

Abstract
New antibiotics that are active against multi-drug-resistant strains and difficult-to-treat bacterial infections are needed. Synthetic modification of spectinomycin, a bacterial protein synthesis inhibitor, has been shown to increase antibacterial activity compared with spectinomycin. Aminomethyl spectinomycins are active against Gram-negative and Gram-positive bacterial pathogens. In this study, the ability of aminomethyl spectinomycins to treat biothreat pathogens is examined by MIC profiling, synergy testing, and in vivo efficacy experiments. Compound 1950 exhibited potent antibacterial activity against Gram-negative pathogens Brucella spp., Burkholderia mallei, and Francisella tularensis, but showed little to no growth inhibition against Burkholderia pseudomallei, Bacillus anthracis, and Yersinia pestis. Combination testing in checkerboard assays revealed that aminomethyl spectinomycin-antibiotic combinations had mainly an additive effect against the susceptible biodefense pathogens. The in vivo efficacy of compound 1950 was also demonstrated in mice infected with B. mallei (FMH) or F. tularensis (SchuS4). These results suggest that aminomethyl spectinomycins are promising new candidates for development of therapeutics against biodefense bacterial agents.
Subject terms: Antibiotics, Pathogens
Introduction
The rate of emergence of antibiotic-resistant pathogens is outpacing the development of novel antimicrobial agents [1]. One approach to develop new antibiotics is to synthetically modify existing under-utilized antibiotics, such as spectinomycin. Spectinomycin is an aminocyclitol antibiotic that, although structurally similar to the aminoglycoside class, binds to a unique site in RNA helix 34 of the 30S ribosomal subunit, blocks translocation, and halts protein translation [2–4]. Spectinomycin was successfully used in treatments of Neisseria gonorrhoeae until resistance arose, and it was withdrawn from frontline use in the United States [5]. Nevertheless, spectinomycin demonstrates a high safety margin and has a low-molecular-weight which makes it an ideal antibiotic for synthetic modification.
Previous work has demonstrated that spectinomycins with amide modifications on the 3′ position, also called “spectinamides” (Fig. 1a), have increased activity against multi-drug-resistant strains of Mycobacterium tuberculosis. Critical to this series is the addition of a pyridyl side-chain motif that enables the spectinamide to avoid the Rv1258c efflux pump [6, 7]. Recently, novel N-benzyl aminomethyl spectinomycins (amSPCs) demonstrated broad spectrum in vitro activity against Gram-positive and Gram-negative bacterial pathogens. The amSPCs were active against multi-drug-resistant strains of Streptococcus pneumoniae and N. gonorrhoeae and were also able to protect mice from a lethal pulmonary challenge with S. pneumoniae [8, 9]. Although the new spectinomycin analogs had improved activity, they were not active against all drug-resistant strains of N. gonorrhoeae. This clearly demonstrates that modification of spectinomycin can result in more potent broad-spectrum antibiotics, and that both the amSPCs and spectinamides are efficacious in treating pulmonary infections in animal models.
Fig. 1.

a General structures of modified spectinomycin analogs; b Structures of the aminomethyl spectinomycins examined in this study. Note: AmSPCs 1950, 1951, and 2241 exist as the trihydrochloride salts; 2324 exists as the trihydrobromide salt. See the Materials and methods section for details
There are bacteria of importance to biodefense with natural and acquired antibiotic resistance. Burkholderia pseudomallei, in particular, is resistant to penicillins, cephalosporins, quinolones, rifampin, and aminoglycosides [10–12]. While Burkholderia mallei is resistant to fewer antibiotics than B. pseudomallei, it is also naturally resistant to multiple classes of antibiotics [10, 13, 14]. Treatment of Francisella tularensis is limited to the use of three antibiotic classes: fluoroquinolones, tetracyclines, and aminoglycosides. Although F. tularensis strains resistant to these antibiotics have not yet been reported, treatment failures in up to 25% of cases have occurred [15–17]. Facing the threat that these dangerous pathogens pose, new antibiotics that are active against these critical agents are needed.
Here, we report the testing of amSPCs against biothreat bacterial pathogens B. mallei, B. pseudomallei, F. tularensis, Brucella spp., Bacillus anthracis, and Yersinia pestis. Lead amSPCs possess improved activity against several of these pathogens compared with spectinomycin. In addition, treatment with an amSPC led to an increased survival rate of mice infected with B. mallei or F. tularensis.
Materials and methods
Synthesis of the amSPCs
Compounds 1950, 1951, 2241, and 2324 were synthesized according to the procedure reported previously [8]. The purity and identity of the compounds was determined using reverse phase Ultra-Performance Liquid Chromatography (UPLC) analysis with Evaporating Light Scattering Detector MS (ELSD/MS) detection and by 1H and 13C NMR spectroscopy. All compounds were confirmed to be >95% pure before being submitted for biological testing. Elemental analysis of select compounds was performed by Atlantic Microlabs to confirm salt composition.
3′-(R)-3′-(N-(4-fluorobenzyl)aminomethyl)dihydrospectinomycin trihydrochloride (1950)
1H NMR (D2O, 400 MHz): δ 7.34 (dd, J = 3.6, 10.3 Hz, 2H), 7.12–7.07 (m, 2H), 4.51 (s, 1H), 4.48 (s, 1H), 3.98 (t, J = 10.5 Hz, 1H), 3.90 (d, J = 13.4 Hz, 1H), 3.86–3.76 (m, 2H), 3.73–3.73 (m, 2H), 2.99 (d, J = 13.5 Hz, 1H), 2.88–2.79 (m, 3H), 2.57 (s, 3H), 2.42 (s, 3H), 1.75–1.60 (m, 2H), 1.13 (d, J = 6.0 Hz, 3H). 13C NMR (126 MHz, D2O): δ 163.20(d), 132.25, 132.18, 125.96, 116.24, 116.06, 93.17, 92.31, 72.16, 69.59, 67.31, 65.87, 65.28, 61.45, 59.46, 58.43, 50.74, 48.88, 40.00, 30.55, 30.29, 19.84. High-resolution electrospray ionization mass spectrometry (HRMS-ESI) calculated for C22H34FN3O7 [M+H]+ 472.2459, found 472.2459. Elemental analysis theoretical composition for empirical formula C22H34FN3O7∙3 HCl∙2.5 H2O: C, 42.46; H, 6.74; N, 6.75; Cl, 17.09; F, 3.05. Found formula for C22H34FN3O7∙3 HCl∙2.5 H2O: C, 42.21; H, 6.86; N, 6.52; Cl, 16.73; F, 2.85.
3′-(R)-3′-(N-(4-ethylbenzyl)aminomethyl)dihydrospectinomycin trihydrochloride (1951)
1H NMR (D2O, 400 MHz): δ 7.44 (d, J = 7.6 Hz, 2H), 7.40 (d, J = 7.6 Hz, 2H), 4.74 (s, 1H), 4.33–4.20 (m, 3H), 4.01 (t, J = 10 Hz, 1H), 3.94 (t, J = 10 Hz, 1H), 3.69 (m, 1H), 3.50 (m, 1H), 3.38 (ABq, J = 14.0 Hz, 1H), 3.17 (ABq, J = 14.0 Hz, 1H), 2.81 (s, 3H), 2.76 (s, 3H), 2.68 (m, 2H), 1.82 (m, 2H), 1.22 (m, 8H). 13C NMR (126 MHz, D2O): δ 146.68, 130.10, 128.77, 127.39, 93.20, 92.35, 72.12, 69.80, 67.27, 66.01, 65.48, 61.58, 59.60, 58.54, 51.11, 48.76, 40.06, 30.54, 30.35, 27.97, 19.84, 14.84. HRMS-ESI calculated for C24H39N3O7 [M+H]+ 482.2788, found 482.2873.
3′-(R)-3′-(N-(3-phenylpropyl)aminomethyl)dihydrospectinomycin trihydrochloride (2241)
1H NMR (D2O, 400 MHz): δ 7.40 (m, 2H), 7.32 (m, 3H), 4.89 (s, 1H), 4.67 (t, J = 4 Hz, 1H), 4.15 (t, J = 12.0 Hz, 1H), 4.01 (t, J = 8.0 Hz, 1H), 3.90 (t, J = 8.0 Hz, 1H), 3.84 (m, 1H), 3.43 (ABq, J = 14.0 Hz, 1H), 3.22 (br, 2H), 3.10 (m, 3H), 2.77 (s, 3H), 2.74 (m, 2H), 2.63 (s, 3H), 2.05 (m, 2H), 1.88 (d, J = 13.2 Hz, 1H), 1.78 (d, J = 13.2 Hz, 1H), 12.5 (d, J = 6.0 Hz, 3H). 13C NMR (126 MHz, D2O): δ 128.80, 128.53, 128.47, 126.50, 96.35, 93.23, 70.02, 67.32, 66.19, 65.72, 61.69, 59.77, 58.64, 50.22, 48.83, 48.15, 42.79, 40.04, 31.84, 30.61, 30.46, 26.59, 23.19, 19.88. HRMS-ESI calculated for C24H39N3O7 [M+H]+ 482.2866, found 482.2860. Elemental analysis theoretical composition for the empirical formula C24H39N3O7∙3 HCl∙2.3 H2O: C, 45.58; H, 7.43; N, 6.64; Cl, 16.82. Found formula for C22H34FN3O7∙3 HCl∙2.3 H2O: C, 45.36; H, 6.96; N, 6.54; Cl, 16.66.
3′-(R)-3′-(N-(4-chlorobenzyl)aminomethyl)dihydrospectinomycin trihydrobromide (2324)
1H NMR (D2O, 400 MHz): δ 7.48–7.37 (m, 4H), 4.30–4.16 (m, 3H), 3.97 (t, J = 9.9 Hz, 1H), 3.88 (t, J = 10.0 Hz, 1H), 3.73–3.62 (m,1H), 3.50–3.45 (m, 1H), 3.33 (d, J = 13.6 Hz, 1H), 3.26 (s, 2H), 3.21–3.12 (m, 2H), 2.75 (s, 6H), 1.83–1.67 (m, 2H), 1.15 (d, J = 6.0 Hz, 3H). 13C NMR (126 MHz, D2O): δ 135.28, 131.57, 129.34, 128.53, 93.16, 92.32, 72.18, 69.56, 67.33, 65.86, 65.28, 61.43, 59.45, 58.43, 50.76, 49.03, 40.02, 30.60, 30.37, 19.87. HRMS-ESI calculated for C22H34ClN3O7 [M+H]+ 488.2164, found 488.2169.
MIC assay
The MICs were determined as recommended by CLSI in 96-well plates with compound at an initial concentration of 200 µg ml−1 and serially diluted twofold. Approximately 5 × 104 CFU of bacteria in CAMHB (or CAMHB supplemented with 2% IsoVitalex for F. tularensis) were added to each well and plates were incubated at 37°C for 24–48 h. The MIC was determined as the lowest concentration of the compound that prevented growth of the bacterial strain. The MIC50 and MIC90 values were determined as the concentration of the amSPCs at which growth was inhibited in 50 and 90%, respectively, of the strains that were tested.
The MICs of spectinomycin and compound 1950 were also tested in combination with other antibiotics or polymyxin B nonapeptide (PMBN). For the dual antibiotic studies, a checkerboard serial dilution of antibiotics was completed in 96-well plates so that, when possible, the MIC of an individual antibiotic was in the middle of the dilutions (i.e., there were at least two dilutions above and below the MIC in the checkerboard). The bacteria were applied to the plates and incubated as described above. The fractional inhibitory concentration (FIC) index was calculated with the following formula: FIC = (A/MICA) + (B/MICB), where MICA and MICB are the MIC of each drug individually and A and B are the MICs of each antibiotic in combination. Effects were determined to be synergistic (FIC ≤ 0.5), additive (0.5 < FIC ≤ 1.0) indifferent (1.0 < FIC ≤ 4.0), or antagonistic (FIC > 4.0).
For the PMBN combination studies, a checkerboard serial dilution of spectinomycin or compound 1950 (initial concentration 64 µg ml−1) and PMBN (initial concentration 32 µg ml−1) were completed in a 96-well plate. F. tularensis LVS or B. pseudomallei strain Bp82 were applied to the plate as described above and incubated at 37°C with 5% CO2 for 40–48 h. The MIC of spectinomycin or 1950 was recorded for each concentration of PMBN.
Time-kill assay
An overnight culture of B. mallei strain FMH in CAMHB was diluted to an OD600 of 0.01 and incubated, with shaking, at 37 °C for 6 h to allow the culture to reach the mid-exponential phase of growth. The culture was then diluted to a concentration of 5 × 105 CFU ml−1. Compound 1950, spectinomycin, or an equal volume of DMSO/diluent was added such that the final concentration of compound/antibiotic was 50 µg ml−1 and DMSO was 0.5%. The samples were incubated at 37°C with aeration. A sample was removed from the culture at 0, 1.25, 2.5, 4, 6, and 24 h for serial dilution and enumeration of the bacteria on sheep blood agar (SBA) plates. Significant differences between the groups at each time point were determined by multiple t tests with the Holm-Šídák method to correct for multiple comparisons in GraphPad Prism 7.
Mouse infection and colonization
Research was conducted under an IACUC approved protocol in compliance with the Animal Welfare Act, PHS Policy, and other Federal statutes and regulations relating to animals and experiments involving animals. The facility where this research was conducted is accredited by the Association for Assessment and Accreditation of Laboratory Animal Care, International and adheres to principles stated in the Guide for the Care and Use of Laboratory Animals, National Research Council, 2011. Six- to eight-week-old BALB/c mice (Charles River Laboratories/NCI, Frederick, MD) were used for all experiments.
Mice received an intranasal (i.n.) instillation of 3 × 104 CFU B. mallei strain FMH in 20 µl. Treatment with amSPC 1950 (25 or 50 mg kg−1) or saline was initiated at 1 h post-infection. Treatments were administered twice daily (BID) via s.c. injection in 200 µl for 14 days. Mice were closely monitored for morbidity and mortality. On day 40, five mice were removed from the 1950-treated groups (25 and 50 mg kg−1) for necropsy and were marked as censored subjects for the survival curve. Mice that survived until day 60 were euthanized and necropsy was completed. The lungs, livers, and spleens of mice were removed, homogenized, and CFU g−1 of tissue was determined.
To determine if a delay in initiation of treatment had an effect on survival of mice post-infection, mice received an i.n. inoculum of 2 × 104 CFU B. mallei strain FMH in 20 µl. Treatment with 1950 (50 mg kg−1, BID), administered s.c. in 200 µl, was initiated at 1, 6, 24, or 48 h post infection and continued for 14 days. Control mice were administered saline or spectinomycin (50 mg kg−1, BID) starting at 1 h post infection. Mice were closely monitored for morbidity and mortality for 42 days.
Mice were infected i.n. with 1 × 103 CFU F. tularensis strain SchuS4 in 20 µl. Treatment with compound 1950 (50 or 75 mg kg−1) or spectinomycin (50 mg kg−1) in 200 µl was initiated at 1 h post-infection and continued BID for 14 days. Mice were closely monitored for morbidity and mortality for 55 days. In a second experiment, mice were infected with 900 CFU F. tularensis SchuS4 and s.c. administration of saline, spectinomycin (50 mg kg−1), or 1950 (50 mg kg−1) in 200 µl was initiated at 1 h post-infection and continued three times daily (TID) for 14 days. Mice were closely monitored for morbidity and mortality for 37 days.
Significant differences in survival between groups were determined by log-rank (Mantel–Cox) test in GraphPad Prism 7. Significant differences in bacterial load were determined by Mann–Whitney test in GraphPad Prism 7.
Results
AmSPCs are active against B. mallei, Brucella spp., and F. tularensis
The MICs were determined for four amSPCs (structures shown in Fig. 1b) against panels of representative isolates of B. mallei, Brucella spp., F. tularensis, B. anthracis, and B. pseudomallei. The four amSPCs investigated had lower MIC50 and MIC90 values than those observed for spectinomycin for all species tested, except B. pseudomallei (Table 1). The Brucella spp. (Brucella suis, Brucella abortus, and Brucella melitensis) were most susceptible to the amSPCs; the 29 strains tested had an MIC range of 0.4–12.5 µg ml−1 (Table 1; Table S1). An increase in potency of the amSPCs over spectinomycin was also observed in the 29 strains of B. mallei; the MICs ranged from 1.56 to 50 µg ml−1 (Table 1; Table S2). Twenty-eight B. mallei strains had MICs that were ≤12.5 µg ml−1 and only one strain of B. mallei had an MIC value of 50 µg ml−1 for all four amSPC compounds that were tested (Table S2). F. tularensis showed a broad range of sensitivity to the amSPCs; the MICs ranged from 1.56 to 200 µg ml−1 across 26 strains (Table 1; Table S3). B. anthracis was less susceptible to the amSPCs with an MIC range of 3.13–100 µg ml−1 (Table 1; Table S4). For B. anthracis, the MIC50 values for the compounds were 12.5 or 25 µg ml−1, compared with the MIC50 for spectinomycin, 64 µg ml−1. Y. pestis strain CO92 and B. pseudomallei strain K96243 were less sensitive to the amSPCs, with MICs ≥25 µg ml−1 or ≥200 µg ml−1, respectively (Table 1).
Table 1.
Antibacterial activity of amSPC compounds
| MIC (µg ml-1) | ||||||
|---|---|---|---|---|---|---|
| Pathogen | 1950 | 1951 | 2241 | 2324 | SPTa | |
| B. mallei | MIC Rangeb | 3.13–50 | 3.13–50 | 3.13–50 | 1.56–50 | 8–16 |
| MIC50 | 6.3 | 6.3 | 6.3 | 6.3 | 8 | |
| MIC90 | 12.5 | 12.5 | 12.5 | 12.5 | 16 | |
| Brucella spp.c | MIC Rangeb | 0.4–12.5 | 0.4–12.5 | 0.4–12.5 | ≤0.2–6.3 | 1–32 |
| MIC50 | 1.56 | 3.13 | 1.56 | 1.56 | 4 | |
| MIC90 | 3.13 | 6.25 | 3.13 | 6.25 | 8 | |
| F. tularensis | MIC Rangeb | 1.56–200 | 3.13–200 | 3.13–200 | 1.56–200 | 1->64 |
| MIC50 | 12.5 | 6.25 | 12.5 | 6.25 | 64 | |
| MIC90 | 100 | 50 | 50 | 50 | >64 | |
| B. anthracis | MIC Rangeb | 12.5–100 | 6.25–50 | 3.13–25 | 12.5–50 | 16->64 |
| MIC50 | 25 | 25 | 12.5 | 25 | 64 | |
| MIC90 | 50 | 50 | 12.5 | 25 | 64 | |
| Y. pestis CO92 | MIC | 50 | 50 | 25 | 50 | 32 |
| B. pseudomallei d | MIC | >200 | >200 | >200 | >200 | >64 |
aSPT: spectinomycin
bThe range of MIC values for 29 strains of B. mallei, 29 strains of Brucella spp., 26 strains of F. tularensis, and 30 strains of B. anthracis
cBrucella species tested: B. abortus, B. suis, and B. melitensis
dMIC was the same for all 10 B. pseudomallei strains tested
Activity of amSPCs in combination with other antibiotics
Prior in vitro studies have shown that spectinomycins and spectinamides exhibit synergistic activity with different classes of antibiotics [18, 19]. To investigate possible synergistic effects, spectinomycin and amSPC 1950 were tested in combination with at least three antibiotics against each of the six biodefense pathogens, and the FIC index was determined for each combination (Table 2). Ten combinations for spectinomycin and 12 combinations for Lee 1950 had additive effects. Eight combinations for spectinomycin and nine combinations with Lee 1950 had indifferent combinations. Only three combinations for spectinomycin possessed FIC indices that would classify the combination as synergistic: spectinomycin with doxycycline or trimethoprim against B. mallei and with ciprofloxacin against Y. pestis. None of the combinations with 1950 had an FIC that would indicate synergy. There was one antagonistic combination observed with 1950 and streptomycin against B. anthracis.
Table 2.
FIC indices for spectinomycin and compound 1950 in combination with other antibiotics
| Bacterial strain | B. pseudomallei K96243 | B. mallei FMH | B. anthracis Ames | B. suis | F. tularensis SchuS4 | Y. pestis CO92 | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| MIC used for FIC calculationa | 0.5 X MIC | 0.25 X MIC | 0.5 X MIC | 0.25 X MIC | 0.5 X MIC | 0.25 X MIC | 0.5 X MIC | 0.25 X MIC | 0.5 X MIC | 0.25 X MIC | 0.5 X MIC | 0.25 X MIC |
| Spectinomycin FIC | ||||||||||||
| Doxycycline | 1.5 | 1.25 | 0.62 | 0.5 | 0.75 | 0.75 | 0.74 | 1.25 | 0.625 | 0.75 | 0.75 | 0.75 |
| Ciprofloxacin | – | – | – | – | 1.5 | 1.25 | – | – | 1.5 | 2.25 | 0.74 | 0.49 |
| Streptomycin | – | – | – | – | 2.5 | 2.25 | 1.5 | 1.25 | 2.5 | 2.25 | 1 | 1.25 |
| Chloramphenicol | – | – | – | – | – | – | – | – | 0.75 | 0.75 | 1 | 1.25 |
| Clarithromycin | – | – | – | – | 1 | 0.75 | – | – | – | – | – | – |
| Trimethoprim | 1.5 | 1.25 | 0.50 | 0.375 | – | – | 1.5 | 1.25 | – | – | n.d.b | n.d.b |
| Ceftazidime | 1.5 | 0.75 | 1 | 0.75 | – | – | – | – | – | – | – | – |
| 1950 FIC | ||||||||||||
| Doxycycline | 2.5 | 1.25 | 0.62 | 0.75 | 0.75 | 0.75 | 0.74 | 1.25 | 0.75 | 0.75 | 0.625 | 0.75 |
| Ciprofloxacin | – | – | – | – | 2.58 | 2.33 | – | – | 1.5 | 1.25 | 1 | 1.25 |
| Streptomycin | – | – | – | – | 4.5 | 4.25 | 2.5 | 1.25 | 2.5 | 2.25 | 1.5 | 1.25 |
| Chloramphenicol | – | – | – | – | – | – | – | – | 1.0 | 0.75 | 0.75 | 0.75 |
| Clarithromycin | – | – | – | – | 1 | 0.75 | – | – | – | – | – | – |
| Trimethoprim | 1.5 | 1.25 | 0.56 | 1.25 | – | – | 1.5 | 1.25 | – | – | 0.75 | 0.75 |
| Ceftazidime | 2.5 | 2.25 | 0.75 | 1.25 | – | – | – | – | – | – | – | – |
aMIC of Spectinomycin or compound 1950 used for FIC calculation
bCould not be calculated
We also tested polymyxin B nonapeptide (PMBN) in combination with spectinomycin and 1950 against F. tularensis live vaccine strain (LVS) and B. pseudomallei Bp82 (Table 3). PMBN binds to the bacterial cell membrane and can lower the MIC values of antibiotics [20]. PMBN was unable to reduce the MIC for spectinomycin or 1950 against B. pseudomallei Bp82; the MIC remained >64 µg ml−1 at a concentration of 32 µg ml−1 of PMBN. F. tularensis LVS was susceptible to PMBN at a concentration of 32 µg ml−1. When spectinomycin was combined with either 16 or 8 µg ml−1 PMBN, the MIC was reduced from 16 (spectinomycin alone) to 8 µg ml−1. The addition of 16 µg ml−1 PMBN reduced the MIC of 1950 from 4 to 1 µg ml−1. These data indicate that there are possible combination therapies that can improve the efficacy of amSPCs against biodefense pathogens.
Table 3.
MICs of spectinomycin and compound 1950 in combination with PMBN
| Bacteria Strain | MIC with PMBN (µg ml−1) | PMBNa | ||||
|---|---|---|---|---|---|---|
| 0 | 4 | 8 | 16 | 32 | ||
| Spectinomycin | ||||||
| B. pseudomallei Bp82 | >64 | >64 | >64 | >64 | >64 | >32 |
| F. tularensis LVS | 16 | 16 | 8 | 8 | 0.125 | 32 |
| Compound 1950 | ||||||
| B. pseudomallei Bp82 | >64 | >64 | >64 | >64 | >64 | >32 |
| F. tularensis LVS | 4 | 4 | 4 | 1 | 0.125 | 32 |
aMIC (µg ml-1) of PMBN without antibiotic
AmSPC 1950 is bactericidal against B. mallei
The ability of amSPC 1950 and spectinomycin to kill B. mallei in vitro was assayed over a 24 h time frame (Fig. 2). By t1.25, there was a greater than 10-fold decrease and a greater than 100-fold decrease in the amount of viable bacteria in the 1950-treated and spectinomycin-treated groups, respectively, compared with that of the DMSO control group. There was a decline in the number of bacteria present at each time point in the 1950-treated group until no bacteria were detected at t24. Bacteria were not detected in the spectinomycin-control group from t2.5 through the end of the assay. There were statistically fewer bacteria in both the 1950-treated group and the spectinomycin-treated group from t1.25 to t24 (P < 0.0004). These data indicate that amSPC 1950 is bactericidal against B. mallei.
Fig. 2.
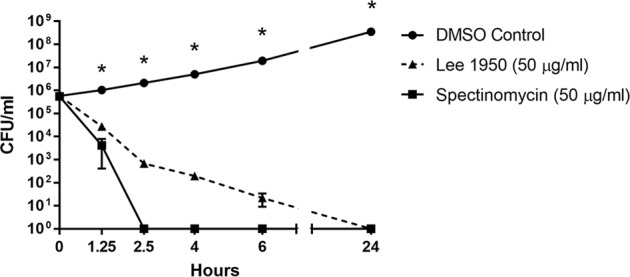
Bactericidal activity of 1950 against B. mallei. B. mallei FMH was cultured in the presence of 50 µg ml−1 compound 1950, spectinomycin, or an equal dilution of DMSO (control). Enumeration of bacteria was completed at 0, 1.25, 2.5, 4, 6, and 24 h after initiation of the assay. *Significantly different from the untreated control group (P < 0.0004) as determined by multiple t tests with the Holm–Šídák method
AmSPC 1950 improves B. mallei infection outcome
In addition to its in vitro potency, amSPC 1950 was selected for murine studies due to its improved half-life compared with spectinomycin and previously demonstrated efficacy against other pathogens [8, 9]. We determined whether 1950 could protect BALB/c mice from an i.n. infection with 3 × 104 CFU B. mallei strain FMH (10 x LD50). Treatment with 1950 (25 or 50 mg kg−1, BID, s.c.) was initiated at 1 h post-inoculation and continued for 14 days. The mice that received no treatment succumbed to infection by day 5 post-infection. Treatment with 1950 resulted in an increased survival of infected mice; 10/10 mice that received 1950 (50 mg kg−1, BID) and 8/10 of mice that received 1950 (25 mg kg−1, BID) survived until day 40 post-infection (Fig. 3a). On day 40, five mice from each treatment group were euthanized to determine the bacterial load within different tissues and the remaining mice were monitored until day 60. Three mice that received 1950 (50 mg kg−1, BID) and one mouse that received 1950 (25 mg kg−1, BID) survived until day 60 post-challenge. The survival of mice that received either dose of 1950 was significantly different from the survival of the untreated control mice (P < 0.0001), but there was no difference in survival between the two treatment groups.
Fig. 3.
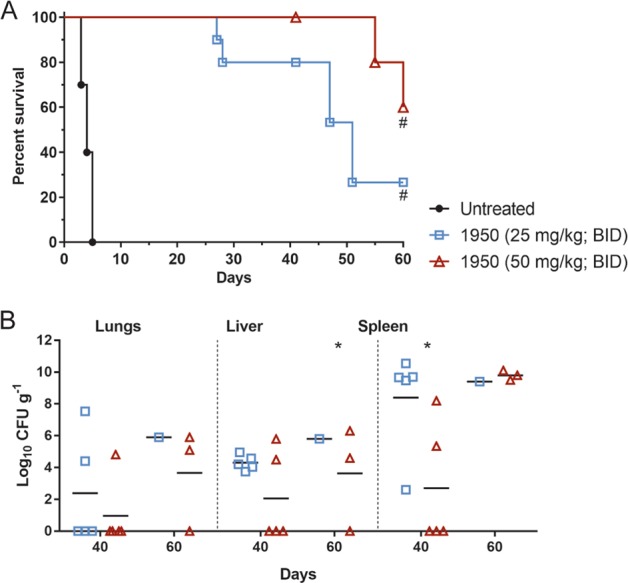
Effect of compound 1950 on B. mallei infection. a Survival of BALB/c mice (n = 10) infected i.n. with 3 x 104 CFU B. mallei strain FMH. Treatment with saline (untreated) or 1950 (25 or 50 mg kg−1, BID) administered s.c. was initiated at 1 h post-infection and continued for 14 days. Mice were monitored for morbidity and mortality for 60 days. #Significantly different from untreated control mice (P < 0.0001). b Bacterial load in the lungs, livers, and spleens of mice on day 40 (n = 5) or day 60 (n = 3, 50 mg kg−1; n = 1; 25 mg kg−1) post infection. Each symbol represents the CFU g−1 tissue for a mouse and the bars represent the mean. The five mice removed for determination of bacterial load on day 40 were censored on the survival curve on that date. *Significantly different from 1950 (50 mg kg−1) spleen, day 60 (P ≤ 0.0357) as determined by Mann–Whitney test
The bacterial load in the lungs, livers, and spleens were determined in mice from the 1950 treatment groups (25 and 50 mg kg−1, BID) on day 40. Only two mice that received 1950 (50 mg kg−1, BID) had detectable levels of bacteria in their lungs, livers, and spleens (Fig. 3b). In contrast, all five mice that received 1950 (25 mg kg−1, BID) had bacteria in their livers and spleens while two of these mice also had bacteria in their lungs. The decreased bacterial load in the mice that received 1950 (50 mg kg−1, BID) is indicative of a dose-dependent response. Furthermore, the bacterial load in the spleens of mice that received 1950 (50 mg kg−1, BID) was significantly different from those that received 1950 (25 mg kg−1, BID) (P = 0.0317). The tissue burden in the mice that survived until day 60 post-infection in the 1950 (50 mg kg−1, BID) group (n = 3) and the 1950 (25 mg kg−1, BID) group (n = 1) was also determined (Fig. 3b). Of the three mice that survived the infection after treatment with 1950 (50 mg kg−1, BID), two mice had bacteria in their lungs, livers, and spleens and one mouse only had bacteria in its spleen on day 60. In the one mouse that survived in the 1950 (25 mg kg−1, BID) group, bacteria were present in its lungs, liver, and spleen. There was a trend that the bacterial load in the mice increased from day 40 post infection to day 60 post infection for the 1950 (25 mg kg−1, BID) and 1950 (50 mg kg−1, BID) treatment groups, although the only significant difference was in the spleen between the 1950 (50 mg kg−1, BID) day 40 and 1950 (50 mg kg−1, BID) day 60 groups (P = 0.0357). This suggests that there is an expansion of the bacteria that survived the 14-day treatment regimen.
We also tested whether the time of treatment initiation post-infection altered the outcome in a B. mallei mouse infection (Fig. 4). Mice were infected i.n. with 2 x 104 CFU B. mallei strain FMH and s.c. treatment with 1950 (50 mg kg−1) was initiated at 1, 6, 24, or 48 h post infection and continued for 14 days (BID). Untreated control mice had 2/10 mice survive until day 42 post-infection. Initiation of treatment at 24 or 48 h post-infection resulted in survival that was similar to the untreated control mice, 1/10 and 2/10, respectively. In comparison, treatment initiation at 1 or 6 h post-infection resulted in survival of 6/8 or 6/10 mice, respectively. The mice that received 1950 at 1 or 6 h post-infection had a significant increase in survival compared with the untreated control mice (P < 0.05). Initiation of spectinomycin (50 mg kg−1, BID) at 1 h post-infection resulted in the survival of 6/10 mice, which was similar to the survival observed after 1950 treatment. These data indicate that 50 mg kg−1 is an effective dose of 1950 when administered up to 6 h post-infection.
Fig. 4.
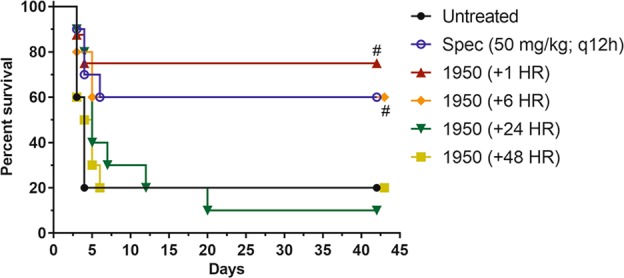
Survival of BALB/c mice infected i.n. with 2 × 104 CFU B. mallei FMH. Treatment with 1950 (50 mg kg−1, BID) was initiated at 1, 6, 24, or 48 h post-infection and was administered s.c. for 14 days. Control mice were treated with saline (untreated) or spectinomycin (50 mg kg−1, BID) starting at 1 h post-infection. 1950 (1 h) group: n = 8; all other groups: n = 10. #Significantly different from untreated control group (P < 0.05)
AmSPC 1950 increases mice survival after F. tularensis infection
We determined whether treatment with 1950 could protect BALB/c mice from an i.n. infection with 1 x 103 CFU F. tularensis strain SchuS4 (10 x LD50). Treatment with 1950 (50 or 75 mg kg−1) or spectinomycin (50 mg kg−1), administered s.c., was initiated at 1 h post-infection and continued for 14 days (BID). Untreated mice (n = 10) succumbed to the infection by day 5 and the spectinomycin-treated (n = 9, 50 mg kg−1, BID) mice succumbed to the infection by day 8 (Fig. 5a). Treatment with 1950 at either dose (50 or 75 mg kg−1, BID) resulted in survival of 3/10 mice at day 55 post-infection which was significantly different from the untreated control (P < 0.0001) and the spectinomycin control (P ≤ 0.0013; Fig. 5a). No bacteria were detected in the lung, spleen, or liver tissue isolated from the mice that survived the infection (data not shown).
Fig. 5.
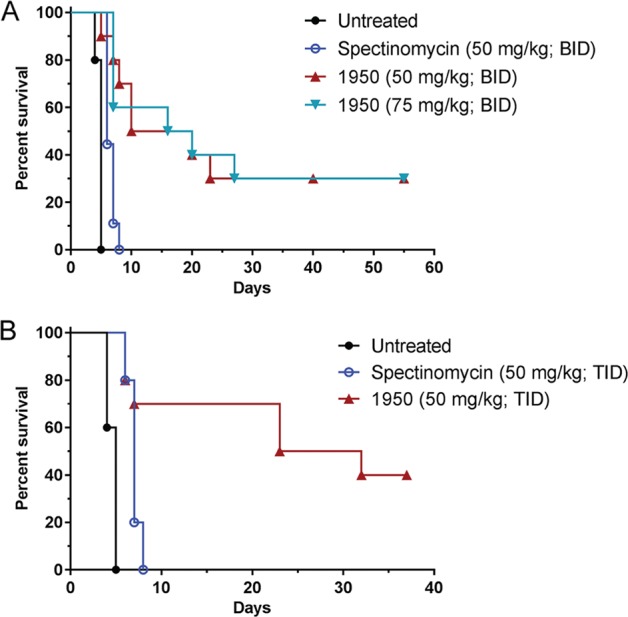
Effect of compound 1950 on F. tularensis infection. a Survival of mice (n = 10) infected i.n. with 1 × 103 CFU F. tularensis strain SchuS4 (10 x LD50). Treatment with saline (untreated), spectinomycin (50 mg kg−1, BID), or 1950 (50 or 75 mg kg−1, BID) administered s.c. started at 1 h post-infection and continued for 14 days. Mice were monitored for morbidity and mortality for 55 days. #Significantly different from untreated control (P < 0.0001) and spectinomycin control (P ≤ 0.0013). b Survival of mice infected i.n. with 900 CFU F. tularensis strain SchuS4 (10 x LD50). Treatment with saline (untreated), spectinomycin (50 mg kg−1, TID), or 1950 (50 mg kg−1, TID) administered s.c. was initiated at 1 h post-infection and continued for 14 days. Mice were monitored for morbidity and mortality for 37 days. #Significantly different from untreated control (P < 0.0001) and the spectinomycin control mice (P = 0.0315)
Since treatment with 1950 (75 or 50 mg kg−1, BID) protected less than half of the mice from F. tularensis infection, we also tested whether administration of 1950 (50 mg kg−1, TID) could improve survival (Fig. 5b). After infection with 900 CFU F. tularensis strain SchuS4, untreated control mice (n = 10) succumbed to infection by day 5 and mice that received spectinomycin (n = 10; 50 mg kg−1, TID) succumbed to infection by day 8. Administration of 1950 (50 mg kg−1, TID) resulted in the survival of 4/10 mice, which was significantly increased compared with the untreated control mice (P < 0.0001) and the spectinomycin control mice (P = 0.0315). Together, these data suggest that amSPC 1950 is a more effective treatment than spectinomycin in the in vivo F. tularensis model.
Discussion
This study demonstrates that amSPCs possess useful antibacterial activity against multiple biothreat pathogens. The modifications to the spectinomycin core rendered these compounds more active than spectinomycin against the Gram-negative pathogens Brucella spp., B. mallei, and F. tularensis. Although there was a slight improvement of activity against the Gram-positive pathogen B. anthracis compared with spectinomycin, the MIC values were higher than those which would be preferred for a B. anthracis therapeutic. Of the antibiotic combination therapies tested, we determined that most possessed additive or indifferent rather than synergistic effects with compound 1950, and that the previously noted synergy with macrolides and spectinamides was not analogous to the amSPCs [18]. It is worth noting that streptomycin was antagonistic when combined with compound 1950, but not with spectinomycin, against B. anthracis. This effect was not observed when tested in Gram-negative bacteria, so the issue may be related to the differences between Gram-positive and Gram-negative susceptibilities and uptake of these antibiotics or specific to B. anthracis itself. In addition, while streptomycin and spectinomycin bind to the 16S rRNA, they have distinct binding sites, so competition is not expected to be the cause [21].
AmSPC compound 1950 has an improved half-life compared with spectinomycin, and was able to protect mice from S. pneumoniae or N. gonorrhoeae infections [8, 9]. Here, we investigated the efficacy of amSPC 1950 against two intracellular pathogens, B. mallei and F. tularensis, which have innate antibacterial resistance [10, 13, 14, 16]. Since amSPCs were able to control the intracellular pathogen Chlamydia trachomatis in cell culture, we anticipated that these compounds could gain access to the intracellular B. mallei or F. tularensis in vivo [8, 9]. While the MIC90 was lower for B. mallei than for F. tularensis, the challenge strains for the two species had the same MIC (12.5 µg ml−1). Despite these similar MIC values, compound 1950 had a greater efficacy against B. mallei than it did against F. tularensis, even when treatment initiation was delayed from 1 to 6 h post-infection. The majority of mice that succumbed to B. mallei did so after the treatment with 1950 ended; this corroborates with the increase in bacterial load from day 40 to day 60 post infection in 1950-treated mice. However, almost half of the mice that succumbed to F. tularensis infection did so in the first 14 days post-infection, the time frame for 1950 treatment. These data indicate that compound 1950 reached therapeutic levels in B. mallei-infected cells and was able to reduce, but not eliminate, the B. mallei infection while the compound was unable to effectively lower F. tularensis below lethal quantities. A longer treatment regimen may have improved the survival of B. mallei-infected mice, but would likely have no effect on the survival of F. tularensis-infected mice.
Compound 1950 had a greater improvement in efficacy in vivo compared with spectinomycin in F. tularensis-infected mice than in B. mallei-infected mice. The survival of mice administered compound 1950 after F. tularensis infection was significantly better than the survival of mice that received spectinomycin, the dose was administered BID or TID. The difference in the MICs of spectinomycin (64 µg ml−1) and 1950 (12.5 µg ml−1) against the challenge F. tularensis strain, SchuS4, explains this difference in survival of mice. The in vitro time-kill assay demonstrated that spectinomycin killed B. mallei faster than compound 1950, despite similar MIC values (12.5 µg ml−1 for 1950, 16 µg ml−1 for spectinomycin). However, the survival of mice administered 1950 at 1 h post-infection with B. mallei FMH was not significantly different from that of the mice that received spectinomycin, although the 1950-treated mice had a slightly higher survival percentage (75% compared with 60%). This demonstrates amSPCs may have improved activity compared with spectinomycin against some pathogens, like F. tularensis.
Overall, we demonstrated that amSPCs have some useful activity against Gram-negative biothreat pathogens in vitro and in vivo. We further demonstrated the potential for spectinomycin analogs as broad-spectrum antibiotics that function against Gram-negative pathogens of clinical and biodefense importance. Efforts are ongoing to improve the potency of the amSPCs, such as boosting their ability to penetrate Gram-negative pathogens which would lead to a greater level of protection against biothreat pathogens.
Supplementary information
Acknowledgements
Opinions, interpretations, conclusions, and recommendations are those of the authors and are not necessarily endorsed by the U.S. Army. Funding for this research is supported by the National Institutes of Health grant AI111449, and ALSAC, St. Jude Children’s Research Hospital.
Compliance with ethical standards
Conflict of interest
The authors declare that they have no conflict of interest.
Footnotes
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
The online version of this article (10.1038/s41429-019-0194-8) contains supplementary material, which is available to authorized users.
References
- 1.Silver LL. Challenges of antibacterial discovery. Clin Microbiol Rev. 2011;24:71–−109. doi: 10.1128/CMR.00030-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Carter AP, Clemons WM, Brodersen DE, Morgan-Warren RJ, Wimberly BT, Ramakrishnan V. Functional insights from the structure of the 30S ribosomal subunit and its interactions with antibiotics. Nature. 2000;407:340–8. doi: 10.1038/35030019. [DOI] [PubMed] [Google Scholar]
- 3.Wirmer J, Westhof E. Molecular contacts between antibiotics and the 30S ribosomal particle. Methods Enzymol. 2006;415:180–202. doi: 10.1016/S0076-6879(06)15012-0. [DOI] [PubMed] [Google Scholar]
- 4.Borovinskaya MA, Shoji S, Holton JM, Fredrick K, Cate JHD. A steric block in translation caused by the antibiotic spectinomycin. ACS Chem Biol. 2007;2:545–52. doi: 10.1021/cb700100n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Boslego JW, Tramont EC, Takafuji ET, Diniega BM, Mitchell BS, Small JW, et al. Effect of spectinomycin use on the prevalence of spectinomycin-resistant and of penicillinase-producing Neisseria gonorrhoeae. N Engl J Med. 1987;317:272–8. doi: 10.1056/NEJM198707303170504. [DOI] [PubMed] [Google Scholar]
- 6.Lee RE, Hurdle JG, Liu J, Bruhn DF, Matt T, Scherman MS, et al. Spectinamides: a new class of semisynthetic antituberculosis agents that overcome native drug efflux. Nat Med. 2014;20:152–8. doi: 10.1038/nm.3458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Liu J, Bruhn DF, Lee RB, Zheng Z, Janusic T, Scherbakov D, et al. Structure-activity relationships of spectinamide antituberculosis agents: a dissection of ribosomal inhibition and native efflux avoidance contributions. Acs Infect Dis. 2017;3:72–88. doi: 10.1021/acsinfecdis.6b00158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bruhn DF, Waidyarachchi SL, Madhura DB, Shcherbakov D, Zheng Z, Liu J, et al. Aminomethyl spectinomycins as therapeutics for drug-resistant respiratory tract and sexually transmitted bacterial infections. Sci Transl Med. 2015;7:288ra75. doi: 10.1126/scitranslmed.3010572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Butler MM, Waidyarachchi SL, Connolly KL, Jerse AE, Chai W, Lee RE, et al. Aminomethyl spectinomycins as therapeutics for drug-resistant gonorrhea and Chlamydia coinfections. Antimicrob Agents Chemother. 2018;62:e00325–−18. doi: 10.1128/AAC.00325-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Thibault FM, Hernandez E, Vidal DR, Girardet M, Cavallo JD. Antibiotic susceptibility of 65 isolates of Burkholderia pseudomallei and Burkholderia mallei to 35 antimicrobial agents. J Antimicrob Chemother. 2004;54:1134–8. doi: 10.1093/jac/dkh471. [DOI] [PubMed] [Google Scholar]
- 11.Wuthiekanun V, Peacock SJ. Management of melioidosis. Expert Rev Anti Infect Ther. 2006;4:445–55. doi: 10.1586/14787210.4.3.445. [DOI] [PubMed] [Google Scholar]
- 12.Viktorov DV, Zakharova IB, Podshivalova MV, Kalinkina EV, Merinova OA, Ageeva NP, et al. High-level resistance to fluoroquinolones and cephalosporins in Burkholderia pseudomallei and closely related species. Trans R Soc Trop Med Hyg. 2008;102(Suppl 1):S103–−10. doi: 10.1016/S0035-9203(08)70025-7. [DOI] [PubMed] [Google Scholar]
- 13.Kenny DJ, Russell P, Rogers D, Eley SM, Titball RW. In vitro susceptibilities of Burkholderia mallei in comparison to those of other pathogenic Burkholderia spp. Antimicrob Agents Chemother. 1999;43:2773–5. doi: 10.1128/AAC.43.11.2773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Heine HS, England MJ, Waag DM, Byrne WR. In vitro antibiotic susceptibilities of Burkholderia mallei (causative agent of glanders) determined by broth microdilution and E-test. Antimicrob Agents Chemother. 2001;45:2119–21. doi: 10.1128/AAC.45.7.2119-2121.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Perez-Castrillon JL, Bachiller-Luque P, Martin-Luquero M, Mena-Martin FJ, Herreros V. Tularemia epidemic in northwestern Spain: clinical description and therapeutic response. Clin Infect Dis. 2001;33:573–6. doi: 10.1086/322601. [DOI] [PubMed] [Google Scholar]
- 16.Boisset S, Caspar Y, Sutera V, Maurin M. New therapeutic approaches for treatment of tularaemia: a review. Front Cell Infect Microbiol. 2014;4:40. doi: 10.3389/fcimb.2014.00040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Caspar Y, Maurin M. Francisella tularensis susceptibility to antibiotics: a comprehensive review of the data obtained in vitro and in animal models. Front Cell Infect Microbiol. 2017;7:122. doi: 10.3389/fcimb.2017.00122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bruhn DF, Scherman MS, Liu J, Scherbakov D, Meibohm B, Bottger EC, et al. In vitro and in vivo evaluation of synergism between anti-tubercular spectinamides and non-classical Tuberculosis Antibiotics. Sci Rep. 2015;5:13985. doi: 10.1038/srep13985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ramon-Garcia S, Ng C, Anderson H, Chao JD, Zheng X, Pfeifer T, et al. Synergistic drug combinations for tuberculosis therapy identified by a novel high-throughput screen. Antimicrob Agents Chemother. 2011;55:3861–9. doi: 10.1128/AAC.00474-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tsubery H, Ofek I, Cohen S, Fridkin M. Structure-function studies of polymyxin B nonapeptide: implications to sensitization of Gram-negative bacteria. J Med Chem. 2000;43:3085–92. doi: 10.1021/jm0000057. [DOI] [PubMed] [Google Scholar]
- 21.Hong W, Zeng J, Xie J. Antibiotic drugs targeting bacterial RNAs. Acta Pharm Sin B. 2014;4:258–65. doi: 10.1016/j.apsb.2014.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.