

Abstract
Complement activation plays an important role in pharmacokinetic and performance of intravenously administered nanomedicines. Significant efforts have been directed toward engineering of nanosurfaces with low complement activation, but due to promiscuity of complement factors and redundancy of pathways, it is still a major challenge. Cell membrane-anchored Decay Accelerating Factor (DAF, a.k.a. CD55) is an efficient membrane bound complement regulator that inhibits both classical and alternative C3 convertases by accelerating their spontaneous decay. Here we tested the effect of various short consensus repeats (SCRs, “sushi” domains) of human CD55 on nanoparticle-mediated complement activation in human sera and plasma. Structural modeling suggested that SCR-2, SCR-3 and SCR-4 are critical for binding to the alternative pathway C3bBb convertase, whereas SCR-1 is dispensable. Various domains were expressed in E.coli and purified by an affinity column. SCRs were added to lepirudin plasma or sera from different healthy subjects, to monitor nanoparticle-mediated complement activation as well as C3 opsonization. Using superparamagnetic iron oxide nanoworms (SPIO NWs), we found that SCR-2-3-4 was the most effective inhibitor (IC50 ~0.24 µM for C3 opsonization in sera), followed by SCR-1-2-3-4 (IC50 ~0.6 µM), whereas shorter domains (SCR-3, SCR-2-3, SCR-3-4) were ineffective. SCR-2-3-4 also inhibited C5a generation (IC50 ~0.16 µM in sera). In addition to SPIO NWs, SCR-2-3-4 effectively inhibited C3 opsonisation and C5a production by clinically approved nanoparticles (Feraheme, LipoDox and Onivyde). SCR-2-3-4 inhibited both lectin and alternative pathway activation by nanoparticles. When added to lepirudin-anticoagulated blood from healthy donors, it significantly reduced the uptake of SPIO NWs by neutrophils and monocytes. These results suggest that soluble domains of membrane bound complement inhibitors are potential candidates for preventing nanomedicine- mediated complement activation in human subjects.
Keywords: liposome, SPIO, complement, inhibitor, leukocyte
Graphical Abstract

INTRODUCTION
Complement activation presents a significant challenge for clinical development of nanomedicines. The exposure of a nanosurface to serum or plasma may result in a rapid (within minutes) generation of C3 and C5 convertases that promote opsonization events (e.g., through covalent attachment of C3b) and anaphylatoxins (e.g., C4a, C3a and C5a) release [1]. C3b and its further cleavage products iC3b, C3dg, C3d are recognized by variety of complement receptors, leading to a robust uptake by neutrophils, monocytes, eosinophils, lymphocytes, and resident tissue macrophages [2–4]. Mechanisms of complement activation and evolvement are complex, but in general they begin with deposition of antibodies, C1q, mannose-binding lectin or nascent reactive C3 [C3b or meta-stable hydrated C3 (H2O)], on a foreign surface [5]. Recently we demonstrated that the key steps such as C3 deposition and convertase assembly on many nanoparticles and nanomedicines are far more complex than thought and efficient complement activation may require the presence of biomolecular corona and deposition of natural antibodies on the latter [6, 7]. One of the conclusions was that preventing biomolecular corona and immunoglobulin absorption could be an effective way to prevent complement activation. Unfortunately, no straightforward strategies exist to completely block deposition of blood factors on nanoparticles, although dense brush coating of polyethylene glycol (PEG) does appear to exclude some of the blood proteins and lipoproteins from the biomolecule corona [8]. Indeed, as exemplified by PEGylated liposomal doxorubicin (Doxil and follow-on products such as LipoDox), there are fewer C3 molecules deposited per vesicle compared with a minimally PEGylated liposomal nanomedicine (Onivyde) or dextran-stabilized superparamagnetic iron oxide nanoworms (SPIO NW) [9]. Nevertheless, Doxil and other PEGylated nanoparticles still show potent complement activation in mice and humans [10, 11], with potentially deleterious effects such as anaphylatoxin generation and inflammation-triggered tumor progression [12–14].
In view of the daunting challenges in preventing complement activation through nanosurface engineering, we reasoned that complement inhibitors could be a viable strategy to block nanomedicine- induced complement activation, at least at the initial stage of bolus intravenous injection. Complement therapeutics is a highly active field in pharmaceutical development, due to the ubiquitous role of complement in many diseases, and abundance of druggable proteolytic targets in the cascade [15, 16]. Several small molecules inhibitors and antibodies have been developed to block downstream steps, including cleavage of C5 into C5a and C5b by the monoclonal antibody eculizimab [17] and binding of C5a to its receptor by small molecules [18]. Equally important is to prevent complement C3 opsonization, which is the upstream step of the complement cascade and one of the main triggers of nanoparticle clearance, and this has been achieved with synthetic C3 small inhibitors such as Compstatin [19, 20]. However, there are a number of naturally occurring complement regulators and inhibitors. Indeed, complement control proteins (CCPs) are well characterized regulators expressed on the surface of most blood cell types and erythrocytes as well as in the fluid phase, that inhibit different stages of complement activation [21]. CD55 (DAF) is a glycosylphosphatidylinositol (GPI)-anchored, and CD35 (complement receptor 1 a.k.a. CR1) a transmembrane protein present on the surface of blood cells, including erythrocytes [22]. These proteins work in tandem with plasma factor H in limiting spontaneous complement activation (tick-over) both in the fluid phase and on the cell surface. CCPs consist of multiple domains called short consensus repeats (SCRs) of about 60–70 amino acids each. Some of the SCRs (SCR-1-2-3 of CR1, SCR-2-3-4 of CD55 and SCR-1-2-3-4 of FH) directly bind to the alternative convertase C3bB and induce irreversible disassembly (decay accelerating activity or DAA) [21], whereas others SCRs of CD35 and FH bind factor I resulting in irreversible inactivation of C3b through cleavage into iC3b, C3dg and C3d (cofactor activity) [23, 24]. In addition, SCRs 2 and 3 of CD55 has appreciable DAA against the classical pathway convertase [25, 26], which potentially can block complement activation via both the lectin and the classical pathways. We hypothesized that SCR-2-3-4 of CD55 would be a minimal inhibitory unit that contains all the necessary functions to inhibit complement deposition on nanoparticle surface. Although DAF has been cloned and characterized earlier [26–30], to the best of our knowledge there are no reports on the use of DAF in prevention of complement activation by nanoparticles. Since the applicability of animal models to predicting complement activation by nanomedicines in humans is not a settled matter [31], we tested different SCRs derived from human CD55 in vitro in sera, plasma and blood of healthy human donors. We demonstrate that soluble SCR-2-3-4 efficiently blocks complement C3 opsonization and C5a release in middle to high nanomolar range, using various preclinical and clinical nanoparticles. In addition, SCR-2-3-4 blocks complement mediated immune cell uptake of nanoparticles. While the development of clinically viable inhibitors of nanomedicine- induced complement activation is still a long way ahead, we hypothesize that soluble SCRs may be used to inhibit complement activation by nanomedicines in patients.
MATERIALS AND METHODS
Materials
Linear dextran (15–25 kDa molecular weight) and iron salts were purchased from Sigma-Aldrich (Saint Louis, MO). Purified C3b, iC3b and C1 inhibitor were from Complement Technology (Tyler, TX). Soluble CD55 was purchased from Sino Biological (Wayne, PA). cP40 (compstatin) was generously provided by Dr. John Lambris, University of Pennsylvania. cDNA for soluble CD55 (GenBank ID: AAP36604.1, residues 1 to 382) was optimized for bacterial expression and synthesized by Epoch Life Sciences (Missouri City, TX). All proteins were aliquoted at 1 mg/mL and stored in −80°C. Every protein was exposed to no more than 2 freeze thaw cycles. Mouse anti-human C3 was from Quidel Corporation (San Diego, CA). Secondary goat anti mouse IgG labelled with IRDye 800CW was from Li-COR Biosciences (Lincoln, NE). Feraheme (lot 10021802 and 10051302) was generously provided by Dr. Natalie Serkova, Department of Radiology UC Denver. Onivyde (lot 1501279A) and LipoDox (lots 500592 and 500546) were provided as sterile leftovers after administration to patients (free of charge) by the UC Denver Cancer Center Pharmacy. All liposomes were stored in original sterile vials at 4°C prior to use. Anonymous human sera, plasma and blood from healthy donors (males and females, average age 47 ± 16 years) were obtained from the University of Colorado Blood Donation Center using “no anticoagulant” Z vacutainer tubes (BD) for sera, or Multiplate® Hirudin (lepirudin) blood tubes (DiaPharma) for plasma and blood, respectively. Sera were processed as described by us previously [9] and stored in frozen aliquots at −80 ºC. Plasma was obtained by centrifugation of lepirudin tubes at 5,000g for 20 min at room temperature immediately after the collection and stored in frozen aliquots at −80 °C.
Modeling of complement proteins
Crystal structure coordinates for the C3bBb convertase (PDB ID: 2WII, 2WIN) [32], Factor H and C3b complex (PDB ID: 2WII), CD55 (PDB ID: 1OJV) and CD35 (PDB ID: 1GKG) were downloaded from the Protein Data Bank. Protein structures were prepared, measured, and visualized using Biovia Discovery Studio 4.5 (Biovia, Inc., San Diego, CA).
Cloning and expression
Full length human CD55 was modified via PCR to add an EcoRI restriction site and cysteine to the 5ʹ terminus, and to add a GALT-6His tag and NotI restriction site to the 3ʹ terminus, for each SCR construct. The primers used were as follows:
CATAGCGAATTCATGTGCCGTAGCTGCGAGGTGCCAAC-Forward for SCR-2-3 and SCR-2-3-4;
CATAGCGAATTCATGTGCAAATCATGCCCTAATCCGGGAG-Forward for SCR-3 and SCR-3-4;
CCTCGAGCGGCCGCTCAATGGTGGTGGTGATGATGTGTAAGGGCACCTTCTCT GCACTCTGGCAACGG-Reverse for SCR-3 and SCR-2-3;
CCTCGAGCGGCCGCTCAATGGTGGTGGTGATGATGTGTAAGGGCACCTTTTCC TCTGCATTCAGGTGGTGGG-Reverse for SCR-2-3 and SCR-2-3-4.
All constructs were then restriction digested and ligated into pET21a+ plasmid (Novagen) downstream of the T7 promoter using T4 ligase (New England Biolabs), and verified using nucleotide sequence analysis. All constructs were trans formed into OneShot BL21(DE3) E. Coli(Invitrogen) for subsequent expression. Expression was performed by growing an overnight 100 mL starter culture of transformed bacteria in Modified Terrific Broth (Fisher)-Ampicillin at 37°C. The following morning, the starter culture was added to 900 mL of MTB-Amp and grown for 1 hour at 37 °C to an OD600 ~ 0.6, and induced by adding 1mL of 0.5 M IPTG (Invitrogen), to a final concentration of 0.5 mM IPTG. The culture was then grown at 37 °C for 6 h and pelleted at 8,000g for 15 min, after which the pellet was frozen for later processing.
Isolation of inclusion bodies and refolding was performed in a process similar to the one described before [28]. The his-tagged protein was purified by affinity chromatography using N i2+ NTA agarose (Invitrogen) and desalted in a 7 kDa Zeba column (Thermo Fisher) to remove the remaining imidazole. The concentration of the protein was determined by running reducing SDS-PAGE along with bovine serum albumin standard protein curve. Proteins were aliquoted and stored at −80°C.
Complement activation assays
for the inhibition curves, SCR construct comparisons, and complement pathway inhibition experiments C3 on particles and C5a in fluid phase was determined as described below. Human serum or plasma (20 μL) was mixed with protein samples (10 µL), and then nanoparticles (10 µL at 1mg/mL Fe or drug) were added and mixed. The tubes were placed in a 37 °C water bath and incubated for 30 min. After the incubation, the particles were pelleted at 450,000–550,000g in a Beckman Optima ultracentrifuge for 5 min. 30 µL of the supernatant was collected, mixed with 10 µL of 100 mM EDTA and stored at 4 °C for C5a ELISA analysis. The pellet was resuspended in 200 µL of Dulbecco Phosphate Buffered Saline (DPBS) and sonicated briefly in a bath sonicator to break up the pellets, then ultracentrifuged for 5 min again. This washing step was repeated 3 times, followed by a final wash in 500 µL of DPBS. After the final wash, the supernatant was removed and the pellet was resuspended in 30 µL DPBS and sonicated. Particles in 2 µL triplicates were blotted on nitrocellulose membrane (0.45 μm, Bio-Rad). After drying, the membrane was blocked for 1 h at room temperature in 5% (v/v) milk/PBS-Tween 0.1% (v/v), and then probed with goat anti-human C3 antibody at a concentration of 1:1000 for 1 h at room temperature. The membrane was then washed for 15 min in PBS-Tween 0.1% (v/v), and then probed with anti-goat secondary antibody. This was followed by a wash in PBS-Tween 0.1% (v/v) and the membrane was scanned using a LICOR Odyssey scanner. SDS-PAGE was run using the Mini-Protean Tetra System (Bio-Rad) with Mini-Protean TGX 4–20% gels (Bio-Rad) and preformulated 10X Tris/Glycine/SDS and Tris/Glycine buffers (Bio-Rad). Western blot transfers were performed with Tris/10%Glycine/20%Methanol transfer buffer pre-chilled to 4 °C, using 65V for 90 min in a cold room. Western blotting was performed using the same protocol as for dot blotting. C5a ELISA was performed using the DuoSet C5a sandwich ELISA kit (R&D Systems) per manufacturer’s instructions.
Nanoparticle uptake
Lepirudin anticoagulated donor blood was obtained fresh and processed within 1 h. EDTA-anticoagulated blood was also collected from the same donor as a negative control. Blood (500 µL) was mixed with either PBS or SCR-2-3-4 at a final concentration of 1.6 µM. SPIO NWs at a final concentration of 0.2 mg Fe/mL was then added to the blood samples, and incubated at 37 °C in a Thermomixer (Eppendorf) at 600 rpm for 1 h. During the incubation, MACS MIDI magnetic columns (Miltenyi Biotec) were equilibrated with 2 column volumes of PBS, and then coated with 1 column volume of 20% (v/v) fetal bovine serum in PBS. After the incubation, each blood sample was applied to a magnetic column and allowed to flow through, and then each column was washed with 10 column volumes of PBS. Cells were eluted using 1ml of 1% BSA/PBS, and then 200 µL of eluted cells from each sample was applied to a slide using a Cytospin 3 centrifuge. The nuclei were then fixed with 4% v/v formalin, stained with Hoechst and analyzed by fluorescent microscopy. The remaining 800 µL of eluted cells further diluted 2.5 times and analyzed by forward and side scattering using Guava easyCyte™ flow cytometer (cell counting mode, 35 µL per sample) in order to compare the numbers of magnetic cells between the samples.
RESULTS
Membrane bound CD55 and CD35 and soluble FH consist of short consensus repeats (SCRs, or “sushi” domains) (Fig. 1A). Previous studies suggested that GPI anchor and O-linked glycosylation are not required for the complement inhibition function of DAF [25, 26]. According to the structural modeling (Fig. 1B), DAF forms a tight complex with the alternative convertase C3bBb, thereby accelerating the decay of the convertase through irreversible dissociation of C3b and Bb. At the same time, only SCR-2-3-4 appear to form the inhibiting surface, consistent with studies showing that SCR1 and N-glycosylation site between SCR-1 and SCR-2 are not required for DAA and are actually needed for enterovirus and lymphocyte binding [29, 33]. In view of the dispensable glycosylation, SCR-2-3-4 of DAF appears to be suitable for bacterial expression. Previous studies demonstrated feasibility and functionality of expression of SCR-1-2-3 of CR1 and DAF in E. coli. [28, 34]. At the same time, it is still not clear whether individual SCRs (e.g., SCR-3, Fig 1B) would be sufficient to inhibit C3 deposition on nanoparticles. We expressed His-tagged SCR-3, SCR-2-3, SCR-2-3-4 and SCR-3-4 in E. coli and purified them by nickel chromatography (Fig. 1C-D and Methods). In order to test whether SCRs could inhibit C3 opsonization, we used 110 nm diameter SPIO NWs. These particles are excellent magnetic resonance imaging contrast agents [35], but show potent complement activation primarily via lectin and alternative pathways in human sera and plasma [36]. Intravenously injected SPIO NWs show very rapid clearance from the blood in mice with the half-life of few minutes [35]. Nanoparticles were incubated in lepirudin anticoagulated plasma obtained from healthy volunteers, with or without addition of inhibitors, washed by ultracentrifugation and the amount of bound C3 was detected with dot blot and/or Western blotting (Fig. 2A). The addition of SCR-2-3-4 to 3 plasma samples at 810 nM (18.6 µg/mL) led to between 71 and 99% inhibition of C3 deposition on SPIO NWs. As positive control, EDTA at 25 mM completely inhibited C3 binding in all 3 plasma samples. At the same time, none of the single and double SCR constructs showed significant inhibition of C3 deposition, suggesting that SCRs-2, 3, and 4 are required for nanoparticle-mediated complement activation. Similar outcomes were reported in cell lysis inhibition studies [27, 37].
Fig. 1. Short consensus repeats (SCRs) that we re used in the study:

A) Schematic representation of membrane-bound complement control proteins (CCPs) CD55 and CD35, and plasma factor H. CCPs consist of multiple short consensus repeats (sushi domains). SCRs primarily responsible for decay accelerating activity are shown; B) structural model of complement convertase C3bBb forming a complex with CD55, leading to the decay; C) open view of SCR-1-2-3-4 (shown in different colors, from top to bottom) of DAF interacting with C3b (green). SCR-1 does not interact with C3b and is not needed for decay activity; D) different constructs containing single or multiple SCRs and His-tags. Soluble CD55 was obtained commercially, all other constructs were cloned and expressed as described in Methods; E) representative SDS-PAGE of the purified constructs.
Fig. 2. C3 opsonization of SPIO NWs is inhibited by SCR-2-3-4.

A) a schematic representation of procedures for measuring C3 deposition on SPIO NWs; B) SPIO NWs were incubated in human plasma and washed of excess proteins. Bound C3 was analyzed with SDS-PAGE. Lane labels: 1) C3b; 2) iC3b; 3) no inhibitor; 4) 20 mM EDTA; 5) 81.5 nM SCR-2-3-4; 6) 410 nM SCR-2-3-4; 7) 815 nM SCR-2-3-4; 8) 4100 nM SCR-2-3-4. The experiment was done in 3 different plasma donors [legends show gender (f/m) and age (number) of each donor]. Individual data points of 3 technical replicates for each donor are shown.
We next analyzed the level of C3 deposition and the C3 cleavage pattern with Western blotting. As shown in Fig. 2B, C3 deposited on nanoparticles was mostly in the iC3b form, but there was also non-cleaved α-chain (120kDa), which possibly corresponds to metastable C3 (C3(H2O)) that can be deposited on a foreign surface [38, 39]. Addition of increasing amounts of SCR-2-3-4 to plasma led to gradual disappearance of C3 bands, including the non-cleaved α-chain. The reason why the addition of SCRs led to the disappearance of non-cleaved C3 is not clear, in view of other studies that did not show decrease in C3(H2O) deposition on liposomes after complement inhibition [38], and warrants future investigation. However, it is plausible that nanoparticle bound C3(H2O) undergoes a conformational change thereby exposing epitopes that allow SCR-2-3-4 binding. Accordingly, on SCR-2-3-4 binding, C3(H2O) may be released from the nanoparticle surface.
After confirming a robust activity of SCR-2-3-4, and eliminating other SCR constructs that did not show any activity, we measured inhibitory concentrations (IC50) of SCR-2-3-4 following addition of SPIO NWs to sera and matched plasma from 4 different donors. As shown in Fig. 3A, 3C, and Table 1, there was a dose dependent inhibition of C3 deposition on NWs (average of 4 donors: IC50 of 243 ± 62 nM and 287 ± 114 nM for sera and plasma samples, respectively). The inhibition was sometimes incomplete even at high plasma concentrations of SCR-2-3-4 (average of 12.8 ± 9% of remaining C3 at 4.1 µM for the four donors, compared with an average of 3.4 ± 2.6% of remaining C3 at 25 mM EDTA, data from Table 1). We also tested the effect of SCR2-3-4 on C5a generation, which is the downstream product of complement activation, in the same sera and plasma samples. According to Fig. 3B, 3D, and Table 1, there was a dose dependent inhibition (average of 4 donors: IC50 of 156 ± 92 nM and 155 ± 84 nM for sera and plasma samples, respectively, data from Table 1). Next, we measured IC50 of C3 inhibition using commercially available full length soluble CD55 (SCR-1-2-3-4 plus C-terminal spacer, Fig. 1C), in 3 plasma samples. This construct showed lower activity than SCR-2-3-4 (average of 3 donors: 603 ± 107 nM, Fig. 3E and Table 1). The reason for the lower activity is not clear, but could be due to the limited accessibility of the bulkier sCD55 to complement convertases on the nanoparticle surface. Lastly, we determined IC50 of SCR-2-3-4 to that of a small molecule C3 inhibitor, 1.78 kDa peptide compstatin (cP40), which is currently in clinical development [19]. CP40 binds to C3 molecules, preventing their cleavage by convertases. CP40 showed an average IC50 of 5.3 µM, Fig 3F and Table 1) in two plasma samples, but the inhibition level at high concentrations was more complete (less than 1% of residual C3 at 8.7 µM of cP40, Table 1). The direct comparison between these two inhibitors is not straightforward, because of different mechanisms of action.
Fig. 3. Log inhibition curves (IC50) of C3 opsonization and C5a release in sera and plasma by SPIO NWs.
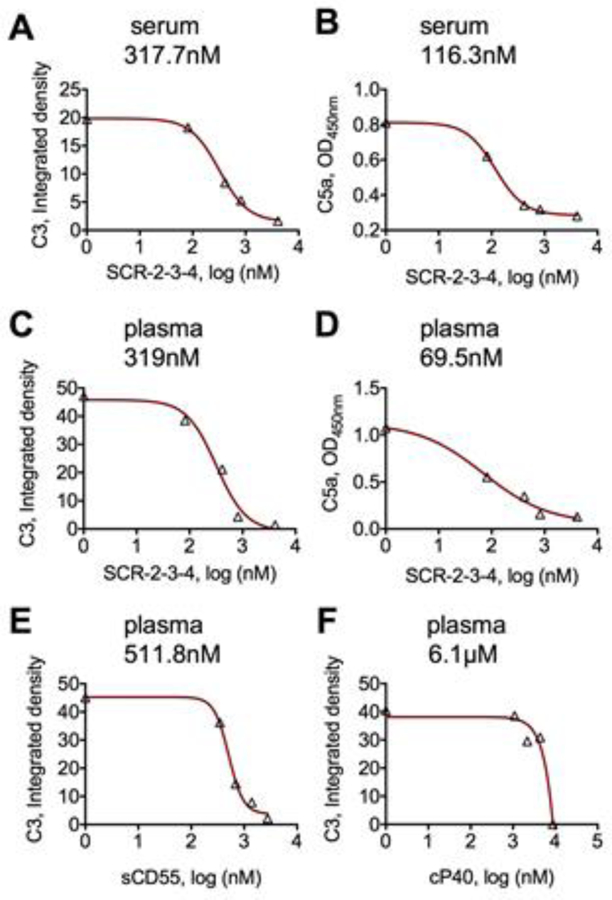
SPIO NWs were incubated in sera and plasma in the presence or absence of increasing concentration of inhibitors as described in Methods. A) C3 deposition, SCR-2-3-4 in serum; B) C5a levels, SCR-2-3-4 in serum; C) C3 deposition, SCR-2-3-4 in plasma; D) C5a levels, SCR-2-3-4 in plasma; E) C3 deposition, soluble CD55 in serum; F) C3 deposition, SCR-2-3-4 and cP40 (compstatin) in plasma. Each graph shows a corresponding IC50 value above. Typical inhibition curve is shown; multiple donors are compiled in Table 1. All graphs show raw values on Y-axis. Each point is the average of 3 technical replicates.
Table 1. Inhibition of complement activation by SPIO NWs in sera or plasma from different healthy donors.
Each inhibitor was tested with 2–4 sera/plasma.
| Medium/construct | C3 IC50, nM | C5a IC50, nM | C3 at 4.1 µM* (% of control) | C3 at 25 mM EDTA (% of control) |
|---|---|---|---|---|
| Serum/SCR-2-3-4 | 271.0 | n/m | 26.9 | 0.1 |
| 190.1 | 91.2 | 13.4 | 0.0 | |
| 317.7 | 116.3 | 0.1 | 0.0 | |
| 191.7 | 261.4 | 0.2 | 0.0 | |
| Plasma/SCR-2-3-4 | 167.2 | 157.3 | 24.0 | 7.2 |
| 430.1 | 236.5 | 15.5 | 2.4 | |
| 319.2 | 69.6 | 3.0 | 1.6 | |
| 231.7 | n/m | 8.8 | 2.3 | |
| Plasma/sCD55 | 578.0 | n/m | 3,4 | 1.1 |
| 721.5 | n/m | 5.7 | 0.0 | |
| 511.8 | n/m | 5.6 | 2.8 | |
| Plasma/cP40 | 6134.0 | n/m | <1% | 0.0 |
| 4468.0 | n/m | <1% | 0.0 |
Concentration was 2.7 µM for sCD55 and 8.74 µM for cP40. n/m = either not measured or cannot be determined by curve fitting.
Next, we measured the ability of SCR-2-3-4 to block complement activation by clinically approved nanomedicines: 30 nm carboxymethyl dextran iron oxide Feraheme, 90 nm PEGylated liposomal doxorubicin LipoDox and 110 nm minimally-PEGylated liposomal irinotecan Onivyde in 3 plasma samples (Fig. 4A). The characterization of these particles has been reported before [7, 9]. Feraheme and Doxil circulate for a few days in humans [40, 41], whereas Onivyde is rapidly cleared and metabolized by macrophages [42]. As shown in Figure 4B-G (representative curves), and summarized in Table 2, SCR-2-3-4 inhibited C3 deposition and C5a generation by all the particles in 3 plasma samples. The average IC50 values of C3 deposition and C5a release for Feraheme were 165 ± 167 nM and 172 ± 171 nM, respectively. The corresponding the IC50 values for LipoDox, were 396 ± 52 nM and 383 ± 5 nM, respectively, and for Onivyde the IC50 values of C3 and C5a were 204 ± 101 nM and 107 ± 18 nM, respectively (Table 2). Similar to SPIO NWs, there was some residual C3 left on all nanoparticles after incubation with 4.1 µM of SCR-2-3-4 (Table 2).
Fig. 4. Log inhibition curves (IC50) of C3 opsonization and C5a release in plasma by clinically approved nanomedicines.

A) Nanoparticles used were Feraheme, LipoDox and Onivyde. Nanoparticles were incubated in plasma in the presence or absence of increasing concentration of SCR-2-3-4 as described in Methods. B) C3 deposition, Feraheme; B) C5a levels, Feraheme; C) C3 deposition, LipoDox; D) C5a levels, LipoDox; E) C3 deposition, Onivyde; F) C5a levels, Onivyde. Each graph shows a corresponding IC50 value above. Representative inhibition curve is shown for one donor; multiple donors are compiled in Table 2. All graphs show raw values on Y-axis. Each point is the average of 3 technical replicates.
Table 2. Inhibition of complement activation by clinically approved nanomedicines with SCR-2-3-4 in plasma of different healthy donors.
Each nanoparticle type was tested in 3 different plasma donors.
| Particle | C3 IC50, nM | C5a IC50, nM | C3 at 4.1 µM (% of control) | C3 at 25 mM EDTA (% of control) |
|---|---|---|---|---|
| Feraheme | 64.3 | 48.6 | 0.0 | 0.0 |
| 72.9 | 366.9 | 0.1 | 0.0 | |
| 357.1 | 101.7 | 18.4 | 0.9 | |
| LipoDox | 399.6 | 377.1 | 0.9 | 0.0 |
| 343.4 | 385.8 | 2.5 | 0.0 | |
| 446.9 | 385.8 | 11.5 | 2.0 | |
| Onivyde | 239.6 | 95.0 | 0.5 | 0.0 |
| 90.2 | 96.7 | 3.3 | 0.0 | |
| 282.7 | 127.7 | 1.1 | 0.0 |
Nanoparticles activate complement via different pathways [1]. Earlier, PEGylated liposomes and iron oxide nanoparticles have been shown to trigger lectin and alternative pathways in human sera and plasma [43, 44], whereas some nanoparticles coated with alternative polymers have triggered the classical pathway of the complement system on direct binding of C1q [2]. In order to determine which pathways in plasma were inhibited by SCR-2-3-4, we added these 4 nanoparticle types to normal plasma from healthy individuals and measured C3 deposition in the absence or presence of 125 µg/mL C1INH (classical pathway (CP) inhibitor), 13 mM EGTA/Mg2+ (CP and LP inhibitor) or 4.1 µM SCR-2-3-4. According to Fig. 5, C1INH did not inhibit complement activation by any of the tested nanoparticles. On the other hand, nanoparticle0mediated complement activation was inhibited ~50% on EGTA/Mg2+ treatment. Addition of SCR-2-3-4 to C1INH reduced C3 deposition on nanoparticles by 90%, and addition to EGTA/Mg2+ incubations reduced C3 deposition by over 99%. These results suggest that all the tested nanoparticles activate complement via LP and AP without involvement of the CP, and that SCR-2-3-4 inhibits the AP convertase more efficiently than convertases assembled from calcium-dependent pathways.
Fig. 5. Mechanisms of C3 deposition inhibition by SCR-2-3-4:

A) a general (simplified) scheme of C3 deposition on pathogen surface. The deposition starts with generation of C3b through lectin pathway (LP) or classical pathway (CP), or with spontaneous addition of C3/C3b through various mechanisms.[59] Following the initial deposition step, there is an amplification step via AP. CP activation can be inhibited by C1 inhibitor, and LP and CP activation can be inhibited by EGTA/Mg2+; B) SPIO NWs, Feraheme, LipoDox and Onivyde were incubated in plasma in the presence or absence of SCR-2-3-4 (4.1 µM), EGTA/Mg2+, or 125 µg/mL C1 inhibitor as described in Methods. C1INH did not block C3 deposition, whereas EGTA/Mg2+ decreased deposition by 50%. The data demonstrate that all the particles activate complement via the AP and LP, but not the CP. SCR-2-3-4 appears to inhibit C3 deposition via both AP and LP. Individual data points of 3 technical replicates for each donor are shown.
Complement opsonization can prime nanomedicines for uptake by blood leukocytes through complement receptors. SPIO NWs are very convenient for study of the leukocyte uptake, since cells that take up NWs become magnetically labeled and can be easily isolated and counted [3]. In order to test the ability of SCR-2-3-4 to block the uptake by leukocytes, SPIO NWs were incubated with whole lepirudin blood treated with or without SCR-2-3-4 (0.2 mg/mL Fe; 30 µg/mL SCR2-3-4). Magnetically labeled cells were isolated over MACS MIDI columns and applied to a slide. Nuclei were visualized with Hoechst stain and counted (Fig 6A). As shown in Fig. 6, for all 6 donors (5 females and 1 male) there were a large number of magnetically labeled leukocytes eluted from MACS column, and these were predominantly neutrophils (high magnification images in Supplementary Figure 1). Addition of SCR-2-3-4 dramatically decreased the uptake in all tested blood samples (Fig. 6B). SCR-2-3-4 almost completely inhibited C3 opsonization in all tested blood samples. Notably, the levels of uptake were variable and did not correlate with the level of opsonization. Thus, sample F53 and F42 showed very different levels of uptake (Fig 6B), but a similar level of C3 opsonization (Fig 6C). There were some leukocytes left after treatment with SCR-2-3-4, suggesting that some of the uptake was complement-independent. We further analyzed the eluted cells from samples F25 and F58 by flow cytometry. Side scattering- forward scattering dot-plot shows that majority of cells were granulocytes and minority was monocytes and lymphocytes, and SCR-2-3-4 decreased the number of leukocytes by 90% and 96%, respectively (Fig. 6D).
Fig. 6. Uptake of SPIO NWs by leukocytes is inhibited by SCR-2-3-4.

A) SPIO NWs were incubated with lepirudin blood in presence of 30 µg/mL (1.3µM) of SCR-2-3-4; B) Magnetically labeled cells were concentrated on a slide and stained with Hoechst. High-magnification images showing nuclear morphology are in Supplement. Most of the cells were neutrophils. The uptake was inhibited by SCR-2-3-4. Letters (f/m) and numbers are gender and age of human blood donors; C) C3 deposition in plasma of donors used in the study, and inhibition of the deposition by SCR-2-3-4. Individual data points of 3 technical replicates for each donor are shown; D) magnetic cells from F38 and F53 samples were eluted, diluted 10 times and analyzed with flow cytometry (forward scattering- light scattering) using the same volume of eluted material (20µL) for each sample. Regions of interest are leukocytes (right) and RBCs (lower left). Boxed in red are absolute numbers of leukocytes in 20 µL. SCR-2-3-4 inhibits the binding and uptake by ~90%, but does not affect the number of magnetically labeled RBCs. EDTA effect was variable.
DISCUSSION
In this study we used complement therapeutics approach in order to inhibit the initial steps in complement activation by preclinical and clinical nanomedicines. Animal models for testing the hypothesis of whether complement activation by nanomedicines contributes to cardiopulmonary distress are notoriously unreliable, and are either insensitive (mouse) or hypersensitive (pig) [31, 45]. We recently outlined a strategy to inhibit complement activation by nanomedicines and to test the hypothesis on the possible involvement of complement system in infusion reactions [46]. We suggested that one of the ways to test the role of complement in infusion reactions is to conduct a clinical trial in which complement inhibitors are administered systemically along with nanomedicines. In view of the numerous complement inhibitors at different stages of preclinical and clinical development [15], it is imperative to first prescreen the inhibitors in vitro in human sera, plasma and blood, in order to select the best performing inhibitors that can be moved further to preclinical studies in non-human primates and next to the clinical level.
Our results demonstrate that soluble SCR-2-3-4 of DAF efficiently prevents complement C3 opsonization and C5a release by various preclinical and clinical nanoparticles in human sera and plasma. Despite the variability of complement reactivity towards these nanomedicines in general population [9] the inhibition was robust, with IC50 in the mid-nanomolar range. With some nanoparticles (e.g., SPIO NWs and Onivyde) IC50 values for C5a was somehow lower than for C3 opsonization, but for others (e.g., Feraheme and LipoDox) the values were comparable. These observations may be a reflection of quantitative differences in initial C3b deposition [7] and subsequent assembly of C3 convertases (e.g., C3bBb) and C5 convertase (C3bBbC3b) between different nanoparticle types.
We further established that SCR-2-3-4 was approximately two-fold more potent than soluble CD55, and about twenty-fold more potent than compstatin, a peptide that is further along in clinical development. The IC50 value for cP40 was similar to the one reported in the paroxysmal nocturnal haemoglobinuria model [19]. One of the reasons for higher molar IC50 than for SCR-2-3-4 could be the requirement to bind to all C3 molecules, which is present at ~1.5 mg/mL in serum. Other molecules may also be used for inhibition of C3 opsonization, including recently developed inhibitor of factor D [47] and PEGylated version of cP40, which are both undergoing clinical development. The choice of the inhibitor will eventually be determined by a combination of the inhibitor’s pharmacokinetics, immunogenicity, and safety profile. The majority of the complement inhibitors cannot achieve 100% inhibition in vivo [48], likely due to rapid turnover of the complement factors and vigorous complement activation in the diseased state. There is also a concern that systemic and prolonged complement inhibition is associated with infections and weakened innate immune response [16]. We believe that for inhibition of C3 opsonization and C5a release by nanoparticles, the administration of complement inhibitors should be short-term in order to minimize possible complement-mediated adverse events. While the effective dose and safety of the complement inhibitors in combination with nanomedicines in human patients remains to be determined, in our study 30µg/mL effectively inhibited leukocyte uptake and C3 deposition, which translates to ~2mg/kg in an adult. Previous clinical trial with the derivative of CR1 (TT30) that used 3mg/kg intravenously showed good safety and lack of immunogenicity. [49]. It also must be noted that safety and anti- inflammatory properties of mouse variants of complement control proteins have been established in animal models [22, 50–53]. At the same time, if the purpose is to delay complement activation upon nanomedicine infusion, there is probably a lower tolerance threshold for toxicity and efficacy, so that a temporary inhibition by 80– 90% may be adequate to achieve the desired effect.
It is possible that other SCRs, for example those derived from CR1, will show a similar, if not higher activity. For example, soluble CR1 contains both decay accelerating and cofactor activities for all three pathways [23], which could further decrease the IC50. Furthermore, it was shown that targeting the SCRs to the membrane (their physiological site of action) enhances potency and inhibitory effect [28]. Various strategies have been tried to bring SCRs to the activating surface, e.g., by fusion with lipids [28, 50] or with complement receptor 2 [54, 55]. The latter has shown a better potency than soluble SCRs due to its ability to bind to surface deposited C3dg (cleavage product of C3b). Anecdotally, simple precoating of nanoparticles with Factor H also can render some complement inhibitory activity [56]. Lastly, SCRs could be covalently anchored to the surface, which is the ongoing effort in our laboratory. It appears that for covalent nanoparticle modification with SCRs, a better understanding of the structural requirements and function on the nanosurface is needed. However, covalent surface modification may add unnecessary complexity to the nanoparticle design, which could further complicate scalability and decrease the chances for translation [57]. Therefore, the premise of our study was to test the efficiency of SCRs in the soluble form, before conjugating these inhibitors directly to the nanoparticle surface.
In conclusion, this study demonstrated the robust effect of soluble complement inhibitory domains on C3 deposition, C5a release and immune uptake of nanoparticles. Considering the disparities in nanoparticle-mediated complement activation and C3 opsonization processes between animal models and humans [2], and the ongoing debate on the role of complement in nanomedicine-mediated infusion reactions [31, 45], the described inhibitors either in soluble form or in their surface-bound derivatives, may provide a direct approach for assessing the role of complement system in nanomedicine uptake by immune cells and hypersensitivity reactions in human subjects.
Supplementary Material
ACKNOWLEDGEMENTS
The study was supported by the NIH grants EB022040 and CA194058 to DS, and AR51749 to VMH and NKB. Authors thank Dr. Lambris for a gift of cP40 and for advice in writing the paper. SMM acknowledges financial support by International Science and Technology Cooperation of Guangdong Province (reference 2015A050502002) and Guangzhou City (reference 2016201604030050) with RiboBio Co, Ltd., China.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- [1].Moghimi SM, Andersen AJ, Ahmadvand D, Wibroe PP, Andresen TL, Hunter AC, Material properties in complement activation, Adv. Drug Delivery Rev 63 (2011) 1000–1007. [DOI] [PubMed] [Google Scholar]
- [2].Tavano R, Gabrielli L, Lubian E, Fedeli C, Visentin S, Polverino De Laureto P, Arrigoni G, Geffner-Smith A, Chen F, Simberg D, Morgese G, Benetti EM, Wu L, Moghimi SM, Mancin F, Papini E, C1q-mediated complement activation and C3 opsonization trigger recognition of stealth poly(2-methyl-2-oxazoline)-coated silica nanoparticles by human phagocytes, ACS Nano, 12 (2018) 5834–5847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Inturi S, Wang G, Chen F, Banda NK, Holers VM, Wu L, Moghimi SM, Simberg D, Modulatory role of surface coating of superparamagnetic iron oxide nanoworms in complement opsonization and leukocyte uptake, ACS Nano 9 (2015) 10758–10768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Dobrovolskaia MA, Aggarwal P, Hall JB, McNeil SE, Preclinical studies to understand nanoparticle interaction with the immune system and its potential effects on nanoparticle biodistribution, Mol. Pharm 5 (2008) 487–495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Ricklin D, Hajishengallis G, Yang K, Lambris JD, Complement: a key system for immune surveillance and homeostasis, Nat. Immunol 11 (2010) 785–797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Chen F, Wang G, Griffin JI, Brenneman B, Banda NK, Holers VM, Backos DS, Wu L, Moghimi SM, Simberg D, Complement proteins bind to nanoparticle protein corona and undergo dynamic exchange in vivo, Nat. Nanotechnol 12 (2017) 387–393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Vu VP, Gifford GB, Chen F, Benasutti H, Wang G, Groman EV, Scheinman R, Saba L, Moghimi SM, Simberg D, Immunoglobulin deposition on biomolecule corona determines complement opsonization efficiency of preclinical and clinical nanoparticles, Nat. Nanotechnol 14 (2019) 260–268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Karmali PP, Simberg D, Interactions of nanoparticles with plasma proteins: implication on clearance and toxicity of drug delivery systems, Expert Opin. Drug Deliv 8 (2011) 343–357. [DOI] [PubMed] [Google Scholar]
- [9].Benasutti H, Wang G, Vu VP, Scheinman R, Groman E, Saba L, Simberg D, Variability of Complement Response toward Preclinical and Clinical Nanocarriers in the General Population, Bioconjug. Chem 28 (2017) 2747–2755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Szebeni J, Baranyi L, Savay S, Milosevits J, Bodo M, Bunger R, Alving CR, The interaction of liposomes with the complement system: in vitro and in vivo assays, Method. Enzymol 373 (2003) 136–154. [DOI] [PubMed] [Google Scholar]
- [11].Chanan-Khan A, Szebeni J, Savay S, Liebes L, Rafique NM, Alving CR, Muggia FM, Complement activation following first exposure to pegylated liposomal doxorubicin (Doxil): possible role in hypersensitivity reactions, Ann. Oncol 14 (2003) 1430–1437. [DOI] [PubMed] [Google Scholar]
- [12].Petersen GH, Alzghari SK, Chee W, Sankari SS, La-Beck NM, Meta-analysis of clinical and preclinical studies comparing the anticancer efficacy of liposomal versus conventional non-liposomal doxorubicin, J. Control. Release, 232 (2016) 255–264. [DOI] [PubMed] [Google Scholar]
- [13].Gabizon AA, Patil Y, La-Beck NM, New insights and evolving role of pegylated liposomal doxorubicin in cancer therapy, Drug Resist. Updat 29 (2016) 90–106. [DOI] [PubMed] [Google Scholar]
- [14].Moghimi SM, Cancer nanomedicine and the complement system activation paradigm: anaphylaxis and tumour growth, J. Control. Release, 190 (2014) 556–562. [DOI] [PubMed] [Google Scholar]
- [15].Morgan BP, Harris CL, Complement, a target for therapy in inflammatory and degenerative diseases, Nat. Rev. Drug Discov 14 (2015) 857–877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Barnum SR, Therapeutic inhibition of complement: well worth the risk, Trend. Pharmacol. Sci 38 (2017) 503–505. [DOI] [PubMed] [Google Scholar]
- [17].Woodruff TM, Nandakumar KS, Tedesco F, Inhibiting the C5-C5a receptor axis, Mol. Immunol 48 (2011) 1631–1642. [DOI] [PubMed] [Google Scholar]
- [18].Short A, Wong AK, Finch AM, Haaima G, Shiels IA, Fairlie DP, Taylor SM, Effects of a new C5a receptor antagonist on C5a- and endotoxin-induced neutropenia in the rat, Br. J. Pharmacol 126 (1999) 551–554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Risitano AM, Ricklin D, Huang YJ, Reis ES, Chen H, Ricci P, Lin ZE, Pascariello C, Raia M, Sica M, Del Vecchio L, Pane F, Lupu F, Notaro R, Resuello RRG, DeAngelis RA, Lambris JD, Peptide inhibitors of C3 activation as a novel strategy of complement inhibition for the treatment of paroxysmal nocturnal hemoglobinuria, Blood 123 (2014) 2094–2101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Ordóñez-Gutiérrez L, Posado-Fernández A, Ahmadvand D, Lettiero B, Wu L, Antón M, Flores O, Moghimi SM, Wandosell F, ImmunoPEGliposome-mediated reduction of blood and brain amyloid levels in a mouse model of Alzheimer’s disease is restricted to aged animals, Biomaterials 112 (2017) 141–152. [DOI] [PubMed] [Google Scholar]
- [21].Forneris F, Wu J, Xue X, Ricklin D, Lin Z, Sfyroera G, Tzekou A, Volokhina E, Granneman JCM, Hauhart R, Bertram P, Liszewski MK, Atkinson JP, Lambris JD, Gros P, Regulators of complement activity mediate inhibitory mechanisms through a common C3b-binding mode, EMBO J 35 (2016) 1133–1149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Smith GP, Smith RA, Membrane-targeted complement inhibitors, Mol. Immunol 38 (2001) 249–255. [DOI] [PubMed] [Google Scholar]
- [23].Krych M, Clemenza L, Howdeshell D, Hauhart R, Hourcade D, Atkinson JP, Analysis of the functional domains of complement receptor-type-1 (C3b/C4b receptor, CD35) by substitution mutagenesis, J. Biol. Chem 269 (1994) 13273–13278. [PubMed] [Google Scholar]
- [24].Alsenz J, Schulz TF, Lambris JD, Sim RB, Dierich MP, Structural and functional analysis of the complement component factor-H with the use of different enzymes and monoclonal-antibodies to factor-H, Biochem. J 232 (1985) 841–850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Brodbeck WG, Liu D, Sperry J, Mold C, Medof ME, Localization of classical and alternative pathway regulatory activity within the decay-accelerating factor, J. Immunol 156 (1996) 2528–2533. [PubMed] [Google Scholar]
- [26].Brodbeck WG, Kuttner-Kondo L, Mold C, Medof ME, Structure/function studies of human decay-accelerating factor, Immunology 101 (2000) 104–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Coyne KE, Hall SE, Thompson S, Arce MA, Kinoshita T, Fujita T, Anstee DJ, Rosse W, Lublin DM, Mapping of epitopes, glycosylation sites, and complement regulatory domains in human decay accelerating factor, J. Immunol 149 (1992) 2906–2913. [PubMed] [Google Scholar]
- [28].White J, Lukacik P, Esser D, Steward M, Giddings N, Bright JR, Fritchley SJ, Morgan BP, Lea SM, Smith GP, Smith RA, Biological activity, membrane-targeting modification, and crystallization of soluble human decay accelerating factor expressed in E. coli, Protein Sci 13 (2004) 2406–2415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Karnauchow TM, Dawe S, Lublin DM, Dimock K, Short consensus repeat domain 1 of decay-accelerating factor is required for enterovirus 70 binding, J. Virol 72 (1998) 9380–9383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Lukacik P, Roversi P, White J, Esser D, Smith GP, Billington J, Williams PA, Rudd PM, Wormald MR, Harvey DJ, Crispin MD, Radcliffe CM, Dwek RA, Evans DJ, Morgan BP, Smith RA, Lea SM, Complement regulation at the molecular level: the structure of decay-accelerating factor, Proc. Natl. Acad. Sci. U. S. A 101 (2004) 1279–1284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Moghimi SM, Nanomedicine safety in preclinical and clinical development: focus on idiosyncratic injection/infusion reactions, Drug Discov Today 23 (2018) 1034–1042.. [DOI] [PubMed] [Google Scholar]
- [32].Forneris F, Ricklin D, Wu J, Tzekou A, Wallace RS, Lambris JD, Gros P, Structures of C3b in complex with factors B and D give insight into complement convertase formation, Science 330 (2010) 1816–1820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Hamann J, Vogel B, van Schijndel GM, van Lier RA, The seven-span transmembrane receptor CD97 has a cellular ligand (CD55, DAF), J . Exp. Med 184 (1996) 1185–1189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Dodd I, Mossakowska DE, Camilleri P, Haran M, Hensley P, Lawlor EJ, McBay DL, Pindar W, Smith RA, Overexpression in Escherichia coli, folding, purification, and characterization of the first three short consensus repeat modules of human complement receptor type 1, Protein Expr. Purif 6 (1995) 727–736. [DOI] [PubMed] [Google Scholar]
- [35].Wang G, Inturi S, Serkova NJ, Merkulov S, McCrae K, Russek SE, Banda NK, Simberg D, High-relaxivity superparamagnetic iron oxide nanoworms with decreased immune recognition and long-circulating properties, ACS Nano, 8 (2014) 12437–12449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Banda NK, Mehta G, Chao Y, Wang G, Inturi S, Fossati-Jimack L, Botto M, Wu L, Moghimi S, Simberg D, Mechanisms of complement activation by dextran-coated superparamagnetic iron oxide (SPIO) nanoworms in mouse versus human serum, Part. Fibre Toxicol 11 (2014) 64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Brodbeck WG, Liu DC, Sperry J, Mold C, Medof ME, Localization of classical and alternative pathway regulatory activity within the decay-accelerating factor, J. Immunol 156 (1996) 2528–2533. [PubMed] [Google Scholar]
- [38].Klapper Y, Hamad OA, Teramura Y, Leneweit G, Nienhaus GU, Ricklin D, Lambris JD, Ekdahl KN, Nilsson B, Mediation of a non-proteolytic activation of complement component C3 by phospholipid vesicles, Biomaterials 35 (2014) 3688–3696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Bexborn F, Andersson PO, Chen H, Nilsson B, Ekdahl KN, The tick-over theory revisited: formation and regulation of the soluble alternative complement C3 convertase (C3(H2O)Bb), Mol. Immunol 45 (2008) 2370–2379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Harisinghani M, Ross RW, Guimaraes AR, Weissleder R, Utility of a new bolus-injectable nanoparticle for clinical cancer staging, Neoplasia 9 (2007) 1160–1165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Gabizon AA, Pegylated liposomal doxorubicin: metamorphosis of an old drug into a new form of chemotherapy, Cancer Invest 19 (2001) 424–436. [DOI] [PubMed] [Google Scholar]
- [42].Kang MH, Wang J, Makena MR, Lee JS, Paz N, Hall CP, Song MM, Calderon RI, Cruz RE, Hindle A, Ko W, Fitzgerald JB, Drummond DC, Triche TJ, Reynolds CP, Activity of MM-398, nanoliposomal irinotecan (nal-IRI), in Ewing’s family tumor xenografts is associated with high exposure of tumor to drug and high SLFN11 expression, Clin. Cancer. Res 21 (2015) 1139–1150. [DOI] [PubMed] [Google Scholar]
- [43].Moghimi SM, Hamad I, Andresen TL, Jorgensen K, Szebeni J, Methylation of the phosphate oxygen moiety of phospholipid-methoxy(polyethylene glycol) conjugate prevents PEGylated liposome-mediated complement activation and anaphylatoxin production, FASEB J 20 (2006) 2591–2593. [DOI] [PubMed] [Google Scholar]
- [44].Cunningham CM, Kingzette M, Richards RL, Alving CR, Lint TF, Gewurz H, Activation of human complement by liposomes: a model for membrane activation of the alternative pathway, J. Immunol 122 (1979) 1237–1242. [PubMed] [Google Scholar]
- [45].Moghimi SM, Simberg D, Translational gaps in animal models of human infusion reactions to nanomedicines, Nanomedicine (Lond.) 13 (2018) 973–975. [DOI] [PubMed] [Google Scholar]
- [46].Szebeni J, Simberg D, Gonzales-Fernandez A, Barenholz Y, Dobrovolskaia MA, Roadmap and strategy for overcoming infusion reactions to nanomedicines, Nat. Nanotechnol 13 (2018) 1100–1108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Yuan X, Gavriilaki E, Thanassi JA, Yang G, Baines AC, Podos SD, Huang Y, Huang M, Brodsky RA, Small-molecule factor D inhibitors selectively block the alternative pathway of complement in paroxysmal nocturnal hemoglobinuria and atypical hemolytic uremic syndrome, Haematologica 102 (2017) 466–475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Harder MJ, Kuhn N, Schrezenmeier H, Hochsmann B, von Zabern I, Weinstock C, Simmet T, Ricklin D, Lambris JD, Skerra A, Anliker M, Schmidt CQ, Incomplete inhibition by eculizumab: mechanistic evidence for residual C5 activity during strong complement activation, Blood 129 (2017) 970–980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Risitano AM, Storek M, Sahelijo L, Doyle M, Dai Y, Weitz IC, Marsh JCW, Elebute M, O’Connell CL, Kulasekararaj AG, Ramsingh G, Marotta S, Hellmann A, Lundberg AS, Safety and Pharmacokinetics of the complement inhibitor TT30 in a phase I trial for untreated PNH patients, Blood 126(23) (2015) 2137. [Google Scholar]
- [50].Souza DG, Esser D, Bradford R, Vieira AT, Teixeira MM, APT070 (Mirococept), a membrane-localised complement inhibitor, inhibits inflammatory responses that follow intestinal ischaemia and reperfusion injury, Br. J. Pharmacol 145 (2005) 1027–1034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Rinder CS, Rinder HM, Smith BR, Fitch JC, Smith MJ, Tracey JB, Matis LA, Squinto SP, Rollins SA, Blockade of C5a and C5b-9 generation inhibits leukocyte and platelet activation during extracorporeal circulation, J. Clin. Invest 96 (1995) 1564–1572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Alawieh A, Langley EF, Tomlinson S, Targeted complement inhibition salvages stressed neurons and inhibits neuroinflammation after stroke in mice, Sci. Transl. Med 10 (2018) doi: 10.1126/scitranslmed.aao6459 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Song H, Qiao F, Atkinson C, Holers VM, Tomlinson S, A complement C3 inhibitor specifically targeted to sites of complement activation effectively ameliorates collagen-induced arthritis in DBA/1J mice, J. Immunol 179 (2007) 7860–7867. [DOI] [PubMed] [Google Scholar]
- [54].Holers VM, Rohrer B, Tomlinson S, CR2-Mediated Targeting of Complement Inhibitors: Bench-to-Bedside Using a Novel Strategy for Site-Specific Complement Modulation, Complement Therapeutics 735 (2013) 137–154. [DOI] [PubMed] [Google Scholar]
- [55].Holers M, Banda N, Mehta G, Fridkis-Hareli M, Or E, Storek M, Altman R, Johnson K, Katti S, The human complement receptor type 2 (CR2)/CR1 fusion protein TT32, a targeted inhibitor of the classical and alternative pathway C3 convertases, prevents arthritis in active immunization and passive transfer models and acts by CR2-dependent targeting of CR1 regulatory activity, Immunobio l 217 (2012) 1210–1210. [Google Scholar]
- [56].Belling JN, Jackman JA, Avsar SY, Park JH, Wang Y, Potroz MG, Ferhan AR, Weiss PS, Cho NJ, Stealth immune properties of graphene oxide enabled by surface-bound complement factor H, ACS Nano, 10 (2016) 10161–10172. [DOI] [PubMed] [Google Scholar]
- [57].Hare JI, Lammers T, Ashford MB, Puri S, Storm G, Barry ST, Challenges and strategies in anti-cancer nanomedicine development: an industry perspective, Adv. Drug Delivery Rev 108 (2017) 25–38. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.