

Abstract
For over a century, psychiatric disorders have been defined by expert opinion and clinical observation. The modern DSM has relied on a consensus of experts to define categorical syndromes based on clusters of symptoms and signs, and, to some extent, external validators such as longitudinal course and response to treatment. In the absence of an established etiology, psychiatry has struggled to validate these descriptive syndromes, and to define the boundaries between disorders and between normal and pathologic variation. Recent advances in genomic research, coupled with large-scale collaborative efforts like the Psychiatric Genomics Consortium, have identified hundreds of common and rare genetic variations that contribute to a range of neuropsychiatric disorders. At the same time, they have begun to address deeper questions about the structure and classification of mental disorders: To what extent do genetic findings support or challenge our clinical nosology? Are there genetic boundaries between psychiatric and neurologic illness? Do the data support a boundary between disorder and normal variation? Is it possible to envision a nosology based on genetically informed disease mechanisms? This review provides an overview of conceptual issues and genetic findings that bear on the relationships among and boundaries between psychiatric disorders and other conditions. We highlight implications for the evolving classification of psychopathology and the challenges for clinical translation.
Keywords: psychiatric genetics, nosology, heritability, cross-disorder, GWAS
The fifth edition of the Diagnostic and Statistical Manual of Mental Disorders (DSM51) is the latest update of a prevailing diagnostic approach to psychiatric illness. The DSM consists of symptoms and signs of illness, often complemented by requirements for a particular duration and associated distress or disability. Diagnoses are arrived at by checklists, with diseases defined by presence of some minimal number of criteria, often leaving substantial clinical heterogeneity within disorders. While useful clinically, the validity of the DSM boundaries is uncertain. The underlying genetics do not appear to precisely support such definitions. The genetic overlap among clinically-defined psychiatric syndromes was first demonstrated by family and twin studies. More recently, the work of large-scale collaborations, most notably the Psychiatric Genomics Consortium (PGC), has enabled DNA-level studies of genetic relationships among psychiatric disorders and related traits. Here, we review the state of the genetic structure of psychiatric disorders and its implications for psychiatric nosology.
What does Genetic Epidemiology tell us about the Structure of Psychopathology?
While the observation that psychiatric disorders ran in families can be traced back to antiquity, psychiatric genetics as a scientific discipline arose in the 20th century with the first major family, twin, and adoption studies being published, respectively, in 1916 2, 19283 and 19664. Family studies using modern methodology show substantial familial aggregation for all major psychiatric disorders including depression, schizophrenia, bipolar disorder, and alcohol dependence, as well as many other syndromes such as panic disorder, ADHD, drug abuse, autism, and obsessive-compulsive disorders5. Early twin studies, based on hospital or national twin registries, showed evidence of strong heritable effects for schizophrenia and bipolar disorder. Later, population-based twin studies showed substantial genetic influences on conditions such as major depression, eating and anxiety disorders, alcoholism, ADHD and personality disorders6, 7. Adoption studies are more difficult to perform, but show heritable risks for schizophrenia (e.g. 8), ADHD (e.g. 9) and alcohol dependence (e.g. 10).
An important question first addressed by family studies was whether there was familial overlap among different disorders. When examined with sufficient power, the answer was nearly always that a range of psychiatric disorders cluster together in families. Examples include bipolar disorder, ADHD and depression; depression and anxiety; mood disorders, ADHD and substance abuse; and schizophrenia and schizotypal personality disorder. When this question was examined in twin and adoption studies, similar overlaps were found, and could be attributed to heritable factors that at least partially overlap (Table 1). One population-based twin study showed that common axis-I and axis-II (personality) psychiatric disorders had a coherent underlying genetic structure that reflected just two major dimensions, illustrating the extensive sharing of heritable risk factors across disorders11. In sum, family and twin studies have long suggested that heritable influences on psychopathology transcend diagnostic boundaries; our genes don’t seem to have read the DSM.
Table 1.
Heritability and Genetic Correlation Estimates for Selected Psychiatric Disorders
| Disorder | ASD | ADHD | SCZ | BD | MDD | AN | OCD | PTSD |
|---|---|---|---|---|---|---|---|---|
| ASD | 0.17(0.12-0.22)† | −0.13 (−0.38-0.12)# | 0.18 (0.08-0.28)# | 0.06 (−0.09-0.22)# | 0.14 (−0.06 0.34)# | 0.039 (−0.11-0.13)# | 0.07 (−0.13-0.27)+ | N/A |
| 0.74 (0.70-0.87)¢ | ||||||||
| ADHD | .87 (0.77-1.0)* | 0.28 (0.23-0.33)† | 0.18 (0.02-0.34)# | 0.53 (0.28-0.78)# | 0.43 (0.06-0.80)# | 0.17 (−0.05-0.39)# | −0.07 (−0.24-0.10)+ | N/A |
| 0.79 (0.61-0.88)* | ||||||||
| SCZ | N/A | N/A | 0.23 (0.21-0.25)† | 0.83 (0.76-0.90)# | 0.46 (0.34-0.58)# | 0.19 (0.10-0.28)# | 0.34 (0.22-0.46)+ | 0.33 (0.17-0.49)^ |
| 0.77 (0.67-0.87)¥ | ||||||||
| BD | 0.24 (0.24-0.291)& | 0.33 (0.32-0.39)& | 0.68 (0.67-0.73)! | 0.25 (0.23-0.27)† | 0.64 (0.48-0.80)# | 0.11 (−0.05-0.27)# | 0.31 (0.18-0.44)+ | 0.16 (−0.06-0.38)^ |
| 0.68 (0.64-0.72)¥ | ||||||||
| MDD | N/A | N/A | N/A | 0.65 (0.58-0.751)$ | 0.21(0.17-0.25)† | 0.14 (−0.11-0.341)# | 0.25 (0.07-0.431)+ | 0.34 (0.10-0.58)^ |
| 0.45 (035-0.55)¥ | ||||||||
| AN | N/A | N/A | N/A | N/A | N/A | 0.56(0.50.0.62)# | 0.53 (0.19-0.86)+ | N/A |
| 0.57 (0.0-0.81)¶ | ||||||||
| OCD | N/A | 0.63 (0.39-0.87)% | N/A | N/A | N/A | 0.52 (0.26-0.81)@ | 0.37 (023-0.51)£ | N/A |
| 0.45 (0.30-0.60)¥ | ||||||||
| PTSD | N/A | N/A | N/A | N/A | 1.0$ | N/A | N/A | 0.18 (0.06-0.30)^ |
| 0.38 (0.25-0.52)√ |
On the diagonal: Heritability estimates (and 95% CI) from pedigree/twin studies (lower, shaded orange) and genomewide SNP analyses (upper, shaded light blue). The SNP-based estimates reflect only the common variant component of heritability. Also shown are pairwise genetic correlations from pedigree/twin studies (below diagonal) and genomewide SNP analyses (above diagonal). Bolded entries: 95% CI does not include 0.0. ASD: autism spectrum disorder; ADHD: attention deficit-hyperactivity disorder; SCZ: schizophrenia; BD: bipolar disorder; MDD: major depressive disorder; AN: anorexia nervosa; OCD: obsessive-compulsive disorder; PTSD: posttraumatic stress disorder; N/A: not available. Sources:
109.
How Does Molecular Genetics Confirm and Extend Genetic Epidemiologic Findings?
Studies in the early years of psychiatric molecular genetics tested the parsimonious hypothesis that single variants of large effect would underlie the etiology of psychiatric disorders, but these were not realized except for rare cases. The method of genome-wide association studies (GWAS) gave us a tool for identifying common DNA risk variants (single nucleotide polymorphisms, SNPs) along with rare copy-number variants (CNVs). This method began to produce results with schizophrenia in 2008 and 2009 12–15 Since then, an avalanche of molecular genetic data has generated trustworthy findings for schizophrenia, bipolar disorder, major depressive disorder, ADHD and autism. A broad picture of the genetic architecture of these disorders is emerging. A large portion of the genetic risk for each disorder appears to result from many common SNPs of individually small effect size, with additional contributions from relatively rare (but somewhat more penetrant) CNVs. With the advent of full genome and exome sequencing, rare single-nucleotide variants (SNVs) are also being discovered. One lesson from this work is that very large studies and mega-analyses are required to find these variants. Such analyses have become possible because of a sea change in the culture of psychiatric genomic research. Investigators across the world have come together to share, and often jointly analyze, genetic data through large-scale collaborations and consortia, most notably the PGC 16 (http://www.med.unc.edu/pgc) and iPSYCH (http://ipsych.au.dk).
The identification by GWAS of common variants reliably associated with psychiatric disorders has followed a trajectory similar to that of other common complex biomedical disorders17. Early efforts from individual research teams studying relatively small samples yielded little fruit, power analyses suggesting the need for much larger samples. Consortia were formed to aggregate samples sufficient to reliably detect small effects of common variants. The value of such efforts has been made clear by the growing catalogue of robustly associated common variants, including more than 100 associated with schizophrenia in a PGC analysis of nearly 37,000 cases and more than 113,000 controls18. Individual SNPs associated with these diseases explain only a tiny fraction of the heritable variance for psychiatric disorders, with most individual risk-predisposing alleles associated with odds ratios of 1.1 or less. However, the aggregation of these effects into polygenic risk scores (PRSs)19 that capture the additive effects of thousands of SNPs accounts for substantially larger fractions of the heritable variance. A range of statistical methods allow the estimation of heritability based on genomewide SNPs, often referred to as SNP-chip heritability (h2SNP)19. Applied to psychiatric disorders, these methods show that the genetic architecture includes a substantial contribution of common variation, though the estimates vary from less than 10% (for anxiety disorders and PTSD)20, 21 to more than 20% (for ADHD, bipolar disorder and schizophrenia) 22, 23 (see Table 1). These estimates are lower than those derived from twin studies, in part because h2SNP only includes effects due to common variants.
Genomic studies also show that rare, de novo variations (arising in the gametes or embryo) contribute to psychiatric disorders. Thus far, this has most convincingly been demonstrated for autism spectrum disorder and schizophrenia. For example, the largest analysis of CNV data, by the PGC’s CNV and Schizophrenia Workgroups, comprising more than 40,000 subjects, identified eight CNV loci associated with schizophrenia risk24. Although these loci were rare (most with a frequency of <0.5% among cases), their effects were much larger (ORs ranging from 3.8 to more than 67) than those seen with individual common variants. They include a deletion on chromosome 22 which is the cause of 22qdel syndrome (velocardiofacial syndrome) and has long been recognized as carrying a high risk for psychosis. The contribution of specific rare SNVs has been harder to establish, though large-scale sequencing of the exome has shown that the aggregate burden of rare protein-altering variants is elevated in schizophrenia25, autism, 26 and Tourette syndrome. 27 In addition, mutations in several genes have been associated with autism spectrum disorders (e.g. CHD8, SCN2A, SHANK3, GRIN2B)28, 29 and, to a lesser extent, schizophrenia (SETD1A)30.
Genome-wide studies have documented that psychiatric disorders are highly polygenic, reflecting a combination of thousands of common variants of individually small effect and rarer variants of larger effect. With the disorders best characterized, especially schizophrenia, it is increasingly clear that a substantial fraction of genetic risk is the result of common SNP variation. As sample sizes grow, additional variants are expected to be found. With these discoveries, however, has arisen the challenge of translating these molecular findings into insights about etiologic pathways.
Cross-disorder studies
Similar to family and twin studies, GWAS first focused on single disorders and then examined if risk-conferring variants for one disorder affect risks for others; i.e., pleiotropy. (Figure 1 depicts common approaches for evaluating cross-disorder genetic effects). Given the large number of human phenotypes and the limited number of genes, it is not surprising that pleiotropy is a common phenomenon for many traits and disorders, by no means specific to psychiatry31. Among the earliest evidence of shared molecular genetic influences across neuropsychiatric disorders were findings that rare CNVs are associated with multiple disorders including autism, ADHD, and schizophrenia as well as epilepsy and intellectual disability 32. GWAS data have also been used to examine the cross-disorder sharing of common variants. For example, the International Schizophrenia Consortium reported that a PRS derived from GWAS of schizophrenia was strongly associated with risk for bipolar disorder.12. Subsequently, PRS have been used to demonstrate cross-disorder genetic overlap of a wide range of psychiatric phenotypes. For example, schizophrenia PRS has been associated with psychotic experience 33 and schizoaffective disorder 33 as well as related phenotypes such as cognitive ability 34, 35, sensory motor gating 36, working memory brain activation (fMRI signal) 37, childhood neurodevelopmental impairments38, major depressive disorder39, ADHD and conduct/oppositional defiant disorder40, adolescent anxiety disorder41, and PTSD21.
Figure 1. Methods commonly used to evaluate genetic overlap between phenotypes.
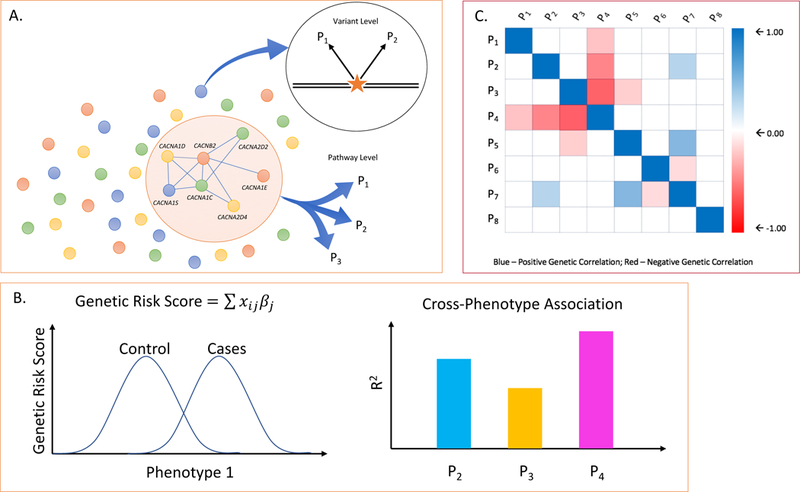
A. At the DNA variant level, individual loci (e.g. SNPs, rare mutations, or CNVs) may show evidence of pleiotropic association with two (e.g. P1, P2) or more phenotypes. At the level of biological pathways, gene sets assigned to a pathway may be enriched in association signals beyond chance expectation across multiple phenotype (e.g. P1, P2, P3). B. Genetic risk scores (or polygenic risk scores, PRS) are developed in a “discovery“ GWAS sample and computed for each individual in an independent “target” sample. The genetic risk score for each individual (i) in the target sample is computed as the product of the number of risk alleles (X) at each SNP (j) multiplied by that SNP’s association effect size (βj) and summing over all SNPs. The left hand plot shows the distribution of genetic risk scores in cases and controls in an independent target set for the same phenotype as that of the discovery sample. To examine cross-phenotype overlap, the discovery risk score is applied to target samples of other phenotypes. The proportion of variance explained by the discovery GWAS (R2) for each target phenotype (e.g. P2, P3, P4). is shown in the plot on the right hand side. C. Genetic correlation between phenotypes (ranging from −1.0 to +1.0) can be estimated using multiple methods that compare genetic and phenotypic similarity among unrelated individuals. The figure shows a hypothetical genetic correlation matrix between multiple pairs of phenotypes (P1 – P8).
SNP-chip heritability methods have been extended to estimate the genetic correlation between pairs of disorders averaged over all common SNPs. For example, the Cross-Disorder Group of the PGC reported significant genetic correlations of schizophrenia with bipolar disorder (0.68), major depressive disorder (0.43) and autism (0.16), and between major depressive disorder and bipolar disorder (0.47) and ADHD (0.32)22 (see Table 1). A recent analysis of of the Big Five personality traits further demonstrates specific patterns of genetic correlation with psychiatric disorders 42. For a database presenting the many statistically significant molecular genetic correlations among psychiatric disorders and other phenotypes see LD Hub.43
Individual common variants have also shown pleiotropic effects across clinically distinct disorders39, providing initial clues to a shared biology. Network and pathway analyses have pointed to biological pathways that show genetic association across diagnostic groups. For example, genes related to calcium channel signaling, histone methylation, synaptic function, and immune function have been implicated in both mood disorders and schizophrenia.39, 44
These results suggest that susceptibility to each psychiatric disorder, as currently defined by DSM, is influenced by many genetic risk factors and that any given psychiatric disorder will share some genetic risk factors with others. This risk-sharing extends beyond the genome to the environment, with early developmental insults and trauma implicated in risk for several disorders as well. This knowledge has large implications for understanding the pathology of psychiatric disorders. However, advances in our statistical methods and a range of in vitro and in vivo experimental studies are needed to translate the shared genetic and environmental association signals into molecular genetic mechanisms.
What does it all mean?
Psychiatric disorders are highly polygenic
Family and twin studies, now complemented by molecular genetics, show that genetic variation accounts for a substantial portion of the risk for psychiatric disorders, and that some of the heritability is shared among disorders. However, many variants remain to be discovered, and this will require much larger sample sizes, and additional cross-disorder studies. The history of schizophrenia genetics provides a compelling example that simply expanding sample size is a winning strategy for driving discovery18. But there are obstacles. To make more progress, funders will have to accept the drudgery of “normal science” rather than uncritically requiring innovative methods of gene discovery.
Discovering biological mechanisms from common variants will be challenging.
Going from a GWAS-associated locus to identifying causal variation underlying the association is a challenge, but one that can be overcome with significant effort. A good example is Sekar et al.’s 45 study of common variation associated with schizophrenia in the major histocompatibility complex, which identified functional alleles of the complement component 4 (C4) genes and implicated their role in microglia-mediated synaptic pruning. Identifying the causal alleles required complex fine-mapping followed by functional studies of gene expression in mouse and human brain, and high-resolution immunohistochemistry.
Another issue is that most common risk variants individually contribute only a very small portion of overall susceptibility to common complex psychiatric disorders. Mechanistic studies must eventually integrate the effects of many risk loci. Fortunately, powerful new technologies for studying systems genomics and neuroscience (including the use of induced pluripotent stem cells, gene-editing methods, and optogenetic interrogation of brain circuits) provide new opportunities for dissecting the functional effects of risk variants and pathways discovered from them. 46, 47 As the growing catalog of established genetic variants converge on biological pathways, new opportunities arise for identifying treatment targets. For decades, psychiatric drug development has focused on pathways modulated by drugs that had been serendipitously discovered. By moving the focus of drug discovery to genetically validated functional pathways, GWAS can point to new “druggable” targets, enabling the development of novel therapeutics for specific or cross-diagnostic clinical characteristics.48
Discovering biological mechanisms from rare variants of larger-effect may be easier. Unlike common SNPs, rare disease-associated mutations often have direct functional consequences, making them especially suitable for mechanistic studies. Some have argued the mechanisms discovered via rare variants will not be relevant to the large majority of patients, but empirical examples show that a biological pathway implicated by a rare variant is relevant to mechanisms implicated by common variation. For example, rare variants of PCSK9 cause a rare autosomal dominant familial hypercholesterolemia. Drugs that inhibit PCSK9 protein lower cholesterol and are a viable treatment for common forms of hypercholesterolemia and atherosclerosis.49
Genetic studies challenge the DSM paradigm.
The pervasive cross-disorder heritability of psychiatric disorders challenges the DSM paradigm which, from its inception, emphasized hierarchical diagnoses disallowing diagnoses of some disorders in the presence of others. Twin data point to genetic hierarchies with a general psychopathology factor, internalizing and externalizing factors, along with unique sources of heritability for each disorder50. Twin studies have also reported genetic correlations between disorders and the normal range of personality variation (e.g. 51) and support the view that some disorders are the extreme of a continuous trait in the population 52, 53. Genomic studies are also questioning the idea that psychiatric conditions are discrete entities by demonstrating substantial molecular genetic correlations across diagnostic categories and between disorders and normal ranges of phenotypic variation in the population54–57. In some cases, these findings challenge fundamental assumptions of our clinical nosology. For example, the separation of schizophrenia and bipolar disorder has been a foundational distinction for psychiatric classification, dating to Kraepelin’s work more than a century ago. The modern DSM defines these disorders as mutually exclusive, belonging to different classes of mental illness. However, genomic studies have shown that, at a genetic level, these conditions are highly overlapping, with a genetic correlation of nearly 0.70.
Taken together, genetic findings suggest that the structure of much psychopathology is defined by dimensional variation in the population, as is the case for hypertension and hypercholesterolemia, where the same genes often contain variants that cause diagnosable disorders and other variants that impact on variation in the “normal” range. By contrast, schizophrenia may be more complex as the disorder may represent a concatenation of several dimensions of risk including liability to psychosis, cognitive difficulties and social dysfunction. Although diagnostic categories will continue to be needed from a practical standpoint, future iterations of psychiatric nosology may be usefully informed and refined by incorporating our emerging understanding of the etiologic overlap among clinical syndromes. This is, indeed, a premise of NIMH’s nascent Research Domain Criteria (RDoC) initiative 58. A former NIMH Director created controversy when he described the DSM as lacking validity and that NIMH would be “re-orienting its research away from DSM categories” 59. Others (e.g. 60) have called for a shift from what Kendler61 called the ‘soft’ symptom-based, etiologically blind diagnoses of the DSM to ‘hard’ diagnoses based on etiologically based biological features. Will genetic data lead to ‘hard’ empirically derived diagnoses? In isolation, probably not, but they, and mechanisms learned from them, may aid in the revision or formulation of novel diagnostic criteria in the future. They may also suggest novel hypotheses about the biological basis of psychiatric disorders. For example, a recent large genomic analysis found substantial genetic correlations between anorexia nervosa and a range of metabolic traits (including measures of cholesterol and lipids, fasting insulin, fasting glucose, insulin resistance, and leptin levels), suggesting that the disorder might be both a psychiatric and metabolic syndrome. 62
The idea that genetic data should inform diagnostic nosologies is not new. Robins and Guze63 included family history as one criterion of a validation method that stimulated the design of structured diagnostic criteria. Kendler 64 cautioned that purely data-driven scientific nosologies could not address fundamental issues facing nosologists; e.g., how to integrate information from different types of “validators”. Tsuang et al.65 suggested that psychiatric genetics could play a limited role by creating a nosology for genetic research aimed at better defining distinct genetic entities. Although such approaches are useful for researchers, the idea that psychiatric genetic findings will revolutionize the clinical psychiatric nosology has been questioned66. This view suggests that, rather than leading to breakthroughs in genetic nosology, genetic data will, as it has in the past, incrementally help the DSM evolve via a process that Kendler 67 described as ‘epistemic iteration,’ whereby the evidence base sequentially iterates with the acquisition of new data to provide a better approximation of the latent, but unknown, structure of psychopathology. Alternatively, a more data-driven DSM process might consider a revolutionary recasting of diagnostic categories as dimensional entities with well-defined thresholds demarcating wellness from subthreshold and clinically significant disorders. This would provide clinicians the categories they need within a framework that better corresponds to the latent structure of psychopathology. However, the study of risk genes, which impact only on the liability to illness, will not, of itself, permit us to empirically define the boundaries between illness and health or between closely related disorders. Doing so would require including other empirical evidence regarding both diagnostic validity and clinical utility.
Genetic complexity is a challenge for identifying clinically relevant biomarkers.
In 1980, when DSM-III was released, 21 papers were published on biomarkers in psychiatry. Thirty-five years later, that number had grown to 1,555. This search for objective measures for defining disorders and their underlying pathophysiological processes reflects the field’s growing discomfort with the subjectively assessed signs and symptoms that define DSM disorders. Although a Google patent search yields over 8,000 relevant patents, with the exception of mutations causing rare, syndromal forms of psychiatric disorders and some useful genetic predictors of pharmacokinetics, this intensive search for biomarkers has not been impacted clinical practice.
Genomic studies indicate that there are many causal genes and pathways, which implies that there will be many peripheral markers of disease, as has been shown for ADHD68. This suggests that multifactorial biomarkers will be needed. Initial attempts to do this with genomic studies (e.g. 69) have been intriguing, but their interpretation is clouded by methodological concerns70. Thus, the current use of biomarkers to aid in diagnosis or genetic counseling in psychiatry has been limited to rare syndromic forms of disorders. In the future, PRS may become useful biomarkers if their precision and predictive value can be improved. By aggregating genetic effects across many genes (and, indirectly, biological pathways) improved PRS may provide an informative summary marker despite the underlying genetic complexity.
Where Do We Go From Here?
The first and most obvious agenda for future research is to pursue ever-larger genomewide common and rare variant studies of psychiatric disorders. Empirical evidence and simulations show that for GWAS there is a sample-size-dependent inflection point beyond which the number of genomewide significant loci increases linearly. 71 For SCZ, the inflection point was seen at about 15,000 cases, but for other, perhaps more complex or heterogenous disorders such as MDD and SUDs, the inflection point may not be reached until as many as 75,000 to 100,000 cases are examined. 71 The numbers needed depend on a phenotype’s heritability and polygenicity and the effect sizes of the contributing SNPs 72. In the case of rare variants, effect sizes may be larger, but their rarity again requires large sample sizes (more than 25,000 cases) to allow detection of a sufficient number of risk variant carriers. 73, 74 The third wave of the PGC (PGC3), now underway, aims to enable this next generation of larger-scale GWAS and pathway analyses. 75 Currently, the PGC encompasses ten disorders and aims for 100,000 cases each. Though some have questioned the value of continuing to pursue larger GWAS, the evidence to date for psychiatric disorders and other complex diseases suggests that such efforts will continue to be important for identifying additional genes and through them the biological pathways that underlie disease. This will provide a foundation for novel diagnostic and therapeutic approaches, and for realizing the hope for personalized treatment. 17, 76
Expanding the size and scope of genomewide common and rare variant studies will also help elucidate the genetic architecture of disorder-specific and cross-disorder genetic effects. Ongoing analyses by the PGC Cross-Disorder Workgroup are focusing on characterizing the association and functional significance of specific variants and pathways related to the nine disorders enumerated above, as well as looking for pleiotropic effects of variants detected. In addition, the PGC3’s Brainstorm initiative is linking the PGC with other GWAS consortia to examine the genetic relationships among psychiatric disorders, neurologic disorders, and dimensional measures of personality, cognition, and brain structure and function. Initial Brainstorm analyses across 23 brain disorders (with a total N = 842, 820) indicate that genetic overlap is stronger among traditionally defined psychiatric disorders than among neurologic disorders (e.g. multiple sclerosis, stroke, migraine) or between psychiatric and neurologic disorders 77, supporting the clinical demarcation between neurologic and psychiatric disorders. The average genetic correlation based on genomewide SNP data was +0.21 among eight psychiatric disorders (autism spectrum disorder, schizophrenia, bipolar disorder, major depressive disorder, ADHD, anorexia nervosa, obsessive-compulsive disorder, and Tourette syndrome), substantially higher than that observed among the ten neurologic disorders (0.06). Schizophrenia showed broad sharing with the other psychiatric disorders while autism and Tourette syndrome appeared to be the most genetically distinct. Strikingly, none of the neurologic disorders were significantly genetically correlated with the psychiatric disorders with the exception of migraine. 77 However, the low genetic correlation between the psychiatric and neurologic disorders could in part result from diagnostic differences as the psychiatric disorders were assessed using symptoms and signs and the neurological disorders often utilized imaging and/or neurophysiologic measures.
We can also now capitalize on growing resources in electronic health records (EHR) and population-based registries linked to genomic data to examine the pleiotropic effects of psychiatric genetic risk variants across the phenome. 78 For example, the eMERGE network, a consortium of biobanks linked to EHR data, and large-scale cohort studies like the UK Biobank 79, which recently released genomewide data for 500,000 participants, and the recently launched All of Us Program of the NIH Precision Medicine Initiative80, offer opportunities to conduct well-powered phenomewide association studies (“PheWas”) of common and rare variants. These analyses may reveal unexpected etiologic relationships between psychiatric and other diseases or traits.81 The All of Us Research Program seeks to collect broad and deep phenotypic data (using EHRs, mobile technologies, surveys, and biospecimens) along with genomic data in a longitudinal cohort of 1 million or more Americans, and will offer an even larger resource for testing hypotheses about the spectrum of genotype-phenotype relationships for medical and psychiatric illness. Longitudinal population-based cohorts with genomic data can identify trajectories of disorder risk and gene-environment interplay. 82 Therefore, the ability to recall participants in biobanks or large-scale cohorts for further deep phenotyping based on genotype is critical.
To date, most genomic studies have been restricted to cross-sectional, case-control analyses. The focus has been on phenotypic characterization, with little emphasis on the collection of high quality information on critical environmental risk factors. A more complete understanding of the etiology and structure of psychopathology will require an understanding of how genetic and environmental risk factors act and interact across development. To answer these questions will require study designs that attend to environmental risks and/or take an explicitly developmental perspective. For example, when in human development do risk alleles and pathways exert their effects? 83, 84 Might some pathways confer risk to a broad vulnerability to psychopathology and others to more differentiated disorders? Are there sensitive periods of development when environmental risk factors act or interact more potently to confer risk? What are the molecular or cellular mechanisms by which genetic variation and environmental exposures confer risk over the lifespan? These and other questions may be addressed with data from epidemiological birth cohorts (e.g., the Avon Longitudinal Study of Parents and Children, ALSPAC85), population-based registries (e.g., those available in Scandinavian countries 86) as well as large cohorts consented for follow-up (e.g. the UK Biobank79, All of Us80)
Polygenic methods (e.g. PRS) may capture more complex combinations of disorder-specific and cross-disorder variants as indices of genetic vulnerability, and be very useful in longitudinal and developmental studies. The PGC3 analytic plan includes PRS analyses of nine disorders in a longitudinal sample of nearly 14,000 twins followed from age 9 to age 24. PGC analyses will also examine how environmental exposures modify genetic risk (PRS x environment interaction studies) to influence risk of a range of disorders. Although allelic-additive models are the most impervious to model misspecification, it should ultimately be possible to account for additional missing heritability in psychiatry by appropriately modeling dominant and recessive alleles, as well as gene-gene and gene-environment interactions.
Incorporating data beyond the categorical diagnostic variables that have been the predominant focus of psychiatric genetic studies to date will allow better characterization of the structure of psychopathology. To this end, investigators are undertaking studies of dimensional traits (e.g., RDoC measures) and incorporating neuroimaging phenotypes that may identify genetic underpinnings of neural, cognitive, affective and social phenotypes that transcend diagnostic boundaries. For example, recent analyses have demonstrated genetic overlaps between psychiatric disorders and measures of brain structure, including shared genetic influences for schizophrenia and thickness of the left superior frontal gyrus, a region where thinning and volume loss have previously been associated with the disorder 87. The international ENIGMA consortium has brought together genomic and brain imaging data for more than 30,000 individuals spanning a broad range of psychiatric disorders, enabling a growing catalogue of discoveries about the genetic basis of brain structure and function and their relationship to psychopathology. 88 Other studies have examined the genetic relationship between disorder and normal variation in quantitative traits. 42 For example, common and rare genetic variants associated with autism spectrum disorder influence dimensions of social cognition, cognition/intelligence, and communication abilities in the general population 54, 56. Similarly, genetic risk scores for ADHD predict attention problems in population-based samples of children 55.
Efforts to dissect the fundamental intermediate phenotypes underlying risk of psychiatric disorder face important challenges. Most importantly, we still do not know which are the most relevant levels of analyses and which of the large number of possible intermediate traits are causally related to mental illness. The domains enumerated in the RDoC framework89 represent one of several possible approaches. Genetic data may help resolve the causal status of putative intermediate phenotypes. For example, a candidate intermediate phenotype for which robust genetic associations are known can be analyzed using Mendelian randomization (which uses the associated variants as instrumental variables) to determine whether it is causally linked to a disorder 90. This approach has been used successfully in other areas, providing evidence, for example, that central adiposity is causally related to coronary heart disease while HDL is not. 91, 92 In the realm of psychiatry, recent analyses have suggested that cannabis use maybe a causal risk factor for schizophrenia. 93 Because this approach is biased when there is pleiotropy (which is widespread in psychiatric genetics), other methods that can evaluate causality in presence of genetic correlation may be needed. 94
Finally, having a more complete understanding of the genetic basis of psychiatric disorders will allow us to determine whether genetic variation, in concert with other more traditional variables, can improve prediction of clinically relevant outcomes. To date, the variance explained by common and rare variants, individually or as aggregate genetic risk scores, has been insufficient to be clinically useful for diagnosis or for prediction of clinical course. An exception is autism spectrum disorder, where genetic evaluation, including testing for structural and rare mutations, is recommended as part of the diagnostic process by the American College of Medical Genetics and Genomics95. In general, genetic biomarkers will only be clinically useful if they add value or efficiency beyond established non-genetic diagnostic procedures or risk factor profiles. As the power and precision of polygenic risk profiles improve and as more powerful rare variant studies allow us to fractionate disorder heterogeneity, this may be achievable for some disorders. For example, recent analyses support the existence of autism spectrum disorder subtypes that differ by their genetic architecture96, 97. Specific, highly penetrant structural and rare exonic mutations may represent genetic subtypes of heterogeneous disorders. In addition, a more complete characterization of the pleiotropic effects of psychiatric risk variants may have implications for genetic counseling. Numerous rare structural and single nucleotide variants have already been shown to influence a range of psychiatric disorders98. This catalogue of pleiotropic variants is expected to increase as the PGC3 conducts whole-genome sequencing analyses of pedigrees densely affected by multiple psychiatric disorders to identify rare variants of strong effect. Given the substantial co-morbidity and overlap in genetic contributions to psychiatric disorders, studies should invest in phenotyping across disorders. Some, such as substance use disorders and depression, likely contribute to many common medical diseases, so studies of those diseases (e.g., liver disease, heart disease, cancers) should also gather information on lifetime patterns of substance use, abuse and dependence and symptoms of depression. We will also need to convince funders that GWAS will lead to a better mechanistic understanding and, ultimately, better prevention and treatment.
Conclusions
A substantial and growing body of genetic research has begun to elucidate the underlying structure of psychopathology. Before the modern era of genomic research, family and twin studies demonstrated that all major psychiatric disorders aggregate in families and are heritable. Over the past decade, the success of large-scale genomic studies has confirmed several key principles: 1) psychiatric disorders are highly polygenic, reflecting the contribution of hundreds to thousands of common variants of small effect and rare (often de novo) SNVs and CNVs; 2) the delineation of disease mechanisms from variant discovery is a difficult problem that requires integrating genetic information across multiple levels of functional data from “omics” to neurobiology to clinical phenotypes; and 3) genetic influences on psychopathology commonly transcend the diagnostic boundaries of our clinical DSM nosology. At the level of genetic etiology, there are no sharp boundaries between diagnostic categories or between disorder and normal variation. In the coming years, ever-larger studies incorporating DNA sequencing, environmental exposures, and phenome-wide analyses will facilitate a more granular understanding of the genetic etiology and phenotypic spectrum of mental illness.
The highly interconnected nature of cellular networks in the brain suggests that many, perhaps most, variants that regulate or participate in these pathways have some non-zero effect across psychiatric disorders (a concept called “network pleiotropy” 76) Thus, as naïve single gene and oligogenic models for psychiatric disorders have been replaced by the reality of hundreds if not thousands of risk variants, the field is facing a problem of daunting complexity. However, it is better to confront directly this challenging reality than comfort ourselves with unrealistically simplistic models. Improvements in our ability to link genetic variation to gene function, sort out signal from noise with evolving statistical methods, and build in vitro and in vivo biological models to explore the “curse of polygenicity” should help with the translation from molecular genetic discoveries into etiologic insights. Progress might be slow, but molecular genetic methods remain one of our few tools that can provide us direct insight into etiological pathways that underlie the structure of psychopathology.
Acknowledgments
Supported in part by NIH awards K24MH094614 (JWS), the Research Council of Norway (223273) and KG Jebsen Stiftelsen (OAA). Dr. Smoller is a Tepper Family MGH Research Scholar. “All of Us” is a service mark of the U.S. Department of Health and Human Services. The authors thank Nicholas Merriam for assistance with producing the figure.
Footnotes
Disclosures:
Dr. Smoller is an unpaid member of the Scientific Advisory Board of PsyBrain Inc. and the Bipolar/Depression Research Community Advisory Panel of 23andMe. Dr. Andreassen has received speaker’s honorarium from Lundbeck. The remaining authors have no disclosures.
References
- 1.American Psychiatric Association. Diagnostic and statistical manual of mental disorders : DSM-5. 5th edn American Psychiatric Association: Arlington, VA, 2013, xliv, 947 p.pp. [Google Scholar]
- 2.Rudin E Studien uber Vererbung und entstehung geistiger Storungen. I. Zur vererbung und neuentstehung der Dementia praecox (Studies on the inheritance and origin of mental illness. I. The problem of the inheritance and primary origin of dementia praecox). Monographien aus dem Gesamtgebiet der Neurologie und Psychiatrie, Number 12 Berlin: Springer; 1916. [Google Scholar]
- 3.Luxenburger H Vorlaufiger Bericht uder psychiatrische Serienuntersuchungen und Zwillingen. Z Gesamte Neurol Psychiatr 1928; 116: 297–326. [Google Scholar]
- 4.Heston LL. Psychiatric disorder in foster home reared children of schizophrenic mothers. British Journal of Psychiatry 1966; 112: 819–825. [DOI] [PubMed] [Google Scholar]
- 5.Kendler KS, Eaves LJ. Psychiatric Genetics, Review of Psychiatry, Vol 24, vol. American Psychiatric Publishing, Inc: Arlington,VA, 2005. [Google Scholar]
- 6.Kendler KS. Twin studies of psychiatric illness: an update. Arch Gen Psychiatry 2001; 58(11): 1005–1014. [DOI] [PubMed] [Google Scholar]
- 7.Kendler KS, Prescott CA, Myers J, Neale MC. The structure of genetic and environmental risk factors for common psychiatric and substance use disorders in men and women. Arch Gen Psychiatry 2003; 60(9): 929–937. [DOI] [PubMed] [Google Scholar]
- 8.Kety SS. The significance of genetic factors in the etiology of schizophrenia: results from the national study of adoptees in Denmark. J Psychiatr Res 1987; 21(4): 423–429. [DOI] [PubMed] [Google Scholar]
- 9.Sprich S, Biederman J, Crawford MH, Mundy E, Faraone SV. Adoptive and biological families of children and adolescents with ADHD. J Am Acad Child Adolesc Psychiatry 2000; 39(11): 1432–1437. [DOI] [PubMed] [Google Scholar]
- 10.Verhulst B, Neale MC, Kendler KS. The heritability of alcohol use disorders: a meta-analysis of twin and adoption studies. Psychol Med 2015; 45(5): 1061–1072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kendler KS, Aggen SH, Knudsen GP, Roysamb E, Neale MC, Reichborn-Kjennerud T. The structure of genetic and environmental risk factors for syndromal and subsyndromal common DSM-IV axis I and all axis II disorders. The American journal of psychiatry 2011; 168(1): 29–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.International Schizophrenia Consortium Purcell SM, Wray NR Stone JL, Visscher PM O’Donovan MC et al. Common polygenic variation contributes to risk of schizophrenia and bipolar disorder. Nature 2009; 460(7256): 748–752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Stefansson H, Ophoff RA, Steinberg S, Andreassen OA, Cichon S, Rujescu D et al. Common variants conferring risk of schizophrenia. Nature 2009; 460(7256): 744–747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Stefansson H, Rujescu D, Cichon S, Pietilainen OP, Ingason A, Steinberg S et al. Large recurrent microdeletions associated with schizophrenia. Nature 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.International Schizophrenia Consortium. Rare chromosomal deletions and duplications increase risk of schizophrenia. Nature 2008; 455(7210): 237–241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Psychiatric GWAS Consortium Coordinating Committee. Genomewide association studies: history, rationale, and prospects for psychiatric disorders. Am J Psychiatry 2009; 166(5): 540–556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Visscher PM, Wray NR, Zhang Q, Sklar P, McCarthy MI, Brown MA et al. 10 Years of GWAS Discovery: Biology, Function, and Translation. Am J Hum Genet 2017; 101(1): 5–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Schizophrenia Working Group of the Psychiatric Genomics C. Biological insights from 108 schizophrenia-associated genetic loci. Nature 2014; 511(7510): 421–427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wray NR, Lee SH, Mehta D, Vinkhuyzen AA, Dudbridge F, Middeldorp CM. Research review: Polygenic methods and their application to psychiatric traits. J Child Psychol Psychiatry 2014; 55(10): 1068–1087. [DOI] [PubMed] [Google Scholar]
- 20.Otowa T, Hek K, Lee M, Byrne EM, Mirza SS, Nivard MG et al. Meta-analysis of genome-wide association studies of anxiety disorders. Mol Psychiatry 2016; 21(10): 1485. [DOI] [PubMed] [Google Scholar]
- 21.Duncan LE, Ratanatharathorn A, Aiello AE, Almli LM, Amstadter AB, Ashley-Koch AE et al. Largest GWAS of PTSD (N=20 070) yields genetic overlap with schizophrenia and sex differences in heritability. Mol Psychiatry 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cross-Disorder Group of the Psychiatric Genomics C, Lee SH, Ripke S, Neale BM, Faraone SV, Purcell SM et al. Genetic relationship between five psychiatric disorders estimated from genome-wide SNPs. Nat Genet 2013; 45(9): 984–994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Charney AW, Ruderfer DM, Stahl EA, Moran JL, Chambert K, Belliveau RA et al. Evidence for genetic heterogeneity between clinical subtypes of bipolar disorder. Transl Psychiatry 2017; 7(1): e993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.CNV Schizophrenia Working Groups of the Psychiatric Genomics Consortium. Contribution of copy number variants to schizophrenia from a genome-wide study of 41,321 subjects. Nat Genet 2017; 49(1): 27–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Genovese G, Fromer M, Stahl EA, Ruderfer DM, Chambert K, Landen M et al. Increased burden of ultra-rare protein-altering variants among 4,877 individuals with schizophrenia. Nat Neurosci 2016; 19(11): 1433–1441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Deciphering Developmental Disorders S. Prevalence and architecture of de novo mutations in developmental disorders. Nature 2017; 542(7642): 433–438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Willsey AJ, Fernandez TV, Yu D, King RA, Dietrich A, Xing J et al. De Novo Coding Variants Are Strongly Associated with Tourette Disorder. Neuron 2017; 94(3): 486–499 e489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Vorstman JA, Parr JR, Moreno-De-Luca D, Anney RJ, Nurnberger JI Jr., ,Hallmayer JF. Autism genetics: opportunities and challenges for clinical translation. Nat Rev Genet 2017. [DOI] [PubMed] [Google Scholar]
- 29.de la Torre-Ubieta L, Won H, Stein JL, Geschwind DH. Advancing the understanding of autism disease mechanisms through genetics. Nat Med 2016; 22(4): 345–361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Singh T, Kurki MI, Curtis D, Purcell SM, Crooks L, McRae J et al. Rare loss-of-function variants in SETD1A are associated with schizophrenia and developmental disorders. Nat Neurosci 2016; 19(4): 571–577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Solovieff N, Cotsapas C, Lee PH, Purcell SM, Smoller JW. Pleiotropy in complex traits: challenges and strategies. Nat Rev Genet 2013; 14(7): 483–495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Malhotra D, Sebat J. CNVs: harbingers of a rare variant revolution in psychiatric genetics. Cell 2012; 148(6): 1223–1241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tesli M, Espeseth T, Bettella F, Mattingsdal M, Aas M, Melle I et al. Polygenic risk score and the psychosis continuum model. Acta psychiatrica Scandinavica 2014; 130(4): 311–317. [DOI] [PubMed] [Google Scholar]
- 34.Hatzimanolis A, Bhatnagar P, Moes A, Wang R, Roussos P, Bitsios P et al. Common genetic variation and schizophrenia polygenic risk influence neurocognitive performance in young adulthood. Am J Med Genet B Neuropsychiatr Genet 2015; 168(5): 392–401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.McIntosh AM, Gow A, Luciano M, Davies G, Liewald DC, Harris SE et al. Polygenic risk for schizophrenia is associated with cognitive change between childhood and old age. Biol Psychiatry 2013; 73(10): 938–943. [DOI] [PubMed] [Google Scholar]
- 36.Roussos P, Giakoumaki SG, Zouraraki C, Fiullard JF, Karagiorga V-E, Tsapakis E-M et al. The relationship of common risk variants and polygenic risk for schizophrenia to sensorimotor gating Biological Psychiatry In Press. [DOI] [PubMed] [Google Scholar]
- 37.Kauppi K, Westlye LT, Tesli M, Bettella F, Brandt CL, Mattingsdal M et al. Polygenic risk for schizophrenia associated with working memory-related prefrontal brain activation in patients with schizophrenia and healthy controls. Schizophr Bull 2015; 41(3): 736–743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Riglin L, Collishaw S, Richards A, Thapar AK, Maughan B, O’Donovan MC et al. Schizophrenia risk alleles and neurodevelopmental outcomes in childhood: a population-based cohort study. Lancet Psychiatry 2017; 4(1): 57–62. [DOI] [PubMed] [Google Scholar]
- 39.Cross Disorder Group of the Psychiatric GWAS Consortium. Identification of risk loci with shared effects on five major psychiatric disorders: a genome-wide analysis. Lancet 2013; 381(9875): 1371–1379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Nivard MG, Gage SH, Hottenga JJ, van Beijsterveldt CE, Abdellaoui A, Bartels M et al. Genetic Overlap Between Schizophrenia and Developmental Psychopathology: Longitudinal and Multivariate Polygenic Risk Prediction of Common Psychiatric Traits During Development. Schizophr Bull 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Jones HJ, Stergiakouli E, Tansey KE, Hubbard L, Heron J, Cannon M et al. Phenotypic Manifestation of Genetic Risk for Schizophrenia During Adolescence in the General Population. JAMA Psychiatry 2016; 73(3): 221–228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lo MT, Hinds DA, Tung JY, Franz C, Fan CC, Wang Y et al. Genome-wide analyses for personality traits identify six genomic loci and show correlations with psychiatric disorders. Nat Genet 2017; 49(1): 152–156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zheng J, Erzurumluoglu AM, Elsworth BL, Kemp JP, Howe L, Haycock PC et al. LD Hub: a centralized database and web interface to perform LD score regression that maximizes the potential of summary level GWAS data for SNP heritability and genetic correlation analysis. Bioinformatics 2017; 33(2): 272–279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Network and Pathway Analysis Subgroup of Psychiatric Genomics Consortium. Psychiatric genome-wide association study analyses implicate neuronal, immune and histone pathways. Nat Neurosci 2015; 18(2): 199–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sekar A, Bialas AR, de Rivera H, Davis A, Hammond TR, Kamitaki N et al. Schizophrenia risk from complex variation of complement component 4. Nature 2016; 530(7589): 177–183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Devor A, Andreassen OA, Wang Y, Maki-Marttunen T, Smeland OB, Fan CC et al. Genetic evidence for role of integration of fast and slow neurotransmission in schizophrenia. Mol Psychiatry 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Gandal MJ, Leppa V, Won H, Parikshak NN, Geschwind DH. The road to precision psychiatry: translating genetics into disease mechanisms. Nat Neurosci 2016; 19(11): 1397–1407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Breen G, Li Q, Roth BL, O’Donnell P, Didriksen M, Dolmetsch R et al. Translating genome-wide association findings into new therapeutics for psychiatry. Nat Neurosci 2016; 19(11): 1392–1396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kathiresan S Developing Medicines That Mimic the Natural Successes of the Human Genome: Lessons From NPC1L1, HMGCR, PCSK9, APOC3, and CETP. Journal of the American College of Cardiology 2015; 65(15): 1562–1566. [DOI] [PubMed] [Google Scholar]
- 50.Lahey BB, Applegate B, Hakes JK, Zald DH, Hariri AR, Rathouz PJ. Is there a general factor of prevalent psychopathology during adulthood? J Abnorm Psychol 2012; 121(4): 971–977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kerekes N, Brandstrom S, Lundstrom S, Rastam M, Nilsson T, Anckarsater H. ADHD, autism spectrum disorder, temperament, and character: phenotypical associations and etiology in a Swedish childhood twin study. Compr Psychiatry 2013; 54(8): 1140–1147. [DOI] [PubMed] [Google Scholar]
- 52.Plomin R, Haworth CM, Davis OS. Common disorders are quantitative traits. Nat Rev Genet 2009; 10(12): 872–878. [DOI] [PubMed] [Google Scholar]
- 53.Larsson H, Anckarsater H, Rastam M, Chang Z, Lichtenstein P. Childhood attention-deficit hyperactivity disorder as an extreme of a continuous trait: a quantitative genetic study of 8,500 twin pairs. J Child Psychol Psychiatry 2012; 53(1): 73–80. [DOI] [PubMed] [Google Scholar]
- 54.Clarke TK, Lupton MK, Fernandez-Pujals AM, Starr J, Davies G, Cox S et al. Common polygenic risk for autism spectrum disorder (ASD) is associated with cognitive ability in the general population. Mol Psychiatry 2016; 21(3): 419–425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Groen-Blokhuis MM, Middeldorp CM, Kan KJ, Abdellaoui A, van Beijsterveldt CE, Ehli EA et al. Attention-deficit/hyperactivity disorder polygenic risk scores predict attention problems in a population-based sample of children. J Am Acad Child Adolesc Psychiatry 2014; 53(10): 1123–1129 e1126. [DOI] [PubMed] [Google Scholar]
- 56.Robinson EB, St Pourcain B, Anttila V, Kosmicki JA, Bulik-Sullivan B, Grove J et al. Genetic risk for autism spectrum disorders and neuropsychiatric variation in the general population. Nat Genet 2016; 48(5): 552–555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Germine L, Robinson EB, Smoller JW, Calkins ME, Moore TM, Hakonarson H et al. Association between polygenic risk for schizophrenia, neurocognition and social cognition across development. Transl Psychiatry 2016; 6(10): e924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Insel T, Cuthbert B, Garvey M, Heinssen R, Pine DS, Quinn K et al. Research domain criteria (RDoC): toward a new classification framework for research on mental disorders. Am J Psychiatry 2010; 167(7): 748–751. [DOI] [PubMed] [Google Scholar]
- 59.Insel TR. Director’s Blog: Transforming diagnosis. https://www.nimh.nih.gov/about/directors/thomas-insel/blog/2013/transforming-diagnosis.shtml 2013.
- 60.Hyman SE. The diagnosis of mental disorders: the problem of reification. Annu Rev Clin Psychol 2010; 6: 12.11–12.25. [DOI] [PubMed] [Google Scholar]
- 61.Kendler KS. Levels of explanation in psychiatric and substance use disorders: implications for the development of an etiologically based nosology. Mol Psychiatry 2012; 17(1): 11–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Duncan L, Yilmaz Z, Gaspar H, Walters R, Goldstein J, Anttila V et al. Significant Locus and Metabolic Genetic Correlations Revealed in Genome-Wide Association Study of Anorexia Nervosa. Am J Psychiatry 2017: appiajp201716121402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Robins E, Guze S. Establishment of diagnostic validity in psychiatric illness: its application to schizophrenia. Am J Psychiatry 1970; 126: 983–987. [DOI] [PubMed] [Google Scholar]
- 64.Kendler KS. Toward a scientific psychiatric nosology. Strengths and limitations. Archives of general psychiatry 1990; 47(10): 969–973. [DOI] [PubMed] [Google Scholar]
- 65.Tsuang M, Faraone S, Lyons M. Identification of the phenotype in psychiatric genetics. European archives of psychiatry and clinical neuroscience 1993; 243: 131–142. [DOI] [PubMed] [Google Scholar]
- 66.Kendler KS. Reflections on the relationship between psychiatric genetics and psychiatric nosology. Am J Psychiatry 2006; 163(7): 1138–1146. [DOI] [PubMed] [Google Scholar]
- 67.Kendler KS. An historical framework for psychiatric nosology. Psychol Med 2009; 39(12): 1935–1941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Scassellati C, Bonvicini C, Faraone SV, Gennarelli M. Biomarkers and attention-deficit/hyperactivity disorder: a systematic review and meta-analyses. J Am Acad Child Adolesc Psychiatry 2012; 51(10): 1003–1019 e1020. [DOI] [PubMed] [Google Scholar]
- 69.Okser S, Pahikkala T, Airola A, Salakoski T, Ripatti S, Aittokallio T. Regularized machine learning in the genetic prediction of complex traits. PLoS Genet 2014; 10(11): e1004754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Wray NR, Yang J, Hayes BJ, Price AL, Goddard ME, Visscher PM. Pitfalls of predicting complex traits from SNPs. Nat Rev Genet 2013; 14(7): 507–515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Levinson DF, Mostafavi S, Milaneschi Y, Rivera M, Ripke S, Wray NR et al. Genetic studies of major depressive disorder: why are there no genome-wide association study findings and what can we do about it? Biol Psychiatry 2014; 76(7): 510–512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Holland D, Wang Y, Thompson WK, Schork A, Chen CH, Lo MT et al. Estimating Effect Sizes and Expected Replication Probabilities from GWAS Summary Statistics. Front Genet 2016; 7: 15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Zuk O, Schaffner SF, Samocha K, Do R, Hechter E, Kathiresan S et al. Searching for missing heritability: designing rare variant association studies. Proc Natl Acad Sci U S A 2014; 111(4): E455–464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Moutsianas L, Agarwala V, Fuchsberger C, Flannick J, Rivas MA, Gaulton KJ et al. The power of gene-based rare variant methods to detect disease-associated variation and test hypotheses about complex disease. PLoS Genet 2015; 11(4): e1005165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Sullivan PF, Agrawal A, Bulik CM, Andreassen OA, Borglum A, Breen G et al. Psychiatric genomics: An update and an agenda. BiorXiv 2017; 10.1101/115600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Boyle EA, Li YI, Pritchard JK. An Expanded View of Complex Traits: From Polygenic to Omnigenic. Cell 2017; 169(7): 1177–1186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Anttila V, Bulik-Sullivan B, Finucane H, Bras J, Duncan L, Escott-Price V et al. Analysis of shared heritability in common disorders of the brain. BioRXiv 2016; 10.1101/048991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Smoller JW. The use of electronic health records for psychiatric phenotyping and genomics. Am J Med Genet B Neuropsychiatr Genet 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Sudlow C, Gallacher J, Allen N, Beral V, Burton P, Danesh J et al. UK biobank: an open access resource for identifying the causes of a wide range of complex diseases of middle and old age. PLoS Med 2015; 12(3): e1001779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Precision Medicine Initiative (PMI) Working Group. The Precision Medicine Initiative Cohort Program – Building a Research Foundation for 21st Century Medicine2015 September 17, 2015. [Google Scholar]
- 81.Bush WS, Oetjens MT, Crawford DC. Unravelling the human genome-phenome relationship using phenome-wide association studies. Nat Rev Genet 2016; 17(3): 129–145. [DOI] [PubMed] [Google Scholar]
- 82.Riglin L, Collishaw S, Thapar AK, Dalsgaard S, Langley K, Smith GD et al. Association of Genetic Risk Variants With Attention-Deficit/Hyperactivity Disorder Trajectories in the General Population. JAMA Psychiatry 2016; 73(12): 1285–1292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Gulsuner S, Walsh T, Watts AC, Lee MK, Thornton AM, Casadei S et al. Spatial and temporal mapping of de novo mutations in schizophrenia to a fetal prefrontal cortical network. Cell 2013; 154(3): 518–529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Willsey AJ, Sanders SJ, Li M, Dong S, Tebbenkamp AT, Muhle RA et al. Coexpression networks implicate human midfetal deep cortical projection neurons in the pathogenesis of autism. Cell 2013; 155(5): 997–1007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Niarchou M, Zammit S, Lewis G. The Avon Longitudinal Study of Parents and Children (ALSPAC) birth cohort as a resource for studying psychopathology in childhood and adolescence: a summary of findings for depression and psychosis. Soc Psychiatry Psychiatr Epidemiol 2015; 50(7): 1017–1027. [DOI] [PubMed] [Google Scholar]
- 86.Mors O, Perto GP, Mortensen PB. The Danish Psychiatric Central Research Register. Scand J Public Health 2011; 39(7 Suppl): 54–57. [DOI] [PubMed] [Google Scholar]
- 87.Lee PH, Baker JT, Holmes AJ, Jahanshad N, Ge T, Jung JY et al. Partitioning heritability analysis reveals a shared genetic basis of brain anatomy and schizophrenia. Mol Psychiatry 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Bearden CE, Thompson PM. Emerging Global Initiatives in Neurogenetics: The Enhancing Neuroimaging Genetics through Meta-analysis (ENIGMA) Consortium. Neuron 2017; 94(2): 232–236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Cuthbert BN. Research Domain Criteria: toward future psychiatric nosologies. Dialogues Clin Neurosci 2015; 17(1): 89–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Evans DM, Davey Smith G. Mendelian Randomization: New Applications in the Coming Age of Hypothesis-Free Causality. Annu Rev Genomics Hum Genet 2015; 16: 327–350. [DOI] [PubMed] [Google Scholar]
- 91.Emdin CA, Khera AV, Natarajan P, Klarin D, Zekavat SM, Hsiao AJ et al. Genetic Association of Waist-to-Hip Ratio With Cardiometabolic Traits, Type 2 Diabetes, and Coronary Heart Disease. JAMA 2017; 317(6): 626–634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Voight BF, Peloso GM, Orho-Melander M, Frikke-Schmidt R, Barbalic M, Jensen MK et al. Plasma HDL cholesterol and risk of myocardial infarction: a mendelian randomisation study. Lancet 2012; 380(9841): 572–580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Vaucher J, Keating BJ, Lasserre AM, Gan W, Lyall DM, Ward J et al. Cannabis use and risk of schizophrenia: a Mendelian randomization study. Mol Psychiatry 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Pickrell JK, Berisa T, Liu JZ, Segurel L, Tung JY, Hinds DA. Detection and interpretation of shared genetic influences on 42 human traits. Nat Genet 2016; 48(7): 709–717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Schaefer GB, Mendelsohn NJ, Professional P, Guidelines C. Clinical genetics evaluation in identifying the etiology of autism spectrum disorders: 2013 guideline revisions. Genet Med 2013; 15(5): 399–407. [DOI] [PubMed] [Google Scholar]
- 96.Samocha KE, Robinson EB, Sanders SJ, Stevens C, Sabo A, McGrath LM et al. A framework for the interpretation of de novo mutation in human disease. Nat Genet 2014; 46(9): 944–950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Robinson EB, Samocha KE, Kosmicki JA, McGrath L, Neale BM, Perlis RH et al. Autism spectrum disorder severity reflects the average contribution of de novo and familial influences. Proc Natl Acad Sci U S A 2014; 111(42): 15161–15165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Zhu X, Need AC, Petrovski S, Goldstein DB. One gene, many neuropsychiatric disorders: lessons from Mendelian diseases. Nat Neurosci 2014; 17(6): 773–781. [DOI] [PubMed] [Google Scholar]
- 99.Davis LK, Yu D, Keenan CL, Gamazon ER, Konkashbaev AI, Derks EM et al. Partitioning the heritability of Tourette syndrome and obsessive compulsive disorder reveals differences in genetic architecture. PLoS Genet 2013; 9(10): e1003864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Lichtenstein P, Carlstrom E, Rastam M, Gillberg C, Anckarsater H. The genetics of autism spectrum disorders and related neuropsychiatric disorders in childhood. The American journal of psychiatry 2010; 167(11): 1357–1363. [DOI] [PubMed] [Google Scholar]
- 101.Polderman TJ, Benyamin B, de Leeuw CA, Sullivan PF, van Bochoven A, Visscher PM et al. Meta-analysis of the heritability of human traits based on fifty years of twin studies. Nat Genet 2015; 47(7): 702–709. [DOI] [PubMed] [Google Scholar]
- 102.Song J, Bergen SE, Kuja-Halkola R, Larsson H, Landen M, Lichtenstein P. Bipolar disorder and its relation to major psychiatric disorders: a family-based study in the Swedish population. Bipolar Disord 2015; 17(2): 184–193. [DOI] [PubMed] [Google Scholar]
- 103.Cardno AG, Rijsdijk FV, Sham PC, Murray RM, McGuffin P. A twin study of genetic relationships between psychotic symptoms. Am J Psychiatry 2002; 159(4): 539–545. [DOI] [PubMed] [Google Scholar]
- 104.McGuffin P, Rijsdijk F, Andrew M, Sham P, Katz R, Cardno A. The heritability of bipolar affective disorder and the genetic relationship to unipolar depression. Arch Gen Psychiatry 2003; 60(5): 497–502. [DOI] [PubMed] [Google Scholar]
- 105.Bulik CM, Thornton LM, Root TL, Pisetsky EM, Lichtenstein P, Pedersen NL. Understanding the relation between anorexia nervosa and bulimia nervosa in a Swedish national twin sample. Biological psychiatry 2010; 67(1): 71–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Mathews CA, Grados MA. Familiality of Tourette syndrome, obsessive-compulsive disorder, and attention-deficit/hyperactivity disorder: heritability analysis in a large sib-pair sample. Journal of the American Academy of Child and Adolescent Psychiatry 2011; 50(1): 46–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Cederlof M, Thornton LM, Baker J, Lichtenstein P, Larsson H, Ruck C et al. Etiological overlap between obsessive-compulsive disorder and anorexia nervosa: a longitudinal cohort, multigenerational family and twin study. World Psychiatry 2015; 14(3): 333–338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Tick B, Bolton P, Happe F, Rutter M, Rijsdijk F. Heritability of autism spectrum disorders: a meta-analysis of twin studies. J Child Psychol Psychiatry 2016; 57(5): 585–595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Stein MB, Jang KL, Taylor S, Vernon PA, Livesley WJ. Genetic and environmental influences on trauma exposure and posttraumatic stress disorder symptoms: a twin study. Am J Psychiatry 2002; 159(10): 1675–1681. [DOI] [PubMed] [Google Scholar]