

Abstract
Cancer stem cells (CSCs) subpopulation within the tumour is responsible for metastasis and cancer relapse. Here we investigate in vitro and in vivo the effects of a pancreatic (pro)enzyme mixture composed of Chymotrypsinogen and Trypsinogen (PRP) on CSCs derived from a human pancreatic cell line, BxPC3. Exposure of pancreatic CSCs spheres to PRP resulted in a significant decrease of ALDEFLUOR and specific pancreatic CSC markers (CD 326, CD 44 and CxCR4) signal tested by flow cytometry, further CSCs markers expression was also analyzed by western and immunofluorescence assays. PRP also inhibits primary and secondary sphere formation. Three RT2 Profiler PCR Arrays were used to study gene expression regulation after PRP treatment and resulted in, (i) epithelial-mesenchymal transition (EMT) inhibition; (ii) CSCs related genes suppression; (iii) enhanced expression of tumour suppressor genes; (iv) downregulation of migration and metastasis genes and (v) regulation of MAP Kinase Signalling Pathway. Finally, in vivo anti-tumor xenograft studies demonstrated high anti-tumour efficacy of PRP against tumours induced by BxPC3 human pancreatic CSCs. PRP impaired engrafting of pancreatic CSC’s tumours in nude mice and displayed an antigrowth effect toward initiated xenografts. We concluded that (pro)enzymes treatment is a valuable strategy to suppress the CSC population in solid pancreatic tumours.
Subject terms: Cancer stem cells, Drug development
Introduction
Improving therapies against hematopoietic malignancies and solid tumours are making cancer a chronic disease and represent a short-term success. Nevertheless, among cancers, pancreatic ductal adenocarcinoma still has a very poor prognosis with short life expectancy. To decrease cancer mortality rate, strategies to prevent cancer recurrence are needed.
The discovery of a subpopulation of cells with stem cell-like features, called cancer stem cells (CSCs), in a wide spectrum of solid tumours1 has led to a new model of tumorigenesis, the CSCs hypothesis. This model suggests that the intrinsic characteristics of these cells, such as self-renewal and the ability to differentiate, drive tumour initiation and progression, and the subsequent chance of developing metastasis and tumour recurrence2. The CSCs hypothesis is based on the following findings: (i) CSCs represent a small fraction of cancer cells within a tumour; CSCs have tumorigenic potential when transplanted into immunodeficient mice; (ii) CSCs can be isolated by specific surface markers; (iii) tumours resulting from the CSCs contain mixed tumorigenic and non-tumorigenic cells of the original tumour; and (iv) CSCs can be serially transplanted through multiple generations, indicating that it is a self-renewing population3,4. An additional and very important feature of the CSCs population is their ability to remain quiescent for extended periods of time, and therefore evade conventional therapies (chemo- and radiotherapy) that are targeted to highly proliferative cells5. Consequently, a priority for improving cancer treatment and reducing the risk of cancer relapse is to develop new strategies that selectively target CSC eradication while sparing normal stem cells.
One of the biggest challenges in CSCs research has been the development of methods for culture, expansion, and analyses of undifferentiated cancer cells in vitro. In this respect, our group has experience in CSCs enrichment from cancer cell lines or from cancer cells isolated from patients. We have recently patented a method to isolate and culture CSCs and we have also implemented different studies to test drugs directed against CSCs6.
Previous research has suggested a putative utility of pancreatic (pro)enzymes in cancer treatment7,8. In agreement with these studies, we have shown in vivo and in vitro the anti-tumour efficacy of a novel pro-enzyme formulation consisting of a combination of Trypsinogen and Chymotrypsinogen A (PRP), that proves to be an effective and non-toxic treatment, able to induce lineage specific cellular differentiation, inhibit angiogenesis, tumour growth, cancer cell migration and invasiveness. Furthermore, a suppository formulation containing both pancreatic pro-enzymes increased the life expectancy of advanced cancer patients9,10. Based on these findings, we postulated that pancreatic pro-enzymes may induce differentiation of CSCs and reduce recurrence and metastatic spread. Here we have tested, in vitro and in vivo, the effect of the formulation PRP on CSCs isolated from the human pancreatic cancer cell line BxPC3. To do so, we first determined the effect of the anti-cancer drug on cell viability, proliferation and sphere formation capacity of CSCs isolated from BxPC3. Second, we analysed changes in pancreatic CSCs markers after treatment. Third, we have analysed with RT2 Profiler PCR Arrays changes of expression of genes involved in EMT or related to CSCs and MAP Kinase Signalling Pathway induced by PRP treatment. Four, we have studied expression of mirRNAs related to CSCs and to characterize the mechanism of action of the pro-enzyme formulation we have studied protein expression. Finally, we have conducted a xenograft study to assess the effect of PRP in suppressing tumour growth in vivo.
We can conclude that PRP reduces the CSC population, tumour initiation and decreases fibrotic tissue in tumours.
Results
PRP treatment reduces CSCs population in pancreatic cancer
We have previously described the anti-tumor effect of PRP in some pancreatic, colon, oesophagus and ovarian cancer cell lines9,10. To test the selective action of PRP against pancreatic CSCs enriched subpopulations we first determined the IC50 by MTT assay (Fig. 1A). ALDH has been described as a marker for the identification of CSCs11. Here we detected ALDH positive cells by ALDEFLUOR assay using flow cytometry (Fig. 1B) and proved that treatment with PRP significantly reduces the population of ALDH positive cells (from 47.2% to 17.5%) in pancreatic CSCs. Furthermore, Fig. 1B shows that the percentage of cells that express specific pancreatic CSCs markers CD 44, CD 326 and CxCR4 significantly decreased after PRP treatment from 75.1% to 9.5%; from 51.2% to 5.5% and from 63.1% to 42.4% respectively, which signifies that PRP treatment reduces CSCs population in pancreatic cancer cells. Finally, we further determine the expression of the pancreatic cancer markers CD44, CD326 and CxCR4 by immunofluorescence (Fig. 1C) and by western blotting (Fig. 1D and Supplementary Fig. S1). Both assays demonstrate that PRP reduced the expression of specific pancreatic CSCs markers by a decrease in fluorescence red signal shown by treated cells when compared with control cells, which was corroborated by western blotting analysis.
Figure 1.

In vitro analysis of tumorigenic capacity of BXPC3 CSCs after PRP treatment. (A) Antiproliferative activity of PRP against CSCs from pancreatic BXPC3 cell line. (B) Decreased ALDH1 activity and CSCs markers expression: CD44, CD326 and CXCR4 in BXPC3 CSCs. (C) Representative confocal images of CD44, CxCR-4 and CD326 expression in BxPC3 treated and control CSCs. CD44, CD326 and CxCR-4 expression were detected in red and nuclei were counterstained with DAPI (blue). Scale bar represents 40 μm. (D) Western blot analysis of CD44, CD326 and CxCR-4 in BXPC3 CSCs treated with PRP versus non-treated. Β-actin and GAPDH were used as a internal controls. (E) Representative images of BXPC-3 primary and secondary spheres treated with PRP (T/C 0.07/0.42 mg/mL) at 48 and 72 hours after treatment. Scale bar represents 50 μm. (F) In vitro proliferation assay on BXPC-3 CSCs after treatment with PRP and gemcitabine. BXPC-3 primary and secondary spheres were incubated with PRP, gemcitabine (0,01 μM treatment) or with a combination of PRP/gemcitabine 72 h. The PRP, gemcitabine and combination of PRP and gemcitabine treatment resulted in a statistically significant decrease in primary and secondary CSCs spheres compared to control. Statistical significance indicated **p < 0.01. vs. samples not treated.
PRP inhibits spheroid formation by CSCs
We next tested if PRP could suppress sphere formation by pancreatic CSCs. To do so, PRP, was added to the medium at day 0, on cancer cells cultured under sphere conditions (conditioned medium and low attached culture wells) and compared with the effect of gemcitabine, one of the main chemotherapy drugs used to treat pancreatic cancer. Figure 1E shows images of cell cultures on day 2 and day 3. On day 2 it is clear that the addition of PRP to the medium significantly reduces the number and size of CSC spheres. In addition, the morphology of the spheres formed is markedly different, being regular in the control non-treated cells while it was quite irregular in PRP treated cells. On day 3, PRP treated cells do not form spheres and showed the appearance of non-healthy disaggregated cells. BXPC-3 primary spheres treated with PRP demonstrated inhibition of proliferation significantly different from the control group and similar to gemcitabine treatment. The combination treatment of PRP and gemcitabine resulted in a statistically significant decrease in primary CSCs compared to both PRP treatment alone (p < 0.01) and gemcitabine treatment alone (p < 0.01) (Fig. 1F). Moreover, PRP abolished secondary sphere-formation (Fig. 1E) an effect comparable to treatment with gemcitabine. There were statistically significant differences between formed secondary spheres when cells were treated with PRP or gemcitabine alone compared with the control group and between the PRP/gemcitabine combined treatment and all other experimental groups (p < 0.01). The highest rate of secondary sphere inhibition was observed in the group treated with PRP and gemcitabine (p < 0.01) (Fig. 1F).
PRP treatment induces genome-wide gene expression changes in pancreatic CSCs
To understand the molecular changes underlying PRP exposure to pancreatic CSCs, we performed three PCR microarray analysis to measure regulation of the EMT process, CSCs characteristics and variations in Kinase Signalling Pathway related to the treatment. Figure 2A shows up regulation of selective genes that are down regulated during the EMT process. It is important to note that E-cadherin displayed a fold regulation of 4.3, indicating that PRP treatment clearly induces its expression. Furthermore, Kruppel-like-factor 17 (KLF17) a negative regulator of metastasis and EMT12 was also up-regulated by PRP treatment, reaching a fold regulation of 5.9. Other genes such as MST1R (1.6 folds); TFPI2 (4.4 folds) and NUDT13 (2.3 folds), significantly increased their expression after pro-enzyme treatment, implying potent anti-EMT effects of the PRP formulation. Genes that are up regulated during EMT were analysed and the results are summarized in Fig. 2B. Remarkably, 13 genes that have been described to induce EMT were down regulated with PRP treatment. Of interest, the transcription factor Snail which is up-regulated in metastatic cells that are undergoing EMT, was down regulated (SNAI1: −1.9 folds and SNAI2: −1.86 folds). In addition, the serine protease inhibitor SERPINE1, a poor prognostic biomarker in various cancers that promotes tumour progression13, presented a noticeable fold down regulation of −4.6. Furthermore, gene expression of the integrins ITGA5 (−2.5 folds) and ITGAV (−1.02 folds) were also down regulated.
Figure 2.
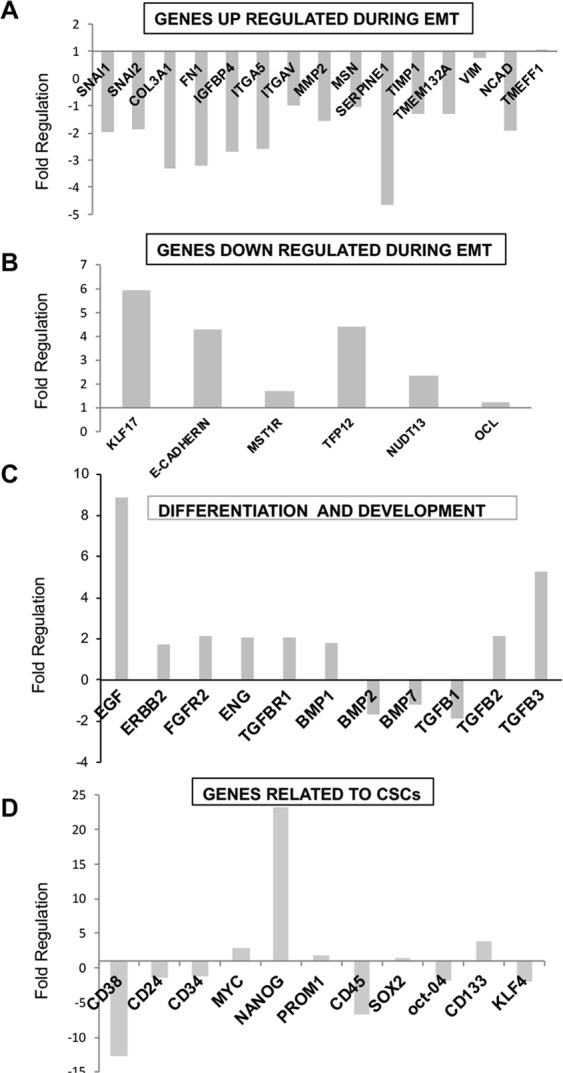
Gene expression profiling of BxPC3 CSCs treated with PRP using RNA Human EMT and CSCs RT2 Profiler PCR Array. BxPC3 CSCs were treated with PRP on day 2 and on day 4. On day 5, total RNA from treated and non-treated CSCs was extracted. A pool of three total RNAs extracted from three independent experiments was used for first strand cDNA synthesis. Gene expression was determined using RT2 Profiler PCR Array of EMT and CSCs. (A) Expression of genes up regulated during EMT. (B) Expression of genes down regulated during EMT. (C) Expression of genes related to differentiation and development. (D) Expression of genes related to CSCs. All gene expressions were normalised to the untreated control.
In previous studies, we proved that PRP induces cell differentiation in the pancreatic cancer cell line Panc19. Here, 7 genes related to cell differentiation were up-regulated after PRP treatment (Fig. 2C). Also, the expression of epidermal growth factor (EGF), related to beta cell differentation14, increased 8.9 folds when compared with control cells. Furthermore, pro-enzyme treatment of pancreatic CSCs induced down-regulation of 6 genes recognized as CSCs markers (Fig. 1D). Although we found a high up-regulation of Nanog (23 folds), interestingly other genes directly involved in the maintenance or acquisition of stemness such as Oct4 and KLF4 were down-regulated due to PRP addition. In addition, the specific CSCs markers CD38, CD45, CD24 and CD34 were down regulated (−12.8 folds; −6.7 folds; −1.4 folds and −1.2 folds respectively); while CD133 was up-regulated (3.9 folds) (Fig. 2D).
Figure 3A shows that 8 genes implicated in metastasis and cell invasion were down-regulated by PRP treatment (PLAUR; BMP7; DKK1; ITGB1; PLAT; FN1; PDGFRB and SPP1) Furthermore, the expression of seven genes related to cell adhesion were up-regulated (Fig. 3B). It is important to note that the expression of E-cadherin (4.3 folds) and β-catenin (4 folds) was enhanced by pro-enzyme treatment in agreement with our previous studies9. In addition, 11 genes with a tumour suppressor role were up-regulated by pro-enzyme treatment, results are summarized in Fig. 3C. Remarkably there was enhanced expression of the transcription factor FOXP1 (6.04 folds), TWIST2 (5.2 folds) and THY1 (3.622 folds). In addition, SPARC, an extracellular protein involved in the deposition and modelling of the extracellular matrix that is downregulated in many tumour types was found to be up-regulated (3.6 folds) by PRP treatment. Furthermore, SIRT1, a member of the sirtuin family of proteins proven to inhibit proliferation of pancreatic cancer cells15, increased the expression in 2 folds in treated cells.
Figure 3.
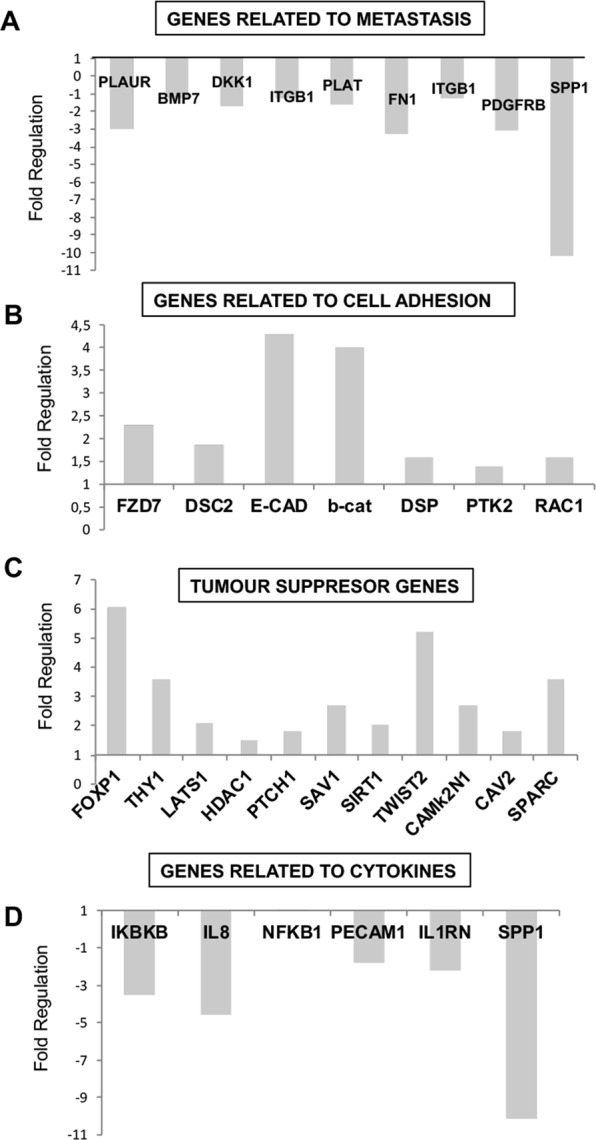
Human EMT and CSCs RT2 Profiler PCR Array of BxPC3 CSCs treated with PRP. BxPC3 CSCs were treated with PRP on day 2 and on day 4. On day 5, total RNA from treated and non-treated CSCs was extracted. A pool of three total RNAs extracted from three independent experiments was used for first strand cDNA synthesis. Gene expression was determined using RT2 Profiler PCR Array of EMT and CSCs. (A) Expression of genes genes related to metastasis. (B) Expression of genes related to cell adhesion. (C) Expression of tumour suppresor genes. (D) Expression of genes related to cytokines. All gene expressions were normalised to the untreated control.
The expression of genes encoding cytokines was investigated after PRP treatment and compared with untreated pancreatic CSCs. Figure 3D shows down-regulation of IKBKB; IL8; PECAM1; IL1RN and SPP1. Remarkably, IL-8 (−4.5 folds) and Secreted phosphoprotein-1 (SPP1, −10 folds).
Finally, the Human MAP Kinase Signaling Pathway RT2 Profiler PCR Array was used to profile the expression of 84 genes related to the MAP kinase (MAPK) signalling pathway. As can be observed in Fig. 4A,B, genes related to JNK and ERK pathways are down-regulated as a consequence of PRP treatment, for instance relevant oncogenic transcription factors JUN (−7 folds), FOS (−7.3 folds) and MEK1 (MAP2K1) (−4.7 folds). Furthermore, PRP induced the up-regulation of cyclin-dependent kinase inhibitor genes (CDKN2A, 3.9 folds; CDKN2B, 2 folds and CDKN2C, 3.8 folds) and decreased the expression of DLK1 (−8.5 folds). In addition, SFN-1 (YWHAZ) gene was down regulated by PRP treatment (−5.2 folds). Moreover, we found p16; SMAD4 and TP53 to be up-regulated Fig. 4A.
Figure 4.

Screening of MAP Kinase Signalling Pathway genes by RT2 Profiler PCR Array. (A) BxPC3 CSCs were treated with PRP on day 2 and on day 4. On day 5, total RNA from treated and non-treated CSCs were extracted. A pool of three total RNAs extracted from three independent experiments was used for first strand cDNA synthesis. Gene expression was determined using MAP Kinase Signalling Pathway RT2 Profiler PCR Array. Sixteen genes were up-regulated, whereas twelve were downregulated compared to the controls. (B) Functional in-silico analysis of proteins related to the c-Jun N-terminal kinase (JNK) pathway. Grey lines represent protein-protein associations, line thickness indicates the strength of data support (active interaction sources from experimental and database only). Red circles highlights genes coding for those proteins that were downregulated by PRP treatment.
Figure 4B summarizes in an in-silico analysis the relationship between some of the genes that were downregulated after PRP treatment, which are involved in the TGFβ SMAD-independent pathway, such as mitogen-activated protein kinases (MAPKs) including c-Jun N-terminal kinase (JNK), p38, and extracellular signal-regulated kinase (ERK) (reviewed by Solomon, 2014)16.
The expression levels of other genes in the microPCR panels did not significantly change after PRP treatment as is shown in Supplementary Fig S2.
Q-PCR analyssis of Rac1, Rac1b and Smad7 gene expression in pancreatic CSCs treated with PRP
Since Rac1 is a key mediator of EMT in pancreatic cancer, and it has been shown that the Rac1-related isoform, Rac1b, acts as an endogenous inhibitor of Rac1 in TGFβ signalling, we analyzed Rac1b and Rac1 + Rac1b expression in PRP treated cells by RTqPCR. As demonstrated in Fig. 5A, Rac1 + Rac1b and Rac1b gene expression were significantly up-regulated following PRP treatment (2.1-fold and 3.4-fold respectively, p < 0.01) indicating that (pro)enzymes treatment may favours decreased expression of TGFβ1-induced tumorigenic events, including EMT, invasion, and metastasis.
Figure 5.

PRP treatment downregulated both Smad-dependent and Smad-independent pathways in TGFβ signaling. (A) RT-qPCR analysis of Rac1b; Rac1 + Rac1b and Smad 7 after PRP treatment. All gene expressions were normalised to the untreated control. (B) RT-qPCR analysis show the fold change of miR-21-5p, miR-182-3p and miRNA-7. Values were normalized using the UniSp6 RNA Spike-in control primer set. (C) Treated and not treated BxPC3 CSCs were incubated with TGFβ1 (5 ng/mL) for 2 h, 4 h and 10 h. The phosphorylation levels of Smad2/3 and total Smad2/3 were analyzed by western blotting at indicated time points. (D) The relative expression of p-Smad2/3 and total Smad2/3 normalized to β-Actin was quantified using Image-J software NIH and is shown as a ratio of p-Smad2/3 versus Smad2/3. (E) Western blotting analysis of p-p38 and total p38 expression in CSCs treated with PRP versus non-treated. The relative expression of p-p38 and total p38 normalized to GAPDH was quantified using Image-J software NIH and is shown as a ratio p-p38/p38 Different letters stand for significant differences (p < 0.05).
In addition the expression of Smad7 (Fig. 5A) was significantly downregulated after PRP treatment (approximately 1 fold).
PRP treatment change miRNA expression
To further explore the effect of PRP formulation on pancreatic CSCs, we analysed known pancreatic cancer miRNAs. Interestingly, we observed that miR-21-5p and miR-182-3p levels were significantly down-regulated after pancreatic CSCs were treated with PRP (Fig. 5B) (p < 0.01). In addition, results showed that treated CSCs increased expression of miRNA-7 (Fig. 5B) (p < 0.05).
PRP treatment downregulates both Smad-dependent and Smad-independent pathways in TGFβ signaling
To support the claim that PRP regulates TGFβ pathway the expression of phosphorylated Smad2/3 was detected under TGFβ stimulation with time intervals in control and treated cells (Fig. 5C, Supplementary Figs S3 and 4 and Fig. 5D). Stimulation of control cells with TGFβ led to an increment of p-Smad2/3 that has a peak at 2 hours of TGFβ induction. On the other hand, cells treated with PRP did not show a significant increment in p-Smad2/3 after TGFβ stimulation in any of the time intervals tested (Fig. 5C,D).
We next studied the effect of PRP on the Smad-independent branch of TGFβ pathway by analyzing the phosphorylation of p38 after PRP treatment. Figure 5E (Supplementary Fig. S5) shows that the amount of phophoyilated p38 significantly decreased after treatment when compared with control cells.
Thus, PRP seams to act throught TGF-β patway, the inhibition of Smad2/3 and p38 phosphorylation may implay that these proteins will not be translocated into the nucleus to regulate multiple EMT-relevant transcription factors as is shown below.
PRP inhibit EMT by directly enhancing E-cadherin and β-catenin expression
We performed immunological analyses to detect the expression of the cell adhesion markers β-catenin and E-cadherin. Our results show that non-treated CSCs displayed distinct membrane localization of β-catenin and weak, diffuse nuclear and cytoplasmic staining while PRP treated CSCs maintain membrane-associated and cytoplasmic staining of β-catenin without nuclear translocation (Fig. 6A). Treated cells maintained E-cadherin expression despite the PRP treatment destroyed the spheroid and many adhesion structures (Fig. 6B).
Figure 6.

Representative confocal images of tumorigenic proteins in PRP treated pancreatic CSCs versus non-treated CSCs. (A) Treatment with PRP maintain membrane-associated β-catenin without nuclear translocation in pancreatic CSCs. (B) PRP treatment increased membrane-associated E-cadherin. Cell surface β-catenin and E-cadherin expression was detected in red and nuclei were counterstained with DAPI (blue). Scale bar represents 20 μm. (C) PRP treatment decreased the protein expression of YAP in BxPC3 CSCs. YAP expression appears green and nuclei stained with DAPI (blue). Scale bar represents 10 μm. (D) Western blot analysis of YAP and p-YAP proteins in CSCs treated with PRP versus non-treated. A negative correlation between YAP and p-YAP expression was found.
Role of PRP in hippo pathway
To corroborate the hypothesis that PRP inhibits CSCs proliferation and tumorigenesis through the Hippo pathway, we further determined the impact of PRP on YAP and P-YAP expression. We found that PRP treatment tended to decrease the expression of YAP on CSCs as judged by immunofluorescence assay Fig. 6C. We then analyzed the expressions of YAP and p-YAP proteins by Western blotting. Analysis confirmed that PRP repressed YAP expression. Moreover, a negative correlation between YAP and p-YAP expression was found (Fig. 6D and Supplementary Fig. S6) with a marked increase in YAP phosphorylation induced by PRP treatment.
PRP inhibits tumour initiation and growth on nude mice
Here we show a xenograft study, with tumours generated from BxPC3 CSCs, to test the effect of PRP as a pre-treatment before tumour induction and as a pre-treatment and continuing treatment after tumour induction (Fig. 7A). 10 animals were used per group in agreement with others17. Nevertheless, one of the main difficulties of interpreting xenograft studies data is the loss of individuals, due to early death, that inevitably occurs during the time-period of the experiment18. In our study, 2 animals from the pre-treatment group and 4 animals from the pre-treatment + treatment group died before completing the experiment (due to anesthesia and ulceration issues). These sudden deaths were not related to an adverse effect of PRP, but rather due to technical problems with the compound administration. Results showed here have been normalized by the final “n” of each experimental group. We found that PRP treatment led to significant reduction in tumour development, thus 9 out of 10 control mice did present palpable tumours; on the contrary, only 3 out of 8 pre-treated mice and 3 out of 6 pre-treated + treated mice developed tumours (Fig. 7B,C). Therefore, tumour incidence (%) represented a 41% in the pre-treated mice and a 50% in the pre-treated + treatment group, indicating a clear suppressive effect of PRP on pancreatic CSC´s tumour engrafting (Fig. 7D). Further, the evolution of tumour growth was evaluated measuring tumour volumes every 2 days for 34 days (Fig. 7E), demonstrating that PRP treatment and the follow up treatment with PRP significantly inhibits tumour growth. Next, we examined the Tumourigenesis Index (TIn) that relates tumour incidence and tumour weight. This index can be used as a measure of malignancy or tumour aggressiveness. Figure 7F shows that significant differences were appreciated in TIn when the mice were pre-treated and treated with PRP, indicating the anti-tumourigenesis effect of PRP. In conclusion, PRP impaired engrafting of pancreatic CSC’s tumours in nude mice and displayed an antigrowth effect toward initiated xenografts.
Figure 7.

PRP reduced tumour growth of pancreatic CSCs in vivo. (A) Scheme of the in vivo study design to determine antitumour activity of PRP against pancreatic CSCs induced tumours. Perpendicular bars stand for PRP bolus injection. BxPC3 CSCs injection into the flank of nude mice is represented by a red arrow. (B) Photographs of the subcutaneous tumors isolated from mice included in the control and PRP treated groups at the end of the experimental protocol. (C) Tumour weight of each single tumour (different colour circles) at Termination (mg), mice with no tumour are represented by a colour circle on the X axis. (D) Tumour incidence. Percentage of mice with tumours respect to control group which represented a 100% of tumour incidence. (E) Tumour size evolution over time. MEM ± SE. *p < 0.05 vs. control group. (F) Tumorigenesis Index (TIn). This index relates tumour incidence and tumour weight. Percentage of mice with tumours respect to control group which represented a 100% of tumour incidence. Mean Tin ± SE at Termination. Different letters stand for significant differences (p < 0.05).
The histological analysis of BxPC-3 tumours sections showed differences in tumour characteristics, particularly in the amount of fibrotic tissue, when comparing tumours of the same size. Large necrotic areas were observed (nude areas) in treated tumours compared to untreated controls (Fig. 8A,B). Hematoxilin and eosin staining showed an increased labelling of stromal proteins in control tumour tissues when compared to tumour sections from the pre-treatment and pre-treatment + treatment groups (Fig. 8A). In adittion, Masson’s Trichrome staining showed that pancreatic CSCs induced tumours form large islets of cohesive cells embedded in an abundant collagenous stroma (green) (Fig. 8B, CTL). Interestingly, the presence of extracellular matrix proteins fibers was strongly reduced in tumours isolated from mice that had been treated with PRP. The presence of abundant lacunae and unhealthy connective tissue can be seen within the fibrotic tissue area (Fig. 8B).
Figure 8.

Effects of PRP on ECM deposition and pancreatic cancer marked CD44 expression. Representative images of tumour sections histologicaly staining with haematoxylin-eosin (H&E) (A) and Masson’s Trichrome (MS). (B) Tumours isolated from treated mice showed less fibrotic tissue (pink staining for the H&E test and green staining for the MS labellin) when compared with tumours isolated from control mice. (C) Immunofluorescence staining of pancreatic tumour sections with CD44 (red channel) demonstrated that tumours from non-treated mice had increased expression of CD44. Magnification, 10×.
Moreover, the tumorigenic stem-like cell marker CD44 expression was analysed in tumour sections by immunofluorescence assay. Control tumour sections exhibited a strongly positive CD44 labelling while tumour sections bellonging to the treatment groups displayed weak CD44 staining consistent with the antitumour effect produced by PRP treatment (Fig. 8C).
Discussion
Cancer stem cells (CSCs) have been characterized as immortal cells (due to their high telomerase activity) with a great tumour-initiating capacity. Furthermore, they can self-renew and they have unique properties, including pluripotent features, high metastatic potential, quiescent capacity and radio- and chemo-resistance1, thus strategies to eradicate CSCs population are needed to prevent cancer recurrence.
Here we have tested the potential therapeutic effect of a pro-enzyme pancreatic preparation, PRP, on pancreatic enriched CSCs population, and shown that PRP treatment in vitro decreased the percentage of ALDH1-positive cells as well as the percentages of cells expressing the pancreatic CSC-specific biomarkers CD326, CD44 and CXCR419. In addition, PRP treatment induced an inhibition of both primary and secondary sphere formation ability of treated pancreatic CSCs, in fact, tumour-derived spheroid structures have been reported to be related to an enrichment of the CSC subpopulation20.
To determine the effect of PRP treatment at mRNA expression level we performed a series of microarray analysis and proved that a wide range of genes closely related to the CSCs phenotype, the EMT process, cell proliferation, metastasis, cell adhesion, tumour suppression mechanisms, cytokine signalling and kinase activities were altered by the treatment. We found that OCT4 which has been reported to play a key role in the maintenance of a stem-like phenotype in pancreatic CSCs was down-regulated by PRP21. Moreover, it is important to highlight the down-regulation of DLK1, induced by PRP treatment, as this factor has been reported to play a significant role in the ability to form spheroid-like structures associated with the CSC phenotype22. These results correlate with the decrease in the CSCs population after PRP treatment shown above.
In addition, PRP treatment induced up-regulation of proven tumour suppressor genes such as the transcription factor FOXP1, a tumour suppressor lost in several tumour types23. and TWIST2 and THY124,25.
Interestingly, the Notch signalling pathway, which is aberrantly activated in many types of malignancies and which is a key regulator of the EMT process26,27, was found to be inhibited by PRP. In fact, the expression of several Notch initiating genes, including NOTCH1, JAG1, FGF and PDGFR was downregulated by PRP treatment. Furtheremore, EMT-related genes, such as Snai1 and Snai2 (also known as Slug) that are target genes of the Notch pathway were down-regulated by PRP treatment. In addition, E-cadherin, a gene suppressed by the Notch pathway, was up-regulated after PRP treatment26.
In studying cytokine expression we found that interleukin-8 (IL-8), a pro-inflammatory factor, belonging to the CXC chemokine family was down-regulated by PRP. Cancer cells secrete IL-8 in an autocrine manner to achieve a proper tumour microenvironment28,29. Particularly, IL-8 and IL-8 receptors have been shown to be over-expressed in pancreatic cancer, but suppressed in normal pancreatic tissues, suggesting that IL-8 plays an important role in the invasiveness of human pancreatic cancer30. In addition, treatment with PRP dramatically decrease the expression of SPP1. This protein plays a crucial role in determining the metastatic potential of various cancers31, promotes cancer cell survival and regulates tumour-associated angiogenesis and inflammation32. Moreover, both JNK and ERK pathways were down-regulated as a consequence of PRP treatment, which is significant since those pathways are crucial in the control of cell proliferation and tumour malignancy. We observed a marked down-regulation of MEK1 (MAP2K1) a cue member of the ERK pathway. Remarkably, MEK inhibitors have been tested in clinical trials as anti-tumour agents, for instance PD0325901, a second generation MEK1/2 inhibitor33. Therefore, PRP seems to control cell proliferation by inhibiting both JNK and ERK pathways through suppression of the expression of two relevant oncogenic transcription factors JUN and FOS.
Furthermore, PRP induced the up-regulation of cyclin-dependent kinase inhibitors genes (CDKN2A; CDKN2, 2 and CDKN2C) which are considered important tumour suppressor genes. Here we found that PRP significantly decreased the expression of DLK1 in treated pancreatic CSCs. Recent studies have shown that DLK1 knockdown could suppress the ability of proliferation, colony formation, spheroid colony formation, and in vivo tumorigenicity of hepatic CSCs22. In addition, the SFN-1 (YWHAZ) gene was dramatically down regulated by PRP treatment. YWHAZ knockdown by specific siRNAs, has been shown to inhibit the proliferation, migration, and invasion of YWHAZ-overexpressing gastric cancer cells34. Feldmann and Maitra (2008)35 have reviewed the molecular genetics of pancreatic ductal adenocarcinomas and presented three important tumour suppressor genes that are deleted in a high percentage of pancreatic cancers: p16; SMAD4 and TP53. These genes inhibit cell proliferation by arresting cell cycle progression. Of interest, PRP up-regulates the expression of these same genes, suggesting the role of PRP in suppressing cell proliferation and malignant transformation.
Some studies state that the TGFβ signalling pathway can exert a tumour suppressive role in earlier stages of tumour development and change to a tumorigenic pathway in the advanced stages after losing Smad436, TGFβR237, TGFβR338 or RAC1b39 expression and gain overexpression of TGFβ139. Interestingly, our results reveal a down-regulation of TGFβ1 and an up-regulation of TGFβ2, TGFβ3, Smad4 and RAC1b in treated cells, suggesting that PRP treatment promotes the TGFβ pathway role via a tumour suppressive route.
In the line of the changing nature of TGFβ pathway during tumour progression, it has been pointed that Smad7, a TGFβ pathway component, can be considered as a “swith” acting as an inhibitor of TGFβ pathway in early stages of tumour progression and as a enhancer of tumour invasion in late stages. Indeed, Smad7 has been shown to be overexpressed in many cancer types and its abundance can be positively correlated with the malignancy potential40. Here we shown a down-regulation of Smad7 in treated cells, supporting the idea that PRP treatment promotes a deep change in TGFβ pathway and decreases tumour malignancy. Additionally, PRP treatment up-regulated KLF17 expression, which enhances TGFβ/Smad-dependent signalling pathway directing cell differentiation41 and not toward cell stemness. Moreover, treated pancreatic CSCs exhibited a decreased activation of both Smad-dependent and Smad-independent branches of TGFβ pathway as shown by the reduction Smad2/3 phosphorylation, even under TGFβ stimulation, and the reduction of p38 phosphorylation. Interestingly, RAC1b has been described to act as a “retaining wall” of TGFβ pathway preventing the hyperactivation of Smad-dependent and Smad-independent branches of TGFβ pathway42,43. Here we have shown that PRP up-regulate RAC1b which matches with the decreased phosphorylation of Smad2/3 and p38 found.
The expression of miR-21-5p, miR-182-3p and miR-7 was analysed after PRP treatment and was found that PRP treatment decreased the expression of miR-21-5p and miR-182-3p and up-regulated the expression of miR-7. FOS and JUN form the protein complex known as Activated Protein 1 (AP1) which plays a key role in cancer development44 and is up-regulated in the CSCs population promoting chemoresistance through miR-21 enhancement in these cells45. Therefore, the down-regulation of FOS and JUN and the down-regulation of miR-21 suggests that PRP treatment may inhibit the activity of AP-1 complex and, subsequently, it may promote the chemo-sensitization of the treated pancreatic CSCs, which is in agreement with the disruption of the CSC phenotype discussed above. Furthermore, miR-21 has been described to suppress Smad4 expression cells46, thus the down-regulation of miR-21 after PRP treatment seems to be linked to the up-regulation of Smad4 and to the alteration of the TGFβ pathway found in treated cells. PRP treatment also diminished the expression of miR-182-3p, others have proposed that up-regulation of miR-182 correlates with the hyperactivation of TGFβ pathway47, therefore, the down-regulation of miR-182 in treated CSCs supports the hypothesis that PRP may be suppressing the hyperactivation of TGFβ pathway. Last, PRP treatment up-regulated the expression of miR-7 which is considered a tumour suppressor since it has been found to suppress the EMT process and the stemness of CSCs in some tumours48,49.
Moreover, mRNA levels of MST1/2, SAV1 and LATS1/2 were found to be upregulated by PRP, together with an increased amount of phospho-YAP protein proved by western blotting. YAP and TAZ belong to the Hippo signalling pathway and are accumulated in the cytoplasm by phosphorylation prompted by MST1/2, SAV1 and LATS1/2. Thus, phosphorylation secuestrates these proteins in the cytoplasm, preventing migration to the nucleus and the activation of downstream genes involved in cell proliferation50,51. Likewise, the inhibition of the activation (phosphorylation) of p38 shown here is relevant since p38 pathway has been described as blocking the phosphorylation of YAP, which correlates with the increment of phosphorylated YAP shown here after PRP treatment of pancreatic CSCs52. It appears that PRP treatment inhibits the uncontrolled proliferation promoted by an aberrant hyperactivation (dephosphorylation) of YAP, which is a common event in many types of malignancies51,53,54.
In addition, cytoplasmic phospho-YAP has been reported to promote the phosphorylation and the retention of β-catenin in the cytoplasm55, which correlates with our findings with immunofluorescent assays. β-catenin is a key component of the Wnt signalling pathway, which is over-activated causing a strong enhancement of cell proliferation and tumour progression in many types of malignancies56. In the absence of Wnt-related ligands (Wnt1, Wnt2, Wnt3…), β-catenin is consistently phosphorylated and retained in the cytoplasm and, subsequently, it cannot migrate to the nucleus and activate pro-tumorigenic target genes57. According to these data along with the down-regulation of some Wnt-related initiating ligands such as WNT1 or WNT11 and the up-regulation of several Wnt pathway-suppressors including SOX1058 and SIRT159 shown here, we conclude that PRP treatment strongly inhibits the canonical WYT pathway56. Adittionaly, the increased phosphorylation of YAP after PRP treatment could also explain the inhibition of Notch pathway suggested above54.
Finally, we present a xenograft study to analyse the effect of PRP in vivo. Our results suggest that pre-treatment with PRP might inhibit the production and the action of growth factors and angiogenic stimulating factors that are necessary to prepare the tumour niche and, consequently, it reduces the possibility of tumour engrafting. In fact, the anti-angiogenic effect of PRP has been previously proven by our previous studies9. Interestingly, TGFβ-1, an essential factor to create, maintain and support CSCs60 was strongly down-regulated after PRP treatment, which agrees with other studies that demonstrated oral therapy with proteolytic enzymes decreased TGFβ in human blood (Desser et al. 2001).
Moreover, our histological analysis of the xenografting mice models shows a decreased amount of tumour-surrounding fibrotic tissue of treated mice which can be explained by the strong down-regulation of TGFβ-1 by PRP treatment as TGFβ-1 has been described to be closely related to the corruption of healthy fibroblasts into cancer-associated fibroblasts (CAFs)61,62. Additionally, it has also been reported that the surrounding tumour microenverioment also promotes the appearance of new CSCs derived from non-stem cancerous cells by secreting several factors such as IL6, HGF or TGFβ-163. Consequently, pre-treatment and follow up treatment with PRP might decrease TGFβ-1 and, subsequently, it might impair CSCs tumour engrafting, niche formation and even CSCs subpopulation activation. Further, treated tumours showed a decreased expression of the pancreatic cancer marker CD44. Knockdown of CD44 expression in pancreatic cancer cells has been linked with decreased cellular proliferation, invasiveness and increased sensitivity to gemcitabine64.
In summary, the present study demonstrates the high anti-tumour efficacy of PRP against human pancreatic CSCs, both in vitro and in vivo. The molecular PRP findings presented demonstrates that PRP treatment promotes the up-regulation of RAC1b which avoids the hyper-activation of the p38 pathway induced by the TGFβ pathway leading to the phosphorylation of YAP observed in our results, which also explains the blockage of the canonical Wnt pathway, the inhibition of the Notch pathway as well as the cytoplasmic location of β-catenin in treated cells as discussed above. Morover, the regulation of TGFβ, Hippo, Wnt and Notch pathways could explain the disruption of the CSC phenotype and the EMT process found in treated cells. Finally, we reported that PRP impaired engrafting of pancreatic CSC’s tumors in nude mice and displayed an antigrowth effect toward initiated xenografts, making (pro)enzymes treatment a valuable strategy to reduce CSC population for pancreatic cancer therapy. As cancer treatment moves towards more personalised medicine, proper therapies to target and treat specifically CSCs may prove to be a useful method for reducing recurrence after drug treatment failures.
Material and Methods
Pancreatic pro-enzymes
The pancreatic pro-enzymes Chymotrypsinogen A and Trypsinogen were supplied by AplyChem. A stock solution of 6 mg/ml of Chymotrypsinogen A and 1 mg/ml of Trypsinogen was made dissolving the pancreatic enzymes in PBS and stored at −20 °C. For each experiment, the stock solution was further diluted in a medium to obtain the desired concentrations.
Cell line
The human pancreatic cancer cell lines BxPC3 was obtained from American Type Culture Collection (ATCC) and maintained in RPMI 1640 Medium (Sigma-Aldrich) supplemented with 10% FBS. Cell disassociation for cell passing was done with the trypsin replacement TrypLE reagent (Life Technologies).
Isolation of CSCs
Cancer stem-like cells from BxPC3 pancreatic cancer (adenocarcinoma) cell line were isolated using the ALDEFLUOR assay (StemCell Technologies) by fluorescence-activated cell sorting (FACS) and enriched subpopulation of CSCs were grown with our specific sphere forming medium (PCT/ES2015/070606, Jiménez et al. 2018) in ultralow attachment plates (Corning).
MTT cell proliferation assay
The effect of PRP on cell viability and proliferation was assessed using the MTT (3-(4, 5-dimethylthiazolyl-2)-2, 5-diphenyltetrazolium bromide) assay (Sigma-Aldrich). Briefly, cells (2 × 103 cells/well) were seeded onto 96- well plates and incubated for 24 h and then treated with different PRP concentrations (Fig. 1A). Three days later, treatment was repeated and cells were maintained for 3 additional days. Cells were maintained with the drug for six days. Thereafter, cells were processed as follows, 10 μL of 2 mM MTT reagent was added to each well and cells were incubated at 37 °C for 4 hours. 100 μL detergent reagent was then added and cells were left at room temperature in the dark for 2 hours. Absorbance was recorded at 570 nm. The IC50 values were calculated from four parametric logistic curves by linear interpolation using Sigma Plot software. All of the experiments were plated in triplicate wells and were carried out at least twice.
ALDEFLUOR assay and CSCs markers analysis by flow cytometry
Cancer stem-like cells from BxPC3 cancer cell line were labelled using the ALDEFLUOR assay kit according to the manufacturer’s recommendations (StemCell Technologies). The enriched subpopulation of CSCs was characterized using a FACSAria III flow cytometer (Becton Dickinson).
To analyse CSC markers treated and not treated BxPC3 CSCs were disaggregated by tryple and washed twice in PBS supplemented with 1% bovine serum albumin (Sigma-Aldrich). The cell surface Fc receptor was blocked using IgG (Santa Cruz Biotechnology) on ice for 15 min. Cells were stained for 30 min at 4 °C with anti-CD44-PE and anti-CD326 FITC and CxCR4-APC monoclonal antibodies (BD Biosciences). After washing, cells were analyzed using a FACSAria III flow cytometer (Becton Dickinson).
Effects of PRP and gemcitabine on BXPC-3 CSCs proliferation
Enriched subpopulations of CSCs from formed primary and secondary spheres were seeded in a concentration of 3000 cells/well in 96-well plates in sphere forming medium and treated with PRP (T/C 0.07/0.42 mg/mL), gemcitabine at 0,01 μm and with both PRP (T/C 0.07/0.42 mg/mL) and gemcitabine 0,01 μm. After 72 h, 0.01 ml of Cell Counting Kit-8 (CCK-8) (Dojindo Molecular Technologies), was added to each well and incubated at 37 °C for 2 h. Plates were read at 450 nm on a Bio-Rad plate reader.
PCR microarray analysis of human EMT, CSCs and MAP kinase signalling pathway related genes
BxPC3 enriched CSCs were grown in 6 well plates and treated twice with PRP (T/C 0.07/0.42 mg/mL) on day 2 and on day 4 or non-treated control cells (CTL). On day 5, total cellular RNA was isolated using TRIzol reagent (Life Technologies). A pool of three total RNAs extracted from three independent experiments was used for first strand cDNA synthesis using the kit SuperScript IV First-Strand Synthesis System (ThermoFisher Scientific).
PRP induced changes in expression of genes involved in the EMT process, related to CSCs or involved in MAP Kianse Signalling Pathway were analysed with RT2 Profiler PCR Arrays Human Epithelial to Mesenchymal Transition (PAHS-090Z, SABiosciences); RT2 Profiler PCR Arrays Human Cancer Stem cells (PAHS-176Z, SABiosciences) or RT2 Profiler PCR Arrays Human MAP Kinane Signalling Pathway (PAHS-176Z, SABiosciences) respectively, and processed following the manufacturer’s instructions applied on the ViiA7 fast real-time PCR system (Applied Biosystemsen). A set of controls present on each array enabled data analysis using the ΔΔCT method of relative quantification and assessment of reverse transcription performance, genomic DNA contamination, and PCR performance.
Data analyses were performed using the web-based analysis software (http://pcrdataanalysis.sabiosciences.com/pcr/arrayanalysis.php).
In vivo anti-tumour xenograft studies
To establish xenograft tumours six- to eight-week-old NSG immunodeficient mice were used. All procedures were approved and performed in accordance with the guidelines of the Institutional Animal Care and the Research Ethics Committee of the University of Granada. Mice were housed and maintained at 20 °C to 24 °C, 50% RH, a 14 to 10 h lightdark cycle with food and water ad libitum.
Animals (n = 10 per group) were randomly assigned as control, pre-treatment group and pre-treatment + treatment group. Treated groups received Trypsinogen and Chymotrypsinogen A at a dose of 83.3/500 mg/kg in combination in a single injection administered 3 days per week via intravenous tail vein injection in a dosing volume of 10 mL/kg. The volume of dosing solution administered to each animal was calculated and adjusted based on individual body weight measured immediately prior to dosing. Treatments were administered for 3 weeks before tumour induction (pre-treatment group) and for 3 weeks before tumour induction and continued during 9,5 weeks after tumour induction (pre-treatment + treatment group); control group was inoculated with a physiological saline solution (Fig. 7A).
Tumours were generated by subcutaneous injections of BxPC3 CSCs, to do so, CSCs spheres were disaggregated and 500 viable cells were injected per mouse using 26-gauge needles. Tumour size was calculated according to the formula:
where d and D are the shortest and longest diameters, respectively. Paraffin-embedded blocks of all tumours were sectioned at 5 μm. Each sample was stained with hematoxylin and eosin (H&E) and Mason tricrome for histopathologic analysis. For CD44 staining tumour sections were labelled with HCAM (DF1485) (sc-7297, Sigma-Aldrich), at 4 °C overnight. Following washing with PBS, tumour sections were subsequently incubated with fluorescence conjugated secondary antibody for 2 h at RT. The sections were then mounted with mounting medium containing DAPI (Vector Laboratories). For negative controls, primary antibodies were replaced with PBS. Images were captured using a confocal fluorescence microscope (Leica DM 5500B microscope).
Quantitative real time RT-PCR
ALDEFLUOR sorted BxPC3 pancreatic cancer cells were maintained for 6 days in culture and treated twice (day 2 and day 5) with PRP - Chymotrypsinogen: 0.42 mg/ml - Trypsinogen: 0.07 mg/ml. Total RNA was extracted using the TRIZOL reagent following the instructions of the manufacturer (Life Technologies). cDNA was synthesized by reverse transcription of total RNA using the Reverse Transcription System (Promega) and qRT-PCR assay was done using SYBR Green PCR Master Mix (Promega) and random primers. Each reaction was performed in triplicate from two cDNA dilutions. The comparative threshold cycle (Ct) method was used to calculate the amplification factor as specified by the manufacturer. Human GADPH was used as an internal standard to normalize variations in RNA quality in the quantities of input cDNA. The following primers were used for detection of Rac1 + Rac1b: 5′-ACCATGCAGGCCATCAAGTGTGTGG-3′ and 5′-TTACAACAGCAGGCATTTTCTCTTC-3′. Measurement of Rac1b was performed with exon 3b-specific primers: 5′-GGAGAAACGTACGGTAAGGATATAACC-3′ and 5′-GGCAATCGGCTTGTCTTTGCCC-3′. Smad7 primer was obtained from Sigma. Primer sequences used for GAPDH was: 5′-TGCACCACCAACTGCTTAGC-3′ 5′-GGCATGGACTGTGGTCATGAG-3′.
miRNA real-time reverse transcriptase PCR
To determine the expression of miRNAs (miR-21, miR-182, miRNA-7 and miR-SNORD 44) (Exiqon) in control non-treated cells/PRP treated cells, TRIZOL Reagent (Life Technologies) was used according to the manufacturer’s recommendations. Reverse transcription from miRs was performed using the miRCURY LNATM Synthesis kit II (Exiqon) following the manufacturer’s protocol. qPCR was performed using miRCURY LNA TM EXILENT SYBR Green (Exiqon) in a CFX96 real-time PCR detection system (Bio-Rad). Each reaction was performed in triplicate and the comparative threshold cycle (Ct) method was used to calculate the amplification factor. Human Snord 44 was used as housekeeping.
TGFβ stimulation
BxPC3 enriched CSCs were grown in 6 well plates and treated twice with PRP (T/C 0.07/0.42 mg/mL) on day 2 and on day 4 or non-treated control cells (CTL). On day 5, Recombinant Human TGF-beta 1 Protein (R & D Systems) was added to the cells in a concentration of 5 ng/ml, after 2, 4 and 10 hours of TGFβ stimulation cells were collected and processed for western blotting analysis.
Western blotting
Whole cell lysates were obtained through RIPA lysis buffer (Santa Cruz Biotechnology). The cytosolic and nuclear proteins were extracted with the use of a nuclear protein extraction kit (Beyotime Biotechnology). Protein concentrations were then determined using Bio Rad detergent compatible protein assays (Bio Rad Laboratories). Equal amounts of protein were loaded onto on SDS–PAGE gel, transferred onto nitrocellulose membranes (Bio Rad,162- 0115), and blocked in PBS containing 5% non-fat dry milk for 1 h at room temperature. Primary antibodies used included HCAM (DF1485) (sc-7297, Sigma-Aldrich), CXCR-4 (4G10) (sc-53534, Santa Cruz), Ep-CAM (CD326) (EBA-1) (sc-66020, Santa Cruz), Phospho-Smad2/3 (pThr8, MERCK), Smad2/3 Antibody (07–408, MERCK), Phospho-p38 MAPK ((Thr180/Tyr182), D3F9), (#4511, Cell SignalingTechnology), p38 MAPK Antibody (#9212, Cell SignalingTechnology), GAPDH (#2118, Cell SignalingTechnology), Anti-Yap1(phosphoS127) antibody (ab76252, Abcam), Anti-YAP1 antibody [EP1674Y] (ab52771, Abcam) and β-ACTIN (A2228, Sigma-Aldrich) Secondary antibodies used included anti-rabbit IgG peroxidase conjugate (A0545, Sigma-Aldrich) and anti-mouse IgG peroxidase conjugate (A9044, Sigma-Aldrich). Protein–antibody complexes were visualized by enhanced chemiluminescence (ECL, Bonus) on a LAS 3000 Imaging System (Fujifilm). Protein band Intensities were quantitated using Image-J software (NIH).
Immunofluorescence analysis
Cells were fixed with 4% paraformaldehyde in PBS for 20 min at room temperature (RT), blocked for 1 h at room temperature with 5% BSA, 5% FBS in PBS and incubated with the primary antibody overnight at 4 °C. Primary antibodies used were: HCAM (DF1485) (sc-7297, Sigma-Aldrich), CXCR-4 (4G10) (sc-53534, Santa Cruz), Ep-CAM (CD326) (EBA-1) (sc-66020, Santa Cruz), β-catenin (E-5) (SC-7963, Santa Cruz), E-cadherin (67A4) (SC-21791, Santa Cruz), Sox-2 (H-65) (sc-20088, Santa Cruz) Anti-YAP1 antibody [EP1674Y] (ab52771, Abcam) or p-Smad2/3 (SC11769, Santa Cruz).The next day, samples were washed three times with PBS, incubated with the secondary antibodies (Alexa) for 1 h at RT, washed three times with PBS and mounted with 4′,6-diamidino-2-phenylindole (DAPI)-containing mounting medium. Images were acquired with a Leica DM 5500B microscope.
Supplementary information
Acknowledgements
Pablo Hernández-Camarero is supported by a FPU grant from the Ministry of Education, Culture and Sport.
Author Contributions
P.H.C. and E.L.R. data acquisition, data analysis and interpretation, drafting the article. C.G.L., M.A.G. and C.C.W. data collection, analysis and interpretation. J.A.M. design of the study, data analysis and article review. M.P. conception and design of the study, data analysis and interpretation, drafting the article and final approval of submitted manuscript. J.K. conception and design of the study, and final approval of submitted manuscript. All authors: final approval of submitted manuscript.
Competing Interests
Dr. Julian Kenyon is the Founder and Scientific Director of Propanc Biopharma Inc. and owns stock in the company. Propanc Biopharma has funded the University of Jaen to carry out this research.
Footnotes
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Pablo Hernández-Camarero and Elena López-Ruiz contributed equally.
Contributor Information
Julian Kenyon, Email: jnkenyon@doveclinic.com.
Macarena Perán, Email: mperan@ujaen.es.
Supplementary information
Supplementary information accompanies this paper at 10.1038/s41598-019-47837-7.
References
- 1.Reya T, Morrison SJ, Clarke MF, Weissman IL. Stem cells, cancer, and cancer stem cells. Nature. 2001;414:105–11. doi: 10.1038/35102167. [DOI] [PubMed] [Google Scholar]
- 2.Tang DG. Understanding cancer stem cell heterogeneity and plasticity. Cell Res. 2012;22:457–72. doi: 10.1038/cr.2012.13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Prince MEP, Ailles LE. Cancer stem cells in head and neck squamous cell cancer. J. Clin. Oncol. 2008;26:2871–5. doi: 10.1200/JCO.2007.15.1613. [DOI] [PubMed] [Google Scholar]
- 4.Krishnamurthy S, Nör JE. Head and neck cancer stem cells. J. Dent. Res. 2012;91:334–40. doi: 10.1177/0022034511423393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chikamatsu K, et al. Resistance to apoptosis-inducing stimuli in CD44+ head and neck squamous cell carcinoma cells. Head Neck. 2012;34:336–43. doi: 10.1002/hed.21732. [DOI] [PubMed] [Google Scholar]
- 6.Ramírez A, et al. HER2-signaling pathway, JNK and ERKs kinases, and cancer stem-like cells are targets of Bozepinib small compound. Oncotarget. 2014;5:3590–606. doi: 10.18632/oncotarget.1962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wald M, et al. Mixture of trypsin, chymotrypsin and papain reduces formation of metastases and extends survival time of C57Bl6 mice with syngeneic melanoma B16. Cancer Chemother. Pharmacol. 2001;47(Suppl):S16–22. doi: 10.1007/s002800170004. [DOI] [PubMed] [Google Scholar]
- 8.Saruc M, et al. Pancreatic enzyme extract improves survival in murine pancreatic cancer. Pancreas. 2004;28:401–12. doi: 10.1097/00006676-200405000-00009. [DOI] [PubMed] [Google Scholar]
- 9.Perán M, Marchal JA, García MA, Kenyon J, Tosh D. In vitro treatment of carcinoma cell lines with pancreatic (pro)enzymes suppresses the EMT programme and promotes cell differentiation. Cell. Oncol. (Dordr). 2013;36:289–301. doi: 10.1007/s13402-013-0134-8. [DOI] [PubMed] [Google Scholar]
- 10.Perán M, et al. A formulation of pancreatic pro-enzymes provides potent anti-tumour efficacy: a pilot study focused on pancreatic and ovarian cancer. Sci. Rep. 2017;7:13998. doi: 10.1038/s41598-017-14571-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Huang EH, et al. Aldehyde Dehydrogenase 1 Is a Marker for Normal and Malignant Human Colonic Stem Cells (SC) and Tracks SC Overpopulation during Colon Tumorigenesis. Cancer Res. 2009;69:3382–3389. doi: 10.1158/0008-5472.CAN-08-4418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Liu F-Y, et al. Down-regulated KLF17 expression is associated with tumor invasion and poor prognosis in hepatocellular carcinoma. Med. Oncol. 2013;30:425. doi: 10.1007/s12032-012-0425-3. [DOI] [PubMed] [Google Scholar]
- 13.Annecke K, et al. uPA and PAI-1 in breast cancer: review of their clinical utility and current validation in the prospective NNBC-3 trial. Adv. Clin. Chem. 2008;45:31–45. doi: 10.1016/S0065-2423(07)00002-9. [DOI] [PubMed] [Google Scholar]
- 14.Baeyens L, et al. In vitro generation of insulin-producing beta cells from adult exocrine pancreatic cells. Diabetologia. 2005;48:49–57. doi: 10.1007/s00125-004-1606-1. [DOI] [PubMed] [Google Scholar]
- 15.Publishing Asia B, et al. Association Pancreatic adenocarcinoma up-regulated factor (PAUF), a novel up-regulated secretory protein in pancreatic ductal adenocarcinoma. Cancer Sci. 2009;100:828–836. doi: 10.1111/j.1349-7006.2009.01106.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Salomon D. Transforming Growth Factor in Cancer: Janus, the Two-Faced God. JNCI J. Natl. Cancer Inst. 2014;106:djt441–djt441. doi: 10.1093/jnci/djt441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hather G, et al. Growth Rate Analysis and Efficient Experimental Design for Tumor Xenograft Studies. Cancer Inform. 2014;13s4:CIN.S13974. doi: 10.4137/CIN.S13974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sausville, E. A. & Burger, A. M. Contributions of human tumor xenografts to anticancer drug development. Cancer Res. 66, 3351–4, discussion 3354 (2006). [DOI] [PubMed]
- 19.Bünger S, et al. Pancreatic carcinoma cell lines reflect frequency and variability of cancer stem cell markers in clinical tissue. Eur. Surg. Res. 2012;49:88–98. doi: 10.1159/000341669. [DOI] [PubMed] [Google Scholar]
- 20.Ishiguro T, et al. Tumor-derived spheroids: Relevance to cancer stem cells and clinical applications. Cancer Sci. 2017;108:283–289. doi: 10.1111/cas.13155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Herreros-Villanueva M, Bujanda L, Billadeau DD, Zhang J-S. Embryonic stem cell factors and pancreatic cancer. World J. Gastroenterol. 2014;20:2247–54. doi: 10.3748/wjg.v20.i9.2247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Xu X, et al. DLK1 as a Potential Target against Cancer Stem/Progenitor Cells of Hepatocellular Carcinoma. Mol. Cancer Ther. 2012;11:629–638. doi: 10.1158/1535-7163.MCT-11-0531. [DOI] [PubMed] [Google Scholar]
- 23.Banham AH, et al. The FOXP1 winged helix transcription factor is a novel candidate tumor suppressor gene on chromosome 3p. Cancer Res. 2001;61:8820–9. [PubMed] [Google Scholar]
- 24.Ishikawa T, et al. Twist2 functions as a tumor suppressor in murine osteosarcoma cells. Cancer Sci. 2013;104:880–888. doi: 10.1111/cas.12163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lung HL, Lung ML. Abstract 1150: The role of the tumor suppressor gene THY1 in suppression of epithelial-mesenchymal transition (EMT) in nasopharyngeal carcinoma. Cancer Res. 2014;74:1150–1150. [Google Scholar]
- 26.Wang Z, Li Y, Kong D, H. Sarkar F. The Role of Notch Signaling Pathway in Epithelial-Mesenchymal Transition (EMT) During Development and Tumor Aggressiveness. Curr. Drug Targets. 2010;11:745–751. doi: 10.2174/138945010791170860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yuan X, et al. Notch signaling and EMT in non-small cell lung cancer: biological significance and therapeutic application. J. Hematol. Oncol. 2014;7:87. doi: 10.1186/s13045-014-0087-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Konno H, Ohta M, Baba M, Suzuki S, Nakamura S. The role of circulating IL-8 and VEGF protein in the progression of gastric cancer. Cancer Sci. 2003;94:735–40. doi: 10.1111/j.1349-7006.2003.tb01511.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chen Y, et al. Interleukin-8, a promising predictor for prognosis of pancreatic cancer. World J. Gastroenterol. 2012;18:1123–9. doi: 10.3748/wjg.v18.i10.1123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kuwada Y, et al. Potential involvement of IL-8 and its receptors in the invasiveness of pancreatic cancer cells. Int. J. Oncol. 2003;22:765–71. [PubMed] [Google Scholar]
- 31.Rangaswami H, Bulbule A, Kundu GC. Osteopontin: role in cell signaling and cancer progression. Trends Cell Biol. 2006;16:79–87. doi: 10.1016/j.tcb.2005.12.005. [DOI] [PubMed] [Google Scholar]
- 32.Psallidas I, et al. Secreted phosphoprotein-1 directly provokes vascular leakage to foster malignant pleural effusion. Oncogene. 2013;32:528–535. doi: 10.1038/onc.2012.57. [DOI] [PubMed] [Google Scholar]
- 33.Rinehart J, et al. Multicenter Phase II Study of the Oral MEK Inhibitor, CI-1040, in Patients With Advanced Non-Small-Cell Lung, Breast, Colon, and Pancreatic Cancer. J. Clin. Oncol. 2004;22:4456–4462. doi: 10.1200/JCO.2004.01.185. [DOI] [PubMed] [Google Scholar]
- 34.Nishimura Y, et al. Overexpression of YWHAZ relates to tumor cell proliferation and malignant outcome of gastric carcinoma. Br. J. Cancer. 2013;108:1324–1331. doi: 10.1038/bjc.2013.65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Feldmann G, Maitra A. Molecular genetics of pancreatic ductal adenocarcinomas and recent implications for translational efforts. J. Mol. Diagn. 2008;10:111–22. doi: 10.2353/jmoldx.2008.070115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Schniewind B, et al. Dissecting the role of TGF-beta type I receptor/ALK5 in pancreatic ductal adenocarcinoma: Smad activation is crucial for both the tumor suppressive and prometastatic function. Oncogene. 2007;26:4850–4862. doi: 10.1038/sj.onc.1210272. [DOI] [PubMed] [Google Scholar]
- 37.Biswas S, et al. Mutational inactivation ofTGFBR2 in microsatellite unstable colon cancer arises from the cooperation of genomic instability and the clonal outgrowth of transforming growth factor β resistant cells. Genes, Chromosom. Cancer. 2008;47:95–106. doi: 10.1002/gcc.20511. [DOI] [PubMed] [Google Scholar]
- 38.Jovanović B, et al. TβRIII Expression in Human Breast Cancer Stroma and the Role of Soluble TβRIII in Breast Cancer Associated Fibroblasts. Cancers (Basel). 2016;8:100. doi: 10.3390/cancers8110100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ungefroren H, et al. Rac1b negatively regulates TGF-&#x3B2;1-induced cell motility in pancreatic ductal epithelial cells by suppressing Smad signalling. Oncotarget. 2014;5:277–90. doi: 10.18632/oncotarget.1696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Luo L, Li N, Lv N, Huang D. SMAD7: a timer of tumor progression targeting TGF-β signaling. Tumor Biol. 2014;35:8379–8385. doi: 10.1007/s13277-014-2203-7. [DOI] [PubMed] [Google Scholar]
- 41.Ali A, et al. KLF17 empowers TGF-β/Smad signaling by targeting Smad3-dependent pathway to suppress tumor growth and metastasis during cancer progression. Cell Death Dis. 2015;6:e1681. doi: 10.1038/cddis.2015.48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ungefroren H, et al. Rac1b negatively regulates TGF-B-induced cell motility in pancreatic ductal epithelial cells by suppressing Smad signalling. Oncotarget. 2014;5:277–290. doi: 10.18632/oncotarget.1696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Witte D, et al. Negative regulation of TGF-β1-induced MKK6-p38 and MEK-ERK signalling and epithelial-mesenchymal transition by Rac1b. Sci. Rep. 2017;7:17313. doi: 10.1038/s41598-017-15170-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Jochum W, Passegué E, Wagner EF. AP-1 in mouse development and tumorigenesis. Oncogene. 2001;20:2401–2412. doi: 10.1038/sj.onc.1204389. [DOI] [PubMed] [Google Scholar]
- 45.Misawa A, et al. AP-1-Dependent miR-21 expression contributes to chemoresistance in cancer stem cell-like SP cells. Oncol. Res. 2010;19:23–33. doi: 10.3727/096504010X12828372551759. [DOI] [PubMed] [Google Scholar]
- 46.Pfeffer SR, Yang CH, Pfeffer LM. The Role of miR-21 in Cancer. Drug Dev. Res. 2015;76:270–277. doi: 10.1002/ddr.21257. [DOI] [PubMed] [Google Scholar]
- 47.Song L, et al. TGF-β induces miR-182 to sustain NF-κB activation in glioma subsets. J. Clin. Invest. 2012;122:3563–78. doi: 10.1172/JCI62339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zhang H, et al. MiR-7, Inhibited Indirectly by LincRNA HOTAIR, Directly Inhibits SETDB1 and Reverses the EMT of Breast Cancer Stem Cells by Downregulating the STAT3 Pathway. Stem Cells. 2014;32:2858–2868. doi: 10.1002/stem.1795. [DOI] [PubMed] [Google Scholar]
- 49.Chang Y-L, et al. MicroRNA-7 inhibits the stemness of prostate cancer stem-like cells and tumorigenesis by repressing KLF4/PI3K/Akt/p21 pathway. Oncotarget. 2015;6:24017–24031. doi: 10.18632/oncotarget.4447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kim N-G, Koh E, Chen X, Gumbiner BM. E-cadherin mediates contact inhibition of proliferation through Hippo signaling-pathway components. Proc. Natl. Acad. Sci. 2011;108:11930–11935. doi: 10.1073/pnas.1103345108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Moroishi T, Hansen CG, Guan K-L. The emerging roles of YAP and TAZ in cancer. Nat. Rev. Cancer. 2015;15:73–79. doi: 10.1038/nrc3876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Huang D, et al. Regulation of Hippo signalling by p38 signalling. J. Mol. Cell Biol. 2016;8:328–337. doi: 10.1093/jmcb/mjw036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kapoor A, et al. Yap1 Activation Enables Bypass of Oncogenic Kras Addiction in Pancreatic. Cancer. Cell. 2014;158:185–197. doi: 10.1016/j.cell.2014.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Bae JS, Kim SM, Lee H. The Hippo signaling pathway provides novel anti-cancer drug targets. Oncotarget. 2017;8:16084–16098. doi: 10.18632/oncotarget.14306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Attisano L, Wrana JL. Signal integration in TGF-β, WNT, and Hippo pathways. F1000Prime Rep. 2013;5:17. doi: 10.12703/P5-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Klaus A, Birchmeier W. Wnt signalling and its impact on development and cancer. Nat. Rev. Cancer. 2008;8:387–98. doi: 10.1038/nrc2389. [DOI] [PubMed] [Google Scholar]
- 57.MacDonald BT, Tamai K, He X. Wnt/β-Catenin Signaling: Components, Mechanisms, and Diseases. Dev. Cell. 2009;17:9–26. doi: 10.1016/j.devcel.2009.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Tong X, et al. SOX10, a novel HMG-box-containing tumor suppressor, inhibits growth and metastasis of digestive cancers by suppressing the Wnt/&#x3B2;-catenin pathway. Oncotarget. 2014;5:10571–83. doi: 10.18632/oncotarget.2512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Cho I-R, et al. SIRT1 inhibits proliferation of pancreatic cancer cells expressing pancreatic adenocarcinoma up-regulated factor (PAUF), a novel oncogene, by suppression of β-catenin. Biochem. Biophys. Res. Commun. 2012;423:270–275. doi: 10.1016/j.bbrc.2012.05.107. [DOI] [PubMed] [Google Scholar]
- 60.Plaks V, Kong N, Werb Z. The Cancer Stem Cell Niche: How Essential Is the Niche in Regulating Stemness of Tumor Cells? Cell Stem Cell. 2015;16:225–238. doi: 10.1016/j.stem.2015.02.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.De Wever O, Van Bockstal M, Mareel M, Hendrix A, Bracke M. Carcinoma-associated fibroblasts provide operational flexibility in metastasis. Semin. Cancer Biol. 2014;25:33–46. doi: 10.1016/j.semcancer.2013.12.009. [DOI] [PubMed] [Google Scholar]
- 62.Heneberg P. Paracrine tumor signaling induces transdifferentiation of surrounding fibroblasts. Crit. Rev. Oncol. Hematol. 2016;97:303–311. doi: 10.1016/j.critrevonc.2015.09.008. [DOI] [PubMed] [Google Scholar]
- 63.Hernández-Camarero P, et al. Revisiting the dynamic cancer stem cell model: Importance of tumour edges. Crit. Rev. Oncol. Hematol. 2018;131:35–45. doi: 10.1016/j.critrevonc.2018.08.004. [DOI] [PubMed] [Google Scholar]
- 64.Zhao S, et al. CD44 Expression Level and Isoform Contributes to Pancreatic Cancer Cell Plasticity, Invasiveness, and Response to Therapy. Clin. Cancer Res. 2016;22:5592–5604. doi: 10.1158/1078-0432.CCR-15-3115. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.