

Abstract
Differentiation of T follicular helper (TFH) cells is regulated by a complex transcriptional network, with mutually antagonistic Bcl6-Blimp1 as a core regulatory axis. It is well-established that Tcf1 acts upstream of Bcl6 for its optimal induction to program TFH cell differentiation. Here we show that whereas genetic ablation of Tcf1 in mice greatly diminished TFH cells in response to viral infection, compound deletion of Blimp1 with Tcf1 restored TFH cell frequency, numbers and generation of germinal center B cells. Aberrant upregulation of T-bet and Id2 in Tcf1-deficient TFH cells was also largely rectified by ablating Blimp1. Tcf1 ChIP-seq in TFH cells identified two strong Tcf1 binding sites in the Blimp1 gene at a 24-kb upstream and an intron-3 element. Deletion of the intron-3 element, but not the 24-kb upstream element, impaired production of TFH cells. Our data demonstrate that Tcf1-mediated Blimp1 repression is functionally critical for safeguarding TFH cell differentiation.
Introduction
In response to infections by intracellular pathogens including bacteria and viruses, CD4+ T cells are activated, and a portion of the activated CD4+ T cells differentiate into T follicular helper (TFH) cell lineage by upregulating Bcl6 transcription factor and CXCR5 chemokine receptor. CXCR5 expression directs the early/nascent TFH cells to follicles in secondary lymphoid organs where they have the initial contact with B cells. The TFH and B cells then undergo intra-dependent maturation processes, with fully matured TFH cells providing both contact-dependent and soluble factors to activated B cells. The proliferating B cells form germinal center (GC), a micro-anatomical structure in the follicles, where they continue to receive help from GC-TFH cells to undergo class-switch recombination, affinity maturation, and give rise to plasma cells and long-lived memory B cells (1-3). TFH cells are therefore essential for mounting protective humoral responses to pathogens.
At the center of TFH cell differentiation lies the mutually antagonistic Bcl6-Blimp1 axis, where Bcl6 is both necessary and sufficient for generation of TFH cells, while Blimp1 promotes a T helper 1 (TH1) fate in type 1 immune responses to viral infection (4-6). Recent progress indicates that transcription factors from multiple families form a complex regulatory network, integrating signals derived from TCR stimulation and cytokines and ultimately converging on the Bcl6-Blimp1 axis. An important node in the TFH-programming network consists of T cell factor 1 (Tcf1) and its homologue, lymphoid enhancer factor binding 1 (Lef1), transcription factors in the Tcf/Lef family (7-10). Tcf1 and Lef1 have versatile functions in regulating T cell development in the thymus and mature T cell functions in the periphery (11-13). We and others have shown that Tcf1 and Lef1 are essential for generation of TFH cells in response to viral infection, by regulating optimal induction of Bcl6 through direct binding to the Bcl6 transcription start site (TSS) (7, 8). Our follow-up studies further reveal that Ezh2 is rapidly induced in activated CD4+ T cells and specifically phosphorylated at its Ser21 residue in nascent and mature TFH cells (14). Importantly, the Ser21-phosphorylated form of Ezh2 is recruited to the Bcl6 TSS by Tcf1 through direct protein-protein interaction, and functions as a coactivator of Tcf1 to induce Bcl6 expression (14). Furthermore, forced expression of Bcl6 in Tcf1-deficient or Ezh2-deficient CD4+ T cells restored TFH cell differentiation (8, 9, 14). These observations establish the importance of Tcf1/Ezh2-Bcl6 axis in promoting TFH cell differentiation elicited by viral infection.
It has been observed that Tcf1 deficiency is associated with increased expression of Blimp1 in several types of T cells including mature CD8+ thymocytes (15) and CD4+ regulatory T cells (16). This association is also true in TFH cells, with Tcf1- or Tcf1/Lef1-deficient TFH cells exhibiting elevated Blimp1 expression (7-10). Tcf1-mediated Blimp1 repression appears to be a regulatory circuit conserved in multiple cell contexts; however, the functional requirements for the Tcf1-Blimp1 axis in TFH cells remain unknown. In this study, we investigated the interplay between Tcf1 and Blimp1 in TFH responses to viral infection.
Materials and Methods
Mice and CRISPR/Cas9-mediated genome editing
Tcf7fl/fl and hCD2-Cre mice were previously described (7, 17), and Prdm1fl/fl mice were from the Jackson Laboratory (18). To target Bcl6- and/or Tcf1-bound regions in intron 3 of the Prdm1 gene (encoding Blimp1), we used the CRISPR/Cas9 approach. In brief, three CRISPR sgRNAs (5’-CGCGCAAACTTCCAGGAACA, 5’-GCAGCCTGCGAGAGCTTCCG and 5’-ACACTCCTACTGAAACCAAG were designed using MIT’s CRISPR design tool (CRISPR.MIT.EDU), and synthesized by ThermoFisher’s in vitro transcription service (Fig. S1A). The sgRNAs (20 ng/μl each) were co-microinjected with Cas9 mRNA (100 ng/μl) into fertilized eggs collected from C57BL/6N mice. The injected embryos were cultured overnight in M16 medium, and those embryos that reached 2-cell stage of development were implanted into oviducts of pseudo-pregnant foster mothers. The offspring were then genotyped by PCR, and DNA sequencing was performed on positive founder mice to identify the following mutations, as illustrated in Fig. S1A: the first line contained deletion of an 102-bp segment harboring a Bcl6 motif and was named Prdm1(int3-Bcl6)M allele; the second line contained deletion of a 435-bp segment harboring two Tcf/Lef motifs and was named Prdm1(int3-Tcf)M allele; and the third line contained deletion of a 470-bp segment harboring all Bcl6 and Tcf/Lef motifs and was named Prdm1(int3)M allele.
Prdm1(−24kb)M allele was previously reported (16). The Prdm1(int3)M/M mice were used to target the −24 kb element again in this study to generate Prdm1(int3)MPrdm1(−24kb)M allele using the same targeting and verification approaches as described (16). Both male and female mice were used between 6-12 weeks of age for all experiments. All mouse experiments were performed under protocols approved by the Institutional Animal Use and Care Committee of the University of Iowa and the National Heart, Lung, and Blood Institute.
Adoptive transfer, viral infection, and flow cytometry,
Naïve CD45.2+ Smarta CD4+ T cells were isolated from the lymph nodes (LNs) of donor mice of various genotypes, and 1 × 104 Vα2+ Smarta CD4+ T cells were intravenously (i.v.) injected into CD45.1+ B6.SJL recipient mice and infected intraperitoneally (i.p) with 2 × 105 PFU of lymphocytic choriomeningitis virus, Armstrong strain (LCMV-Arm). Single-cell suspensions of spleens were prepared for surface or intracellular staining as previously described (10, 14). Data were collected on a FACSVerse (BD Biosciences) and were analyzed with FlowJo software (TreeStar).
Quantitative RT-PCR
In Smarta CD4+ T cell adoptive transfer and recipient infection experiments described above, CXCR5+SLAMlo TFH cells were sort-purified from CD45.2+CD4+ splenocytes on 8 dpi. The total RNA was extracted, reverse-transcribed and analyzed by quantitative PCR as previously described (10, 14). The primers used for Tbx21 were 5’-tcaaccagcaccagacagag and 5’-ccacatccacaaacatcctg, those for Id2 were 5’-catcagcatcctgtccttgc and 5’-gtgttctcctggtgaaatgg, and those for Dnmt3b were 5’-gctgggtacagtggtttggt and 5’-gtgtggtacatggccttcct.
Results and Discussion
Ablation of Blimp1 rectifies Tcf1 deficiency in TFH cell differentiation.
To investigate functional requirements for the Tcf1-Blimp1 axis in TFH cell differentiation, we used human CD2 promoter-driven Cre recombinase (hCD2-Cre) transgenic mice to ablate Tcf7 and Prdm1 genes (encoding Tcf1 and Blimp1, respectively) in mature T cells (7, 18), so as to avoid potential impact on thymocyte development. We also used the Rosa26-STOP-GFP (Rosa26GFP) allele, where GFP expression marked cells in which the floxed genomic sequences have been deleted by the hCD2-Cre (7, 19). By crossing hCD2-Cre+Rosa26GFP to Tcf7- and/or Prdm1-floxed alleles, we generated hCD2-Cre+Rosa26GFPTcf7FL/FL, hCD2-Cre+Rosa26GFPPrdm1FL/FL, and hCD2-Cre+Rosa26GFPTcf7FL/FLPrdm1FL/FL mice (called Tcf7−/−, Prdm1−/−, and Tcf7−/−Prdm1−/− hereafter), along with control littermates. The mice were i.p. infected with LCMV-Arm to elicit acute viral infection. On 8th day post-infection (dpi), the deletion efficiency by hCD2-Cre, as marked by GFP positivity, was in the range of 85-92% in splenic CD4+ T cells (Fig. S1B), and polyclonal CD4+ T cell responses were then analyzed in GFP+ CD44hiCD62L− activated CD4+ T cells. Consistent with previous reports, Tcf7−/− mice showed greatly diminished frequency and numbers of SLAMloCXCR5+ TFH cells, while Prdm1−/− mice showed predominantly TFH response to LCMV infection (Fig. 1A, 1B). Interesting, CD4+ T cell responses in Tcf7−/−Prdm1−/− mice were skewed toward the TFH lineage, similar to those in Prdm1−/− mice (Fig. 1A, 1B). The similar trend was observed in generation of PD-1hiCXCR5+ GC-TFH cells, which was diminished in Tcf7−/− mice but enhanced in Prdm1−/− and Tcf7−/−Prdm1−/− mice (Fig. 1C, 1D). In addition, generation of Fas+GL7hi GC-B cells was compromised in Tcf7−/− mice, consistent with impaired capacity of Tcf7−/− TFH cells in conferring B cell help; in contrast, the GC-B cell defect was no longer observed in Tcf7−/−Prdm1−/− mice (Fig. 1E, 1F). It is of note that there were approximately 10-15% undeleted, GFP-negative TFH cells in all genotypes, but their absolute counts were low, in particular in Tcf7−/− mice (Fig. S1C), and may not provide adequate help to B cells. These observations collectively suggest that Tcf1 deficiency-derived TFH defects are largely rectified by ablating Blimp1.
Figure 1. Ablation of Blimp1 rectified Tcf1 deficiency in polyclonal TFH and B cell responses.

A–B. Detection of SLAMloCXCR5+ TFH cells. Mice of indicated genotypes were infected with LCMV-Arm, and on 8 dpi, TFH cells were detected in GFP+ CD44+CD62Llo activated CD4+ T cells in the spleens. Representative contour plots (A) are from ≥3 independent experiments, with values denoting frequency of TH1 and TFH cells, and cumulative data of frequency (left panel) and numbers (right panel) of TFH cells are in B.
C–D. Detection of PD-1hiCXCR5+ GC-TFH cells in LCMV-Arm-infected mice, with representative and cumulative data in C and D, respectively.
E–F. Detection of Fas+GL7hi GC- B cells. On 8 dpi, GC-B cells were detected in B220+CD19+ B cells in the spleens, with representative and cumulative data in E and F, respectively. Data in B, D, and F are means ± s.d, with each open circle representing one mouse analyzed. Statistical significance was determined by one-way ANOVA, followed by Student’s t-test for indicated pairwise comparisons. ns, not statistically significant; *, p<0.05; ** p <0.01; ***, p<0.001, unless an actual p value is specified. This annotation applies to all figures in this paper.
To further substantiate this conclusion on a per cell basis, we crossed the various gene-targeted mouse strains with a Smarta TCR transgene, which encodes a TCR specific for the LCMV glycoprotein 61-80 epitope. We then adoptively transferred the same numbers of naïve Smarta CD4+ T cells into CD45 allele-disparate recipient mice, followed by LCMV-Arm infection. Analysis of Smarta CD4+ T cells in the recipient spleens on 8 dpi showed diminished TFH response due to loss of Tcf1 but enhanced TFH response upon ablation of Blimp1, and importantly, ablating Blimp1 in Tcf1-deficient Smarta cell potently restored TFH differentiation (Fig. 2A). Consistent with reported roles of Tcf1 and Blimp1 in promoting and antagonizing Bcl6 induction in TFH cells, respectively, the portion of Bcl6hiCXCR5+ GC-TFH cells was diminished in Tcf7−/− but was elevated in Prdm1−/− as well as Tcf7−/−Prdm1−/− Smarta cells (Fig. 2B). Thus, our data in both polyclonal and monoclonal CD4+ T cell responses consistently indicate that ablation of Blimp1 is adequate to rectify TFH defects caused by Tcf1 deficiency. These observations further suggest that Tcf1-mediated repression of Blimp1 constitutes a functionally important branch in promoting TFH cell differentiation, in addition to Tcf1-dependent Bcl6 induction.
Figure 2. Ablation of Blimp1 rectified Tcf1 deficiency in TFH cell responses on per cell basis.

A. Detection of SLAMloCXCR5+ TFH cells. Naïve CD45.2+ Smarta CD4+ T cells were isolated from LNs of mice of indicated genotypes, adoptively transferred into CD45.1+ recipients, followed by LCMV-Arm infection. On 8 dpi, TFH cells were detected in GFP+ CD45.2+ Smarta CD4+ T cells in the spleens.
B. Detection of Bcl6hiCXCR5+ GC-TFH cells by intracellular staining of GFP+ CD45.2+ Smarta CD4+ T cells on 8 dpi. Representative contour plots (left panels) are from ≥3 independent experiments, and cumulative data on frequency of Smarta TFH or GC-TFH cells are in right panels.
Identification of Blimp1-dependent and –independent Tcf1 targets in TFH cells.
Previous transcriptomic analyses revealed that Tcf1 deficiency had at least two most apparent effects, resulting in compromised activation of genes in the TFH program and aberrant induction of genes in the TH1 program (7-9). In the TFH program, besides Bcl6, Tcf1/Lef1-deficient TFH cells showed diminished expression of ICOS, a key costimulatory molecule, and gp130, a receptor chain in the IL-6 receptor complex (7). This was validated on protein level by cell surface staining in Tcf7−/− Smarta TFH cells (Fig. 3A-B). While only minimally affected in Prdm1−/− TFH cells, the ICOS and gp130 expression in Tcf7−/−Prdm1−/− TFH cells remained as low as that in Tcf7−/− TFH cells (Fig. 3A-B). This observation suggests that Tcf1 is directly responsible for optimal expression of ICOS and gp130, independent of Blimp1. In line with this notion, our recent study showed that Tcf1 recruits Ezh2 to the Icos TSS for its induction (14), and our genome-wide mapping of Tcf1 occupancy in TFH cells by ChIP-seq identified strong Tcf1 binding peaks at the TSSs and intronic regions of Bcl6 and Icos genes, and multiple regulatory regions upstream of Il6st (encoding gp130) (Fig. S2A)(14).
Figure 3. Tcf1 has Blimp1-dependent and –independent downstream targets.
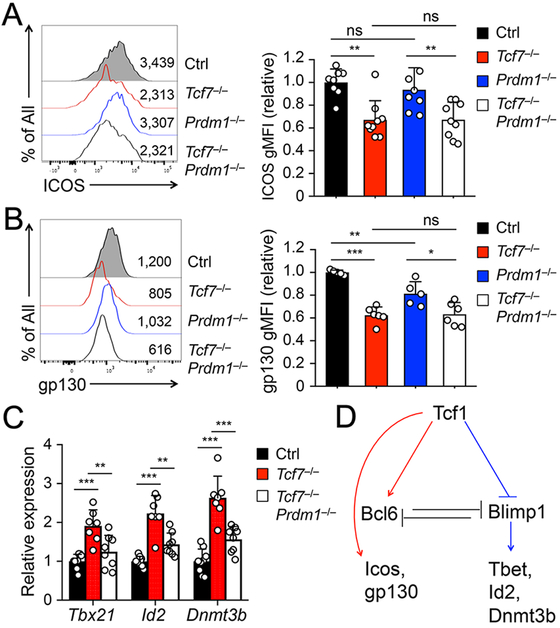
A–B. Detection of ICOS (A) and gp130 (B) expression on GFP+ SLAMloCXCR5+ Smarta TFH cells by flow cytometry on 8 dpi. Representative histograms in left panels are from ≥3 independent experiments, with values denoting geometric mean fluorescence intensity (gMFI). Cumulative data in right panels are pooled data of relative expression of ICOS or gp130 with the gMFI values all normalized to those of control TFH cells in each independent experiment.
C. Detection of Tbx21, Id2, and Dnmt3b expression in SLAMloCXCR5+ Smarta TFH cells. GFP+ Smarta TFH cells were sort-purified on 8 dpi, and analyzed for indicted transcripts by qRT-PCR. The relative expression of each target transcript (after normalization to the house-keeping Hprt gene) in control TFH cells was set at 1, and that in cells of other genotypes was normalized accordingly. Cumulative data are from ≥ 3 experiments.
D. Proposed model of Tcf1-mediated regulation of downstream targets in TFH cells. Arrows denote positive regulation, and lines end in bars denote negative regulation.
In the TH1 program, besides Blimp1, Tcf1/Lef1-deficient TFH cells exhibited elevated expression of Tbx21 (encoding T-bet transcription factor), Id2, and Dnmt3b (7), as validated by quantitative RT-PCR in Tcf7−/− TFH cells (Fig. 3C). T-bet is well-established as a master regulator of TH1 cell differentiation (20), and Id2 is necessary for generation of TH1 cells in response to LCMV as well as Toxoplasma gondii infections (21). Interestingly, ablating Blimp1 in Tcf7−/− TFH cells reduced the transcripts of Tbx21, Id2, and Dnmt3b to levels similar to control TFH cells (Fig. 3C). We noted in Tcf1 ChIP-seq analysis that no Tcf1 binding peaks were associated with the Dnmt3b locus, while only weak, solo Tcf1 peak was detected at Tbx21 or Id2 TSS in TFH cells (Fig. S2B). These data suggest that upregulation of the TH1-associated genes in Tcf1-deficient TFH cells is at least partly due to deregulated Blimp1, while Tcf1 contributes to ICOS and gp130 induction independent of Blimp1 (Fig. 3D).
Blimp1 gene regulatory elements influence TFH cell differentiation.
Given the pivotal role of Blimp1 repression during TFH cell differentiation, we then sought to understand how the Prdm1 gene was regulated. Tcf1 ChIP-seq analysis in TFH cells showed that Tcf1 had strong binding to the Prdm1 intron 3 and a 24 kb upstream region (Fig. 4A) (14). Interestingly, these Tcf1 binding events were are also observed in naïve CD4+ and regulatory T cells (16). Tcf1-bound DNA sequence in Prdm1 intron 3 contained two highly conserved Tcf/Lef motifs as well as a Bcl6 motif (Fig. S1A). In fact, Bcl6 also binds to the PRDM1 intron 3 in human GC-TFH cells as determined by BCL6 ChIP-seq (22). Sequence conservation analysis indicates that the Tcf1-occupied site in murine Prdm1 intron 3 overlapped with the BCL6-occupied site in human PRDM1 intron 3. By the CRISPR-cas9 approach, we used a combination of 3 sgRNAs to differentially target the Bcl6 and/or Tcf motifs (Fig. S1A). Indeed, we obtained Prdm1 intron mutant alleles that lacked the Bcl6 motif, the two Tcf/Lef motifs, or both Bcl6 and Tcf/Lef motifs, called Prdm1(int3-Bcl6)M, Prdm1(int3-Tcf)M, or Prdm1(int3)M alleles, respectively (Fig. S1A). After crossing each mutant allele to homozygotes and Smarta TCR transgene, we performed Smarta CD4+ T cell transfer and LCMV-Arm infection. On 8 dpi, TFH cells were detected at similar frequency among control, Prdm1(int3-Bcl6)M/M, and Prdm1(int3-Tcf)M/M Smarta cells, but were diminished by approximately 50% in Prdm1(int3)M/M Smarta cells (Fig. 4B). These data suggest that the presence of intact Bcl6- and Tcf1-binding sites in the Prdm1 intron 3 region is necessary for promoting optimal TFH cell differentiation.
Figure 4. Blimp1 gene regulatory elements in regulation of TFH cell differentiation.

A. Tcf1 binding events at the Prdm1 gene locus in TFH cells. Tcf1 ChIP-seq tracks in WT TFH cells and Tcf1-deficient CD4+ T cells (as negative control) are displayed on the UCSC genome browser. Bars on top of the tracks denote SICER-called Tcf1 peaks, and yellow rectangles mark two Prdm1 regulatory elements targeted in this work.
B–C. TFH response in the absence of Prdm1 regulatory elements. Naïve CD45.2+ Smarta CD4+ T cells were isolated from LNs of mice of indicated genotypes, adoptively transferred and recipients infected by LCMV-Arm. On 8 dpi, Smarta TFH cells were detected in the recipient spleens. Representative contour plots in left panels are from ≥3 independent experiments, and cumulative data of Smarta TFH cell frequency are in right panels.
Using an in vivo dual reporter assay, it was suggested that the Tcf1-bound Prdm1 −24 kb element might mediate Prdm1 repression in TFH cells (8). We have previously generated Prdm1(−24kb)M allele, and indeed regulatory T cells that are homozygous for Prdm1(−24kb)M/M alleles show about 2-fold increase in Prdm1 expression (16). We then tested Smarta+ Prdm1(−24kb)M/M CD4+ T cells in the adoptive transfer and LCMV-Arm infection model in vivo; however, TFH cells were detected at similar frequency in Prdm1(−24kb)M/M and WT Smarta cells on 8 dpi (Fig. 4C). Because TFH differentiation defects in Prdm1(int3)M/M CD4+ T cells were less profound compared with Tcf7−/− CD4+ T cells (compare Fig. 4B with Fig. 2A), a logical deduction dictates that Tcf1 may act simultaneously on multiple regulatory elements to coordinate Prdm1 repression. We therefore re-targeted the −24 kb element in Prdm1(int3)M/M mice and generated Prdm1(int3)M/MPrdm1(−24kb)M/M mice which lacked Tcf1 binding sites in both intron 3 and −24 kb elements, as well as the Bcl6 binding site in intron 3. After crossing to Smarta TCR transgene, we performed adoptive transfer and LCMV-Arm infection as above, and found that Prdm1(int3)M/MPrdm1(−24kb)M/M Smarta CD4+ T cells did exhibit reduced frequency of TFH cells, and these TFH defects, however, were not significantly exacerbated compared with those in Prdm1(int3)M/M Smarta cells (Fig. 4C). These data thus argue against an important contribution by the −24 kb element to Prdm1 gene regulation in TFH cells.
To determine how mutating the intron 3 regulatory element affected Prdm1 expression, we sorted TH1 and TFH cells derived from WT or Prdm1(int3)M/M Smarta CD4+ T cells for gene expression analysis. As expected, WT TH1 cells showed higher Prdm1 expression, while WT TFH cells expressed higher levels of Tcf7 and Bcl6; however, this expression pattern was not significantly affected in Prdm1(int3)M/M TH1 or TFH cells (Fig. S2C). This observation is enigmatic, because Prdm1(int3)M/M did compromise the magnitude of TFH response. A plausible explanation is that the Tcf1 and Bcl6 may have acted at the TFH lineage specification stage within the first few cell divisions after CD4+ T cell activation. At this stage, whereas Ezh2 is rapidly induced in almost all activated CD4+ T cells as we recently demonstrated (14), induction of CD25 and Blimp1 was only observed in a portion of these cells, and the CD25−Blimp1− cells are predisposed to adopt a nascent TFH fate (23). The differential induction of CD25 and Blimp1 among activated CD4+ T cells could have been a result of stochastic events or asymmetric signaling as recently proposed (24). Abrogating Tcf1 and Bcl6 binding to the Prdm1 intron 3 may skew a larger fraction of the activated CD4+ T cells toward Blimp1 induction, resulting in fewer mature TFH cells at the peak response to viral infection. The fact that Prdm1 expression was adequately suppressed in mature TFH cells in the absence of Tcf1 and Bcl6 binding to Prdm1 intron 3 suggests contribution by either additional cis-regulatory elements distal to the Prdm1 locus, or other Tcf1- and/or Bcl6-dependent intermediary protein factors acting in trans on the Prdm1 locus. These possibilities merit future investigations.
Collectively, this study demonstrates that Tcf1-dependent Blimp1 repression is functionally critical for promoting TFH cell differentiation, bearing importance that is no less than Tcf1-mediated activation of the TFH transcriptional program including induction of Bcl6, Icos and IL-6 receptor complexes. In the absence of Tcf1, although Bcl6, Icos and gp130 are induced to a suboptimal level, this is not adequate to counteract against aberrant Blimp1 upregulation, leading to severe TFH defects. On the other hand, our data also suggest that Blimp1 has strong effects on instructing a TH1 fate for activated CD4+ T cells, because ablating Blimp1 alone or even both Blimp1 and Tcf1 diverted CD4+ T cells to predominantly TFH fate. Whereas this work focused on the interplay among these key factors in TFH initiation and commitment during early responses to acute viral infection, an interesting future direction is to determine how their balanced actions regulate sustained GC response and memory B cell formation in longer term. Nonetheless, our in vivo analyses of TFH cells at the effector phase highlight the Tcf1-Blimp1 axis as a conserved regulatory circuit that has important functional impact among T cell subsets.
Supplementary Material
Key points:
Tcf1-mediated repression of Blimp1 is functionally critical for TFH differentiation.
An intron-3 regulatory region in Blimp1 gene is necessary for optimal TFH response.
Acknowledgements.
We thank the University of Iowa Flow Cytometry Core facility for cell sorting, Drs. Jinyong Choi and Shane Crotty for sharing Bcl6 cistrome data. This study is supported in-part by grants from the NIH (AI112579, AI121080 and AI139874) and the Veteran Affairs BLR&D Merit Review Program (BX002903) to H.-H.X.
Footnotes
Disclosures: The authors have no financial conflict of interest.
References
- 1.Crotty S 2015. A brief history of T cell help to B cells. Nat Rev Immunol 15: 185–189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Song W, and Craft J. 2019. T follicular helper cell heterogeneity: Time, space, and function. Immunol Rev 288: 85–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Vinuesa CG, Linterman MA, Yu D, and MacLennan IC. 2016. Follicular Helper T Cells. Annu Rev Immunol 34: 335–368. [DOI] [PubMed] [Google Scholar]
- 4.Johnston RJ, Poholek AC, DiToro D, Yusuf I, Eto D, Barnett B, Dent AL, Craft J, and Crotty S. 2009. Bcl6 and Blimp-1 are reciprocal and antagonistic regulators of T follicular helper cell differentiation. Science 325: 1006–1010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yu D, Rao S, Tsai LM, Lee SK, He Y, Sutcliffe EL, Srivastava M, Linterman M, Zheng L, Simpson N, Ellyard JI, Parish IA, Ma CS, Li QJ, Parish CR, Mackay CR, and Vinuesa CG. 2009. The transcriptional repressor Bcl-6 directs T follicular helper cell lineage commitment. Immunity 31: 457–468. [DOI] [PubMed] [Google Scholar]
- 6.Nurieva RI, Chung Y, Martinez GJ, Yang XO, Tanaka S, Matskevitch TD, Wang YH, and Dong C. 2009. Bcl6 mediates the development of T follicular helper cells. Science 325: 1001–1005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Choi YS, Gullicksrud JA, Xing S, Zeng Z, Shan Q, Li F, Love PE, Peng W, Xue HH, and Crotty S. 2015. LEF-1 and TCF-1 orchestrate T(FH) differentiation by regulating differentiation circuits upstream of the transcriptional repressor Bcl6. Nat Immunol 16: 980–990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Xu L, Cao Y, Xie Z, Huang Q, Bai Q, Yang X, He R, Hao Y, Wang H, Zhao T, Fan Z, Qin A, Ye J, Zhou X, Ye L, and Wu Y. 2015. The transcription factor TCF-1 initiates the differentiation of T(FH) cells during acute viral infection. Nat Immunol 16: 991–999. [DOI] [PubMed] [Google Scholar]
- 9.Wu T, Shin HM, Moseman EA, Ji Y, Huang B, Harly C, Sen JM, Berg LJ, Gattinoni L, McGavern DB, and Schwartzberg PL. 2015. TCF1 Is Required for the T Follicular Helper Cell Response to Viral Infection. Cell reports 12: 2099–2110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gullicksrud JA, Li F, Xing S, Zeng Z, Peng W, Badovinac VP, Harty JT, and Xue HH. 2017. Differential Requirements for Tcf1 Long Isoforms in CD8(+) and CD4(+) T Cell Responses to Acute Viral Infection. J. Immunol. 199: 911–919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Xue HH, and Zhao DM. 2012. Regulation of mature T cell responses by the Wnt signaling pathway. Ann N Y Acad Sci 1247: 16–33. [DOI] [PubMed] [Google Scholar]
- 12.Steinke FC, and Xue HH. 2014. From inception to output, Tcf1 and Lef1 safeguard development of T cells and innate immune cells. Immunol Res 59: 45–55. [DOI] [PubMed] [Google Scholar]
- 13.van Loosdregt J, and Coffer PJ. 2018. The Role of WNT Signaling in Mature T Cells: T Cell Factor Is Coming Home. J. Immunol. 201: 2193–2200. [DOI] [PubMed] [Google Scholar]
- 14.Li F, Zeng Z, Xing S, Gullicksrud JA, Shan Q, Choi J, Badovinac VP, Crotty S, Peng W, and Xue HH. 2018. Ezh2 programs TFH differentiation by integrating phosphorylation-dependent activation of Bcl6 and polycomb-dependent repression of p19Arf. Nat Commun 9: 5452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Xing S, Li F, Zeng Z, Zhao Y, Yu S, Shan Q, Li Y, Phillips FC, Maina PK, Qi HH, Liu C, Zhu J, Pope RM, Musselman CA, Zeng C, Peng W, and Xue HH. 2016. Tcf1 and Lef1 transcription factors establish CD8(+) T cell identity through intrinsic HDAC activity. Nat Immunol 17: 695–703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Xing S, Gai K, Li X, Shao P, Zeng Z, Zhao X, Zhao X, Chen X, Paradee WJ, Meyerholz DK, Peng W, and Xue HH. 2019. Tcf1 and Lef1 are required for the immunosuppressive function of regulatory T cells. J. Exp. Med. 216: 847–866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Steinke FC, Yu S, Zhou X, He B, Yang W, Zhou B, Kawamoto H, Zhu J, Tan K, and Xue HH. 2014. TCF-1 and LEF-1 act upstream of Th-POK to promote the CD4(+) T cell fate and interact with Runx3 to silence Cd4 in CD8(+) T cells. Nat Immunol 15: 646–656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Shapiro-Shelef M, Lin KI, McHeyzer-Williams LJ, Liao J, McHeyzer-Williams MG, and Calame K. 2003. Blimp-1 is required for the formation of immunoglobulin secreting plasma cells and pre-plasma memory B cells. Immunity 19: 607–620. [DOI] [PubMed] [Google Scholar]
- 19.Shan Q, Zeng Z, Xing S, Li F, Hartwig SM, Gullicksrud JA, Kurup SP, Van Braeckel-Budimir N, Su Y, Martin MD, Varga SM, Taniuchi I, Harty JT, Peng W, Badovinac VP, and Xue HH. 2017. The transcription factor Runx3 guards cytotoxic CD8+ effector T cells against deviation towards follicular helper T cell lineage. Nat Immunol 18: 931–939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kallies A, and Good-Jacobson KL. 2017. Transcription Factor T-bet Orchestrates Lineage Development and Function in the Immune System. Trends Immunol 38: 287–297. [DOI] [PubMed] [Google Scholar]
- 21.Shaw LA, Belanger S, Omilusik KD, Cho S, Scott-Browne JP, Nance JP, Goulding J, Lasorella A, Lu LF, Crotty S, and Goldrath AW. 2016. Id2 reinforces TH1 differentiation and inhibits E2A to repress TFH differentiation. Nat Immunol 17: 834–843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hatzi K, Nance JP, Kroenke MA, Bothwell M, Haddad EK, Melnick A, and Crotty S. 2015. BCL6 orchestrates Tfh cell differentiation via multiple distinct mechanisms. J. Exp. Med. 212: 539–553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Choi YS, Kageyama R, Eto D, Escobar TC, Johnston RJ, Monticelli L, Lao C, and Crotty S. 2011. ICOS receptor instructs T follicular helper cell versus effector cell differentiation via induction of the transcriptional repressor Bcl6. Immunity 34: 932–946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Nish SA, Zens KD, Kratchmarov R, Lin WW, Adams WC, Chen YH, Yen B, Rothman NJ, Bhandoola A, Xue HH, Farber DL, and Reiner SL. 2017. CD4+ T cell effector commitment coupled to self-renewal by asymmetric cell divisions. J. Exp. Med. 214: 39–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.