] [Google Scholar]
- 55.Widdowson JP. 1980. The M-associated protein antigens of group A streptococci, p 125–147. In Read SE, Zabriske JB (ed), Streptococcal Diseases and the Immune Response. Academic Press, New York, NY. [Google Scholar]
- 56.Widdowson JP, Maxted WR, Pinney AM. 1971. An M-associated protein antigen (MAP) of group A streptococci. J Hyg (Lond) 69:553–564 10.1017/S0022172400021823. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.van de Rijn I, Zabriskie JB, McCarty M. 1977. Group A streptococcal antigens cross-reactive with myocardium. Purification of heart-reactive antibody and isolation and characterization of the streptococcal antigen. J Exp Med 146:579–599 10.1084/jem.146.2.579. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Goldstein I, Halpern B, Robert L. 1967. Immunological relationship between streptococcus A polysaccharide and the structural glycoproteins of heart valve. Nature 213:44–47 10.1038/213044a0. [DOI] [Google Scholar]
- 59.Dudding BA, Ayoub EM. 1968. Persistence of streptococcal group A antibody in patients with rheumatic valvular disease. J Exp Med 128:1081–1098 10.1084/jem.128.5.1081. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Lyampert IM, Beletskaya LV, Borodiyuk NA, Gnezditskaya EV, Rassokhina II, Danilova TA. 1976. A cross-reactive antigen of thymus and skin epithelial cells common with the polysaccharide of group A streptococci. Immunology 31:47–55. [PMC free article] [PubMed] [Google Scholar]
- 61.Lyampert IM, Borodiyuk NA, Ugryumova GA. 1968. The reaction of heart and other organ extracts with the sera of animals immunized with group A streptococci. Immunology 15:845–854. [PMC free article] [PubMed] [Google Scholar]
- 62.Lyampert IM, Vvedenskaya OI, Danilova TA. 1966. Study on streptococcus group A antigens common with heart tissue elements. Immunology 11:313–320. [PMC free article] [PubMed] [Google Scholar]
- 63.McCarty M. 1964. Missing links in the streptococcal chain leading to rheumatic fever: the Duckett Jones Memorial Lecture. Circulation 29:488–493 10.1161/01.CIR.29.4.488. [PubMed] [DOI] [PubMed] [Google Scholar]
- 64.Sandson J, Hamerman D, Janis R, Rojkind M. 1968. Immunologic and chemical similarities between the streptococcus and human connective tissue. Trans Assoc Am Physicians 81:249–257. [PubMed] [Google Scholar]
- 65.McCarty M. 1956. Variation in the group-specific carbohydrate of group A streptococci. II. Studies on the chemical basis for serological specificity of the carbohydrates. J Exp Med 104:629–643 10.1084/jem.104.5.629. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Cromartie WJ, Craddock JG. 1966. Rheumatic-like cardiac lesions in mice. Science 154:285–287 10.1126/science.154.3746.285. [PubMed] [DOI] [PubMed] [Google Scholar]
- 67.Cromartie WJ, Craddock JG, Schwab JH, Anderle SK, Yang CH. 1977. Arthritis in rats after systemic injection of streptococcal cells or cell walls. J Exp Med 146:1585–1602 10.1084/jem.146.6.1585. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Schwab JH. 1962. Analysis of the experimental lesion of connective tissue produced by a complex of C polysaccharide from group A streptococci. I. In vivo reaction between tissue and toxin. J Exp Med 116:17–28 10.1084/jem.116.1.17. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Schwab JH. 1964. Analysis of the experimental lesion of connective tissue produced by a complex of C polysaccharide from group A streptococci. II. Influence of age and hypersensitivity. J Exp Med 119:401–408 10.1084/jem.119.3.401. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Schwab JH. 1965. Biological properties of streptococcal cell-wall particles. I. Determinants of the chronic nodular lesion of connective tissue. J Bacteriol 90:1405–1411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Schwab JH, Allen JB, Anderle SK, Dalldorf F, Eisenberg R, Cromartie WJ. 1982. Relationship of complement to experimental arthritis induced in rats with streptococcal cell walls. Immunology 46:83–88. [PMC free article] [PubMed] [Google Scholar]
- 72.Cunningham MW, Hall NK, Krisher KK, Spanier AM. 1986. A study of monoclonal antibodies against streptococci and myosin. J Immunol 136:293–298. [PubMed] [Google Scholar]
- 73.Neu N, Rose NR, Beisel KW, Herskowitz A, Gurri-Glass G, Craig SW. 1987. Cardiac myosin induces myocarditis in genetically predisposed mice. J Immunol 139:3630–3636. [PubMed] [Google Scholar]
- 74.Swerlick RA, Cunningham MW, Hall NK. 1986. Monoclonal antibodies cross-reactive with group A streptococci and normal and psoriatic human skin. J Invest Dermatol 87:367–371 10.1111/1523-1747.ep12524838. [PubMed] [DOI] [PubMed] [Google Scholar]
- 75.Antone SM, Cunningham MW. 1992. Cytotoxicity linked to a streptococcal monoclonal antibody which recognizes laminin, p 189–191. In Orefici G (ed), New Perspectives on Streptococci and Streptococcal Infections. Proceedings of the XI Lancefield International Symposium on Streptococci and Streptococcal Diseases, vol. supplement 22. Gustav Fisher Verlag, New York, NY. [Google Scholar]
- 76.Jones TD. 1944. The diagnosis of rheumatic fever. J Am Med Assoc 126:481–484 10.1001/jama.1944.02850430015005. [DOI] [Google Scholar]
- 77.Dajani AS. 1992. Guidelines for the diagnosis of rheumatic fever (Jones criteria, 1992 update). JAMA 268:2069–2073 10.1001/jama.1992.03490150121036. [PubMed] [DOI] [PubMed] [Google Scholar]
- 78.Cunningham MW. 1999. Molecular mimicry between group A streptococci and myosin in the pathogenesis of rheumatic fever, 1st ed, vol. 2. American Registry of Pathology, Armed Forces Institute of Pathology, Washington, DC. [Google Scholar]
- 79.Cunningham MW. 2000. Pathogenesis of group A streptococcal infections. Clin Microbiol Rev 13:470–511 10.1128/CMR.13.3.470-511.2000. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Cunningham MW. 2000. Group A Streptococcal Sequelae and Molecular Mimicry. Lippincott, Williams and Wilkins, Philadelphia, PA. [Google Scholar]
- 81.Cunningham MW. 2000. Pathogenesis of Acute Rheumatic Fever. Oxford Press, New York, NY. [Google Scholar]
- 82.Lange CF. 1994. Localization of [14C] labeled anti-streptococcal cell membrane monoclonal antibodies (anti-SCM mAb) in mice. Autoimmunity 19:179–191 10.3109/08916939408995693. [PubMed] [DOI] [PubMed] [Google Scholar]
- 83.Wu X, Liu B, Van der Merwe PL, Kalis NN, Berney SM, Young DC. 1998. Myosin-reactive autoantibodies in rheumatic carditis and normal fetus. Clin Immunol Immunopathol 87:184–192 10.1006/clin.1998.4531. [PubMed] [DOI] [PubMed] [Google Scholar]
- 84.Carroll P, Stafford D, Schwartz RS, Stollar BD. 1985. Murine monoclonal anti-DNA autoantibodies bind to endogenous bacteria. J Immunol 135:1086–1090. [PubMed] [Google Scholar]
- 85.Lafer EM, Rauch J, Andrzejewski C Jr, Mudd D, Furie B, Furie B, Schwartz RS, Stollar BD. 1981. Polyspecific monoclonal lupus autoantibodies reactive with both polynucleotides and phospholipids. J Exp Med 153:897–909 10.1084/jem.153.4.897. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Andrzejewski C Jr, Rauch J, Lafer E, Stollar BD, Schwartz RS. 1981. Antigen-binding diversity and idiotypic cross-reactions among hybridoma autoantibodies to DNA. J Immunol 126:226–231. [PubMed] [Google Scholar]
- 87.Mertens NM, Galvin JE, Adderson EE, Cunningham MW. 2000. Molecular analysis of cross-reactive anti-myosin/anti-streptococcal mouse monoclonal antibodies. Mol Immunol 37:901–913 10.1016/S0161-5890(01)00007-4. [DOI] [PubMed] [Google Scholar]
- 88.Wedemayer GJ, Patten PA, Wang LH, Schultz PG, Stevens RC. 1997. Structural insights into the evolution of an antibody combining site. Science 276:1665–1669 10.1126/science.276.5319.1665. [PubMed] [DOI] [PubMed] [Google Scholar]
- 89.Gulizia JM, Cunningham MW, McManus BM. 1991. Immunoreactivity of anti-streptococcal monoclonal antibodies to human heart valves. Evidence for multiple cross-reactive epitopes. Am J Pathol 138:285–301. [PMC free article] [PubMed] [Google Scholar]
- 90.Gulizia JM, Cunningham MW, McManus BM. 1992. Anti-streptococcal monoclonal antibodies recognize multiple epitopes in human heart valves: cardiac myosin, vimentin, and elastin as potential valvular autoantigens, p 267–269. In Orefici G (ed), New Perspectives on Streptococci and Streptococcal Infections. Proceedings of the XI Lancefield International Symposium, vol. 22. Zbl Bakt Suppl. Gustav Fisher Verlag, New York, NY. [Google Scholar]
- 91.Köhler G, Milstein C. 1975. Continuous cultures of fused cells secreting antibody of predefined specificity. Nature 256:495–497 10.1038/256495a0. [PubMed] [DOI] [PubMed] [Google Scholar]
- 92.Cunningham MW, Krisher K, Graves DC. 1984. Murine monoclonal antibodies reactive with human heart and group A streptococcal membrane antigens. Infect Immun 46:34–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Barnett LA, Cunningham MW. 1990. A new heart-cross-reactive antigen in Streptococcus pyogenes is not M protein. J Infect Dis 162:875–882 10.1093/infdis/162.4.875. [PubMed] [DOI] [PubMed] [Google Scholar]
- 94.Barnett LA, Ferretti JJ, Cunningham MW. 1992. A 60-kDa acute rheumatic fever associated antigen of Streptococcus pyogenes. In Orefici G (ed), New Perspectives on Streptococci and Streptococcal Infections. Proceedings of the XI Lancefield International Symposium, vol. 22. Zbl Bakt Suppl. Gustav Fisher Verlag, New York, NY. [Google Scholar]
- 95.Kil KS, Cunningham MW, Barnett LA. 1994. Cloning and sequence analysis of a gene encoding a 67-kilodalton myosin-cross-reactive antigen of Streptococcus pyogenes reveals its similarity with class II major histocompatibility antigens. Infect Immun 62:2440–2449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Kaplan MH, Bolande R, Rakita L, Blair J. 1964. Presence of bound immunoglobulins and complement in the myocardium in acute rheumatic fever. Association with cardiac failure. N Engl J Med 271:637–645 10.1056/NEJM196409242711301. [PubMed] [DOI] [PubMed] [Google Scholar]
- 97.Guilherme L, Cunha-Neto E, Coelho V, Snitcowsky R, Pomerantzeff PMA, Assis RV, Pedra F, Neumann J, Goldberg A, Patarroyo ME, Pileggi F, Kalil J. 1995. Human heart-infiltrating T-cell clones from rheumatic heart disease patients recognize both streptococcal and cardiac proteins. Circulation 92:415–420 10.1161/01.CIR.92.3.415. [PubMed] [DOI] [PubMed] [Google Scholar]
- 98.Bronze MS, Dale JB. 1993. Epitopes of streptococcal M proteins that evoke antibodies that cross-react with human brain. J Immunol 151:2820–2828. [PubMed] [Google Scholar]
- 99.Bronze MS, Beachey EH, Seyer JM, Dale JB. 1987. Protective and heart-cross-reactive epitopes of type 19 streptococcal M protein. Trans Assoc Am Physicians 100:80–84. [PubMed] [Google Scholar]
- 100.Beachey EH, Bronze M, Dale JB, Kraus W, Poirier T, Sargent S. 1988. Protective and autoimmune epitopes of streptococcal M proteins. Vaccine 6:192–196 10.1016/S0264-410X(88)80027-6. [DOI] [PubMed] [Google Scholar]
- 101.Sikder S, Williams NL, Sorenson AE, Alim MA, Vidgen ME, Moreland NJ, Rush CM, Simpson RS, Govan BL, Norton RE, Cunningham MW, McMillan DJ, Sriprakash KS, Ketheesan N. 2017. Group G streptococcus induces an autoimmune IL-17A/IFN-gamma mediated carditis in the Lewis rat model of rheumatic heart disease. J Infect Dis 218:324–335. [PubMed] [DOI] [PubMed] [Google Scholar]
- 102.Phillips GN, Flicker PF, Cohen C, Manjula BN, Fischetti VA. 1981. Streptococcal M protein: a-helical coiled-coil structure and arrangements on the cell surface. Proc Natl Acad Sci U S A 78:4698 10.1073/pnas.78.8.4689. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Manjula BN, Fischetti VA. 1980. Tropomyosin-like seven residue periodicity in three immunologically distinct streptococal M proteins and its implications for the antiphagocytic property of the molecule. J Exp Med 151:695–708 10.1084/jem.151.3.695. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Manjula BN, Fischetti VA. 1986. Sequence homology of group A streptococcal Pep M5 protein with other coiled-coil proteins. Biochem Biophys Res Commun 140:684–690 10.1016/0006-291X(86)90786-2. [DOI] [PubMed] [Google Scholar]
- 105.Manjula BN, Trus BL, Fischetti VA. 1985. Presence of two distinct regions in the coiled-coil structure of the streptococcal Pep M5 protein: relationship to mammalian coiled-coil proteins and implications to its biological properties. Proc Natl Acad Sci U S A 82:1064–1068 10.1073/pnas.82.4.1064. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Bronze MS, Beachey EH, Dale JB. 1988. Protective and heart-crossreactive epitopes located within the NH2 terminus of type 19 streptococcal M protein. J Exp Med 167:1849–1859 10.1084/jem.167.6.1849. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Sargent SJ, Beachey EH, Corbett CE, Dale JB. 1987. Sequence of protective epitopes of streptococcal M proteins shared with cardiac sarcolemmal membranes. J Immunol 139:1285–1290. [PubMed] [Google Scholar]
- 108.Bessen D, Jones KF, Fischetti VA. 1989. Evidence for two distinct classes of streptococcal M protein and their relationship to rheumatic fever. J Exp Med 169:269–283 10.1084/jem.169.1.269. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Bessen DE, Fischetti VA. 1990. Differentiation between two biologically distinct classes of group A streptococci by limited substitutions of amino acids within the shared region of M protein-like molecules. J Exp Med 172:1757–1764 10.1084/jem.172.6.1757. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Jones KF, Khan SA, Erickson BW, Hollingshead SK, Scott JR, Fischetti VA. 1986. Immunochemical localization and amino acid sequences of crossreactive epitopes within the group A streptococcal M6 protein. J Exp Med 164:1226–1238 10.1084/jem.164.4.1226. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Jones KF, Fischetti VA. 1988. The importance of the location of antibody binding on the M6 protein for opsonization and phagocytosis of group A M6 streptococci. J Exp Med 167:1114–1123 10.1084/jem.167.3.1114. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Quinn A, Ward K, Fischetti VA, Hemric M, Cunningham MW. 1998. Immunological relationship between the class I epitope of streptococcal M protein and myosin. Infect Immun 66:4418–4424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Bessen DE, Veasy LG, Hill HR, Augustine NH, Fischetti VA. 1995. Serologic evidence for a class I group A streptococcal infection among rheumatic fever patients. J Infect Dis 172:1608–1611 10.1093/infdis/172.6.1608. [PubMed] [DOI] [PubMed] [Google Scholar]
- 114.Mori K, Kamikawaji N, Sasazuki T. 1996. Persistent elevation of immunoglobulin G titer against the C region of recombinant group A streptococcal M protein in patients with rheumatic fever. Pediatr Res 39:336–342 10.1203/00006450-199602000-00024. [PubMed] [DOI] [PubMed] [Google Scholar]
- 115.Jones KF, Whitehead SS, Cunningham MW, Fischetti VA. 2000. Reactivity of rheumatic fever and scarlet fever patients’ sera with group A streptococcal M protein, cardiac myosin, and cardiac tropomyosin: a retrospective study. Infect Immun 68:7132–7136 10.1128/IAI.68.12.7132-7136.2000. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Huber SA, Cunningham MW. 1996. Streptococcal M protein peptide with similarity to myosin induces CD4+ T cell-dependent myocarditis in MRL/++ mice and induces partial tolerance against coxsackieviral myocarditis. J Immunol 156:3528–3534. [PubMed] [Google Scholar]
- 117.Quinn A, Shinnick TM, Cunningham MW. 1996. Anti-Hsp65 antibodies recognize M proteins of group A streptococci. Infect Immun 64:818–824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Huber S, Polgar J, Moraska A, Cunningham M, Schwimmbeck P, Schultheiss P. 1993. T lymphocyte responses in CVB3-induced murine myocarditis. Scand J Infect Dis Suppl 88:67–78. [PubMed] [Google Scholar]
- 119.Huber SA, Moraska A, Cunningham M. 1994. Alterations in major histocompatibility complex association of myocarditis induced by coxsackievirus B3 mutants selected with monoclonal antibodies to group A streptococci. Proc Natl Acad Sci U S A 91:5543–5547 10.1073/pnas.91.12.5543. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Cunningham MW, Antone SM, Gulizia JM, Gauntt CJ. 1992. Common epitopes shared between streptococcal M protein and viruses may be a link to autoimmunity, p 534–536. In Orefici G (ed), New Perspectives on Streptococci and Streptococcal Infections. Proceedings of the XI Lancefield International Symposium, vol. 22. Zbl Bakt Suppl. Gustav Fisher Verlag, New York, NY. [Google Scholar]
- 121.Huber SA, Job LP, Woodruff JF. 1980. Lysis of infected myofibers by coxsackievirus B-3-immune T lymphocytes. Am J Pathol 98:681–694. [PMC free article] [PubMed] [Google Scholar]
- 122.Huber SA, Lodge PA. 1986. Coxsackievirus B-3 myocarditis. Identification of different pathogenic mechanisms in DBA/2 and Balb/c mice. Am J Pathol 122:284–291. [PMC free article] [PubMed] [Google Scholar]
- 123.Gauntt CJ, Tracy SM, Chapman N, Wood HJ, Kolbeck PC, Karaganis AG, Winfrey CL, Cunningham MW. 1995. Coxsackievirus-induced chronic myocarditis in murine models. Eur Heart J 16(Suppl O):56–58 10.1093/eurheartj/16.suppl_O.56. [PubMed] [DOI] [PubMed] [Google Scholar]
- 124.Cohen IR. 1991. Autoimmunity to chaperonins in the pathogenesis of arthritis and diabetes. Annu Rev Immunol 9:567–589 10.1146/annurev.iy.09.040191.003031. [PubMed] [DOI] [PubMed] [Google Scholar]
- 125.Cohen IR, Young DB. 1991. Autoimmunity, microbial immunity and the immunological homunculus. Immunol Today 12:105–110 10.1016/0167-5699(91)90093-9. [DOI] [PubMed] [Google Scholar]
- 126.Schett G, Xu Q, Amberger A, Van der Zee R, Recheis H, Willeit J, Wick G. 1995. Autoantibodies against heat shock protein 60 mediate endothelial cytotoxicity. J Clin Invest 96:2569–2577 10.1172/JCI118320. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.McNamara C, Zinkernagel AS, Macheboeuf P, Cunningham MW, Nizet V, Ghosh P. 2008. Coiled-coil irregularities and instabilities in group A Streptococcus M1 are required for virulence. Science 319:1405–1408 10.1126/science.1154470. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Dale JB, Beachey EH. 1987. Human cytotoxic T lymphocytes evoked by group A streptococcal M proteins. J Exp Med 166:1825–1835 10.1084/jem.166.6.1825. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Pruksakorn S, Galbraith A, Houghten RA, Good MF. 1992. Conserved T and B cell epitopes on the M protein of group A streptococci. Induction of bactericidal antibodies. J Immunol 149:2729–2735. [PubMed] [Google Scholar]
- 130.Pruksakorn S, Currie B, Brandt E, Phornphutkul C, Hunsakunachai S, Manmontri A, Robinson JH, Kehoe MA, Galbraith A, Good MF. 1994. Identification of T cell autoepitopes that cross-react with the C-terminal segment of the M protein of group A streptococci. Int Immunol 6:1235–1244 10.1093/intimm/6.8.1235. [PubMed] [DOI] [PubMed] [Google Scholar]
- 131.Robinson JH, Atherton MC, Goodacre JA, Pinkney M, Weightman H, Kehoe MA. 1991. Mapping T-cell epitopes in group A streptococcal type 5 M protein. Infect Immun 59:4324–4331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Robinson JH, Case MC, Kehoe MA. 1993. Characterization of a conserved helper-T-cell epitope from group A streptococcal M proteins. Infect Immun 61:1062–1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Tomai M, Kotb M, Majumdar G, Beachey EH. 1990. Superantigenicity of streptococcal M protein. J Exp Med 172:359–362 10.1084/jem.172.1.359. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Tomai MA, Aelion JA, Dockter ME, Majumdar G, Spinella DG, Kotb M. 1991. T cell receptor V gene usage by human T cells stimulated with the superantigen streptococcal M protein. J Exp Med 174:285–288 10.1084/jem.174.1.285. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Tomai MA, Schlievert PM, Kotb M. 1992. Distinct T-cell receptor V beta gene usage by human T lymphocytes stimulated with the streptococcal pyrogenic exotoxins and pep M5 protein. Infect Immun 60:701–705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Hutto JH, Ayoub EM. 1980. Cytotoxicity of lymphocytes from patients with rheumatic carditis to cardiac cells in vitro, p 733–738. In Read SE, Zabriskie JB (ed), Streptococcal Diseases and the Immune Response. Academic Press, New York, NY. [Google Scholar]
- 137.Wang B, Schlievert PM, Gaber AO, Kotb M. 1993. Localization of an immunologically functional region of the streptococcal superantigen pepsin-extracted fragment of type 5 M protein. J Immunol 151:1419–1429. [PubMed] [Google Scholar]
- 138.Degnan B, Taylor J, Hawkes C, O’Shea U, Smith J, Robinson JH, Kehoe MA, Boylston A, Goodacre JA. 1997. Streptococcus pyogenes type 5 M protein is an antigen, not a superantigen, for human T cells. Hum Immunol 53:206–215 10.1016/S0198-8859(97)00028-1. [DOI] [PubMed] [Google Scholar]
- 139.Fleischer B, Schmidt K-H, Gerlach D, Köhler W. 1992. Separation of T-cell-stimulating activity from streptococcal M protein. Infect Immun 60:1767–1770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Schmidt K-H, Gerlach D, Wollweber L, Reichardt W, Mann K, Ozegowski J-H, Fleischer B. 1995. Mitogenicity of M5 protein extracted from Streptococcus pyogenes cells is due to streptococcal pyrogenic exotoxin C and mitogenic factor MF. Infect Immun 63:4569–4575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Bisno AL. 1995. Non-suppurative poststreptococcal sequelae: rheumatic fever and glomerulonephritis, p 1799–1810. In Mandell GL, Bennett JE, Dolin R (ed), Principles and Practice of Infectious Diseases, vol 2. Churchill Livingstone, New York, NY. [Google Scholar]
- 142.Guilherme L, Oshiro SE, Faé KC, Cunha-Neto E, Renesto G, Goldberg AC, Tanaka AC, Pomerantzeff PM, Kiss MH, Silva C, Guzman F, Patarroyo ME, Southwood S, Sette A, Kalil J. 2001. T-cell reactivity against streptococcal antigens in the periphery mirrors reactivity of heart-infiltrating T lymphocytes in rheumatic heart disease patients. Infect Immun 69:5345–5351 10.1128/IAI.69.9.5345-5351.2001. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Cunningham MW. 2004. T cell mimicry in inflammatory heart disease. Mol Immunol 40:1121–1127 10.1016/j.molimm.2003.11.023. [PubMed] [DOI] [PubMed] [Google Scholar]
- 144.Cunningham MW. 2003. Autoimmunity and molecular mimicry in the pathogenesis of post-streptococcal heart disease. Front Biosci 8:s533–s543 10.2741/1067. [DOI] [PubMed] [Google Scholar]
- 145.Guilherme L, Cury P, Demarchi LM, Coelho V, Abel L, Lopez AP, Oshiro SE, Aliotti S, Cunha-Neto E, Pomerantzeff PM, Tanaka AC, Kalil J. 2004. Rheumatic heart disease: proinflammatory cytokines play a role in the progression and maintenance of valvular lesions. Am J Pathol 165:1583–1591 10.1016/S0002-9440(10)63415-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Bas HD, Baser K, Yavuz E, Bolayir HA, Yaman B, Unlu S, Cengel A, Bagriacik EU, Yalcin R. 2014. A shift in the balance of regulatory T and T helper 17 cells in rheumatic heart disease. J Investig Med 62:78–83 10.2310/JIM.0000000000000023. [PubMed] [DOI] [PubMed] [Google Scholar]
- 147.Dileepan T, Linehan JL, Moon JJ, Pepper M, Jenkins MK, Cleary PP. 2011. Robust antigen specific th17 T cell response to group A Streptococcus is dependent on IL-6 and intranasal route of infection. PLoS Pathog 7:e1002252 10.1371/journal.ppat.1002252. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Pepper M, Linehan JL, Pagán AJ, Zell T, Dileepan T, Cleary PP, Jenkins MK. 2010. Different routes of bacterial infection induce long-lived TH1 memory cells and short-lived TH17 cells. Nat Immunol 11:83–89 10.1038/ni.1826. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Wang B, Dileepan T, Briscoe S, Hyland KA, Kang J, Khoruts A, Cleary PP. 2010. Induction of TGF-beta1 and TGF-beta1-dependent predominant Th17 differentiation by group A streptococcal infection. Proc Natl Acad Sci U S A 107:5937–5942 10.1073/pnas.0904831107. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Weaver CT, Harrington LE, Mangan PR, Gavrieli M, Murphy KM. 2006. Th17: an effector CD4 T cell lineage with regulatory T cell ties. Immunity 24:677–688 10.1016/j.immuni.2006.06.002. [PubMed] [DOI] [PubMed] [Google Scholar]
- 151.Weaver CT, Hatton RD, Mangan PR, Harrington LE. 2007. IL-17 family cytokines and the expanding diversity of effector T cell lineages. Annu Rev Immunol 25:821–852 10.1146/annurev.immunol.25.022106.141557. [PubMed] [DOI] [PubMed] [Google Scholar]
- 152.Peck A, Mellins ED. 2010. Precarious balance: Th17 cells in host defense. Infect Immun 78:32–38 10.1128/IAI.00929-09. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Cunningham MW. 2006. T regulatory cells: sentinels against autoimmune heart disease. Circ Res 99:1024–1026 10.1161/01.RES.0000250832.30969.6a. [PubMed] [DOI] [PubMed] [Google Scholar]
- 154.Baldeviano GC, Barin JG, Talor MV, Srinivasan S, Bedja D, Zheng D, Gabrielson K, Iwakura Y, Rose NR, Cihakova D. 2010. Interleukin-17A is dispensable for myocarditis but essential for the progression to dilated cardiomyopathy. Circ Res 106:1646–1655 10.1161/CIRCRESAHA.109.213157. [PubMed] [DOI] [PubMed] [Google Scholar]
- 155.Myers JM, Cooper LT, Kem DC, Stavrakis S, Kosanke SD, Shevach EM, Fairweather D, Stoner JA, Cox CJ, Cunningham MW. 2016. Cardiac myosin-Th17 responses promote heart failure in human myocarditis. JCI Insight 1:1 10.1172/jci.insight.85851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Gorton D, Sikder S, Williams NL, Chilton L, Rush CM, Govan BL, Cunningham MW, Ketheesan N. 2016. Repeat exposure to group A streptococcal M protein exacerbates cardiac damage in a rat model of rheumatic heart disease. Autoimmunity 49:563–570 10.1080/08916934.2016.1217999. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Kefalides NA, Pegg MT, Ohno N, Poon-King T, Zabriskie J, Fillit H. 1986. Antibodies to basement membrane collagen and to laminin are present in sera from patients with poststreptococcal glomerulonephritis. J Exp Med 163:588–602 10.1084/jem.163.3.588. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Kefalides NA, Ohno N, Wilson CB, Fillit H, Zabriski J, Rosenbloom J. 1993. Identification of antigenic epitopes in type IV collagen by use of synthetic peptides. Kidney Int 43:94–100 10.1038/ki.1993.16. [PubMed] [DOI] [PubMed] [Google Scholar]
- 159.Michael AF Jr, Drummond KN, Good RA, Vernier RL. 1966. Acute poststreptococcal glomerulonephritis: immune deposit disease. J Clin Invest 45:237–248 10.1172/JCI105336. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Markowitz AS, Lange CF Jr. 1964. Streptococcal related glomerulonephritis. I. Isolation, immunocytochemistry and comparative chemistry of soluble fractions from type 12 nephritogenic streptococci and human glomeruli. J Immunol 92:565–575. [PubMed] [Google Scholar]
- 161.Lange CF. 1969. Chemistry of cross-reactive fragments of streptococcal cell membrane and human glomerular basement membrane. Transplant Proc 1:959–963. [PubMed] [Google Scholar]
- 162.Bisno AL, Wood JW, Lawson J, Roy S, Beachey EH, Stollermann GH. 1978. Antigens in urine of patients with glomerulonephritis and in normal human serum which cross-react with group A streptococci: identification and partial characterization. J Lab Clin Med 91:500–513. [PubMed] [Google Scholar]
- 163.Goroncy-Bermes P, Dale JB, Beachey EH, Opferkuch W. 1987. Monoclonal antibody to human renal glomeruli cross-reacts with streptococcal M protein. Infect Immun 55:2416–2419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Lindberg LH, Vosti KL. 1969. Elution of glomerular bound antibodies in experimental streptococcal glomerulonephritis. Science 166:1032–1033 10.1126/science.166.3908.1032. [PubMed] [DOI] [PubMed] [Google Scholar]
- 165.Barnett LA, Cunningham MW. 1992. Evidence for actinlike proteins in an M protein-negative strain of Streptococcus pyogenes. Infect Immun 60:3932–3936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Malkiel S, Liao L, Cunningham MW, Diamond B. 2000. T-cell-dependent antibody response to the dominant epitope of streptococcal polysaccharide, N-acetyl-glucosamine, is cross-reactive with cardiac myosin. Infect Immun 68:5803–5808 10.1128/IAI.68.10.5803-5808.2000. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Liao L, Sindhwani R, Rojkind M, Factor S, Leinwand L, Diamond B. 1995. Antibody-mediated autoimmune myocarditis depends on genetically determined target organ sensitivity. J Exp Med 181:1123–1131 10.1084/jem.181.3.1123. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Guilherme L, Dulphy N, Douay C, Coelho V, Cunha-Neto E, Oshiro SE, Assis RV, Tanaka AC, Pomerantzeff PM, Charron D, Toubert A, Kalil J. 2000. Molecular evidence for antigen-driven immune responses in cardiac lesions of rheumatic heart disease patients. Int Immunol 12:1063–1074 10.1093/intimm/12.7.1063. [PubMed] [DOI] [PubMed] [Google Scholar]
- 169.Gibofsky A, Kerwar S, Zabriskie JB. 1998. Rheumatic fever: the relationships between host, microbe and genetics. Rheum Dis Clin North Am 24:237–259. [DOI] [PubMed] [Google Scholar]
- 170.Kemeny E, Grieve T, Marcus R, Sareli P, Zabriskie JB. 1989. Identification of mononuclear cells and T cell subsets in rheumatic valvulitis. Clin Immunol Immunopathol 52:225–237 10.1016/0090-1229(89)90174-8. [DOI] [PubMed] [Google Scholar]
- 171.Raizada V, Williams RC Jr, Chopra P, Gopinath N, Prakash K, Sharma KB, Cherian KM, Panday S, Arora R, Nigam M, Zabriskie JB, Husby G. 1983. Tissue distribution of lymphocytes in rheumatic heart valves as defined by monoclonal anti-T cell antibodies. Am J Med 74:90–96 10.1016/0002-9343(83)91124-5. [DOI] [PubMed] [Google Scholar]
- 172.Read SE, Reid HF, Fischetti VA, Poon-King T, Ramkissoon R, McDowell M, Zabriskie JB. 1986. Serial studies on the cellular immune response to streptococcal antigens in acute and convalescent rheumatic fever patients in Trinidad. J Clin Immunol 6:433–441 10.1007/BF00915249. [PubMed] [DOI] [PubMed] [Google Scholar]
- 173.Bhatia R, Narula J, Reddy KS, Koicha M, Malaviya AN, Pothineni RB, Tandon R, Bhatia ML. 1989. Lymphocyte subsets in acute rheumatic fever and rheumatic heart disease. Clin Cardiol 12:34–38 10.1002/clc.4960120106. [PubMed] [DOI] [PubMed] [Google Scholar]
- 174.Chopra P, Narula J, Kumar AS, Sachdeva S, Bhatia ML. 1988. Immunohistochemical characterisation of Aschoff nodules and endomyocardial inflammatory infiltrates in left atrial appendages from patients with chronic rheumatic heart disease. Int J Cardiol 20:99–105 10.1016/0167-5273(88)90319-1. [DOI] [PubMed] [Google Scholar]
- 175.Guilherme L, Kalil J, Cunningham M. 2006. Molecular mimicry in the autoimmune pathogenesis of rheumatic heart disease. Autoimmunity 39:31–39 10.1080/08916930500484674. [PubMed] [DOI] [PubMed] [Google Scholar]
- 176.Martins TB, Hoffman JL, Augustine NH, Phansalkar AR, Fischetti VA, Zabriskie JB, Cleary PP, Musser JM, Veasy LG, Hill HR. 2008. Comprehensive analysis of antibody responses to streptococcal and tissue antigens in patients with acute rheumatic fever. Int Immunol 20:445–452 10.1093/intimm/dxn004. [PubMed] [DOI] [PubMed] [Google Scholar]
- 177.Dinkla K, Rohde M, Jansen WT, Kaplan EL, Chhatwal GS, Talay SR. 2003. Rheumatic fever-associated Streptococcus pyogenes isolates aggregate collagen. J Clin Invest 111:1905–1912 10.1172/JCI17247. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Tandon R, Sharma M, Chandrashekhar Y, Kotb M, Yacoub MH, Narula J. 2013. Revisiting the pathogenesis of rheumatic fever and carditis. Nat Rev Cardiol 10:171–177 10.1038/nrcardio.2012.197. [PubMed] [DOI] [PubMed] [Google Scholar]
- 179.Cunningham MW. 2013. Rheumatic fever revisited. Nat Rev Cardiol 11:123. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Guilherme L, de Barros SF, Kohler KF, Santos SR, Ferreira FM, Silva WR, Alencar R, Postol E, Kalil J. 2017. Rheumatic heart disease: pathogenesis and vaccine. Curr Protein Pept Sci 19:900–908. [PubMed] [DOI] [PubMed] [Google Scholar]
- 181.Guilherme L, Köhler KF, Faé KC. 2015. Editorial: frontiers in autoimmune disease: rheumatic fever and rheumatic heart disease. Front Pediatr 3:91 10.3389/fped.2015.00091. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Guilherme L, Kalil J. 2010. Rheumatic fever and rheumatic heart disease: cellular mechanisms leading autoimmune reactivity and disease. J Clin Immunol 30:17–23 10.1007/s10875-009-9332-6. [PubMed] [DOI] [PubMed] [Google Scholar]
- 183.Guilherme L, Ramasawmy R, Kalil J. 2007. Rheumatic fever and rheumatic heart disease: genetics and pathogenesis. Scand J Immunol 66:199–207 10.1111/j.1365-3083.2007.01974.x. [PubMed] [DOI] [PubMed] [Google Scholar]
- 184.Guilherme L, Faé KC, Oshiro SE, Tanaka AC, Pomerantzeff PM, Kalil J. 2007. T cell response in rheumatic fever: crossreactivity between streptococcal M protein peptides and heart tissue proteins. Curr Protein Pept Sci 8:39–44 10.2174/138920307779941488. [PubMed] [DOI] [PubMed] [Google Scholar]
- 185.Guilherme L, Kalil J. 2013. Rheumatic heart disease: molecules involved in valve tissue inflammation leading to the autoimmune process and anti-S. pyogenes vaccine. Front Immunol 4:352 10.3389/fimmu.2013.00352. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Kowal C, Diamond B. 2012. Aspects of CNS lupus: mouse models of anti-NMDA receptor antibody mediated reactivity. Methods Mol Biol 900:181–206 10.1007/978-1-60761-720-4_9. [PubMed] [DOI] [PubMed] [Google Scholar]
- 187.Huerta PT, Kowal C, DeGiorgio LA, Volpe BT, Diamond B. 2006. Immunity and behavior: antibodies alter emotion. Proc Natl Acad Sci U S A 103:678–683 10.1073/pnas.0510055103. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Brimberg L, Mader S, Fujieda Y, Arinuma Y, Kowal C, Volpe BT, Diamond B. 2015. Antibodies as mediators of brain pathology. Trends Immunol 36:709–724 10.1016/j.it.2015.09.008. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Cox CJ, Sharma M, Leckman JF, Zuccolo J, Zuccolo A, Kovoor A, Swedo SE, Cunningham MW. 2013. Brain human monoclonal autoantibody from sydenham chorea targets dopaminergic neurons in transgenic mice and signals dopamine D2 receptor: implications in human disease. J Immunol 191:5524–5541 10.4049/jimmunol.1102592. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Brimberg L, Benhar I, Mascaro-Blanco A, Alvarez K, Lotan D, Winter C, Klein J, Moses AE, Somnier FE, Leckman JF, Swedo SE, Cunningham MW, Joel D. 2012. Behavioral, pharmacological, and immunological abnormalities after streptococcal exposure: a novel rat model of Sydenham chorea and related neuropsychiatric disorders. Neuropsychopharmacology 37:2076–2087. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Hoffman KL, Hornig M, Yaddanapudi K, Jabado O, Lipkin WI. 2004. A murine model for neuropsychiatric disorders associated with group A beta-hemolytic streptococcal infection. J Neurosci 24:1780–1791 10.1523/JNEUROSCI.0887-03.2004. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Yaddanapudi K, Hornig M, Serge R, De Miranda J, Baghban A, Villar G, Lipkin WI. 2010. Passive transfer of streptococcus-induced antibodies reproduces behavioral disturbances in a mouse model of pediatric autoimmune neuropsychiatric disorders associated with streptococcal infection. Mol Psychiatry 15:712–726 10.1038/mp.2009.77. [PubMed] [DOI] [PubMed] [Google Scholar]
- 193.Lotan D, Benhar I, Alvarez K, Mascaro-Blanco A, Brimberg L, Frenkel D, Cunningham MW, Joel D. 2014. Behavioral and neural effects of intra-striatal infusion of anti-streptococcal antibodies in rats. Brain Behav Immun 38:249–262 10.1016/j.bbi.2014.02.009. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Lotan D, Cunningham M, Joel D. 2014. Antibiotic treatment attenuates behavioral and neurochemical changes induced by exposure of rats to group A streptococcal antigen. PLoS One 9:e101257 10.1371/journal.pone.0101257. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Kirvan CA, Swedo SE, Kurahara D, Cunningham MW. 2006. Streptococcal mimicry and antibody-mediated cell signaling in the pathogenesis of Sydenham’s chorea. Autoimmunity 39:21–29 10.1080/08916930500484757. [PubMed] [DOI] [PubMed] [Google Scholar]
- 196.Singer HS, Mascaro-Blanco A, Alvarez K, Morris-Berry C, Kawikova I, Ben-Pazi H, Thompson CB, Ali SF, Kaplan EL, Cunningham MW. 2015. Neuronal antibody biomarkers for Sydenham’s chorea identify a new group of children with chronic recurrent episodic acute exacerbations of tic and obsessive compulsive symptoms following a streptococcal infection. PLoS One 10:e0120499 10.1371/journal.pone.0120499. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Dale RC. 2003. Autoimmunity and the basal ganglia: new insights into old diseases. QJM 96:183–191 10.1093/qjmed/hcg026. [PubMed] [DOI] [PubMed] [Google Scholar]
- 198.Dale RC, Merheb V, Pillai S, Wang D, Cantrill L, Murphy TK, Ben-Pazi H, Varadkar S, Aumann TD, Horne MK, Church AJ, Fath T, Brilot F. 2012. Antibodies to surface dopamine-2 receptor in autoimmune movement and psychiatric disorders. Brain 135:3453–3468 10.1093/brain/aws256. [PubMed] [DOI] [PubMed] [Google Scholar]
- 199.Cutforth T, DeMille MM, Agalliu I, Agalliu D. 2016. CNS autoimmune disease after Streptococcus pyogenes infections: animal models, cellular mechanisms and genetic factors. Future Neurol 11:63–76 10.2217/fnl.16.4. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Dileepan T, Smith ED, Knowland D, Hsu M, Platt M, Bittner-Eddy P, Cohen B, Southern P, Latimer E, Harley E, Agalliu D, Cleary PP. 2016. Group A Streptococcus intranasal infection promotes CNS infiltration by streptococcal-specific Th17 cells. J Clin Invest 126:303–317 10.1172/JCI80792. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Manjula BN, Acharya AS, Mische SM, Fairwell T, Fischetti VA. 1984. The complete amino acid sequence of a biologically active 197-residue fragment of M protein isolated from type 5 group A streptococci. J Biol Chem 259:3686–3693. [PubMed] [Google Scholar]
- 202.Miller L, Gray L, Beachey E, Kehoe M. 1988. Antigenic variation among group A streptococcal M proteins. Nucleotide sequence of the serotype 5 M protein gene and its relationship with genes encoding types 6 and 24 M proteins. J Biol Chem 263:5668–5673. [PubMed] [Google Scholar]
- 203.Cunningham MW. 1998. Molecular mimicry in autoimmunity and infection, pp85–99. In D.J. LeBlanc, M.S. Lantz, and L.M. Switalski (eds), Microbial Pathogenesis: Current and emerging issues. Proceedings of the 2nd Annual Indiana Conference, Indiana University School of Dentistry, Indianapolis, IN. [Google Scholar]
- 204.Dell A, Antone SM, Gauntt CJ, Crossley CA, Clark WA, Cunningham MW. 1991. Autoimmune determinants of rheumatic carditis: localization of epitopes in human cardiac myosin. Eur Heart J 12(Suppl D):158–162 10.1093/eurheartj/12.suppl_D.158. [PubMed] [DOI] [PubMed] [Google Scholar]
- 205.Cox CJ, Zuccolo AJ, Edwards EV, Mascaro-Blanco A, Alvarez K, Stoner J, Chang K, Cunningham MW. 2015. Antineuronal antibodies in a heterogeneous group of youth and young adults with tics and obsessive-compulsive disorder. J Child Adolesc Psychopharmacol 25:76–85 10.1089/cap.2014.0048. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]