

Abstract
Clostridium difficile is an opportunistic pathogen that establishes in the colon when the gut microbiota is disrupted by antibiotics or disease. C. difficile infection (CDI) is largely caused by two virulence factors TcdA and TcdB. Here, we report a 3.87 Å resolution crystal structure of TcdB holotoxin that captures a unique conformation of TcdB at endosomal pH. Complementary biophysical studies suggest that the CROPs domain of TcdB is dynamic and can sample open and closed conformations that may facilitate modulation of TcdB activity in response to environmental and cellular cues during intoxication. Furthermore, we report three crystal structures of TcdB–antibody complexes that reveal how antibodies could specifically inhibit the activities of individual TcdB domains. Our studies provide novel insights into the structure and function of TcdB holotoxin and identify intrinsic vulnerabilities that could be exploited to develop new therapeutics and vaccines for the treatment of CDI.
Clostridium difficile is classified as one of the top three urgent antibiotic resistance threats by Center for Disease Control and Prevention (CDC). The pathology of CDI is primarily mediated by two homologous exotoxins, TcdA and TcdB, which target and disrupt the colonic epithelium, leading to diarrhea and colitis 1–5. While the relative roles of these two toxins in the pathogenesis of CDI are not completely understood, recent studies showed that TcdB is more virulent than TcdA and more important for inducing the host inflammatory and innate immune responses 5–8. Notably an anti-TcdB neutralizing antibody (bezlotoxumab) was recently approved by the US Food and Drug Administration (FDA) as a prevention against recurrent infection 9. However, more effective therapies for CDI are desperately needed.
TcdA (~308 kDa) and TcdB (~270 kDa) contain four functional domains: an N-terminal glucosyltransferase domain (GTD), a cysteine protease domain (CPD), a central delivery and receptor-binding domain (DRBD), and a C-terminal combined repetitive oligopeptides (CROPs) domain (Fig. 1a). Toxins bind to cell surface receptors via the DRBD and the CROPs, and enter the cells through endocytosis 10–14. Acidification in the endosome triggers conformational changes in the toxins that prompt the DRBD to form a pore and deliver the GTD and the CPD across the endosomal membrane 15–18.
Figure 1.
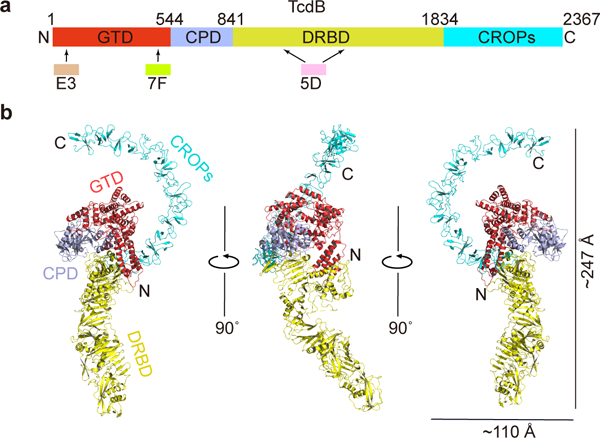
Overall structure of the full length TcdB holotoxin. (a) A schematic diagram showing the domain organization of TcdB and the approximate VHH-binding regions. GTD, glucosyltransferase domain (red); CPD, cysteine protease domain (light blue); DRBD, delivery and receptor-binding domain (yellow); CROPs, combined repetitive oligopeptides domain (blue). (b) Cartoon representations of TcdB holotoxin. The 3 VHHs that were used to facilitate crystallization were omitted for clarity. TcdB domains are colored as in panel (a).
In the cytosol, the CPD is activated by eukaryotic-specific inositol hexakisphosphate (InsP6) and subsequently undergoes autoproteolysis to release the GTD. The GTD then glucosylates small GTPases of the Rho family, including Rho, Rac, and Cdc42, and inhibits their function 19–23. Numerous structures have been reported for fragments of TcdA and TcdB, which have provided tremendous insights into the functions of these toxin domains 10,12,13,24–28. However, it remains unknown how individual domains interact within the supertertiary structure of the holotoxin, and how the holotoxin dynamically responds in a precise stepwise manner to the environmental and cellular cues, such as low pH and InsP6, which lead to intoxication.
Here we report the crystal structure of TcdB holotoxin at 3.87 Å resolution, which was crystallized at an endosomal pH (pH 5.2) and displays an architecture that is distinct from the prior model derived from electron microscopy (EM) 29. To probe the structural dynamics of TcdB in solution, we used a combination of small-angle X-ray scattering (SAXS), single-molecule fluorescence resonance energy transfer (smFRET), and cross-linking mass spectrometry (XL-MS). These experiments consistently demonstrate that the elongated CROPs of TcdB displays pH-dependent structural flexibility, which may help TcdB to modulate its activity in response to environmental pH change. Furthermore, we reveal novel structural mechanisms underlying neutralization of TcdB by three antibodies. These findings collectively reveal new strategies for developing therapeutics and vaccines for the treatment of CDI.
RESULTS
Crystal structure of the full length TcdB
The full length TcdB holotoxin from the M68 strain of C. difficile was expressed using Bacillus megaterium 30. Large molecular weight, multi-domain proteins like TcdB are notoriously difficult to crystallize, and its structural flexibility has also hampered 3D analysis by EM 29. To facilitate crystal packing, we screened a panel of TcdB-binding VHHs, the antigen-binding region (VH) of the heavy-chain-only antibodies (also known as nanobodies or single-domain antibodies, sdAbs). The best X-ray diffraction data were collected at 3.87 Å resolution on a crystal of a heterotetrameric complex composed of TcdB and three neutralizing VHHs (5D, E3, and 7F) 18,31. The TcdB–VHH complex was crystallized at pH 5.2, which is a physiologically relevant pH in an endosome (Fig. 1a–b and Table 1).
Table 1.
Data collection and refinement statistics
| TcdB–5D–E3–7F (PDB 6OQ5) |
TcdB1072–1433–5D (PDB 6OQ6) |
GTD–E3 (PDB 6OQ7) |
GTDVPI10463–7F (PDB 6OQ8) |
|
|---|---|---|---|---|
| Data collection | ||||
| Space group | P212121 | P6122 | P212121 | P43212 |
| Cell dimensions | ||||
| a, b, c (Å) | 149.62, 168.56, 179.92 | 169.82, 169.82, 79.12 | 66.02, 104.10, 113.83 | 114.71, 114.71, 301.01 |
| α, β, γ (°) | 90, 90, 90 | 90, 90, 120 | 90, 90, 90 | 90, 90, 90 |
| Resolution (Å) | 48.91 – 3.87 (3.97 – 3.87) * |
147.07 – 2.97 (3.14 – 2.97) |
43.14 – 2.39 (2.45 – 2.39) |
107.19 – 2.20 (2.26 – 2.20) |
| Rmerge | 0.14 (>1) | 0.15 (>1) | 0.14 (0.5) | 0.11 (0.63) |
| I / σI | 7.7 (0.7) | 12. 5 (1.3) | 14.6 (3.3) | 19.2 (4.6) |
| CC1/2 | 0.998 (0.327) | 0.998 (0.810) | 0.987 (0.927) | 0.999 (0.929) |
| Completeness (%) | 99.4 (99.3) | 99.9 (99.6) | 99.7 (99.9) | 99.6 (99.3) |
| Redundancy | 3.7 (3.6) | 8.6(8.5) | 7.1 (7.1) | 13.4 (13.5) |
| Refinement | ||||
| Resolution (Å) | 48.91 – 3.87 (3.97 – 3.87) |
147.07 – 2.97 (3.05 – 2.97) |
43.14 – 2.39 (2.45 – 2.39) |
107.19 – 2.20 (2.26 – 2.20) |
| No. reflections | 42,865 | 12,367 | 30,060 | 102,252 |
| Rwork / Rfree | 26.3/31.5 | 25.9/27.4 | 19.7/23.6 | 19.7/22.0 |
| No. atoms | ||||
| Protein | 21,503 | 3,719 | 5,229 | 10,757 |
| Ligand/ion | 2 a | - | 39 b | - |
| Water | - | - | 100 | 1,001 |
| B-factors | ||||
| Protein | 169.7 | 57.3 | 50.4 | 32.3 |
| Ligand/ion | 119.8 | - | 47.3 | - |
| Water | - | - | 49.14 | 38.2 |
| R.m.s deviations | ||||
| Bond lengths (Å) | 0.003 | 0.003 | 0.003 | 0.003 |
| Bond angles (°) | 1.097 | 1.216 | 0.998 | 0.898 |
Values in parentheses are for highest-resolution shell.
The 2 atoms are 1 Zn2+ and 1 Mg2+.
The 39 atoms include 1 UDP (25 atoms), 1 Glucose (12 atoms), 1 Mn2+ and 1 Mg2+.
Each dataset was derived from a single crystal.
We solved the crystal structure of the TcdB–VHH complex using molecular replacement (Methods). A complete structure of TcdB holotoxin was built except for two small regions (residues 944–949 and 1032–1047) that have no visible electron density due to high structural flexibility (Fig. 1b and Supplementary Fig. 1a–d). We further validated this structure using an anomalous difference electron density map generated from a crystal of the TcdB–VHH complex soaked in tantalum bromide (Supplementary Fig. 1e). E3 and 7F both bind to the GTD, while 5D binds to the DRBD (Supplementary Fig. 1f). This was further confirmed by three crystal structures of GTD–E3, GTDVPI10463 –7F, and TcdB1072–1433–5D complexes, which we determined at 2.39 Å, 2.20 Å, and 2.97 Å resolution, respectively (Table 1).
The crystal structure reveals that TcdB is composed of three major components. The GTD and CPD form the center piece involving extensive inter-domain interactions. The DRBD forms an extended module, interacting with both the GTD and the CPD on one side and pointing away from GTD/CPD. The most prominent finding is the elongated CROPs domain, which emerges from the junction of the CPD and the DRBD and stretches ~130 Å in the opposite direction to curve around the GTD like a hook (Fig. 1b). The overall architecture of TcdB at endosomal pH is distinct from structural models of TcdB and TcdA that were derived from a negative stain EM study at neutral pH, where the CROPs lies in parallel to and interacts with the DRBD (Supplementary Fig. 1g) 29. Furthermore, we observed that a portion of the pore-forming region in the DRBD (residues 957–1129) adopts a conformation that is different from TcdA at neutral pH 24. This likely represents a rarely seen intermediate state of TcdB in response to endosomal pH, which is “frozen” by a neutralizing antibody (5D) and will be discussed further below.
The unique structure of the CROPs domain
The CROPs of TcdB is composed of two types of repetitive sequences including twenty short repeats (SRs) of 20–23 residues and four long repeats (LRs) of 30 residues (Fig. 2a and Supplementary Fig. 2a). Each SR consists of a β-hairpin followed by a flexible loop, while each LR has three β-strands that form a twisted anti-parallel β-sheet together with the β-hairpin of the preceding SR. Neighboring SRs are packed together into a ~31 screw axis with ~120° rotation between SRs, which creates a left-handed β-solenoid superhelix 32,33. The curvature of the CROPs arises because the straight, rod-like segments of the β-solenoid composed of SRs are interrupted by the interspersed LRs, which cause a ∼132–146° kink (Fig. 2b–c). Structurally, the CROPs could be divided into four equivalent units, and superposition of CROPs I–IV yielded a Cα root-mean square deviation (r.m.s.d.) of ~ 0.9–2.6 Å (Fig. 2c).
Figure 2.
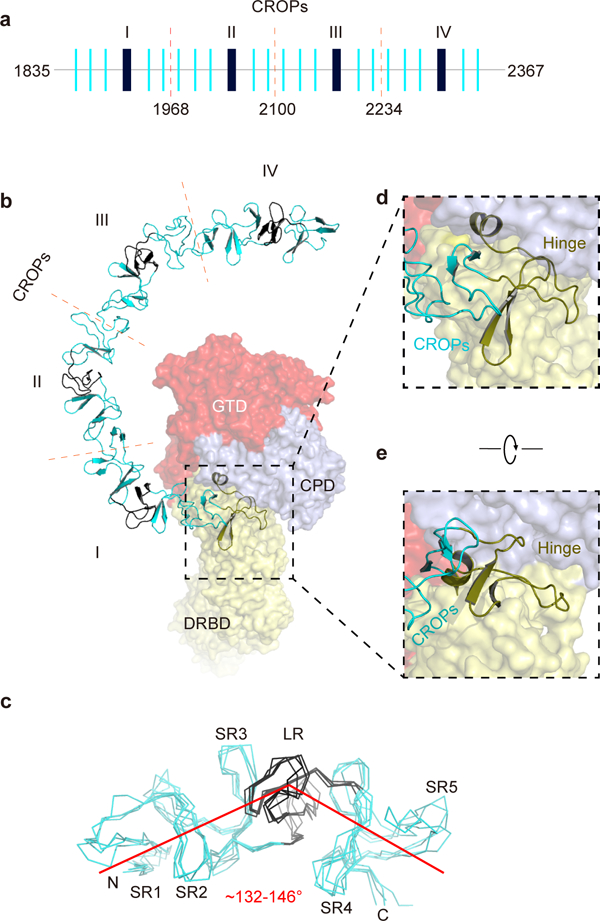
The unique structure of the CROPs of TcdB. (a) A schematic diagram of the CROPs showing the organization of the short repeats (SRs, thin blue bars) and the long repeats (LRs, thick black bars). The dashed red lines indicate the boundaries of four CROPs units (I–IV). (b) A close-up view into the CROPs while the remainder of TcdB is in a surface representation. (c) Superposition of the 4 CROPs units. The LR in each CROPs unit causes a ~132–146° kink. (d, e) The hinge region (colored olive), which connects the CROPs to the rest of the toxin, is located at the center of TcdB and surrounded by the GTD, the CPD, and the DRBD.
Interestingly, we identified an unrecognized SR module (residues 1815–1834) at the C-terminus of the DRBD. This new SR, together with an upstream long loop and a short α helix, form a structurally distinct module (residues 1792–1834), which we refer to as the “hinge” because it connects the DRBD to the elongated CROPs. Furthermore, the hinge directly interacts with a three-stranded β-sheet in the CPD (residues 742–765, termed the β-flap) that is crucial for CPD activation 27, as well as a 3-helical bundle (residues 766–841, referred to as 3-HB) that is located in a crevice surrounded by GTD, CPD, DRBD, and CROPs (Fig. 2d–e and Supplementary Fig. 2c). Because of its strategic location, this hinge is primed to mediate structural communications among all four domains of TcdB. A functional role for this hinge is supported by our earlier studies showing that deletions in this area drastically reduced the toxicity of TcdB 34. Additionally, hypervariable sequences near the hinge may contribute to differences in toxicity and antigenicity displayed by TcdB variants produced by the hypervirulent C. difficile 027 ribotype and other less virulent strains 35–37 (Supplementary Fig. 2b).
Differences in the conformation of TcdB at neutral and acidic pH
To examine the solution structure of TcdB holotoxin, we carried out online size-exclusion chromatography coupled to SAXS (SEC-SAXS) for TcdB at pH 5.0 and pH 7.4 (Supplementary Table 1 and Supplementary Fig. 3a–c). Curve-fit analysis showed that the calculated scattering profile from our crystal structure is well fit to the experimental scattering profile at pH 5.0 (Fig. 3a), suggesting that the structure of TcdB in solution is similar to the crystal structure at pH 5.0. However, the calculated profile for the crystal structure disagreed with the experimental SAXS data at pH 7.4, particularly at the middle-angle (middle q) region of the scattering profile (Fig. 3a), which suggests that TcdB samples different conformations at neutral pH 38.
Figure 3.
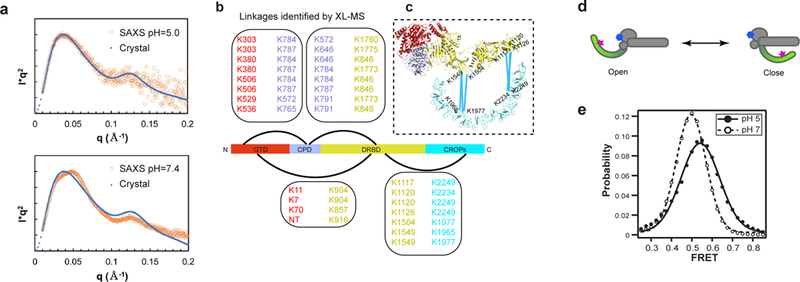
The CROPs undergoes pH-dependent conformational changes. (a) Curve-fit analysis in SAXS studies. The theoretical Kratky plot based on the structure of TcdB holotoxin agreed with the experimental scattering profile at pH 5.0 (upper panel), but differed from that at pH 7.4 (lower panel). (b) Cross-linked peptides between different TcdB domains were identified by XL-MS. (c) The XL-MS results suggest that TcdB could sample a closed conformation at neutral pH, where the central portion and the C-terminal tip of the CROPs move within ~30 Å of the DRBD. (d) A model of the two limiting structure states of TcdB holotoxin. The acceptor dye on the GTD-bound 7F and the donor dye on the CROPs-bound B39 are shown as a blue hexagon and a red star, respectively. (e) Population histogram of unaveraged FRET efficiency from TcdB in complex with dye-labeled VHHs at pH 5.0 (n = 498) and pH 7.0 (n = 594).
Guinier and P(r) analyses showed similar Rg values at pH 5.0 and 7.4. However Dmax at pH 5.0 (~ 233.0 Å) was longer than that at pH 7.4 (~ 205.0 Å). The Dmax at pH 5.0 is comparable to the value predicted from our crystal structure (~247 Å). The shorter Dmax at pH 7.4 is comparable to the value predicted for the TcdB core that is composed of the GTD, CPD, and DRBD (~203 Å) (Supplementary Fig. 3d–e). This suggests that the elongated CROPs may swing towards the TcdB core at neutral pH, which would shorten the maximum dimension.
To better characterize the conformation of the CROPs at pH 7.4, we employed XL-MS to determine inter-domain interactions of TcdB using MS-cleavable cross-linker DSSO (disuccinimidyl sulfoxide) 39 Lysine residues on the surface of TcdB that can approach within 30 Å Cα–Cα distance are preferentially cross-linked and identified by multistage tandem MS 39,40 (Supplementary Fig. 4a–c). In total, we identified 87 intra-molecular cross-links in TcdB at pH 7.4, representing 27 inter-domain and 60 intra-domain interactions (Fig. 3b, Supplementary Note 1, and Supplementary Data 1). When the XL-MS data was mapped to our crystal structure, almost all of these cross-links satisfy the distance cutoff of 30 Å, indicating a good correlation with the crystal structure of TcdB (Supplementary Fig. 4d).
Interestingly, we identified 7 pairs of cross-linked peptides between the CROPs and the DRBD. Based on our crystal structures, the Cα–Cα distances between these cross-linked lysine residues range between 90 Å and 210 Å (Supplementary Fig. 4d). This suggested that the CROPs of TcdB, especially the central portion of the CROPs around residues K1965 and K1977 and the C-terminal tip of the CROPs around residues K2234 and K2249, were able to move within ~30 Å of the DRBD (Fig. 3c). Closing of the CROPs is consistent with the shorter Dmax of TcdB derived from SAXS at pH 7.4, and similar to the closed conformation of TcdB and TcdA observed in the EM study (Supplementary Fig. 1g) 29. Since XL-MS captures dynamic and transient contacts in addition to stable structures, the time that the CROPs spends in a closed conformation at neutral pH remains unknown.
pH-dependent structural flexibility of the CROPs
We used smFRET to further probe the conformation of the CROPs 41. We used three VHHs (7F, B39, and 5D) as molecular tools to label and capture TcdB in order not to modify its endogenous cysteine residues. Specifically, we attached the acceptor dye (Alexa-647) to 7F, which labels the core of TcdB holotoxin, and attached the donor dye (Alexa-555) to B39, which specifically binds to the CROPs IV 25. Biotin-labeled 5D was used for immuno-pulldown of TcdB onto a microscope slide, which has no effect on TcdB conformational change based on an ensemble FRET study (Supplementary Fig. 4e). In the structure of TcdB holotoxin at endosomal pH, the distance between dyes is ~47 Å, which would give FRET efficiency near 0.5. Any movement of the CROPs would affect energy transfer between these two dye-labeled VHHs (Fig. 3d). A structural modeling suggested that we would expect no FRET if TcdB holotoxin stably adopts a closed conformation.
For FRET analysis, we only selected those complexes containing a single donor and acceptor dye that both photobleached to background during observation (Supplementary Fig. 4f and Supplementary Table 2) 42. We observed single FRET peaks for the heterotetrameric TcdB–VHH complexes at both acidic and neutral pH (Fig. 3e and Supplementary Fig. 4g). A simple calculation from the mean FRET efficiency at pH 5.0 (0.532 ± 0.015) gives an estimated distance of 49.9 ± 0.05 Å between the dye-labeled VHHs, which is consistent with the crystal structure of TcdB holotoxin at acidic pH (~47 Å). Similar results were observed at pH 5.5 and pH 5.25 (Supplementary Fig. 4h).
At neutral pH, we observed a slight decrease in mean FRET efficiency (0.484 ± 0.007) and a 25% decrease in the distribution width at pH 7.0 (0.113 ± 0.002) relative to pH 5.0 (0.141 ± 0.026) (Fig. 3e). Simple calculation based on FRET suggests a slight distance increase to 51.5 ± 0.05 Å at neutral pH, which is accompanied by a dramatic increase in the rate of conformational dynamics. We note that a single FRET pair is insufficient to position the CROPs relative to the rest of TcdB, and changes in conformational dynamics could affect the simple conversion of FRET to distance. However, this smFRET data suggests that the CROPs does not stably occupy a closed conformation at neutral pH under the conditions tested.
Thus far, we have identified two limiting structural states for TcdB: an open conformation revealed by the crystal structure at acidic pH and a closed conformation captured by XL-MS at neutral pH (Fig. 3d). Combined with our SAXS and smFRET studies, these data collectively suggest that the CROPs predominantly occupies the open state at acidic pH, but dynamically samples an ensemble of conformations relative to the core of TcdB at neutral pH. Such protein dynamics are faster than our 100 ms integration time in smFRET, so motions between open and closed conformations would be time averaged into a single peak 43. The lack of stabilizing contacts between the CROPs and the TcdB core and the potential structural rearrangement in the hinge that connects the DRBD and the CROPs should permit such conformational sampling.
A pore-forming intermediate state of TcdB at endosomal pH
The DRBD serves to protect the hydrophobic pore-forming region (residues 957–1129), which is predicted to be released upon endosome acidification in order to form a pore that delivers the GTD and the CPD to the cytosol. The pore forming activity of TcdB also contributes to cell necrosis observed in vitro 16,44. A structural comparison between TcdB holotoxin at acidic pH and a TcdA fragment at neutral pH reveals drastic differences in the homologous C-terminal portion of the pore-forming region (residues 1032–1093 in TcdB) (Fig. 4a–b). In TcdA, this region adopts a mixed α/β configuration, where hydrophobic residues are shielded in a continuous groove formed mostly by β-sheets in the DRBD (Fig. 4c–d). However, in the acidic conformation of TcdB, there was no electron density visible for residues 1032–1047, likely due to high flexibility, indicating that these residues unfolded and detached from the toxin core at endosomal pH. Furthermore, TcdB residues equivalent to the α2 in TcdA unfolded into a loop, while TcdB residues equivalent to the β3 and part of the α3 in TcdA assembled into a new helix that occupied the same area as the original α3 in TcdA. Because of this transition, hydrophobic residues in TcdB (residues 1084–1093), which are equivalent to the C-terminal portion of the α3 helix in TcdA, bulged out as an extended loop. Intriguingly, the conformational change did not spread into the region where TcdB is bound by 5D (residues 1094–1134), which maintains a similar conformation as that observed in TcdA.
Figure 4.
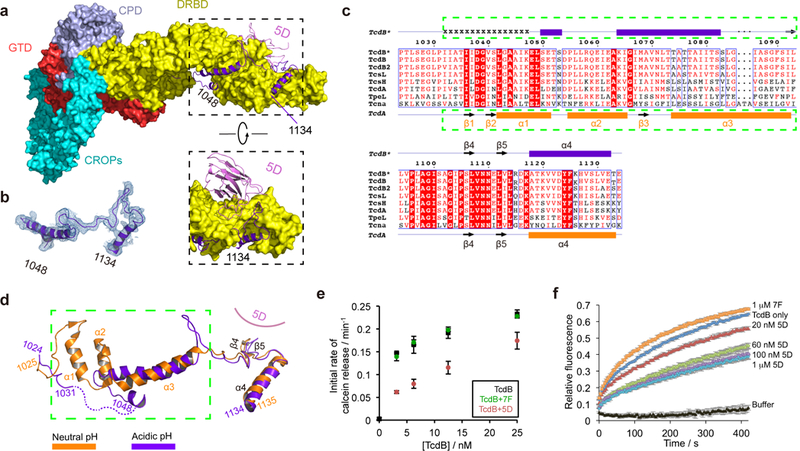
TcdB displays a novel conformation in the pore-forming region at endosomal pH. (a) 5D binds to the DRBD and directly interacts with the pore-forming region. The pore-forming region of TcdB is shown in a purple ribbon model while the rest of the toxin is shown in a surface model. (b) A representative 2Fo-Fc electron density map of a portion of the pore-forming region (residues 1048–1134) contoured at 1.0 σ, which was overlaid with the final refined model. (c) Amino acid sequence alignment of the pore-forming region among different members in the LCGT family. TcdB*, TcdB, and TcdB2 are produced by the M68 strain, the VPI 10463 strain, and the BI/NAP1/027 strain, respectively. Secondary structures of TcdB* and TcdA 24 are shown on the top and the bottom, respectively. Residues 1032–1047 in TcdB* holotoxin that have no visible electron density are indicated by “x”. (d) TcdB at acidic pH (purple) and TcdA at neutral pH (orange) adopt drastically different conformations in the pore-forming region. The two structures were superimposed based on the DRBD. (e) Calcein dye release assay. TcdB (0–25 nM) was tested with liposomes loaded with 50 mM calcein at pH 4.6, in the presence or absence of 5D or 7F. The rate of calcein dye release was determined based on the increase of fluorescence at 525 nm during excitation at 493 nm. (f) Membrane depolarization assay. Liposomes were polarized at a positive internal voltage by adding valinomycin in the presence of a transmembrane KCl gradient. Membrane potential was measured using the voltage-sensitive fluorescence dye ANS (8-anilinonaphthalene-1-sulfonic acid). After 3 min, TcdB with various concentrations of 5D or 7F was added. The data in (e–f) are presented as mean ± SEM, n=3 independent experiments.
We produced TcdB1072–1433 to further examine the effect of acidic pH and 5D on the pore-forming region. Using an environmentally sensitive fluorescent dye 16,35,45, we observed an increase in exposed hydrophobic surface in TcdB1072–1433 upon acidification, which was inhibited by 5D (Supplementary Fig. 5a–b). This is consistent with a pH-triggered conformational change in TcdB that is blocked by 5D. We then determined the crystal structure of TcdB1072–1433 in complex with 5D at pH 8.5 (Table 1), which revealed that its pore-forming region adopts a TcdA-like neutral pH conformation (Supplementary Fig. 5c–d). Together, these findings suggest that the novel conformation observed in the pore-forming region of TcdB likely represents an intermediate state induced by endosomal pH.
Furthermore, we found that the binding mode of 5D to TcdB is almost identical at neutral or acidic pH, in which 5D directly binds P1105, L1107, N1110, and L1112 in the pore-forming region (Supplementary Fig. 5e–f and Supplementary Table 3). Prior mutagenesis studies showed that the pore-forming region around the 5D-binding site in TcdB is crucial for pore formation and cellular toxicity. For example, mutation of L1107 (L1107K), which is located in the β4 and targeted by 5D, caused a >1,000-fold decreased toxicity 16,24. These findings thus suggest that 5D is able to bind to TcdB at both neutral and acidic pH and fix the conformation of β4–β5 in TcdB, which would inhibit the conformational changes necessary for pore formation at endosomal pH. This notion is further supported by observations that TcdB-induced calcein release from liposomes at pH 4.6 was significantly reduced by 5D, and that 5D prevented TcdB from dissipating the valinomycin-induced membrane potential in liposomes (Fig. 4e–f).
Taken together, these findings suggest that 5D neutralizes TcdB 31,46 by preventing the pore-forming region from completing the necessary pH-induced conformational change. Notably, the pore-forming region recognized by 5D is highly conserved among a family of large clostridial glucosylating toxins (LCGTs), which include TcdA and TcdB, C. novyi α-toxin (Tcnα), C. sordellii lethal and hemorrhagic toxins (TcsL and TcsH), and C. perfringens toxin (TpeL) (Fig. 4c) 47. Therefore, this portion of the pore-forming region represents a good target for the development of broad-spectrum vaccines and antibodies targeting TcdA, TcdB, and other LCGTs.
Modulation of autoprocessing of TcdB
Activation of the CPD by InsP6 upon cell entry is a critical step in regulating the pathology of TcdA and TcdB 27,48. The structures of the apo-CPD in TcdB holotoxin and an InsP6-bound CPD fragment are very similar except for the β-flap 11,27,49 (Fig. 5a–b). In the CPD fragment, InsP6 triggers a ~90° rotation of the β-flap (Fig. 5b), which activates the CPD by properly ordering the active site and the substrate pocket 27,49. However, the β-flap partially occupies the P1 substrate pocket of the CPD in TcdB holotoxin, which would prevent substrate binding 50. Furthermore, the InsP6-triggered rotation of the β-flap is prohibited in TcdB holotoxin, because it would otherwise sterically clash with the neighboring 3-HB (Fig. 5c–d).
Figure 5.
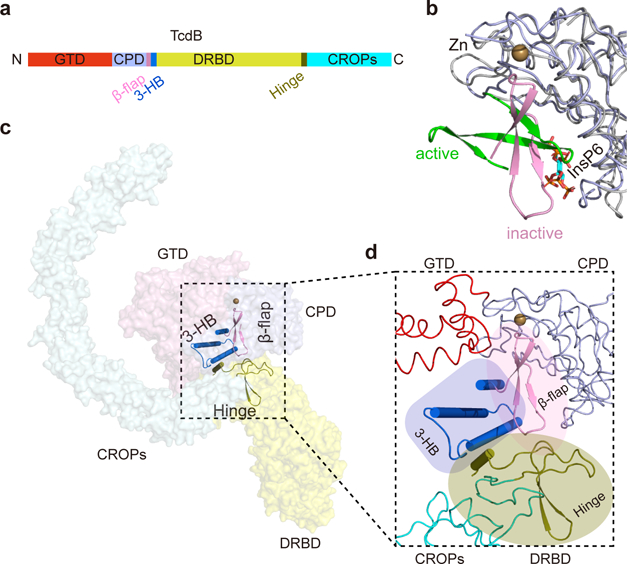
Three modulatory components are clustered at the “heart” of TcdB holotoxin. (a) A schematic diagram showing the locations of the β-flap, the 3-HB, and the hinge in the primary sequence of TcdB. (b) Superposition of the apo CPD in TcdB holotoxin and an InsP6-bound CPD fragment. The overall structure of the CPD remains unchanged (gray coils), while the β-flap displays a ~90° rotation upon InsP6 binding. The zinc atom in the apo CPD is shown as a sphere, and InsP6 is in a stick model. (c, d) The β-flap, the 3-HB, and the hinge co-localize at the center of TcdB, which are well positioned to regulate the interplay among four domains in response to environmental and cellular cues.
Interestingly, we observed a zinc atom in the CPD that simultaneously interacts with the catalytic dyad (H654 and C699), the β-flap (H758), and a residue (D547) near the scissile bond (L544–G545) in the GTD (Supplementary Fig. 6a–c). A similar zinc was observed in TcdA 24, but never seen in any structures of a CPD fragment. As a result, the β-flap in TcdB holotoxin helps to protect the catalytic C699 through Zn–H758 interaction, while the Zn–D547 interaction also prevents the scissile bond from entering the active site. These findings thus provide new insights into how TcdB autoprocessing is inhibited in the holotoxin.
Besides allosteric modulation by InsP6, some studies suggested that the CROPs also affects TcdB autoprocessing 24,51,52. We found that the InsP6-induced cleavage of the GTD was much more efficient in TcdB1–1805, which does not have the hinge or the CROPs, than the holotoxin, suggesting that the CROPs and the hinge help to inhibit the CPD function in TcdB holotoxin (Supplementary Fig. 6d). Furthermore, a previous study showed that TcdA1–1832 that carries the hinge showed a weaker InsP6-dependent cleavage of GTD than TcdA1–1795 without the hinge 24. These data suggest that the hinge is involved in regulation of TcdB and TcdA autoprocessing. Notably, the hinge interacts with the β-flap and the 3-HB in TcdB holotoxin, and they together form the “heart” of TcdB that connects all four domains (Fig. 5c–d). Since the β-flap and the 3-HB are important for coupling between InsP6 binding and CPD activation, structural rearrangement in the hinge, associated with pH-dependent movement of the CROPs, could contribute to the regulation of CPD function.
VHH 7F and E3 reveal two distinct neutralizing epitopes on the GTD
7F inhibits GTD cleavage 18, but does not directly interact with the CPD. Instead, 7F binds to the C-terminus of the GTD, immediately juxtaposed to the cleavage site (Supplementary Fig. 5g–i). Notably, the CDR3 of 7F binds to an α helix upstream of the scissile bond and a neighboring α helix with extensive polar and hydrophobic interactions (Supplementary Table 3). Such interactions interfere with the movement of the scissile bond into the CPD cleavage site and a proper orientation of GTD relative to CPD, and thus inhibit GTD cleavage (Supplementary Fig. 6d).
E3 inhibits Rho glucosylation by targeting the GTD 18,31. In two independently solved crystal structures using the GTD fragment or TcdB holotoxin, E3 binds to the N-terminal four-helix bundle (residues 1–90) in a similar manner, involving extensive polar and hydrophobic interactions (Supplementary Fig. 5j and Supplementary Table 3). Since structure of a GTD–Rho complex has not been reported, it remains unknown how E3 may affect GTD–Rho interactions or the catalysis. The homologous four-helix bundle is also found in other LCGTs, which is believed to be involved in plasma membrane binding of the glucosyltransferase domain 11,53–55. It suggests that E3 may interfere with membrane association of the GTD. The structure of the GTD–E3 complex thus lays the foundation for further validating and exploiting of this mechanism as a new strategy to counteract TcdB and potentially other LCGTs.
DISCUSSION
Prior studies of CDI have provided key insights into the activity and pathogenesis of TcdA and TcdB, and numerous structures have been determined for fragments of these toxins 11. However, knowledge of the holotoxin structures is central to bridge the structure-function gap. Our comprehensive analyses of the structure of TcdB holotoxin collectively demonstrate that TcdB has evolved a delicate mechanism to coordinate its four structurally and functionally distinct domains in order to balance the needs for self-protection and timely activation. Notably, the β-flap, the 3-HB, and the hinge are co-localized at the “heart” of TcdB holotoxin, which are well positioned to modulate structural communications among all four domains and coordinate their activities (Fig. 5c–d and Supplementary Fig. 6b–c). Interestingly, TpeL is the only LCGT member that does not possess a CROPs domain or a hinge-like region 56. Furthermore, TpeL has 13 amino acids deleted near the C-terminus of the 3-HB when compared to other LCGTs, which suggests that TpeL may have a different structure in this modulatory area. The structure of TcdB holotoxin thus provides a blueprint to guide future studies into how TcdB and other LCGTs response to environmental and cellular cues during intoxication. Such mechanistic understanding could help to develop new therapeutics that promote extracellular activation of the CPD and premature cleavage of the GTD before cell entry 57.
The conformational dynamics of the CROPs may impact receptor binding of TcdB and therapy. Chondroitin sulfate proteoglycan 4 (CSPG4) is a TcdB receptor that is believed to interact with the DRBD, the hinge, and the CROPs (Supplementary Fig. 7a) 14,58. Notably, two residues essential for CSPG4 binding (Y1824 and N1839) are located in the hinge and the first SR of CROPs 58. Therefore, conformational flexibility of the CROPs and the hinge may affect the engagement of CSPG4 or the release of CSPG4 as TcdB interacts with multiple host receptors during different stages of cell entry 13,14,58. In contrast, Frizzled proteins (FZDs), which are the major TcdB receptors in the colonic epithelium 59, bound to TcdB holotoxin similarly at pH 5 and pH 7.5 based on an in vitro pull-down assay, and were not affected by conformational dynamics of the CROPs (Supplementary Fig. 7b). FZD binds TcdB in a middle portion of the DRBD, and its binding site is adjacent to, but separate from, the pore-forming region 13 (Supplementary Fig. 7c). Thus, FZD binding will help to orient the pore-forming region of TcdB near the membrane. Interestingly, bezlotoxumab, which is believed to affect TcdB binding to host cells, binds TcdB in the CROPs I and II 28,58. However, in the context of TcdB holotoxin at endosomal pH, there is insufficient space near the CROPs I to allow binding, due to steric clashes with the GTD and the DRBD (Supplementary Fig. 7d–e). Therefore, bezlotoxumab may interfere with the conformational change in the CROPs, and the therapeutic relevance of this novel feature of bezlotoxumab is well worth further studies.
Besides unveiling structural weakness in TcdB holotoxin, our studies reveal distinct mechanisms for antibody neutralization of TcdB by inhibiting autoprocessing and activation of the toxin, glucosylation of Rho GTPases, or transmembrane delivery of the toxin. These findings provide the structural basis for antibody engineering to improve their antitoxin activities, or developing multidomain antibodies that simultaneously target multiple neutralizing epitopes on the toxins 31,46,60. Taken together, our studies expose crucial structural and functional vulnerabilities of TcdB, which provide novel avenues for the development of next generation vaccines and therapeutics that have enhanced potency and broad-reactivity across different C. difficile strains.
METHODS
No statistical method was used to predetermine sample size. The experiments were not randomized and were not performed with blinding to the conditions of the experiments.
Cloning, expression, and purification of recombinant proteins
TcdB produced by the M68 strain of C. difficile was used throughout this study. TcdB holotoxin and its GTD (residues 1–543) were expressed as described previously 61. The gene encoding the four VHHs (5D, E3, 7F, and B39) and the GTD of TcdB produced by the VPI 10463 strain (residues 1–542, termed GTDVPI10463), and a truncated DRBD of TcdB (residues 1072–1433, TcdB1072–1433) were cloned into a modified pET28a vector, which has a 6xHis/SUMO (Saccharomyces cerevisiae Smt3p) tag introduced to the N-terminus of all proteins. A TcdB fragment (residues 1–1805, TcdB1–1805) was cloned into a modified pET22b vector, which has a twin-Strep tag introduced between the SUMO tag and TcdB1–1805 and a C-terminal 6xHis tag. All mutants were generated by two-step PCR and verified by DNA sequencing.
5D, E3, 7F, B39, GTDVPI10463, TcdB1–1805 and TcdB1072–1433 were expressed in Escherichia coli strain BL21-Star (DE3) (Invitrogen). Bacteria were cultured at 37°C in LB medium containing kanamycin or ampicillin. The temperature was reduced to 16°C when OD600 reached ~0.8. Expression was induced with 1 mM IPTG (isopropyl-b-D-thiogalactopyranoside) and continued at 16°C overnight. The cells were harvested by centrifugation and stored at −80°C until use.
The His6-tagged TcdB, GTD, and the His6-SUMO-tagged 5D, E3, 7F, B39, GTDVPI10463, TcdB1–1805, and TcdB1072–1433 were purified using Ni2+-NTA (nitrilotriacetic acid, Qiagen) affinity resins in a buffer containing 50 mM Tris, pH 8.5, 400 mM NaCl, and 10 mM imidazole. The proteins were eluted with a high-imidazole buffer (50 mM Tris, pH 8.5, 400 mM NaCl, and 300 mM imidazole) and then dialyzed at 4°C against a buffer containing 20 mM Tris, pH 8.5, 1 mM TCEP, and 40 mM NaCl. The His6-SUMO tag of 5D, E3, 7F, B39, GTDVPI10463, and TcdB1072–1433 were cleaved by SUMO protease. These proteins, as well as TcdB holotoxin and GTD with un-cleaved His-tag, were further purified by MonoQ ion-exchange chromatography (GE Healthcare) in a buffer containing 20 mM Tris, pH 8.5, and eluted with a NaCl gradient. TcdB1–1805, after cleaved by SUMO protease, was further purified using streptavidin resins.
The TcdB–5D–E3–7F complex was assembled by mixing the purified TcdB holotoxin with the 3 purified VHHs at a molar ratio of 1:2:2:2 for 2 hours on ice. The complex was then purified by MonoQ ion-exchange chromatography in 20 mM Tris, pH 8.5, followed by a Superose 6 size-exclusion chromatography (SEC; GE Healthcare) in 20 mM Tris, pH 8.5, 1 mM TCEP, and 40 mM NaCl. The GTD–E3, GTDVPI10463–7F, and TcdB1072–1433–5D complexes were made by mixing the purified GTD, GTDVPI10463, and TcdB1072–1433 with E3, 7F, and 5D at a molar ratio of 1:2, respectively, for 2 hours on ice, followed by further purification using a MonoQ ion-exchange column (20 mM Tris, pH 8.5) and a Superdex-200 Increase SEC (20 mM Tris, pH 8.5, 1 mM TCEP, and 40 mM NaCl). All protein complexes were concentrated to ~10 mg/ml and stored at −80°C until use.
Crystallization
Initial crystallization screens for all 4 protein complexes were carried out at 20°C with a Gryphon crystallization robot (Art Robbins Instruments) using high-throughput crystallization screening kits (Hampton Research and Qiagen). Extensive manual optimizations were then performed at 20°C using the hanging-drop vapor-diffusion method when proteins were mixed with reservoir solution at 1:1 ratio.
The best crystals for the TcdB–5D–E3–7F complex suitable for X-ray diffraction were obtained in a reservoir containing 0.1 M sodium acetate, 0.1M magnesium acetate, and 5% PEG 8K (final pH 5.2) with a protein concentration of ~4 mg/ml. The crystals were cryo-protected in the mother liquor supplemented with 25% (v/v) ethylene glycol. The tantalum bromide cluster-derivatized crystals were obtained by adding a trace amount of tantalum bromide powder to the crystallization drop. The TcdB–5D–E3–7F crystals were soaked in this solution for 1–3 days until the crystals turned green, which were then cryo-protected similarly as native crystals.
The best crystals for the GTD–E3 complex were obtained in a reservoir containing 0.2 M potassium acetate and 20% PEG 3350 with a protein concentration of ~10 mg/ml, which were then cryo-protected in the same mother liquor supplemented with 25% (v/v) glycerol.
The best crystals for the GTDVPI10463–7F complex were found in a reservoir containing 0.1M Tris, pH 8.8, 0.4 M ammonium sulfate, and 24% PEG 3350 with a protein concentration of ~10 mg/ml. The crystals were cryo-protected in the mother liquor supplemented with 25% (v/v) ethylene glycol.
The best crystals for the TcdB1072–1433–5D complex were obtained in a reservoir containing 0.1 M Tris, pH 8.5, 0.2 M lithium sulfate, 18% PEG 4000 with a protein concentration of ~5 mg/ml, which were cryo-protected in the same mother liquid.
Data collection and structure determination
The X-ray diffraction data for the GTD–E3 complex were collected at 100 K at beam line BL9–2 at Stanford Synchrotron Radiation Lightsource (SSRL). All other data were collected at 100 K at the NE-CAT beamline 24-ID-C, Advanced Photon Source (APS). The data were processed with HKL2000 package 62 or XDS as implemented in RAPD (https://github.com/RAPD/RAPD) 63.
In order to determine the structure of the TcdB–5D–E3–7F complex, we first determined the structures of the GTD–E3 and GTDVPI10463–7F complexes using molecular replacement under PHENIX.Phaser 64. We used structures of the GTD (PDB: 2BVL) 26 and the homology models of E3 or 7F that were built based on a VHH in PDB 3V0A as search models 26,65. The structure of the TcdB–5D–E3–7F complex was solved by 3-rounds of molecular replacement with PHENIX.Phaser. We first located the 3 VHHs and the N-terminal fragment of TcdB (residues 1–1805) using the structures of a truncated TcdA (residues 1–550 and 743–1285, PDB: 4R04), the CPD of TcdB (residues 551–742, PDB: 3PEE), the DRBD of TcdB (residues 1285–1804, PDB: 6C0B), the structures of the GTD–E3 and the GTDVPI10463–7F complexes determined in our lab, and a homology model for 5D (PDB: 3V0A) as models 13,24,27,65. This partial structure was defined as a fixed partial model for a second round of molecular replacement using the CROPs I and II of TcdB (residues 1834–2100, PDB: 4NP4) and the CROPs IV of TcdB (residues 2249–2366, PDB: 4NC2) as search models 25,28. After the structures of the CROPs I, II, and IV were found, the position of the CROPs III was located by another round of molecular replacement using the CROPs I of TcdB (residues 1835–1968, PDB: 4NP4) as a search model. Structural modeling and refinement were carried out iteratively using COOT 66, Prosmart external restraints under Refmac5 67, followed by Phenix-Rosetta 68 and Phenix.Refinement 64 with enforcement of secondary structure restraints. Along this process, the electron density for residues in the hinge region improved steadily, which eventually allowed manual model building in this region. Final refinement was performed by jelly body refinement and Refmac5 67. The model of TcdB holotoxin was additionally validated using anomalous signal from tantalum bromide cluster-derivatized crystals.
The crystals of the TcdB1072–1433–5D complex showed anisotropic diffraction. The diffraction data were scaled using diffraction anisotropy server (https://services.mbi.ucla.edu/anisoscale/), which yielded an ellipsoidal resolution boundary with limits of 3.2, 3.2, and 3.0 Å along the a*, b*, and c* axes, respectively 69. The structure of the TcdB1072–1433–5D complex was solved by molecular replacement with PHENIX.Phaser using a fragment of TcdB holotoxin (residues 1098–1431) as a search model. All the refinement progress was monitored with the free R value using a 5% randomly selected test set 70. The structures were validated using MolProbity 71. The TcdB–5D–E3–7F complex has 85.91%, 13.34%, and 0.75% residues in Ramachandran-favored, allowed, and outlier regions, respectively. All of the other three structures have good geometry without Ramachandran outliers observed. Data collection and structural refinement statistics are listed in Table 1. All structure figures were prepared using Pymol (DeLano Scientific).
SAXS
SEC-SAXS experiments were performed at SSRL beamline 4–2 as described previously (Supplementary Table 1) 45. TcdB holotoxin was exchanged into a buffer containing phosphate-buffered saline (PBS), pH 7.4, and 5 mM DTT, or 20 mM sodium acetate, pH 5.0, 50 mM NaCl, and 5 mM DTT, and then concentrated to 20 mg/ml. SEC-SAXS data were collected at pH 5.0 and 7.4 using Superdex-200 Increase PC 3.2/300 columns (GE Healthcare). A total of 500 images were recorded with 1 sec exposure every 5 sec at 0.05 ml/min flow rate. After background (buffer) data collection, the X-ray shutter was closed until the main elution peak showed up in order to keep the sample cell clean from accumulation of dirt. Data reduction and initial analysis were performed using SECPipe, a real-time data processing and initial analysis pipeline at SSRL beamline 4–2. SECPipe implements the program SASTOOL (https://www-ssrl.slac.stanford.edu/smb-saxs/content/documentation/sastool) and ATSAS AUTORG 72. The first 100 images were used for background images. The data were presented as I(q) versus q, where q = 4πsin(θ)/λ, 2θ is the scattering angle, and λ is the wavelength of the X-ray. After careful manual inspection, the average profiles were generated and used for further analysis (image number 290–304 for pH 5.0 and 280–289 for pH 7.4). The program GNOM was used for the indirect Fourier transform to estimate the distance distribution function P(r) 73. The theoretical scattering profile of the crystal structure was computed and fitted with experimental data using the program CRYSOL 74.
DSSO cross-linking of TcdB
TcdB holotoxin (50 μl, 10 μM) in PBS (pH 7.4) was reacted with DSSO at the molar ratio of 1:100 for 1 hour at room temperature. Cross-linking was quenched by addition of 50-fold excess ammonium bicarbonate for 10 minutes, and the resulting products were subjected to enzymatic digestion using a FASP protocol. Briefly, cross-linked proteins were transferred into Milipore Microcon™ Ultracel PL-30 (30 kDa filters), reduced/alkylated and digested with Lys-C/trypsin sequentially 75. The resulting digests were desalted and fractionated by peptide SEC 76. The fractions containing cross-linked peptides were collected for subsequent MSn analysis 75. Three biological replicates were performed to obtain highly reproducible cross-link data. To confirm intra-TcdB interactions, DSSO cross-linked TcdB was separated by 1-D SDS-PAGE and the cross-linked TcdB monomer band was selected for in-gel digestion 77. The extracted tryptic digest was fractionated by SEC and analyzed by LC MSn.
LC MSn analysis
LC MSn analysis was performed using a Thermo Scientific™ Dionex UltiMate 3000 system online coupled with an Orbitrap Fusion Lumos™ mass spectrometer. A 50 cm x 75 μm Acclaim™ PepMap™ C18 column was used to separate peptides over a gradient of 1% to 25% ACN in 82 mins at a flow rate of 300 nl/min. Two different types of acquisition methods were utilized to maximize the identification of DSSO cross-linked peptides: (1) top 4 data-dependent MS3 and (2) targeted MS3 acquisition optimized for capturing DSSO cross-linked peptides by utilizing the mass difference between characteristic MS2 fragment ions of DSSO cross-linked peptides (α−β) (i.e. Δ = αT−αA = βT−βA = 31.9721 Da) 78.
Data analysis and identification of DSSO cross-linked peptides
MSn data extraction and analysis were performed as previously described 78. MS3 data was subjected to Protein Prospector (v.5.19.1) for database searching, using Batch-Tag against a custom database containing 9 protein entries concatenated with its random version. The mass tolerances were set as ±20 ppm and 0.6 Da for parent and fragment ions, respectively. Trypsin was set as the enzyme with three maximum missed cleavages allowed. Variable modifications included protein N-terminal acetylation, cysteine carbamidomethylation, methionine oxidation, and N-terminal conversion of glutamine to pyroglutamic acid. In addition, three defined modification on un-cleaved lysines were chosen, which included alkene (C3H2O; +54 Da), sulfenic acid (C3H4O2S; +104 Da), and thiol (C3H2SO; +86 Da), representing cross-linker fragment moieties on lysine residues. Only a maximum of 4 modifications on a given peptide was allowed during the search. Initial acceptance criteria for peptide identification at the MS3 level required a reported expectation value ≤0.2, which yielded a false discovery rate >0.5%. The in-house program Xl-tools was used to identify, validate and summarize cross-linked peptides based on MSn data and database searching results 78. Following integration of MSn data, no cross-links involving decoy proteins were identified. Only cross-linked peptides that were identified in all three biological replicates are reported.
Single-molecule FRET analysis of TcdB
A cysteine residue was introduced by mutagenesis into the N-terminus of 7F (at the −1 position) or into a surface-exposed loop in B39 (G42C). Expression and purification of the mutant VHHs were similar to the wild type proteins, except that 5 mM DTT was used in all the buffers during purification. 7F was labeled with Alexa-647 maleimide while B39 was labeled with Alexa-555 maleimide (Thermo Fisher Scientific). The labeling efficiency was determined by UV-Vis spectroscopy to be >90%. 5D was biotinylated using EZ-Link NHS-PEG4-Biotin (Thermo Fisher Scientific) at pH 6.5 to preferentially label the N-terminal amine. TcdB holotoxin in complex with the Alexa-647-labeled 7F, the Alexa-555-labeled B39, and the biotin-labeled 5D was further purified using a Superose 6 SEC to remove the excess VHHs.
Cleaned quartz slides were passivated with biotinylated bovine serum albumin followed by a mixture of 2% Biolipidure 203 and 0.2% Biolipidure 206 (NOF America Corp.) before adding streptavidin. Following this treatment, the preformed TcdB–VHH complex showed no nonspecific binding to the slide at concentrations orders of magnitude higher than the 100 pM concentration used to achieve optical resolution between single molecules. Samples were incubated for 5 minutes, rinsed briefly and exchanged into the indicated buffer supplanted with 0.1 % glucose, 20 U/ml pyranose oxidase, 1000 U/ml catalase and 0.0001 % (vol/vol) cyclooctatetraene to prevent photobleaching and blinking. Using pyranose oxidase and catalase insures pH stability at any arbitrary pH 79. Measurements were made in 50 mM Hepes, pH 7, 100 mM NaCl or 50 mM acetate, pH 5, 100 mM NaCl. For intermediate pH, a mixture of citrate and phosphate was used at 50 mM with 100 mM NaCl.
At such low protein concentrations, the non-covalently bound VHHs partially dissociated so measurements had to be made rapidly, which required seven repeated surface preparations at each pH condition. Samples were imaged using a prism-based Total Internal Reflection Fluorescence microscope constructed on an IX71 base with a 60x, 1.2 NA water-immersion objective (Olympus) 42. Alternating laser excitation, with mechanical shutters (Uniblitz), was used to confirm the presence of both a donor and acceptor dye in all molecules used for analysis. Samples were excited with a laser diode at 637 nm (Coherent Inc.) for Alexa-647 and a diode pumped solid-state laser at 532 nm (Laser Quantum USA) for Alexa-555. Emission from donor and acceptor was separated using an Optosplit ratiometric image splitter (Cairn Research Ltd) containing a 645 nm dichroic mirror with a 585/70 band pass filter for the donor channel and a 670/30 band pass filter for the acceptor channel (IDEX Health & Science). The replicate images were relayed to a single iXon DU-897 EMCCD camera (Andor Technologies) at a frame rate of 10 Hz.
Data was processed in home written MATLAB scripts to cross-correlate the replicate images and extract time traces for diffraction limited spots with intensity above baseline. We selected only those complexes containing a single donor and acceptor dye that showed anti-correlated photobleaching to baseline in a single time step. From the magnitude of the anticorrelated photobleaching event, we can perform per-molecule γ-normalization, which allows us to report the absolute FRET efficiency 42. The FRET efficiency was compiled into histograms, which were fit to Gaussian functions.
Photophysical Controls for the FRET Assays
To insure that FRET changes were not the result of photophysical changes, we measured the relative quantum yield and fluorescence anisotropy for the free dyes, the dye-labeled VHHs, and the individual dye-labeled VHHs in complex with TcdB. All measurements were carried out at a dye concentration of 10 nM using the same buffers as the smFRET at pH 7 and pH 5.
Ensemble fluorescence was recorded on an ISS PC1 photon counting spectrofluorometer using a 2.0 mm excitation slit and a 2.0 mm emission slit. Alexa-555 and Alexa-647 labeled samples were excited at 532 nm at 637 nm respectively. Concentrations of samples used for fluorescence were determined from absorption measurements using the same cuvette. The emission intensity was taken as the sum of a 20 nm window about the emission maxima. Relative quantum yields were calculated by normalizing the intensities to the emission of free dye at pH 7. Anisotropy measurements were collected with 2.0 mm excitation slit and a 2.0 mm emission slit using Glan Thompson polarizers in the L conformation. Emission was recorded at 567 nm and 670 nm for the donor and acceptor, respectively. All measurements were done in triplicate and reported as the mean and standard error (Supplementary Table 2).
The effect of 5D on the fluorescence emission of the TcdB–B39–7F complex was investigated by ensemble FRET study. TcdB (0.5 μM) was incubated with equimolar ratio of the Alexa-555-labeled B39, the Alexa-647-labeled 7F, or both VHHs with or without 5D at room temperature for 15 minutes in a buffer containing 100 mM NaCl, and either 50 mM Hepes (pH 7.0) or 50 mM sodium acetate (pH 5.0). Fluorescence was measured on a Spectramax M2e cuvette module with excitation at 540 nm, cutoff at 550 nm, and fluorescence emission at 550–730 nm. The experiments were performed in duplicate.
ANS (8-anilinonaphthalene-1-sulfonic acid) binding assay
TcdB1072–1433, the TcdB1072–1433–5D complex, or 5D at ~1.2 μM was incubated with 200 μM ANS for 20 minutes at 37°C in a buffer containing 0.1 M NaCl and 0.1 M sodium acetate (pH 4.0–5.8), 0.1 M MES (pH 6.0), or 0.1 M Hepes (pH 7.0). Fluorescence spectra were recorded in a Molecular Devices SpectraMax M2e spectrophotometer with excitation at 366 nm. The emission spectrum was collected from 400 to 640 nm. The fluorescence intensity was corrected by subtraction of background fluorescence from ANS in a buffer without protein. Error bars indicate standard deviation of three replicate measurements.
Calcein dye release assay
Lipids (Avanti Polar Lipid) at the indicated molar ratios were mixed in chloroform and then dried under nitrogen gas and placed under vacuum overnight. The dried lipids were rehydrated and were subjected to five rounds of freezing and thawing cycles. Unilamellar vesicles were prepared by extrusion through a 200 nm pore membrane using an Avanti Mini Extruder.
Dried lipids containing 55% 1,2-dioleoyl-sn-glycero-3-phosphocholine (DOPC), 15% 1,2-dioleoyl-sn-glycero-3-phospho-L-serine (DOPS), and 30 % cholesterol (10 mg/ml) were resuspended in 150 mM NaCl, 20 mM Hepes (pH 7.0), 1 mM EDTA, and 50 mM calcein. Free calcein dye was removed by desalting (Zeba). Fluorescence was measured on a Spectramax M2e cuvette module with excitation at 493 nm and emission at 525 nm. Liposomes were diluted in 150 mM NaCl, 20 mM sodium acetate (pH 4.6), 1 mM EDTA, to give a final concentration of 0.3 mM and incubated until the fluorescence signal was stable. TcdB (0–25 nM), or TcdB pre-incubated with 5D or 7F at a TcdB:VHH=1:2 molar ratio, was added and the fluorescence intensity was recorded for 7 minutes. The reaction was stopped by adding 0.1% Trion X-100. The percentage of fluorescence change was calculated as the ((F − Finitial) / (Ffinal − Finitial)). The initial rate of calcein dye release was deduced from the slope of the linear part of the curve. The experiments were repeated three times independently.
Membrane depolarization assay
Depolarization was measured as previously described 45. Briefly, liposomes composed of 55% DOPC, 15% DOPS, 30% cholesterol were prepared in 200 mM NaCl, 1 mM KCl, and 10 mM Hepes (pH 7.0). To create a trans-positive membrane potential (+135 mV), liposomes were diluted in 200 mM KCl, 1 mM NaCl, 10 mM sodium acetate (pH 4.6) to give a final concentration of 0.1 mM. Membrane potential was monitored using 12 μM ANS. Valinomycin was added at time 0-second to give a final concentration of 30 nM. At 180-second, 100 nM TcdB holotoxin, or TcdB pre-incubated with 0.02–1 μM 5D or 1 μM 7F, was added and the fluorescence intensity at 490 nm was monitored for 7 minutes with excitation at 380 nm. The reaction was stopped by adding 2 μM gramicidin from Bacillus anerinolyticus (Sigma-Aldrich). The fluorescence change relative to the maximal change in the presence of gramicidin was calculated as the ((F − Finitial) / (Ffinal − Finitial)). The experiments were repeated three times independently.
TcdB autoprocessing assay
The autoprocessing assays 36,80 were performed in 25 µl of 20 mM Tris-HCl, pH 8.0, which contained 0.4 µM of TcdB holotoxin or TcdB1–1805, InsP6 at the indicated concentrations, with or without 7F (2 µM). The reaction mixtures were incubated at 37°C for 1 hour, and then boiled for 5 minutes in SDS sample buffer to quench the reaction. The samples were examined by 4–20% SDS-PAGE and visualized by Coomassie blue staining.
Pull-down assay
The purified CRD2 13 was biotinylated using EZ-Link NHS-PEG4-Biotin (Thermo Fisher Scientific) at pH 6.5. The pull-down assays were performed in 1 ml binding buffer, which was composed of 50 mM Tris (pH 7.5) or 20 mM sodium acetate (pH 5.0), together with 400 mM NaCl, 1 mM TCEP, and 0.1% Tween-20. Biotin-labeled CRD2 (~5 µg) was incubated with Strep-Tactin®XT Superflow® resins (IBA Lifesciences) at room temperature for 30 minutes, and the unbound protein was washed away using the binding buffer. The CRD2-bound resins were mixed with a ~2-fold molar excess of TcdB for 30 minutes at room temperature. The resins were then washed twice, and the bound proteins were released from the resins with 50 mM D-biotin and further examined by 4–20% SDS-PAGE.
Supplementary Material
ACKNOWLEGEMENTS
This work was partly supported by National Institute of Health grants R01AI139087, R01AI125704, R21AI123920, R21AI139690, and R21CA235533 to R.J., R01GM074830 and R01GM130144 to L.H., R01DK084509, R01AI088748, R01AI132207, and U19AI109776 to H.F, and R01 MH081923 to M.E.B. NE-CAT at the Advanced Photon Source (APS) is supported by a grant from the National Institute of General Medical Sciences (P30GM124165). The Pilatus 6M detector on 24-ID-C beam line is funded by a NIH-ORIP HEI grant (S10 RR029205). Use of the APS, an Office of Science User Facility operated for the U.S. Department of Energy (DOE) Office of Science by Argonne National Laboratory, was supported by the U.S. DOE under Contract No. DE-AC02–06CH11357. Use of the Stanford Synchrotron Radiation Lightsource, SLAC National Accelerator Laboratory, is supported by the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences under Contract No. DE-AC02–76SF00515. The SSRL Structural Molecular Biology Program is supported by the DOE Office of Biological and Environmental Research, and by the National Institutes of Health, National Institute of General Medical Sciences (including P41GM103393). The contents of this publication are solely the responsibility of the authors and do not necessarily represent the official views of NIH.
Footnotes
COMPETING FINANCIAL INTERESTS
A provisional patent application has been filed by The Regents of the University of California on the use of the structural information described in this manuscript to prevent and/or treat CDI. R.J. is a co-founder of DesignerT Inc. and H.F. is a co-founder of FZata Inc., which had no role in this study.
Data Availability
Atomic coordinates and structure factors of the TcdB–5D–E3–7F, TcdB1072–1433–5D, GTD–E3, and GTDVPI10463–7F complexes have been deposited in the Protein Data Bank under accession codes 6OQ5, 6OQ6, 6OQ7, and 6OQ8, respectively. Source data for Figure 4e–f are available with the paper online. Other data are available upon request.
Reporting Summary. Further information on experimental design is available in the Nature Research Reporting Summary linked to this article.
REFERENCES
- 1.Rupnik M, Wilcox MH & Gerding DN Clostridium difficile infection: new developments in epidemiology and pathogenesis. Nat Rev Microbiol 7, 526–36 (2009). [DOI] [PubMed] [Google Scholar]
- 2.Lessa FC et al. Burden of Clostridium difficile infection in the United States. N Engl J Med 372, 825–34 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Jank T & Aktories K Structure and mode of action of clostridial glucosylating toxins: the ABCD model. Trends Microbiol 16, 222–9 (2008). [DOI] [PubMed] [Google Scholar]
- 4.Seal D et al. Treatment of relapsing Clostridium difficile diarrhoea by administration of a non-toxigenic strain. Eur J Clin Microbiol 6, 51–3 (1987). [DOI] [PubMed] [Google Scholar]
- 5.Kuehne SA et al. The role of toxin A and toxin B in Clostridium difficile infection. Nature 467, 711–3 (2010). [DOI] [PubMed] [Google Scholar]
- 6.Lyras D et al. Toxin B is essential for virulence of Clostridium difficile. Nature 458, 1176–9 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Carter GP et al. Defining the Roles of TcdA and TcdB in Localized Gastrointestinal Disease, Systemic Organ Damage, and the Host Response during Clostridium difficile Infections. MBio 6, e00551 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Carter GP, Rood JI & Lyras D The role of toxin A and toxin B in the virulence of Clostridium difficile. Trends Microbiol 20, 21–9 (2012). [DOI] [PubMed] [Google Scholar]
- 9.Wilcox MH et al. Bezlotoxumab for Prevention of Recurrent Clostridium difficile Infection. N Engl J Med 376, 305–317 (2017). [DOI] [PubMed] [Google Scholar]
- 10.Ho JG, Greco A, Rupnik M & Ng KK Crystal structure of receptor-binding C-terminal repeats from Clostridium difficile toxin A. Proc Natl Acad Sci U S A 102, 18373–8 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Aktories K, Schwan C & Jank T Clostridium difficile Toxin Biology. Annu Rev Microbiol 71, 281–307 (2017). [DOI] [PubMed] [Google Scholar]
- 12.Greco A et al. Carbohydrate recognition by Clostridium difficile toxin A. Nat Struct Mol Biol 13, 460–1 (2006). [DOI] [PubMed] [Google Scholar]
- 13.Chen P et al. Structural basis for recognition of frizzled proteins by Clostridium difficile toxin B. Science 360, 664–669 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yuan P et al. Chondroitin sulfate proteoglycan 4 functions as the cellular receptor for Clostridium difficile toxin B. Cell Res 25, 157–68 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Genisyuerek S et al. Structural Determinants for Membrane Insertion, Pore Formation and Translocation of Clostridium difficile Toxin B. Mol Microbiol (2011). [DOI] [PubMed]
- 16.Zhang Z et al. Translocation domain mutations affecting cellular toxicity identify the Clostridium difficile toxin B pore. Proc Natl Acad Sci U S A 111, 3721–6 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Qa’Dan M, Spyres LM & Ballard JD pH-induced conformational changes in Clostridium difficile toxin B. Infect Immun 68, 2470–4 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Li S et al. Critical roles of Clostridium difficile toxin B enzymatic activities in pathogenesis. Infect Immun 83, 502–13 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sehr P et al. Glucosylation and ADP ribosylation of rho proteins: effects on nucleotide binding, GTPase activity, and effector coupling. Biochemistry 37, 5296–304 (1998). [DOI] [PubMed] [Google Scholar]
- 20.Egerer M, Giesemann T, Jank T, Satchell KJ & Aktories K Auto-catalytic cleavage of Clostridium difficile toxins A and B depends on cysteine protease activity. J Biol Chem 282, 25314–21 (2007). [DOI] [PubMed] [Google Scholar]
- 21.Reineke J et al. Autocatalytic cleavage of Clostridium difficile toxin B. Nature 446, 415–9 (2007). [DOI] [PubMed] [Google Scholar]
- 22.Just I et al. Glucosylation of Rho proteins by Clostridium difficile toxin B. Nature 375, 500–3 (1995). [DOI] [PubMed] [Google Scholar]
- 23.Hofmann F, Busch C, Prepens U, Just I & Aktories K Localization of the glucosyltransferase activity of Clostridium difficile toxin B to the N-terminal part of the holotoxin. J Biol Chem 272, 11074–8 (1997). [DOI] [PubMed] [Google Scholar]
- 24.Chumbler NM et al. Crystal structure of Clostridium difficile toxin A. Nat Microbiol 1, 15002 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Murase T et al. Structural basis for antibody recognition in the receptor-binding domains of toxins A and B from Clostridium difficile. J Biol Chem 289, 2331–43 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Reinert DJ, Jank T, Aktories K & Schulz GE Structural basis for the function of Clostridium difficile toxin B. J Mol Biol 351, 973–81 (2005). [DOI] [PubMed] [Google Scholar]
- 27.Shen A et al. Defining an allosteric circuit in the cysteine protease domain of Clostridium difficile toxins. Nat Struct Mol Biol 18, 364–71 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Orth P et al. Mechanism of action and epitopes of Clostridium difficile toxin B-neutralizing antibody bezlotoxumab revealed by X-ray crystallography. J Biol Chem 289, 18008–21 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Pruitt RN, Chambers MG, Ng KK, Ohi MD & Lacy DB Structural organization of the functional domains of Clostridium difficile toxins A and B. Proc Natl Acad Sci U S A 107, 13467–72 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yang G et al. Expression of recombinant Clostridium difficile toxin A and B in Bacillus megaterium. BMC Microbiol 8, 192 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yang Z et al. A novel multivalent, single-domain antibody targeting TcdA and TcdB prevents fulminant Clostridium difficile infection in mice. J Infect Dis 210, 964–72 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kobe B & Kajava AV When protein folding is simplified to protein coiling: the continuum of solenoid protein structures. Trends Biochem Sci 25, 509–15 (2000). [DOI] [PubMed] [Google Scholar]
- 33.Fernandez-Tornero C, Lopez R, Garcia E, Gimenez-Gallego G & Romero A A novel solenoid fold in the cell wall anchoring domain of the pneumococcal virulence factor LytA. Nat Struct Biol 8, 1020–4 (2001). [DOI] [PubMed] [Google Scholar]
- 34.Zhang Y et al. A segment of 97 amino acids within the translocation domain of Clostridium difficile toxin B is essential for toxicity. PLoS One 8, e58634 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lanis JM, Barua S & Ballard JD Variations in TcdB activity and the hypervirulence of emerging strains of Clostridium difficile. PLoS Pathog 6, e1001061 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lanis JM, Hightower LD, Shen A & Ballard JD TcdB from hypervirulent Clostridium difficile exhibits increased efficiency of autoprocessing. Mol Microbiol 84, 66–76 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Larabee JL, Krumholz A, Hunt JJ, Lanis JM & Ballard JD Exposure of neutralizing epitopes in the carboxyl-terminal domain of TcdB is altered by a proximal hypervariable region. J Biol Chem 290, 6975–85 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Putnam CD, Hammel M, Hura GL & Tainer JA X-ray solution scattering (SAXS) combined with crystallography and computation: defining accurate macromolecular structures, conformations and assemblies in solution. Q Rev Biophys 40, 191–285 (2007). [DOI] [PubMed] [Google Scholar]
- 39.Kao A et al. Development of a novel cross-linking strategy for fast and accurate identification of cross-linked peptides of protein complexes. Mol Cell Proteomics 10, M110 002212 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Yu C & Huang L Cross-Linking Mass Spectrometry: An Emerging Technology for Interactomics and Structural Biology. Anal Chem 90, 144–165 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hellenkamp B et al. Precision and accuracy of single-molecule FRET measurements-a multi-laboratory benchmark study. Nat Methods 15, 669–676 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.McCann JJ, Choi UB, Zheng L, Weninger K & Bowen ME Optimizing methods to recover absolute FRET efficiency from immobilized single molecules. Biophys J 99, 961–70 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Gopich IV & Szabo A FRET efficiency distributions of multistate single molecules. J Phys Chem B 114, 15221–6 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Chumbler NM et al. Clostridium difficile Toxin B causes epithelial cell necrosis through an autoprocessing-independent mechanism. PLoS Pathog 8, e1003072 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lam KH et al. A viral-fusion-peptide-like molecular switch drives membrane insertion of botulinum neurotoxin A1. Nat Commun 9, 5367 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Schmidt DJ et al. A Tetraspecific VHH-Based Neutralizing Antibody Modifies Disease Outcome in Three Animal Models of Clostridium difficile Infection. Clin Vaccine Immunol 23, 774–84 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Just I & Gerhard R Large clostridial cytotoxins. Rev Physiol Biochem Pharmacol 152, 23–47 (2004). [DOI] [PubMed] [Google Scholar]
- 48.Egerer M & Satchell KJ Inositol hexakisphosphate-induced autoprocessing of large bacterial protein toxins. PLoS Pathog 6, e1000942 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lupardus PJ, Shen A, Bogyo M & Garcia KC Small molecule-induced allosteric activation of the Vibrio cholerae RTX cysteine protease domain. Science 322, 265–8 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Puri AW et al. Rational design of inhibitors and activity-based probes targeting Clostridium difficile virulence factor TcdB. Chem Biol 17, 1201–11 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Olling A et al. The combined repetitive oligopeptides of clostridium difficile toxin A counteract premature cleavage of the glucosyl-transferase domain by stabilizing protein conformation. Toxins (Basel) 6, 2162–76 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Zhang Y, Hamza T, Gao S & Feng H Masking autoprocessing of Clostridium difficile toxin A by the C-terminus combined repetitive oligo peptides. Biochem Biophys Res Commun 459, 259–263 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Geissler B, Tungekar R & Satchell KJ Identification of a conserved membrane localization domain within numerous large bacterial protein toxins. Proc Natl Acad Sci U S A 107, 5581–6 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Varela Chavez C et al. The Tip of the Four N-Terminal alpha-Helices of Clostridium sordellii Lethal Toxin Contains the Interaction Site with Membrane Phosphatidylserine Facilitating Small GTPases Glucosylation. Toxins (Basel) 8, 90 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Mesmin B et al. A phosphatidylserine-binding site in the cytosolic fragment of Clostridium sordellii lethal toxin facilitates glucosylation of membrane-bound Rac and is required for cytotoxicity. J Biol Chem 279, 49876–82 (2004). [DOI] [PubMed] [Google Scholar]
- 56.Schorch B et al. LRP1 is a receptor for Clostridium perfringens TpeL toxin indicating a two-receptor model of clostridial glycosylating toxins. Proc Natl Acad Sci U S A 111, 6431–6 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kreimeyer I et al. Autoproteolytic cleavage mediates cytotoxicity of Clostridium difficile toxin A. Naunyn Schmiedebergs Arch Pharmacol 383, 253–62 (2011). [DOI] [PubMed] [Google Scholar]
- 58.Gupta P et al. Functional defects in Clostridium difficile TcdB toxin uptake identify CSPG4 receptor-binding determinants. J Biol Chem 292, 17290–17301 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Tao L et al. Frizzled proteins are colonic epithelial receptors for C. difficile toxin B. Nature 538, 350–355 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Laursen NS et al. Universal protection against influenza infection by a multidomain antibody to influenza hemagglutinin. Science 362, 598–602 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Yang GL et al. Expression of recombinant Clostridium difficile toxin A and B in Bacillus megaterium. Bmc Microbiology 8(2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Otwinowski Z & Minor W Processing of X-ray diffraction data collected in oscillation mode. Macromolecular Crystallography, Pt A 276, 307–326 (1997). [DOI] [PubMed] [Google Scholar]
- 63.Kabsch W Xds. Acta Crystallogr D Biol Crystallogr 66, 125–32 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Adams PD et al. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr D Biol Crystallogr 66, 213–21 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Gu S et al. Botulinum neurotoxin is shielded by NTNHA in an interlocked complex. Science 335, 977–81 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Emsley P, Lohkamp B, Scott WG & Cowtan K Features and development of Coot. Acta Crystallogr D Biol Crystallogr 66, 486–501 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Murshudov GN et al. REFMAC5 for the refinement of macromolecular crystal structures. Acta Crystallographica Section D-Biological Crystallography 67, 355–367 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.DiMaio F et al. Improved low-resolution crystallographic refinement with Phenix and Rosetta. Nature Methods 10, 1102–1104 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Strong M et al. Toward the structural genomics of complexes: crystal structure of a PE/PPE protein complex from Mycobacterium tuberculosis. Proc Natl Acad Sci U S A 103, 8060–5 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Brunger AT Free R value: a novel statistical quantity for assessing the accuracy of crystal structures. Nature 355, 472–5 (1992). [DOI] [PubMed] [Google Scholar]
- 71.Chen VB et al. MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr D Biol Crystallogr 66, 12–21 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Petoukhov MV, Konarev PV, Kikhney AG & Svergun DI ATSAS 2.1 - towards automated and web-supported small-angle scattering data analysis. Journal of Applied Crystallography 40, S223–S228 (2007). [Google Scholar]
- 73.Svergun DI Determination of the Regularization Parameter in Indirect-Transform Methods Using Perceptual Criteria. Journal of Applied Crystallography 25, 495–503 (1992). [Google Scholar]
- 74.Svergun D, Barberato C & Koch MHJ CRYSOL - A program to evaluate x-ray solution scattering of biological macromolecules from atomic coordinates. Journal of Applied Crystallography 28, 768–773 (1995). [Google Scholar]
- 75.Kao AH et al. Development of a Novel Cross-linking Strategy for Fast and Accurate Identification of Cross-linked Peptides of Protein Complexes. Molecular & Cellular Proteomics 10(2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Leitner A, Walzthoeni T & Aebersold R Lysine-specific chemical cross-linking of protein complexes and identification of cross-linking sites using LC-MS/MS and the xQuest/xProphet software pipeline. Nature Protocols 9, 120–137 (2014). [DOI] [PubMed] [Google Scholar]
- 77.Yu C, Kandur W, Kao A, Rychnovsky S & Huang L Developing new isotope-coded mass spectrometry-cleavable cross-linkers for elucidating protein structures. Anal Chem 86, 2099–106 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Yu C et al. Developing a Multiplexed Quantitative Cross-Linking Mass Spectrometry Platform for Comparative Structural Analysis of Protein Complexes. Analytical Chemistry 88, 10301–10308 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Swoboda M et al. Enzymatic oxygen scavenging for photostability without pH drop in single-molecule experiments. ACS Nano 6, 6364–9 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Guttenberg G et al. Inositol hexakisphosphate-dependent processing of Clostridium sordellii lethal toxin and Clostridium novyi alpha-toxin. J Biol Chem 286, 14779–86 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.