

Abstract
The cause of ulcerative colitis still remains unclear. The most popular hypothesis is that colitis develops because of a complex interaction of genetic, microbial, environmental, and immunologic factors. This editorial summarizes the widely accepted hypothesis and comments on a variation of this hypothesis promoted by Dr Roediger.
Keywords: environmental factors, genes, intestinal microbiota, mucosal immunity, ulcerative colitis
The inflammatory bowel disorders, ulcerative colitis and Crohn's disease, markedly increased in incidence and prevalence during the 20th century.1, 2 This was most prominent in North America, Europe, and Oceania, with a peak prevalence similar to or exceeding 0.3% of the population. Incidence and prevalence were lower in Asia, Africa, and South America, but incidence has been rising since 1990 in association with industrialization, affluence, and “westernization.” Currently, the incidence appears to have stabilized in western countries but continues to rise in Asia and South America. The ratio of new cases of ulcerative colitis to Crohn's disease varies in different countries but is often of the order of 1:1.
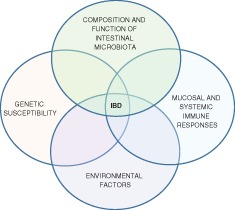
The most popular hypothesis for the etiology of inflammatory bowel disease involves a complex interaction between genetic susceptibility, gastrointestinal microbiota, environmental factors, and mucosal or more generalized immune responses (Fig. 1).3, 4 Using this hypothesis, ulcerative colitis and Crohn's disease are seen as similar inflammatory disorders where the phenotype is determined by variation in one or more susceptibility factors. Consistent with this hypothesis are a similar age of onset for the two disorders, a similar chronic or relapsing course, and a similar response to various drugs including biologics. In addition, some individuals have inflammation that is difficult to characterize as either ulcerative colitis or Crohn's disease (indeterminate colitis), and some affected families have members with ulcerative colitis and other members with Crohn's disease. Those who consider ulcerative colitis and Crohn's disease as entirely separate disorders point to differences in disease location, type of inflammation (mucosal or transmural), and histological features such as the presence of granulomas.
Figure 1.
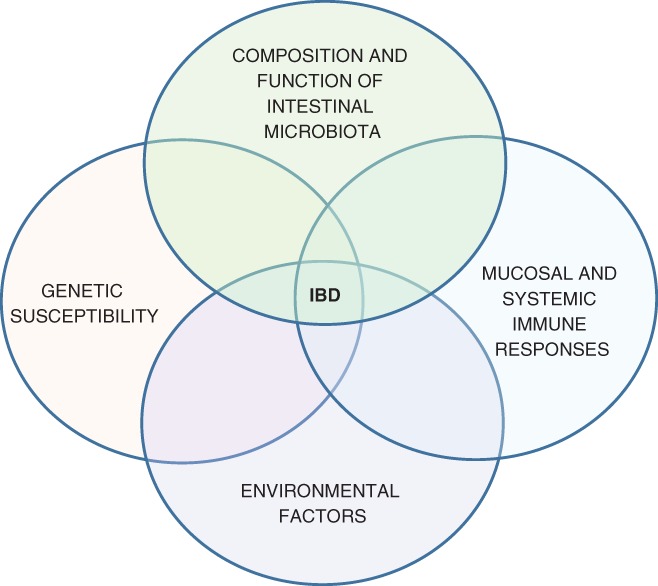
In the most popular hypothesis, inflammatory bowel disease results from the interaction of several factors, including genetic susceptibility, gut microbiota, immune responses, and environmental factors.3
Over the past 40 years, important progress has been made in our understanding of the various susceptibility factors. Arguably, the greatest progress has been made in the area of genetic susceptibility. This began with the identification of the CARD15/NOD2 susceptibility locus in Crohn's disease in 20015, 6 and progressed to the publication of genome‐wide association studies in 2015–2017.7, 8 To date, inflammatory bowel disease has been associated with more than 200 single nucleotide polymorphisms, some specific to Crohn's disease or ulcerative colitis and some common to both disorders. Twin studies have also been informative, with concordance rates in identical twins of 30–35% in Crohn's disease and 10–15% in ulcerative colitis.9 Some of these genes such as CARD15 appear to be involved in host–microbe immune responses, while others such as ATG16LI, LRRK2, and IRGM play a role in intracellular degradation processes known as autophagy. Despite these impressive genetic studies, there is only limited information on the expression of these genes in terms of protein synthesis (proteomics) or end‐point metabolites (metabolomics) or upon modification of gene function by changes in histones or DNA methylation (epigenetics).
Many investigators predicted that important clues to the pathogenesis of inflammatory bowel disease would lie in an analysis of the intestinal microbiota. This prediction is still attractive but has not yet been realized despite the advent of new technologies that identify and quantify gut microbiota by analyzing nucleic acids (DNA and RNA) extracted from feces or colonic biopsies.10, 11, 12 One approach, although perhaps simplistic, anticipated that patients with inflammatory bowel disease would either have an excess of pro‐inflammatory bacteria or a deficiency of anti‐inflammatory bacteria. In clinical studies, patients with inflammatory bowel disease have consistently had a reduced number of bacterial species in feces (reduced bacterial diversity), but whether this feature is primary or secondary to inflammation or diarrhea remains unclear.13 In general, reduction in diversity has been more prominent in Crohn's disease than ulcerative colitis. Another common feature of fecal microbiota in inflammatory bowel disease is a reduced number of bacteria in the phylum Firmicutes, and an increase in the phylum Proteobacteria. The term “dysbiosis” usually encompasses both reduced diversity and an imbalance within intestinal microbiota but currently lacks a strict definition. Overviews of the presence of specific bacterial species in the feces of patients with inflammatory bowel disease are available elsewhere, but thus far, no single organism, groups of organisms, or other components of the microbiota (fungi, archaea, or viruses) have been clearly associated with the inflammatory response.14, 15, 16, 17 On the other hand, there is some evidence that modification of the microbiota is beneficial in at least some affected groups. Examples include induction of remission with longer‐term use of antibiotics in Crohn's disease18 and induction of remission using fecal transplantation in some patients with ulcerative colitis.19, 20
The high incidence of inflammatory bowel disease in industrialized countries has raised the strong possibility of important environmental factors linked to lifestyle and living conditions.21, 22, 23 Whether these exposures are more important in childhood than in adults remains unclear. In childhood, there are minor protective effects with breast feeding; acquisition of Helicobacter pylori; and markers of exposure to greater numbers of microorganisms such as rural living, number of siblings, and access to hot water and animals. The latter factors constitute the “hygiene hypothesis,” although the association is not restricted to inflammatory bowel disease. Antibiotic use in childhood appears to increase the risk of Crohn's disease, particularly when used in the first year of life. In adults, smoking increases the risk of Crohn's disease by 1.7‐fold but decreases the risk of ulcerative colitis by a similar amount. Minor increases in risk have also been associated with oral contraceptives, non‐steroidal anti‐inflammatory drugs, and low levels of vitamin D. An episode of bacterial gastroenteritis also appears to increase risk but only for 1 year after the infection, while appendectomy before the age of 20 years decreases the risk of ulcerative colitis. Although important changes in diet have occurred over the past 100 years, contemporary studies comparing diets in recently diagnosed individuals and control subjects has only demonstrated minor changes of uncertain significance. Most of these environmental associations have been linked to effects on either the intestinal microbiota or immune responses.
The importance of immunological factors is supported by the effectiveness of various immunosuppressive agents in the treatment of inflammatory bowel disease. However, dissection of immunological responses is confounded by issues such as changes in epithelial barrier function, effects on mucosal immunity from changes in intestinal microbiota, and the sheer complexity of the mucosal and systemic immune systems. Potential pathogenic responses include loss of tolerance to intestinal microbiota, impaired tolerance to new microbiota, an overactive mucosal immune system, and the failure of mucosal defense mechanisms to suppress the inflammatory stimulus. All of these pathogenic mechanisms involve the innate and adaptive immune systems with the inclusion of multiple immune cells, immunoglobulins, cytokines, and a variety of other pro‐inflammatory pathways.4, 24, 25 Areas of contemporary interest include the impact of gut microbiota on tolerance induced by subtypes of regulatory T cells26 and the effect of recently discovered proteins, such as the immune sensor NLRP1 that may play a role in the maintenance of “good,” butyrate‐producing bacteria.27
Although the above hypothesis is persuasive, there is always the possibility that alternative hypotheses will prove to be correct. One area of interest is the effect of food additives such as saccharin on the epithelial barrier and gut microbiota.28 Another is the possibility that a currently unrecognized pathogen might be linked to the pathogenesis of inflammatory bowel disease, particularly Crohn's disease. In 1913, Dalziel reported several cases of Crohn's‐like enteritis in humans and noted similarities with Johne's disease in cattle,29 a disease now known to be caused by the Mycobacterium avium subspecies paratuberculosis. Although this issue has been debated for decades, it now seems most unlikely that a mycobacterium is responsible for Crohn's disease, particularly in view of the failure of antimycobacterial therapy.30 Other potential pathogens include a species of Escherichia coli called adherent‐invasive E. coli, Listeria monocytogenes, Mycoplasma fermentans, and others.28, 31
In contrast, there is little evidence of infectious agents in the pathogenesis of ulcerative colitis. In this issue of JGH Open, Roediger highlights a paper published in 1943 that described an ulcerative colitis‐like disease in pigs with pantothenic acid deficiency. Although pantothenic acid deficiency does not occur in humans, the disease in pigs appears to be due to low levels of coenzyme A, an intracellular molecule essential for the oxidation of butyrate and glucose. Low levels of coenzyme A and impaired oxidation of butyrate have also been shown in ulcerative colitis. In the Roediger hypothesis, low levels of coenzyme A in colonic epithelial cells are attributed to toxicity associated with the combined effect of luminal hydrogen sulfide and nitric oxide. By extension, there may be a benefit in diets that limit the sources of sulfur (e.g. meat, eggs, cheese, and whole milk) and nitrogen (vegetables, especially those grown with fertilizer) and perhaps diets that reduce the activity of sulfate‐reducing bacteria. A dietary study based on this hypothesis is currently underway in Australia. We do need some lateral thinking in the area of inflammatory bowel disease, but whether the above hypothesis results in a revolution as occurred with H. pylori and peptic ulceration remains unclear.
Declaration of conflict of interest: Robert V Bryant and Samuel P Costello have received research support and/or speaker fees from the following companies: AbbVie, Emerge Health, Ferring, Janssen, Microbiotica, Shire, and Takeda.
References
- 1. Molodecky NA, Soon IS, Rabi DM et al Increasing incidence and prevalence of the inflammatory bowel diseases with time, based on systematic review. Gastroenterology. 2012; 142: 46–54. [DOI] [PubMed] [Google Scholar]
- 2. Ng SC, Shi HY, Hamidi N et al Worldwide incidence and prevalence of inflammatory bowel disease in the 21st century: a systematic review of population‐based studies. Lancet. 2018; 390: 2769–78. [DOI] [PubMed] [Google Scholar]
- 3. Sator RB. Mechanisms of disease: pathogenesis of Crohn's disease and ulcerative colitis. Nat. Clin. Pract. Gastroenterol. Hepatol. 2006; 3: 390–407. [DOI] [PubMed] [Google Scholar]
- 4. Peloquin JM, Goel G, Villablanca EJ, Xavier RJ. Mechanisms of pediadric inflammatory bowel disease. Annu. Rev. Immunol. 2016; 34: 31–64. [DOI] [PubMed] [Google Scholar]
- 5. Hugot JP, Chamaillard M, Zouali H et al Association of NOD2 leucine‐rich variants with susceptibility to Crohn's disease. Nature. 2001; 411: 599–603. [DOI] [PubMed] [Google Scholar]
- 6. Ogura Y, Bonen DK, Inohara N et al A frameshift mutation in NOD2 associated with susceptibility to Crohn's disease. Nature. 2001; 411: 603–6. [DOI] [PubMed] [Google Scholar]
- 7. Liu JZ, van Sommeren S, Huang H et al Association analyses identify 38 susceptibility loci for inflammatory bowel disease and highlight shared genetic risk across populations. Nat. Genet. 2015; 47: 979–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Luo Y, de Lange KM, Jostins L et al Exploring the genetic architecture of inflammatory bowel disease by whole‐genome sequencing identifies association at ADCY7. Nat. Genet 2017; 49: 186–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Brant SR. Update on the heritability of inflammatory bowel disease: the importance of twin studies. Inflamm. Bowel Dis. 2011; 17: 1–5. [DOI] [PubMed] [Google Scholar]
- 10. Eckburg PB, Bik EM, Bernstein CN et al Diversity of the human intestinal microbial flora. Science. 2005; 308: 1636–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Arumugam M, Raes J, Pelletier E et al Enterotypes of the human gut microbiome. Nature. 2011; 473: 174–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Shendure J, Ji H. Next‐generation DNA sequencing. Nat. Biotechnol. 2008; 26: 1135–45. [DOI] [PubMed] [Google Scholar]
- 13. Ott SJ, Musfeldt M, Wenderoth DF et al Reduction in diversity of the colonic mucosa associated bacterial microflora in patients with active inflammatory bowel disease. Gut. 2004; 53: 685–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Nagalingam NA, Lynch SV. Role of the microbiota in inflammatory bowel diseases. Inflamm. Bowel Dis. 2012; 18: 968–80. [DOI] [PubMed] [Google Scholar]
- 15. Mukhopadhya I, Hansen R, El‐Omar EM, Hold GL. IBD; what role do proteobacteria play? Nat. Rev. Gastroenterol. Hepatol. 2012; 9: 219–30. [DOI] [PubMed] [Google Scholar]
- 16. Haberman Y, Tickle TL, Dexheimer PJ et al Pediatric Crohn's disease patients exhibit specific ileal transcriptome and microbiome signature. J. Clin. Invest. 2014; 124: 3617–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Paramsothy S, Nielsen S, Kamm MA et al Specific bacteria and metabolites associated with response to fecal microbiota transplantation in patients with ulcerative colitis. Gastroenterology. 2019; 156: 1440–54. [DOI] [PubMed] [Google Scholar]
- 18. Ledder O. Antibiotics in inflammatory bowel disease: do we know what we're doing. Transl. Pediatr. 2019; 8: 42–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Paramsothy S, Paramsothy R, Rubin DT et al Faecal microbiota transplantation for inflammatory bowel disease: a systematic review and meta‐analysis. J. Crohns Colitis. 2017; 11: 1180–99. [DOI] [PubMed] [Google Scholar]
- 20. Costello SP, Hughes PA, Waters O et al Effect of fecal microbiota transplantation on 8‐week remission in patients with ulcerative colitis. A randomized clinical trial. JAMA. 2019; 321: 156–6, 164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Molodecky NA, Panaccione R, Ghosh S, Barkema HW, Kaplan GG; Alberta Inflammatory Disease Consortium. Challenges associated with identifying the environmental determinants of the inflammatory bowel diseases. Inflamm. Bowel Dis. 2011; 17: 1792–9. [DOI] [PubMed] [Google Scholar]
- 22. Andersen V, Olsen A, Carbonnel F, Tjonneland A, Vogel U. Diet and risk of inflammatory bowel disease. Dig. Liver Dis. 2012; 44: 185–94. [DOI] [PubMed] [Google Scholar]
- 23. Kimberly W, van der Sloot J, Amini M, Peters V, Dijkstra G, Alizadeh BZ. Inflammatory bowel diseases: review of known environmental protective and risk factors involved. Inflamm. Bowel Dis. 2017; 23: 1499–509. [DOI] [PubMed] [Google Scholar]
- 24. Park JH, Peyrin‐Biroulet L, Eisenhut M, Shin JI. Inflammatory bowel diseases [IBD] immunopathogenesis: a comprehensive review of inflammatory molecules. Autoimmun. Rev. 2017; 16: 416–26. [DOI] [PubMed] [Google Scholar]
- 25. Kmiec Z, Cyman M, Slebioda TJ. Cells of the innate and adaptive immunity and their interactions in inflammatory bowel disease. Adv. Med. Sci. 2017; 62: 1–16. [DOI] [PubMed] [Google Scholar]
- 26. Dominguez‐Villar M, Hafler DA. Regulatory T cells in autoimmune disease. Nat. Immunol. 2018; 19: 665–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Yu CH, Moecking J, Geyer M, Masters SL. Mechanisms of NLRP1‐mediated autoinflammatory disease in humans and mice. J. Mol. Biol. 2018; 430: 142–52. [DOI] [PubMed] [Google Scholar]
- 28. Qin X. Etiology of inflammatory bowel disease: a unified hypothesis. World J. Gastroenterol. 2012; 18: 1708–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Dalziel TK. Chronic interstitial enteritis. Br. Med. J. 1913; 2: 1068–70. [Google Scholar]
- 30. Selby W, Pavli P, Crotty B et al Two‐year combination antibiotic therapy with clarithromycin, rifabutin and clofazimine for Crohn's disease. Gastroenterology. 2007; 132: 2313–9. [DOI] [PubMed] [Google Scholar]
- 31. Roediger B. In Causative factors of ulcerative colitis and Crohn's disease. An exploratory guide. Adelaide: Wakefield Press. [Google Scholar]